

Abstract
BACKGROUND AND PURPOSE
Recent clinical trials report that metformin, an activator of AMP-activated protein kinase (AMPK) used to treat type 2 diabetes, significantly reduces the risk of stroke by actions that are independent of its glucose-lowering effects. However, the underlying molecular mechanisms are not known. Here, we tested the possibility that acute metformin preconditioning confers neuroprotection by pre-activation of AMPK-dependent autophagy in a rat model of permanent middle cerebral artery occlusion (pMCAO).
EXPERIMENTAL APPROACH
Male Sprague-Dawley rats were pretreated with either vehicle, an AMPK inhibitor, Compound C, or an autophagy inhibitor, 3-methyladenine, and were injected with a single dose of metformin (10 mg kg−1, i.p.). Then, AMPK activity and autophagy biomarkers in the brain were assessed. At 24 h after metformin treatment, rats were subjected to pMCAO; infarct volume, neurological deficits and cell apoptosis were evaluated 24 and 96 h later.
KEY RESULTS
A single dose of metformin significantly activated AMPK and induced autophagy in the brain. The enhanced autophagic activity was inhibited by Compound C pretreatment. Furthermore, acute metformin preconditioning significantly reduced infarct volume, neurological deficits and cell apoptosis during a subsequent focal cerebral ischaemia. The neuroprotection mediated by metformin preconditioning was fully abolished by Compound C and partially inhibited by 3-methyladenine.
CONCLUSIONS AND IMPLICATIONS
These results provide the first evidence that acute metformin preconditioning induces autophagy by activation of brain AMPK, which confers neuroprotection against subsequent cerebral ischaemia. This suggests that metformin, a well-known hypoglycaemic drug, may have a practical clinical use for stroke prevention.
Keywords: metformin, autophagy, stroke, preconditioning, AMPK, neuroprotection
Introduction
Metformin is one of the most widely used hypoglycaemic drugs for the treatment of type 2 diabetes (Arunachalam et al., 2014; Schulte et al., 2014). Aside from lowering blood glucose levels, metformin has been found to exert beneficial effects in the prevention of cardiovascular diseases including stroke. Recent clinical trials report that metformin significantly reduces the risk of stroke by actions that are independent of its glucose-lowering effects (Nathan, 1998; Selvin and Hirsch, 2008; Cheng et al., 2014). However, the underlying molecular mechanisms remain largely unknown.
Emerging evidence indicates that most of the beneficial effects of metformin are mediated by activation of AMP kinase (AMPK; see Alexander et al., 2013b), a major regulator of cellular energy homeostasis in the whole body (Russo et al., 2013). AMPK can be phosphorylated and activated in response to an increase in the intracellular AMP-to-ATP ratio (Hardie, 2003). Once activated, AMPK leads to the conservation of intracellular ATP levels via multiple downstream pathways, including autophagy (Hardie, 2011). Autophagy is an evolutionary-conserved process for the degradation and recycling of cellular constituents, participating in bioenergetic management during energy stress (Hale et al., 2013). Several lines of evidence suggest that autophagy can be induced by metformin via an AMPK-dependent manner in peripheral tissues. For example, metformin induces autophagy by activating AMPK in a wide variety of malignant tumours (Harhaji-Trajkovic et al., 2009; Shi et al., 2012). More importantly, acute preconditioning with a single dose of metformin was shown to induce AMPK-mediated autophagy in the heart, which subsequently conferred protection against cardiac dysfunction following diabetes or myocardial ischaemia (Calvert et al., 2008).
Recently, it has been proposed that autophagy has a protective role in cerebral ischaemia (Viscomi et al., 2012; Wang et al., 2012; Papadakis et al., 2013). In particular, pre-activation of autophagy in the brain was found to markedly enhance brain ischaemic tolerance, as it facilitated cellular energy production, limited endoplasmic reticulum stress and prevented neuronal apoptosis during subsequent ischaemic exposure (Sheng et al., 2010; 2012). On consideration of the above evidence, in the present study, we tested the hypothesis that preconditioning with metformin activates autophagy via an AMPK-dependent manner in the brain, thereby conferred neuroprotection against focal cerebral ischaemia in a rat model of permanent middle cerebral artery occlusion (pMCAO).
Methods
Animals
Male Sprague-Dawley rats (250–280 g) were purchased from the Experimental Animals Center of Nanjing Medical University. Animals were maintained in individually ventilated cages in a standard animal room at 20–24°C and relative humidity (30%–70%) with a 12 h light/dark cycle and given free access to food and water. Animal Care and Management Committee of Qingdao Municipal Hospital approved the whole study protocol (permit No.QMHEC-130115). In total 186 rats were included in this study. All animal experiments were conducted in accordance with Guide for the Care and Use of Laboratory Animals of the National Institutes of Health, and were reported in accordance with the ARRIVE guidelines (Kilkenny et al., 2010; McGrath et al., 2010).
Drugs administration and experimental groups
Metformin hydrochloride, 6-[4-(2-piperidin-1-ylethoxy)phenyl]-3-pyridin-4-ylpyrazolo[1,5-a]pyrimidine (Compound C, an AMPK inhibitor, CAS Number: 866405-64-3), and 3-methyladenine (3-MA, an autophagy inhibitor) were purchased from Sigma-Aldrich Inc., St. Louis, MO, USA. Rats were randomly allocated to six groups using a random number table generated by SPSS software 13.0 (IBM Inc., Armonk, NY, USA), and received treatment as shown in Figure 1. In view of the fact that a therapeutic dose of metformin led to the accumulation of lactic acid in the brain (Li et al., 2010), a subtherapeutic dose of metformin (10 mg kg−1, i.p.) was used in this study. The administration route and dose for Compound C were chosen according to a previous study from McCullough et al. (2005). The dose and administration route for 3-MA were determined based on a previous study from Sheng et al. (2010). It should be noted that treatment with Compound C or 3-MA at the doses used did not significantly affect physiological parameters including PaCO2, PaO2, blood pressure and blood glucose levels in rats (Supporting Information Table S1), and no signs of neurotoxicity, such as hind-limb paralysis, vocalization and reduced food intake, were observed. Of note, saline was used as vehicle in this study. All drug and molecular nomenclatures in this article followed Alexander et al. (2013b).
Figure 1.

Schematic illustration of the experimental protocols. Abbreviations: Cpd C, Compound C; Met, Metformin; Veh, Vehicle.
Western blot analysis
At the indicated time after metformin administration, rats were killed. The brain tissue of each rat was homogenized and the total proteins were extracted by RIPA lysis buffer (Beyotime Inc., Shanghai, China). Different samples with an equal amount of protein were separated on 8–12% SDS polyacrylamide gels, transferred to nitrocellulose membranes and blocked in 5% BSA powder in 1 × Tris-buffered saline (TBS) with 0.1% Tween 20 at room temperature for 2 h. Membranes were incubated overnight at 4°C with a mouse monoclonal antibody against p-AMPK (Thr172) (1:600; Cell Signaling Technology Inc., Boston, MA, USA), AMPK (1:800; Cell Signaling Technology Inc.), p-acetyl-CoA carboxylase (ACC, A major downstream target of AMPK activation) (Ser79) (1:1000; Cell Signaling Technology Inc.), ACC (1:1000; Cell Signaling Technology Inc.), microtubule-associated protein 1 light chain 3 (LC3) (1:1000; Sigma-Aldrich Inc.) and P62 (1:1000; Cell Signaling Technology Inc.), then washed with 1 × TBS with 0.1% Tween 20, and incubated with HRP-coupled secondary antibody for 2 h at room temperature. After being washed, protein bands were detected with chemiluminescent HRP substrate (Thermo Scientific Inc., Rockford, IL, USA.) for 5 min at room temperature and exposed to an X-ray film. The band intensity was analysed using Quantity One software 4.6.2 (Bio-Rad Laboratories Inc., Hercules, CA, USA) and normalized to a loading control, β-actin.
Immunofluorescence analysis
Immunofluorescence analysis was carried out as previously described (Gao et al., 2012). Twenty-four hours after metformin administration, rats were deeply anaesthetized (for anaesthetic procedure see later section, pMCAO) and transcardially perfused with PBS, followed by a solution containing 4% paraformaldehyde (PFA). Brains were removed and post-fixed in 4% PFA solution overnight. After being dehydrated in alcohol, the brains were embedded in paraffin and cut into 4–5 μm sections. Afterwards, sections were deparaffinized, hydrated in distilled water, treated with 3% H2O2 for 10 min to remove endogenous peroxidase activity and washed again with PBS. Sections were then permeabilized with 0.1% Triton X-100 for 10 min, blocked with 10% normal goat serum for 2 h at room temperature and incubated in the primary antibodies against LC3 (1:250, Sigma–Aldrich Inc.) 4°C for 24 h. The sections were rinsed with PBS and sequentially incubated, respectively, with tetramethylrhodamine conjugated anti-rabbit IgG (1:1000, Beyotime Inc.) in a humidified container for 1 h at 37°C. Then the sections were further incubated with 0.5 mg mL−1 DAPI for 10 min. After that, the sections were washed with PBS and sealed with a coverslip. The slides were viewed with a fluorescence microscopy (Olympus Inc., Tokyo, Japan), and fluorescence intensity was measured using Image J software 2.1 (Bethesda, MD, USA) by observers who were blinded to the experimental groups. Of note, to ensure the specificity of the immunofluorescence procedure, a control experiment was performed in which the primary antibody was omitted. Under these conditions, no staining for LC3 was observed (Supporting Information Figure S1).
Transmission electron microscopic examination
The transmission electron microscopic examination was performed as previously described (Gao et al., 2012; Jiang et al., 2013b). Briefly, brain tissue was sliced into small sections, immersed in 1% osmium tetroxide for 2 h, dehydrated in graded ethanol and then embedded in epoxy resin. Afterwards, these selections were cut into ultrathin sections (60–70 nm) with an ultramicrotome, poststained with uranyl acetate and lead citrate, and then subsequently examined under a transmission electron microscope. Autophagosome counting was performed by observers who were blinded to the experimental groups, using a protocol described previously (Sheng et al., 2010).
pMCAO
Twenty-four hours after metformin treatment, rats were anaesthetized with urethane (800 mg kg−1) and α-chloralose (40 mg kg−1) i.p. We chose urethane and α-chloralose as anaesthetics because they caused little interference with cardiovascular reflexes, arterial blood pressure or cerebral blood flow (CBF), that might affect stroke outcome (Wyler, 1974; Leoni et al., 2011), and, therefore, were suitable for studies of central cardiovascular regulation and cerebrovascular diseases. However, it is recognized that this combination is not recommended for recovery experiments because of the known toxicity of urethane and the potential for involuntary tremors on recovery from α-chloralose. The depth of anaesthesia was monitored by assessing the withdrawal reflex to footpad pinching. pMCAO was performed by investigators who were blinded to the experimental groups. Briefly, the right common carotid artery, external carotid artery and internal carotid artery (ICA) were isolated through a midline incision. A 30 mm length of nylon filament (Φ 0.26 mm), with its tip rounded by heating near a flame, was inserted from the right external carotid artery into the lumen of ICA, and then advanced to the Circle of Willis to occlude the origin of the right middle cerebral artery. The filament remained there until the rat was killed. The reduction of the CBF was confirmed by a laser-Doppler flow metre (Moor Instruments Inc., Axminster, UK), as described previously (Jiang et al., 2012). Rats in the sham-operated group were subjected to the filament insertion into the ICA but with no reduction in CBF. Our preliminary findings showed that metformin did not significantly affect body temperature (Supporting Information Figure S2), and in order to rule out the influence of hypothermia on stroke outcome, body temperature was maintained in the range of 37.0 ± 0.5°C with a heating pad until rats were killed. In addition, to determine whether metformin preconditioning affects physiological parameters during pMCAO, the left femoral arteries of rats from group 1 and group 2 were cannulated to obtain the values of PaCO2, PaO2, BP and blood glucose levels. These physiological parameters were measured before and at 24 h after pMCAO.
Neurobehavioural testing
Neurobehavioural tests were performed at 24 and 96 h after pMCAO using a 5-point scale (Bederson et al., 1986) by observers who were blinded to the experimental groups: 0, rats extended both forelimbs towards the floor when gently suspended 1 m above the floor and with no other signs of neurological deficit; 1, rats consistently flexed the forelimb contralateral to pMCAO; 2, rats circled towards the contralateral side when the tail was pulled; 3, rats spontaneously circled towards the contralateral side when allowed to move freely; 4, no spontaneous movement with an apparent depressed level of consciousness.
2, 3, 5-triphenyltetrazolium chloride (TTC) staining
TTC staining was performed to evaluate the infarct volume by investigators who were blinded to the experimental groups. Twenty-four hours and 96 h after pMCAO, rats were killed under deep anaesthesia and the brains were rapidly removed, then sectioned coronally into five 3 mm-thick slices using a rat brain matrix. The slices were kept in the dark and stained with 2% TTC for 30 min and then fixed with 4% PFA. The infarct volume was evaluated by Image Pro-Plus 5.1 analysis system (Media Cybernetics Inc., Silver Spring, MD, USA) using Swanson's method, which corrects for oedema (Swanson et al., 1990).
TUNEL assay
Twenty-four hours after pMCAO, rats were deeply anaesthetized and transcardially perfused with PBS, followed by a solution containing 4% PFA. Brains were removed, and the TUNEL assay was performed by using a cell death detection kit (In Situ Cell Death Detection Kit, POD; Roche, Pleasanton, CA, USA) as previously described (Jiang et al., 2013a). Afterwards, sections were costained with DAPI, and were viewed under a fluorescence microscopy. Cells labelled with green fluorescence in the peri-infarct region were identified as TUNEL-positive cells. For each slide, cell counting was performed on three randomly selected non-overlapping fields in the peri-infarct region by observers who were blinded to the experimental groups. Data obtained in every field were added together to make a final data count for each slide and expressed as % of total cell numbers.
Statistical analysis
Statistical analysis was performed by the SPSS software 16.0. Statistically significant differences were evaluated by an independent sample t-test and one- or two-way anova followed by least significant difference post hoc test. For neurological deficits, Mann–Whitney U-test was used for comparisons between two groups. The mortality of rats after pMCAO was assessed with the Chi-squared method. With the exception of mortality and neurological deficit, the data are expressed as mean ± SD. P < 0.05 was considered significant.
Results
Metformin activated AMPK and induced autophagy in the brain
Rats received a single dose of metformin (10 mg kg−1; i.p.), and the ratio of p-AMPK (Thr172)/AMPK was detected 6, 12, 24 and 48 h later by Western blotting. As shown in Figure 2A and B, the p-AMPK (Thr172)/AMPK ratio changed in a monophasic manner: significantly increased from 6 h, peaked at 12 h, started to decrease from 24 h, and was return to the baseline value at 48 h. We subsequently determined the levels of LC3-II, an autophagy biomarker, at 6, 12, 24 and 48 h after metformin treatment. As demonstrated in Figure 2A–C, LC3-II started to increase from 12 h, peaked at 24 h, and began to decrease from 48 h. These findings were confirmed by immunofluorescence analysis using an anti-LC3 antibody with higher selectivity for LC3-II, as rats received metformin showed higher LC3-II immunoreactivity in brain than vehicle-treated rats (Figure 2D). Additionally, transmission electron microscopic examination detected more autophagosomes with double-membrane structure in the brain of metformin-treated rats (Figure 2E). All these findings indicate that metformin activated AMPK and induced autophagy in the brain.
Figure 2.

Metformin (Met) activates AMPK and induces autophagy in brain. (A–C) The ratio of p-AMPK (Thr172)/AMPK and the protein level of LC3-II at 6, 12, 24 and 48 h after metformin treatment were evaluated by Western blotting. n = 6 per group. Data were analysed by two-way anova followed by least significant difference post hoc test (D) Immunofluorescence staining and quantification of LC3-II (an autophagy biomarker) in rat brain at 24 h after Met treatment. Brain sections (distanced −1.6 mm from bregma) were labelled by DAPI (blue) and an anti-LC3 antibody with higher selectivity for LC3-II (red). Note that the LC3-II immunoreactivity in the brain cortex of Met-treated rats was significantly higher than that in vehicle (Veh)-treated rats. Bars: 50 μm. (E) Electron microscope sections obtained from cerebral cortex of vehicle- or Met-treated rat. The red arrows pointed to autophagosomes, and the inset in the represent photo showed an autophagosome magnified by twofold. Bars: 1 μm. Columns represent mean ± SD. *P < 0.05 versus Veh-treated rats.
Inhibition of AMPK by Compound C significantly attenuated metformin-induced autophagy in brain
To further investigate the role of AMPK in metformin-induced autophagy in the brain, rats were pretreated with a single dose of Compound C (20 mg kg−1; i.p.) before they received metformin. The inhibitory effect of Compound C on metformin-induced AMPK activation (indicated by ratios of p-AMPK/AMPK and p-ACC/ACC) is shown in Figure 3A–C. Pretreatment with Compound C markedly inhibited LC3-II levels by 38% (Figure 3D and E, P < 0.05) at 24 h after metformin treatment, and this result was further confirmed by immunofluorescence analysis (Figure 3G). Meanwhile, the metformin-induced reduction of P62 was also reversed by Compound C pretreatment (Figure 3D and F, P < 0.05), further suggesting that inhibition of AMPK by Compound C significantly attenuated the activation of autophagy induced by metformin. It should be noted that Compound C itself did not significantly affect the basal activity of AMPK or autophagy, as both the p-AMPK (Thr172)/AMPK ratio and the LC3-II level in rat brain stayed unchanged after a single dose of Compound C alone (Supporting Information Figure S3).
Figure 3.

Inhibition of AMPK by Compound C significantly attenuates metformin (Met)-induced autophagy in brain. (A–C) The ratios of p-AMPK (Thr172)/AMPK as well as p-ACC (Ser79)/ACC were evaluated by Western blotting. (D–F) The protein levels of LC3-II and P62 were detected by Western blotting. (G) Immunofluorescence staining and quantification of LC3-II (an autophagy biomarker) in rat brain. Brain sections were labelled by DAPI (blue) and an anti-LC3 antibody with higher selectivity for LC3-II (red). Note that pretreatment with Compound C (Cpd C) markedly attenuated the Met-induced increase in LC3-II immunoreactivity. Bars: 50 μm. Columns represent mean ± SD. n = 6 per group. Veh, Vehicle.
Metformin preconditioning reduced infarct volume, neurological deficits and cell apoptosis after pMCAO
Twenty-four hours after metformin treatment, rats were subjected to pMCAO. This time point was selected based on the above results, which showed that autophagic activity in the brain peaked at 24 h after metformin administration.
Five rats died before completion of the experiment and were excluded from the study: one rat (8.3%) in group 1, one rat (8.3%) in group 4, one rat in group 5 (8.3%) and two rats in group 6 (16.7%). Post-mortem examinations did not reveal the occurrence of intracerebral or subarachnoid haemorrhage in any of these animals, and no significant differences were found between the number of deaths in each group. Twenty-four hours after pMCAO, TTC staining, Benderson neurobehavioural tests and TUNEL assay were used to evaluate infarct volume, neurological deficits and cell apoptosis respectively. As indicated by Figure 4A and B, metformin preconditioning markedly decreased the infarct volume following subsequent pMCAO by 29% (P < 0.05). Meanwhile, metformin preconditioning significantly attenuated neurological deficits in rats subjected to pMCAO [median of group 1: 2, median of group 2: 1; P < 0.05] (Figure 4C). In addition, metformin preconditioning also ameliorated cell apoptosis in the peri-infarct region, as the % of TUNEL-positive cells was dramatically reduced by metformin (Figure 4D, 14.2 vs. 25.7% of total cell numbers; P < 0.05). More intriguingly, metformin at the dose used significantly reduced the infarct volume and neurological deficits at 96 h after pMCAO, implying the neuroprotective effects of metformin could last for at least 4 days (Figure 5). It should be noted that metformin, at the dose used, did not significantly affect physiological parameters (including PaCO2, PaO2, BP and blood glucose levels, Table 1) or CBF (Supporting Information Figure S4A) before or after pMCAO when compared with those in vehicle-treated rats. In addition, no difference in brain lactate levels was observed between the vehicle- and metformin-treated rats at 24 h after pMCAO (Supporting Information Figure S4B). Taken together, these results indicate that preconditioning with a single dose of metformin could provide a long-lasting neuroprotection without causing lactate accumulation.
Figure 4.

Metformin (Met) preconditioning reduced infarct volume, neurological deficits and cell apoptosis at 24 h after pMCAO. (A) TTC staining of representative coronal sections at 24 h after pMCAO. (B) Infarct volume was determined at 24 h after pMCAO (n = 10–12 per group). (C) The distribution of neurological deficit score in each group at 24 h after pMCAO (n = 10–12 per group). (D) TUNEL assay was performed at 24 h after Met treatment. Brain sections were costained with DAPI (blue). Note that the number of TUNEL-positive cell (green) in brain of Met-treated rats was higher than that in vehicle (Veh)-treated rats (n = 6 per group). Bars: 25 μm. Columns represent mean ± SD. Cpd C, Compound C.
Figure 5.
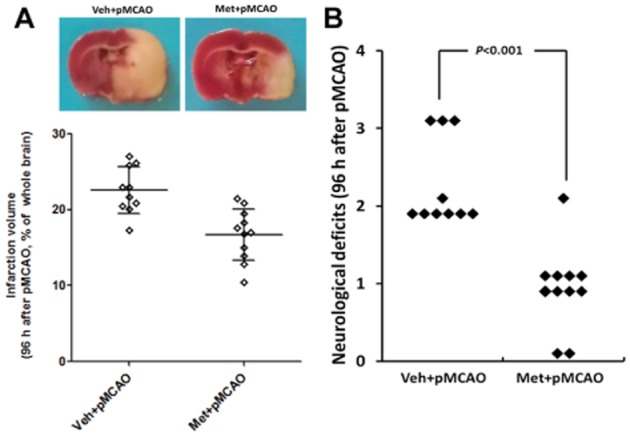
Metformin (Met) preconditioning reduced infarct volume and neurological deficits at 96 h after pMCAO. (A) TTC staining of representative coronal sections at 96 h after pMCAO. White is infarct area and red is normal area. (C) Infarct volume was determined at 96 h after pMCAO (n = 10–11 per group). Columns represent mean ± SD. Veh, Vehicle.
Table 1.
Metformin treatment did not affect physiological parameters
| PaCO2 (mmHg) | PaO2 (mmHg) | BP (mmHg) | GI (g L–1) | |
|---|---|---|---|---|
| Before pMCAO | ||||
| Vehicle | 35.37 ± 3.19 | 149.61 ± 20.81 | 79.17 ± 6.34 | 1.46 ± 0.18 |
| Metformin | 35.93 ± 4.22 | 154.07 ± 22.21 | 80.53 ± 4.61 | 1.41 ± 0.20 |
| After pMCAO | ||||
| Vehicle | 37.28 ± 5.22 | 148.28 ± 24.79 | 81.97 ± 6.99 | 1.57 ± 0.26 |
| Metformin | 38.67 ± 3.94 | 151.34 ± 24.84 | 84.02 ± 6.23 | 1.59 ± 0.23 |
Values were measured before (at 24 h after vehicle or metformin injection) and at 24 h after pMCAO. All values are expressed as mean ± SD. n = 6 per group. It should be noted that metformin treatment did not significantly affect these physiological parameters when compared with vehicle.GI, glucose level.
AMPK-mediated autophagy was involved in the neuroprotection induced by metformin preconditioning
In order to investigate the role of AMPK-mediated autophagy in metformin-induced neuroprotection, rats were pretreated with a single dose of Compound C (20 mg kg−1; i.p.) or 3-MA (200 nmol; i.c.v.) before they received metformin administration. The inhibitory effect of 3-MA on metformin-induced autophagy had been confirmed by Western blotting (Supporting Information Figure S5). Twenty-four hours after pMCAO, the infarct volume and neurological deficits were measured by TTC staining and Benderson neurobehavioural tests respectively. As shown in Figure 4A and B, the metformin-induced reduction in infarct volume was fully abolished by Compound C and partially attenuated by 3-MA. In support of these findings, Benderson neurobehavioural testing demonstrated that the amelioration in neurological deficits induced by metformin was reversed by Compound C or 3-MA (Figure 4C), indicating that inhibition of AMPK or autophagy abolished the neuroprotection provided by metformin preconditioning. It is noteworthy that Compound C or 3-MA at the dose used, when given alone, did not exacerbate the stroke outcome (Supporting Information Figure S6).
Discussion
The primary finding of this study is that acute preconditioning with a single dose of metformin induces autophagy in an AMPK-dependent manner in the brain, which subsequently reduces infarct volume, cell apoptosis and neurological deficits caused by focal cerebral ischaemia (Figure 6).
Figure 6.
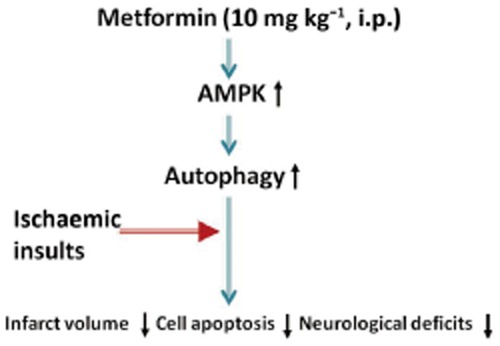
A model illustrating the role of AMPK-mediated autophagy in the neuroprotection mediated by acute metformin preconditioning.
AMPK is a major regulator of cellular and whole-body energy homeostasis, which can be phosphorylated and activated in response to an increase in the intracellular AMP/ATP ratio (Hardie, 2003). Once activated, AMPK leads to the conservation of intracellular ATP levels via multiple downstream pathways, including autophagy. As an AMPK activator, metformin has been revealed to induce autophagy through an AMPK-dependent manner in peripheral tissues including the heart. In diabetic OVE26 mice, Xie et al. (2011) found that metformin activated AMPK and enhanced cardiac autophagy, which was associated with an improvement in heart function. Meanwhile, a recent study from He et al. (2013) revealed that metformin prevented diabetic cardiomyopathy by inducing AMPK-mediated autophagy. Furthermore, Calvert et al. (2008) found that preconditioning with a single dose of metformin induced autophagy in an AMPK-dependent manner, which subsequently provided protection against cardiac dysfunction following myocardial ischaemia. In addition to the heart, AMPK has also been identified in the brain, uniquely localized in neurons and astrocytes (Turnley et al., 1999). In the current study, we demonstrated for the first time that a single dose of metformin activated AMPK and induced autophagy in the brain. In addition, we found that this enhanced autophagic activity was inhibited by Compound C pretreatment, indicating that metformin induced autophagy by activating brain AMPK. It is noteworthy that AMPK can activate autophagy by many signalling pathways. Several lines of evidence suggest that AMPK activation enhances autophagy by inhibiting the mechanistic target of rapamycin (mTOR), a conserved Ser/Thr kinase that negatively regulates autophagy (Zhang et al., 2013). AMPK can also enhance autophagy by directly phosphorylating UNC-51 like kinase 1 (Kim et al., 2011). Hence, the precise pathway by which AMPK activates autophagy in the brain should be investigated further.
The most exciting finding in the present study is that preconditioning with a single dose of metformin significantly reduced infarct volume, cell apoptosis and neurological deficits in the both acute (within 24 h) and subacute phase (within 96 h) of ischaemic stroke. These beneficial effects were reversed by inhibition of AMPK or autophagy, indicating that the neuroprotection of metformin preconditioning was mediated by AMPK-dependent autophagy. To the best of our knowledge, this is the first study to show that acute metformin preconditioning confers neuroprotection in rats with permanent cerebral ischaemia. More importantly, we demonstrated that AMPK-dependent autophagy was involved in the underlying protective mechanisms of metformin preconditioning. In addition, our findings support the hypothesis that pre-activation of autophagy in brain can protect against fatal cerebral ischaemia (Sheng et al., 2010; 2012). Autophagy facilitates cellular energy production by degradation of cellular components, and retards endoplasmic reticulum stress by removing dysfunctional mitochondria and suppressing local inflammatory responses; so increasing the tolerance of the brain to a subsequent ischaemic exposure (Lum et al., 2005; Kim et al., 2007; Sheng et al., 2012; Gao et al., 2013).
However, our findings appear to contradict those of a recent study by Li et al. (2010), as they found that acute preconditioning with metformin worsened the prognosis of stroke. These opposite results can be explained as follows: firstly, the dose of metformin used in our study (10 mg kg−1) was much smaller than that used in the study of Li et al. (100 mg kg−1), and different doses of metformin may lead to variations in the degree of activation of AMPK. Moderate activation of AMPK enhances autophagy as well as astrocytic glycolysis and ketosis to provide energy to neurons in the subsequent ischaemic exposure (Blazquez et al., 1999). However, overactivation of AMPK by a large dose of metformin may lead to prolonged astrocytic glycolysis, which results in a progressive acidosis and inhibits the ability of neurons to use lactate as an energy source, thus contributing to neuronal death (Pellerin and Magistretti, 1994). In addition, AMPK overactivation might result in excessive autophagy, which is also detrimental to neuronal survival (Wen et al., 2008). Secondly, we employed a permanent ischaemic model whereas Li et al. utilized an ischaemia/reperfusion model to investigate the effects of metformin on stroke. The pathophysiology mechanisms underlying these two ischaemic models are quite different, which may account for these opposite findings (Mao et al., 1999).
Lastly, some minor issues should be mentioned here. Firstly, the neuroprotection of metformin preconditioning could be fully inhibited by Compound C while inhibition of autophagy by 3-MA only partly abolished the metformin-induced neuroprotection. This interesting observation implies that, in addition to inducing autophagy, metformin preconditioning might confer neuroprotection through other AMPK-dependent mechanisms; these need to be identified in future studies. Secondly, it is noteworthy that in our study the rodents used were relatively young (12–14 weeks old), whereas ischaemic stroke occurs mainly in the elderly and the responsiveness of AMPK to metformin was revealed to drop sharply with aging (Mennes et al., 2013). Hence, the efficacy of metformin preconditioning on stroke prevention in aged rodents needs to be validated in the future. Thirdly, oral administration of metformin is a more practical and less invasive route, but this might exhibit a different dose-response curve when compared with i.p. injection. Therefore, future investigations are warranted to determine the efficacy of oral administration of metformin on the prevention of stroke.
In summary, our study provides the first evidence that acute preconditioning with a single dose of metformin induces autophagy in an AMPK-dependent manner in the brain, which confers neuroprotection against subsequent focal cerebral ischaemia. These findings reveal the underlying mechanisms by which metformin administration ameliorates the effects of ischaemic stroke, and suggest that metformin may have a practical clinical use for stroke prevention in addition to its hypoglycaemic effects.
Acknowledgments
This work was supported by the grants from the National Natural Science Foundation of China to L.T. (81171209, 81371406) and J-T.Y. (81000544), the grants from the Shandong Provincial Natural Science Foundation to L.T. (ZR2011HZ001) and J-T.Y. (ZR2010HQ004), the Medicine and Health Science Technology Development Project of Shandong Province to L.T. (2011WSA02018) and J-T.Y. (2011WSA02020) and the Innovation Project for Postgraduates of Jiangsu Province to T.J. (CXLX13_561).
Glossary
- ACC
p-acetyl-CoA carboxylase
- AMPK
AMP-activated protein kinase
- CBF
cerebral blood flow
- ICA
internal carotid artery
- 3-MA
3-methyladenine
- LC3
microtubule-associated protein 1 light chain 3
- PFA
paraformaldehyde
- PMCAO
permanent middle cerebral artery occlusion
- TTC
2, 3, 5-triphenyltetrazolium chloride
List of author contributions
J-T Y, Y-D Z and L T conceived and designed the experiments. T J, X-C Z, H-F W and M-S T performed the experiments. Q-Q Z, L C, J-Q S and L G analysed the data. T J wrote the paper.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
Additional Supporting Information may be found in the online version of this article at the publisher's web-site:
http://dx.doi.org/10.1111/bph.12655
Figure S1 Negative control for immunofluorescence. To ensure the specificity of the immunofluorescence procedure, control experiments were performed in which primary antibody was omitted. Note that no staining for LC3 was observed under this condition. Bars: 25 μm.
Figure S2 Metformin (Met) treatment did not significantly affect body temperature. After injection of metformin, body temperature was closely monitored with a rectal thermometer every 3 h until rats were subjected to pMCAO. It should be noted that Met treatment did not significantly affect body temperature when compared with vehicle (Veh). n = 6 per group.
Figure S3 Compound C (Cpd C) alone did not affect basal activity of AMPK and autophagy in brain. Rats received a single dose of Cpd C (20 mg kg−1; i.p.), the ratio of p-AMPK (Thr172)/AMPK and the protein levels of LC3-II were evaluated by Western blotting 24 h later. n = 6 per group. Columns represent mean ± SD. Veh, Vehicle.
Figure S4 Preconditioning with a single dose of metformin (Met; 10 mg kg−1; i.p.) did not affect cerebral blood flow (CBF) and brain lactate levels after pMCAO. (A) a probe was attached to the skull, and CBF was measured in the core region (2 mm caudal to bregma and 6 mm lateral to midline) of the MCA before pMCAO, immediately at the onset of pMCAO, and at 24 h after pMCAO by a laser-Doppler flowmetry (n = 11–12 per group). Data are expressed as % of baseline CBF of vehicle (Veh)-treated rats. Columns represent mean ± SD. (B) Brain lactate levels were evaluated at 24 h after pMCAO by a special kit (Abcam Inc., Cambridge, UK) according to the instructions provided by the manufacturer. n = 6 per group. Columns represent mean ± SD.
Figure S5 The metformin (Met)-induced autophagy was significantly inhibited by pretreatment with 3-MA. Rats were pretreated with a single dose of 3-MA (200 nmol; i.c.v.) before they received metformin administration. Twenty-four hours later, the protein levels of LC3-II was determined by Western blotting. n = 6 per group. Columns represent mean ± SD. Veh, Vehicle.
Figure S6 Compound C (Cpd C) and 3-MA alone did not change infarct volume at 24 h after pMCAO. Rats received a single dose of Compound C (20 mg kg−1; i.p.) or 3-MA (200 nmol; i.c.v.), and were subjected to pMCAO 24 h later. (A) TTC staining of representative coronal sections at 24 h after pMCAO. (B) Infarct volume was determined at 24 h after pMCAO. Columns represent mean ± SD; n = 10–11 per group.
Table S1 Compound C and 3-MA do not affect physiological parameters.
References
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, Peters JA, Harmar AJ, CGTP Collaborators The Concise Guide to PHARMACOLOGY 2013/14: Enzymes. Br J Pharmacol. 2013b;170:1797–1867. doi: 10.1111/bph.12451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arunachalam G, Samuel SM, Marei I, Ding H, Triggle CR. Metformin modulates hyperglycaemia-induced endothelial senescence and apoptosis through SIRT1. Br J Pharmacol. 2014;171:523–535. doi: 10.1111/bph.12496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bederson JB, Pitts LH, Tsuji M, Nishimura MC, Davis RL, Bartkowski H. Rat middle cerebral artery occlusion: evaluation of the model and development of a neurologic examination. Stroke. 1986;17:472–476. doi: 10.1161/01.str.17.3.472. [DOI] [PubMed] [Google Scholar]
- Blazquez C, Woods A, de Ceballos ML, Carling D, Guzman M. The AMP-activated protein kinase is involved in the regulation of ketone body production by astrocytes. J Neurochem. 1999;73:1674–1682. doi: 10.1046/j.1471-4159.1999.731674.x. [DOI] [PubMed] [Google Scholar]
- Calvert JW, Gundewar S, Jha S, Greer JJ, Bestermann WH, Tian R, et al. Acute metformin therapy confers cardioprotection against myocardial infarction via AMPK-eNOS-mediated signaling. Diabetes. 2008;57:696–705. doi: 10.2337/db07-1098. [DOI] [PubMed] [Google Scholar]
- Cheng YY, Leu HB, Chen TJ, Chen CL, Kuo CH, Lee SD, et al. Metformin-inclusive therapy reduces the risk of stroke in patients with diabetes: a 4-year follow-up study. J Stroke Cerebrovasc Dis. 2014;23:e99–e105. doi: 10.1016/j.jstrokecerebrovasdis.2013.09.001. [DOI] [PubMed] [Google Scholar]
- Gao B, Zhang XY, Han R, Zhang TT, Chen C, Qin ZH, et al. The endoplasmic reticulum stress inhibitor salubrinal inhibits the activation of autophagy and neuroprotection induced by brain ischemic preconditioning. Acta Pharmacol Sin. 2013;34:657–666. doi: 10.1038/aps.2013.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao L, Jiang T, Guo J, Liu Y, Cui G, Gu L, et al. Inhibition of autophagy contributes to ischemic postconditioning-induced neuroprotection against focal cerebral ischemia in rats. PLoS ONE. 2012;7:e46092. doi: 10.1371/journal.pone.0046092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hale AN, Ledbetter DJ, Gawriluk TR, Rucker EB., 3rd Autophagy: regulation and role in development. Autophagy. 2013;9:951–972. doi: 10.4161/auto.24273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardie DG. Minireview: the AMP-activated protein kinase cascade: the key sensor of cellular energy status. Endocrinology. 2003;144:5179–5183. doi: 10.1210/en.2003-0982. [DOI] [PubMed] [Google Scholar]
- Hardie DG. AMPK and autophagy get connected. EMBO J. 2011;30:634–635. doi: 10.1038/emboj.2011.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harhaji-Trajkovic L, Vilimanovich U, Kravic-Stevovic T, Bumbasirevic V, Trajkovic V. AMPK-mediated autophagy inhibits apoptosis in cisplatin-treated tumour cells. J Cell Mol Med. 2009;13(9B):3644–3654. doi: 10.1111/j.1582-4934.2009.00663.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He C, Zhu H, Li H, Zou MH, Xie Z. Dissociation of Bcl-2-Beclin1 complex by activated AMPK enhances cardiac autophagy and protects against cardiomyocyte apoptosis in diabetes. Diabetes. 2013;62:1270–1281. doi: 10.2337/db12-0533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang T, Gao L, Guo J, Lu J, Wang Y, Zhang Y. Suppressing inflammation by inhibiting the NF-kappaB pathway contributes to the neuroprotective effect of angiotensin-(1–7) in rats with permanent cerebral ischaemia. Br J Pharmacol. 2012;167:1520–1532. doi: 10.1111/j.1476-5381.2012.02105.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang T, Gao L, Shi J, Lu J, Wang Y, Zhang Y. Angiotensin-(1–7) modulates renin-angiotensin system associated with reducing oxidative stress and attenuating neuronal apoptosis in the brain of hypertensive rats. Pharmacol Res. 2013a;67:84–93. doi: 10.1016/j.phrs.2012.10.014. [DOI] [PubMed] [Google Scholar]
- Jiang T, Gao L, Zhu XC, Yu JT, Shi JQ, Tan MS, et al. Angiotensin-(1–7) inhibits autophagy in the brain of spontaneously hypertensive rats. Pharmacol Res. 2013b;71:61–68. doi: 10.1016/j.phrs.2013.03.001. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG. Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim I, Rodriguez-Enriquez S, Lemasters JJ. Selective degradation of mitochondria by mitophagy. Arch Biochem Biophys. 2007;462:245–253. doi: 10.1016/j.abb.2007.03.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2011;13:132–141. doi: 10.1038/ncb2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leoni RF, Paiva FF, Henning EC, Nascimento GC, Tannús A, de Araujo DB, et al. Magnetic resonance imaging quantification of regional cerebral blood flow and cerebrovascular reactivity to carbon dioxide in normotensive and hypertensive rats. Neuroimage. 2011;58:75–81. doi: 10.1016/j.neuroimage.2011.06.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Benashski SE, Venna VR, McCullough LD. Effects of metformin in experimental stroke. Stroke. 2010;41:2645–2652. doi: 10.1161/STROKEAHA.110.589697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lum JJ, DeBerardinis RJ, Thompson CB. Autophagy in metazoans: cell survival in the land of plenty. Nat Rev Mol Cell Biol. 2005;6:439–448. doi: 10.1038/nrm1660. [DOI] [PubMed] [Google Scholar]
- Mao Y, Yang GY, Zhou LF, Stern JD, Betz AL. Focal cerebral ischemia in the mouse: description of a model and effects of permanent and temporary occlusion. Brain Res Mol Brain Res. 1999;63:366–370. doi: 10.1016/s0169-328x(98)00271-x. [DOI] [PubMed] [Google Scholar]
- McCullough LD, Zeng Z, Li H, Landree LE, McFadden J, Ronnett GV. Pharmacological inhibition of AMP-activated protein kinase provides neuroprotection in stroke. J Biol Chem. 2005;280:20493–20502. doi: 10.1074/jbc.M409985200. [DOI] [PubMed] [Google Scholar]
- McGrath JC, Drummond GB, McLachlan EM, Kilkenny C, Wainwright CL. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mennes E, Dungan CM, Frendo-Cumbo S, Williamson DL, Wright DC. Aging-associated reductions in lipolytic and mitochondrial proteins in mouse adipose tissue are not rescued by metformin treatment. J Gerontol A Biol Sci Med Sci. 2013 doi: 10.1093/gerona/glt156. doi: 10.1093/gerona/glt156. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- Nathan DM. Some answers, more controversy, from UKPDS. United kingdom prospective diabetes study. Lancet. 1998;352:832–833. doi: 10.1016/s0140-6736(98)22937-0. [DOI] [PubMed] [Google Scholar]
- Papadakis M, Hadley G, Xilouri M, Hoyte LC, Nagel S, McMenamin MM, et al. Tsc1 (hamartin) confers neuroprotection against ischemia by inducing autophagy. Nat Med. 2013;19:351–357. doi: 10.1038/nm.3097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellerin L, Magistretti PJ. Glutamate uptake into astrocytes stimulates aerobic glycolysis: a mechanism coupling neuronal activity to glucose utilization. Proc Natl Acad Sci U S A. 1994;91:10625–10629. doi: 10.1073/pnas.91.22.10625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russo GL, Russo M, Ungaro P. AMP-activated protein kinase: a target for old drugs against diabetes and cancer. Biochem Pharmacol. 2013;86:339–350. doi: 10.1016/j.bcp.2013.05.023. [DOI] [PubMed] [Google Scholar]
- Schulte JM, Rothaus CS, Adler JN. Clinical decisions. Management of type 2 diabetes–polling results. N Engl J Med. 2014;370:e2. doi: 10.1056/NEJMclde1314028. [DOI] [PubMed] [Google Scholar]
- Selvin E, Hirsch AT. Contemporary risk factor control and walking dysfunction in individuals with peripheral arterial disease: NHANES 1999–2004. Atherosclerosis. 2008;201:425–433. doi: 10.1016/j.atherosclerosis.2008.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng R, Zhang LS, Han R, Liu XQ, Gao B, Qin ZH. Autophagy activation is associated with neuroprotection in a rat model of focal cerebral ischemic preconditioning. Autophagy. 2010;6:482–494. doi: 10.4161/auto.6.4.11737. [DOI] [PubMed] [Google Scholar]
- Sheng R, Liu XQ, Zhang LS, Gao B, Han R, Wu YQ, et al. Autophagy regulates endoplasmic reticulum stress in ischemic preconditioning. Autophagy. 2012;8:310–325. doi: 10.4161/auto.18673. [DOI] [PubMed] [Google Scholar]
- Shi WY, Xiao D, Wang L, Dong LH, Yan ZX, Shen ZX, et al. Therapeutic metformin/AMPK activation blocked lymphoma cell growth via inhibition of mTOR pathway and induction of autophagy. Cell Death Dis. 2012;3:e275. doi: 10.1038/cddis.2012.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanson RA, Morton MT, Tsao-Wu G, Savalos RA, Davidson C, Sharp FR. A semiautomated method for measuring brain infarct volume. J Cereb Blood Flow Metabolism. 1990;10:290–293. doi: 10.1038/jcbfm.1990.47. [DOI] [PubMed] [Google Scholar]
- Turnley AM, Stapleton D, Mann RJ, Witters LA, Kemp BE, Bartlett PF. Cellular distribution and developmental expression of AMP-activated protein kinase isoforms in mouse central nervous system. J Neurochem. 1999;72:1707–1716. doi: 10.1046/j.1471-4159.1999.721707.x. [DOI] [PubMed] [Google Scholar]
- Viscomi MT, D'Amelio M, Cavallucci V, Latini L, Bisicchia E, Nazio F, et al. Stimulation of autophagy by rapamycin protects neurons from remote degeneration after acute focal brain damage. Autophagy. 2012;8:222–235. doi: 10.4161/auto.8.2.18599. [DOI] [PubMed] [Google Scholar]
- Wang P, Guan YF, Du H, Zhai QW, Su DF, Miao CY. Induction of autophagy contributes to the neuroprotection of nicotinamide phosphoribosyltransferase in cerebral ischemia. Autophagy. 2012;8:77–87. doi: 10.4161/auto.8.1.18274. [DOI] [PubMed] [Google Scholar]
- Wen YD, Sheng R, Zhang LS, Han R, Zhang X, Zhang XD, et al. Neuronal injury in rat model of permanent focal cerebral ischemia is associated with activation of autophagic and lysosomal pathways. Autophagy. 2008;4:762–769. doi: 10.4161/auto.6412. [DOI] [PubMed] [Google Scholar]
- Wyler F. Effect of general anesthesia on distribution of cardiac output and organ blood flow in the rabbit: halothane and chloralose-urethane. J Surg Res. 1974;17:381–386. doi: 10.1016/0022-4804(74)90148-6. [DOI] [PubMed] [Google Scholar]
- Xie Z, Lau K, Eby B, Lozano P, He C, Pennington B, et al. Improvement of cardiac functions by chronic metformin treatment is associated with enhanced cardiac autophagy in diabetic OVE26 mice. Diabetes. 2011;60:1770–1778. doi: 10.2337/db10-0351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Chiu J, Zhang H, Qi T, Tang Q, Ma K, et al. Autophagic cell death induced by resveratrol depends on the Ca(2+)/AMPK/mTOR pathway in A549 cells. Biochem Pharmacol. 2013;86:317–328. doi: 10.1016/j.bcp.2013.05.003. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Negative control for immunofluorescence. To ensure the specificity of the immunofluorescence procedure, control experiments were performed in which primary antibody was omitted. Note that no staining for LC3 was observed under this condition. Bars: 25 μm.
Figure S2 Metformin (Met) treatment did not significantly affect body temperature. After injection of metformin, body temperature was closely monitored with a rectal thermometer every 3 h until rats were subjected to pMCAO. It should be noted that Met treatment did not significantly affect body temperature when compared with vehicle (Veh). n = 6 per group.
Figure S3 Compound C (Cpd C) alone did not affect basal activity of AMPK and autophagy in brain. Rats received a single dose of Cpd C (20 mg kg−1; i.p.), the ratio of p-AMPK (Thr172)/AMPK and the protein levels of LC3-II were evaluated by Western blotting 24 h later. n = 6 per group. Columns represent mean ± SD. Veh, Vehicle.
Figure S4 Preconditioning with a single dose of metformin (Met; 10 mg kg−1; i.p.) did not affect cerebral blood flow (CBF) and brain lactate levels after pMCAO. (A) a probe was attached to the skull, and CBF was measured in the core region (2 mm caudal to bregma and 6 mm lateral to midline) of the MCA before pMCAO, immediately at the onset of pMCAO, and at 24 h after pMCAO by a laser-Doppler flowmetry (n = 11–12 per group). Data are expressed as % of baseline CBF of vehicle (Veh)-treated rats. Columns represent mean ± SD. (B) Brain lactate levels were evaluated at 24 h after pMCAO by a special kit (Abcam Inc., Cambridge, UK) according to the instructions provided by the manufacturer. n = 6 per group. Columns represent mean ± SD.
Figure S5 The metformin (Met)-induced autophagy was significantly inhibited by pretreatment with 3-MA. Rats were pretreated with a single dose of 3-MA (200 nmol; i.c.v.) before they received metformin administration. Twenty-four hours later, the protein levels of LC3-II was determined by Western blotting. n = 6 per group. Columns represent mean ± SD. Veh, Vehicle.
Figure S6 Compound C (Cpd C) and 3-MA alone did not change infarct volume at 24 h after pMCAO. Rats received a single dose of Compound C (20 mg kg−1; i.p.) or 3-MA (200 nmol; i.c.v.), and were subjected to pMCAO 24 h later. (A) TTC staining of representative coronal sections at 24 h after pMCAO. (B) Infarct volume was determined at 24 h after pMCAO. Columns represent mean ± SD; n = 10–11 per group.
Table S1 Compound C and 3-MA do not affect physiological parameters.