

Abstract
BACKGROUND AND PURPOSE
Cerebrovascular remodelling is one of the important risk factors of stroke. The underlying mechanisms are unclear. Integrin β3 and volume-regulated ClC-3 Cl− channels have recently been implicated as important contributors to vascular cell proliferation. Therefore, we investigated the role of integrin β3 in cerebrovascular remodelling and related Cl− signalling pathway.
EXPERIMENTAL APPROACH
Cl− currents were recorded using a patch clamp technique. The expression of integrin β3 in hypertensive animals was examined by Western blot and immunohistochemisty. Immunoprecipitation, cDNA and siRNA transfection were employed to investigate the integrin β3/Src/ClC-3 signalling.
KEY RESULTS
Integrin β3 expression was up-regulated in stroke-prone spontaneously hypertensive rats, 2-kidney 2-clip hypertensive rats and angiotensin II-infused hypertensive mice. Integrin β3 expression was positively correlated with medial cross-sectional area and ClC-3 expression in the basilar artery of 2-kidney 2-clip hypertensive rats. Knockdown of integrin β3 inhibited the proliferation of rat basilar vascular smooth muscle cells induced by angiotensin II. Co-immunoprecipitation and immunofluorescence experiments revealed a physical interaction between integrin β3, Src and ClC-3 protein. The integrin β3/Src/ClC-3 signalling pathway was shown to be involved in the activation of volume-regulated chloride channels induced by both hypo-osmotic stress and angiotensin II. Tyrosine 284 within a concensus Src phosphorylation site was the key point for ClC-3 channel activation. ClC-3 knockout significantly attenuated angiotensin II-induced cerebrovascular remodelling.
CONCLUSIONS AND IMPLICATIONS
Integrin β3 mediates cerebrovascular remodelling during hypertension via Src/ClC-3 signalling pathway.
Keywords: integrin β3, chloride channel, Src, ClC-3, cerebrovascular remodelling
Introduction
Cerebrovascular remodelling is a major risk factor for stroke (Ward et al., 2000). Vascular remodelling with structural and functional changes of cellular and noncellular elements has long been confirmed in a variety of animal models of hypertension, such as stroke-prone spontaneously hypertensive rats (SHRSP; Chillon et al., 1996), 2-kidney 1-clip hypertensive rats (Hocher et al., 1999), Deoxycorticosterone acetate (DOCA)-salt hypertensive mice (Du et al., 2008), Nω-nitro-L-arginine methyl ester (L-NAME)-induced hypertensive rats (Moreau et al., 1995) and angiotensin II (Ang II)-infused hypertensive mice (Ruddy et al., 2009), and experiments in human beings also show that arteries are remodelled in hypertension with a reduction in lumen diameter and increase in media-to-lumen ratio (Heagerty et al., 1993). The latter is an important predictor of cardiovascular events such as myocardial infarction and stroke (De Ciuceis et al., 2007). However, the mechanisms of cerebrovascular remodelling induced by hypertension have not been defined. Although increased haemodynamic stress and hypertrophic stimuli are considered as two main triggers of remodelling (Ward et al., 2000), the downstream signalling is unclear. Interestingly, both mechanical stress and hypertrophic stimuli (Ang II, endothelin-1) activated volume-regulated Cl− currents in cardiomyocytes and vascular smooth muscle cells (VSMCs; Sato and Koumi, 1998; Shi et al., 2007; Ren et al., 2008). Our previous studies demonstrated that ClC-3 (see Alexander et al., 2013) is responsible for volume-regulated Cl− currents in VSMCs (Zhou et al., 2005) and the activity of the volume-regulated ClC-3 Cl− channel displayed a pressure-dependent increase in basilar artery smooth muscle cells (BASMCs) from 2-kidney 2-clip (2K2C) hypertensive rats, and the size of this increase paralleled the severity of cerebrovascular remodelling (Shi et al., 2007). Also, a deficiency of ClC-3 channels attenuated cerebrovascular remodelling during hypertension (Liu et al., 2010; Zheng et al., 2013).
Integrins are transmembrance adhesion proteins that act as mechanotransducers in response to mechanical stresses and convert vascular wall stresses into the intracellular signals (Dahl et al., 2003; Chao and Davis, 2011). Several studies have suggested that integrins played a role in sensing cell volume changes (Pedersen et al., 2001; Dahl et al., 2003) and integrin β1 mediates a stretch-sensitive chloride current in rabbit ventricular myocytes (Browe and Baumgarten, 2003). Other studies have suggested that integrin αVβ3 is required for the proliferation of VSMCs in response to biochemical stimuli (Mawatari et al., 2000; Nguyen Dinh Cat and Touyz, 2011), and is necessary for pressure-induced vascular remodelling (Mawatari et al., 2000; Nguyen Dinh Cat and Touyz, 2011). In this study, we explored the roles of integrin β3 in hypertensive cerebrovascular remodelling and the underlying mechanisms linking this to ClC-3 volume-regulated Cl− channels.
Methods
Animals
Male SHRSP and Wistar Kyoto rats aged 16 weeks were kindly provided by Dr Su D.F. (Second Military Medical University, Shanghai, China). The animals were kept in ventilated isolator cages under specific pathogen-free (SPF) conditions, air-conditioned animal rooms were maintained at 21°C, humidity 50%, with a 12 h light-dark cycle. Animals had free access to regular chow and sterile water.
2K2C hypertensive rats were prepared as previously described (Zeng et al.,1998). Briefly, healthy male Sprague-Dawley rats were anaesthetized by injection of 10% chloral hydrate (3 mg kg−1, i.p.), and ring-shaped silver clips with the internal diameter of 0.3 mm were placed around both right and left renal arteries. The sham-operated rats underwent the same surgical procedure except for the placement of silver clips and served as control. In this and the following procedures the depth of anaesthesia was assessed by absence of hind limb withdrawal in response to paw pinch as well as an absence of a corneal blink reflex.
Ang II-infused hypertensive mice were prepared as previously described (Daugherty et al., 2000). Briefly, 20–25 g mice (C57BL/6J mice, ClC-3+/+ wild type (wt) and ClC-3−/− mice) were anaesthetized with pentobarbital (60 mg kg−1, i.p.). Alzet osmotic minipumps (model 1002; ALZA Scientific Products, Mountain View, California, USA) were implanted into the s.c. space of these anaesthetized mice through a small incision in the back of the neck that was closed with surgical glue. Pumps were filled with saline vehicle or solutions of angiotensin II (1.5 mg·kg day−1, 14 day release, Sigma Chemical Co., St. Louis, MO, USA).
All animals used in the study received humane care in compliance with institutional guidelines for the health and care of experimental animals and conformed to the ‘Guide for the Care and Use of Laboratory Animals’ of the National Institute of Health In China, and were approved by Animal Ethical and Welfare Committee of Sun Yat-sen University (Guangdong, Guangzhou, China). All studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (Kilkenny et al., 2010; McGrath et al., 2010).
Immunohistochemistry
Rats were anaesthetized with 10% chloral hydrate and were perfused intracardiacly with 0.1 mol L−1 phosphate buffer containing heparin (100 U kg−1) and nitroglycerol (0.3 μg kg−1), followed by 4°C fixative solution containing 4% freshly depolymerized paraformaldehyde in 0.1 mol L−1 phosphate buffer for 15 min, the pressure was controlled ≈100 mmHg. The rat brain was carefully removed and sections (8 μm) were prepared from freshly frozen rat basilar arteries as previously described (Shi et al., 2007; Liu et al., 2010)2. Randomly selected 3 sections in each group were pretreated with the solution of 3% hydrogen peroxide and methanol at the ratio of 1:50 for 30 min at room temperature, and then blocked with 5% BSA in PBS for 30 min. Then the sections were exposed to α-actin monoclonal antibody (Sigma; dilution 1:400), ClC-3 rabbit polyclonal antibody (Alomone Labs; dilution 1:100) or integrin β3 antibody(1:10050; Cell Signaling Technology, Danvers, MA, USA) at 4°C overnight and then were treated with goat FITC-conjugated secondary antibody, goat anti-mouse antibody or goat anti-rabbit antibody. Positive immunostaining was determined with Boster SABC kit( Boster, Wuhan, Hubei, China) and visualized with diaminobenzidine substrate, followed by counterstaining with haematoxylin (Sigma Chemical Co., St. Louis, Missouri, USA). (Cell Signaling Technology; dilution 1:400) or Alexa Fluor® 488 goat anti-rabbit IgG (Cell Signaling Technology; dilution 1:400 ) at room temperature for 30 min. The quantification of α-SM-actin staining was analysed using confocal system (OLYMPUS, FV500-IX 81, magnification 400×) and Image-Pro Plus 5.0. For each tested group at each time point, 6 rat brains were taken for the experiment, respectively. A total of 8 randomly selected sections from the area of basilar artery per brain were quantitated for each type of labelling.
Electron microscopy
Electron microscopy were prepared as previously described (Zheng et al., 2013), mice were anaesthetized with 10% chloral hydrate and perfused intracardiacly with 0.1 mol L−1 phosphate buffer containing heparin (100 U kg−1) and nitroglycerol (0.3 μg kg−1), followed by 4°C fixative solution containing 4% paraformaldehyde, 0.25% glutaral, 15% saturated trinitrophenol in 0.1 mol L−1 phosphate buffer for 15 min. The brains were removed, and the tissue blocks containing the basilar artery at midpoint were cut into cubes of 1 mm × 1 mm × 3 mm, and then immersed into fixative solution at 4°C overnight. After further fixed in 2% osmic acid for 2 h, the tissue blocks were dehydrated via graded alcohols and embedded in Epon 812. Ultrathin sections with thickness of 80–100 nm were prepared and stained with uranyl acetate and lead citrate, and then were viewed under a transmission electron microscope (FEI TECNAI spirit G2, USA).
Immunoprecipitation
Cells were lysed on ice in Nonidet P-40 buffer containing 50 mM Tris-HCl, pH 7.6, 150 mM NaCl, 1%Nonidet P-40, Leupeptin (1 μg·mL−1), aprotinin (1 μg·mL−1), and PMSF (400 μM). After clarification, 500 μg of protein were immunoprecipitated with anti-HA (Cell Signaling Technology) or IgG (Cell Signaling Technology) at 4°C overnight, then with 50 μg of Protein G Plus-Agarose beads (Santa Cruz Biotechnology Inc; USA) for 90 min at 4°C. Protein complexes were subjected to Western blotting with the indicated antibodies. For comparison, 40 μg of cell lysate were applied to adjacent lanes.
Electrophysiological experiments
The Cl− current was recorded with an Axopatch 200B Amplifier (Axon Instruments, Foster City, CA, USA) using amphotericin-perforated patch recording technique. Amphotericin-B was freshly dissolved in DMSO at a concentration of 60 mg·mL−1, and then 20 μL of this solution was mixed with 5 mL of pipette solution by vortexing. The cell was held at −40 mV, and test potentials were applied from −100 mV to +120 mV for 400 ms in +20 mV increments at an interval of 5 s. Currents were filtered at frequency of 2 kHz and digitized at 5 kHz using pCLAMP8.0 software (Axon Instruments).
Details of the methods concerning the cell culture, Western blot analysis, electrophysiological experiments, cell transfection and generation of stable ClC-3 mutant-expressing cell lines are presented in the Supporting Information.
Statistical analysis
All data are expressed as means ± SEM. Statistical analyses were performed using Student's t-test or anova followed by a post hoc comparison using the least significant difference test. ‘n’ represents the number of independent tests. Values of P < 0.05 were considered statistically significant.
Results
Integrin β3 was involved in cerebrovascular remodelling in hypertension
In rabbit ventricular myocytes, direct and specific stretch of integrin β1 activated an outwardly rectifying, tamoxifen-sensitive Cl− current, which resembled the volume-regulated chloride channel (VRAC, the currents, ICl.vol; Browe and Baumgarten, 2003). Meanwhile, other studies suggested that integrin β3 was involved in hypertensive vascular remodelling (Han et al., 2007; Panchatcharam et al., 2010). So, we determined the expression of integrin β1 and β3 in BASMCs. To our surprise, the expression of integrin β1 was almost non-existent in BASMCs, whereas integrin β3 was highly expressed (see Supporting Information Figure S1). In 2K2C hypertensive rats, the BP increased progressively after the operation (Supporting Information Figure S2) and integrin β3 expression was up-regulated significantly in basilar arteries from these rats as shown by Western blot (Figure 1A); this was positively correlated with BP, r = 0.8112 (Supporting Information Figure S3a). The increases in integrin β3 expression were further confirmed by immunohistochemical staining of integrin β3 (Supporting Information Figure S4). Our previous studies showed that smooth muscle α-actin staining was increased time-dependently in basilar arteries from 2K2C hypertensive rats. The mean values of medial cross-sectional area (CSA) increased in the hypertensive group after 4 weeks post-operatively compared with those in corresponding control groups (Liu et al., 2010). The present study showed that integrin β3 expression was positively correlated with the medial CSA of the basilar artery from 2K2C hypertensive rats, r = 0.6025 (Figure 1D). This increased expression of integrin β3 in basilar arteries during hypertension was further confirmed in two other animal models, SHRSP and Ang II-infused hypertensive mice (Figure 1B and C). Next, we investigated the functional role of integrin β3 in the proliferation of BASMCs. Ang II (200 nM) increased BrdU incorporation in BASMCs by 1.57-fold (P < 0.05, n = 5). Integrin β3 knockdown by siRNA (see Supporting Information Figure S5) significantly inhibited the proliferation of BASMCs induced by Ang II (Figure 1E).
Figure 1.
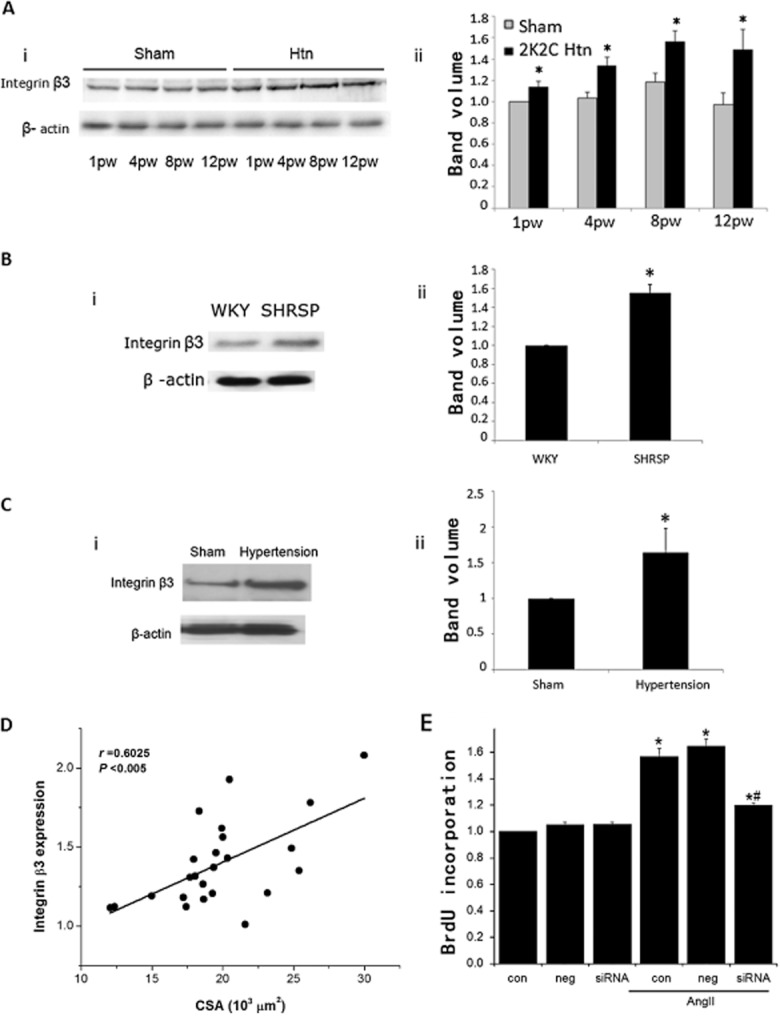
Integrin β3 expression in different hypertensive animal models. (A) (i) Expression of integrin β3 in basilar artery from 2K2C hypertensive groups and corresponding control groups. (ii) Densitometric analysis of integrin β3 expression. Values are mean ± SEM. n = 6 from 72 separate rats. (*P < 0.05 compared with sham in each group.). (B) (i) Expression of integrin β3 in basilar artery from 16-week-old WKY rats and age-matched SHRSP rats. (ii) Densitometric analysis of integrin β3 expression, n = 4, from eight separate rats. (*P < 0.01 compared with WKY). (C) (i) Expression of integrin β3 in basilar artery from Ang II hypertensive mice. (ii) Densitometric analysis of integrin β3 expression, n = 4, from 20 separate rats. (*P < 0.01 compared with sham). (D) Correlative analysis shows that there was a positive correlation between integrin β3 expression and medial CSA of basilar artery from 2K2C hypertensive rats (data from 24 rats and three independent experiments, r = 0.6025 P < 0.005). (E) Effect of integrin β3 knockdown on Ang II-induced BASMCs proliferation. Increase in cell growth induced by 200 nM Ang II was determined by assaying BrdU incorporation. Incubation of integrin β3 siRNA for 48 h significantly decreased BrdU incorporation (n = 5; *P < 0.05 vs. control, # P < 0.05 vs. control AngII).
Our previous study demonstrated that the expression of ClC-3 in basilar arteries was increased gradually with the development of cerebrovascular remodelling induced by hypertension (Liu et al., 2010), and a deficiency of ClC-3 attenuated cerebrovascular remodelling in DOCA-salt hypertension (Zheng et al., 2013). In the present study we showed that integrin β3 expression was positively correlated with ClC-3 expression in basilar arteries from 2K2C hypertensive rats, r = 0.641 (see Supporting Information Figure S3b). These findings suggest that integrin β3 is involved in cerebrovascular remodelling induced by hypertension and the volume-regulated ClC-3 chloride channel might be one of the mechanisms underlying this effect.
Integrin β3 mediated VRAC through the Src/ClC-3 signalling pathway in BASMCs
Src kinase activation is required for integrin-mediated osmosensing (Häussinger and Reinehr, 2011). Phosphorylation of tyrosine 416 (Tyr416) is a critical step leading to full activation of Src kinase (Thomas and Brugge, 1997). We determined the role of integrin β3 in Src kinase activation induced by hypotonic solution. Figure 2A shows that hypotonic solution increased phospho-Src (pY416) in a time-dependent manner. The increase in phospho-Src (pY416) induced by hypotonic solution treatment for 30 s was significantly enhanced by integrin β3 overexpression (Figure 2B), but reduced by integrin β3 knockdown (Figure 2C). These results suggest that integrin β3 mediates the Src phosphorylation induced by hypotonic solution.
Figure 2.
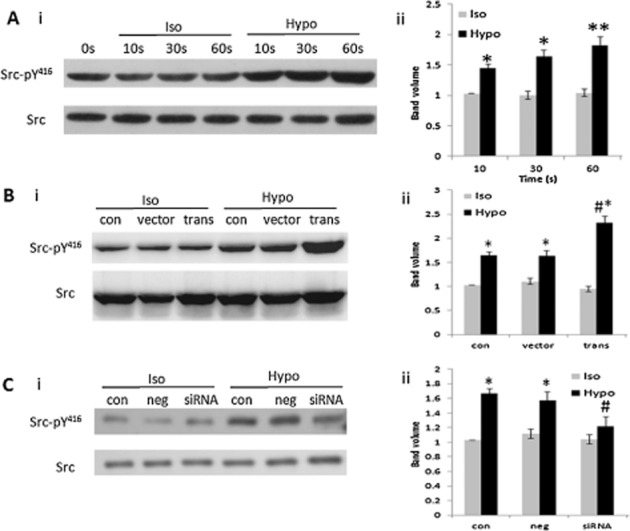
Integrin β3 mediated Src phosphorylation induced by hypotonic solution. (A) Phospho-Src (pY416) stimulated by hypotonic solution from 0 to 60 s. (i) Representative Western blot is shown. (ii) Densitometric analysis shows that phospho-Src (pY416) induced by hypotonic solution was time dependent (*P < 0.05 vs. isotonic (Iso) group, **P < 0.01 vs. Iso group; data from five different experiments). (B) Effect of integrin β3 cDNA transfection on phospho-Src (pY416). (i) Representative Western blot. (ii) Densitometric analysis. (*P < 0.05 vs. Iso group, #P < 0.05 vs. vector group; data from five different experiments). (C) Effect of integrin β3 siRNA transfection on phospho-Src (pY416) induced by hypotonic solution. (i) Representative Western blot. (ii) Densitometric analysis. (*P < 0.05 vs. Iso group, #P < 0.05 vs. negative group; data from five different experiments). Iso, isotonic solution; Hypo, hypotonic solution.
Previous studies in ventricular myocytes demonstrated that stretch of integrin β1 activates an outwardly rectifying Cl− current, of which the characteristics resembled the ICl.vol (Browe and Baumgarten, 2003). Our group and others recently demonstrated cell swelling and pressure-induced membrane stretch stimulated VRAC, and the currents were dependent on ClC-3 expression (Duan et al., 1997; Zhou et al., 2005; Qian et al., 2009). Here, we further investigated the signalling mechanisms linking integrin β3 and the ClC-3 chloride channel.
Hypotonic solution activated ICl.vol in wild-type (wt) BASMCs with current densities of −13.2 ± 1.4 pA·pF−1 at −80 mV and 24.9 ± 6.7 pA·pF−1 at +80 mV. Knockdown of integrin β3 by siRNA almost completely blocked the currents, and current densities of ICl.vol in integrin β3 siRNA-transfected cells were −2.4 ± 3.8 pA·pF−1 at −80 mV and 3.7 ± 6.4 pA·pF−1 at +80 mV (n = 6; Figure 3).
Figure 3.
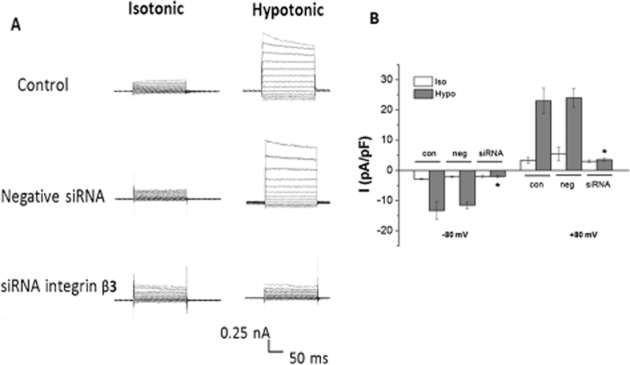
Effect of integrin β3 siRNA transfection on ICl.vol in BASMCs. (A) Representative traces of Cl− currents induced by isotonic and hypotonic solution in control, negative siRNA-transfected group and integrin β3 siRNA-transfected groups. (B) Mean current densities measured at −80 mV (downward bars) and +80 mV (upward bars). The cells were transfected for 48 to 72 h (*P < 0.05 vs.corresponding control group, n = 6). Iso, isotonic solution; Hypo, hypotonic solution.
Previous studies demonstrated that Src kinase was required for integrin-mediated VRAC activation (Browe and Baumgarten, 2003; Barfod et al., 2005). We previously found that the Src kinase inhibitor SU6656 inhibited ICl.vol in a concentration-dependent manner and Src kinase regulated ICl.vol by interacting with ClC-3 protein and phosphorylating the channel at tyrosine 284 (Tyr284). The phosphomimetic mutation, Y284D, significantly increased ClC-3-mediated Cl− current and Cl− efflux, while the non-phosphorylating mutation, Y284F, almost completely abolished the ClC-3-mediated increased Cl− current and Cl− efflux induced by hypotonic solution (Wang et al., 2013). We hypothesized that integrin β3 mediates ICl.vol through Src and ClC-3. We firstly tested the interaction of Src with integrin β3 and ClC-3 by using co-immunoprecipitation. We found that immunoprecipitation of HA-Src coprecipitated integrin β3 and ClC-3, and immunoprecipitation of ClC-3 coprecipitated integrin β3 and HA-Src (Figure 4A).
Figure 4.
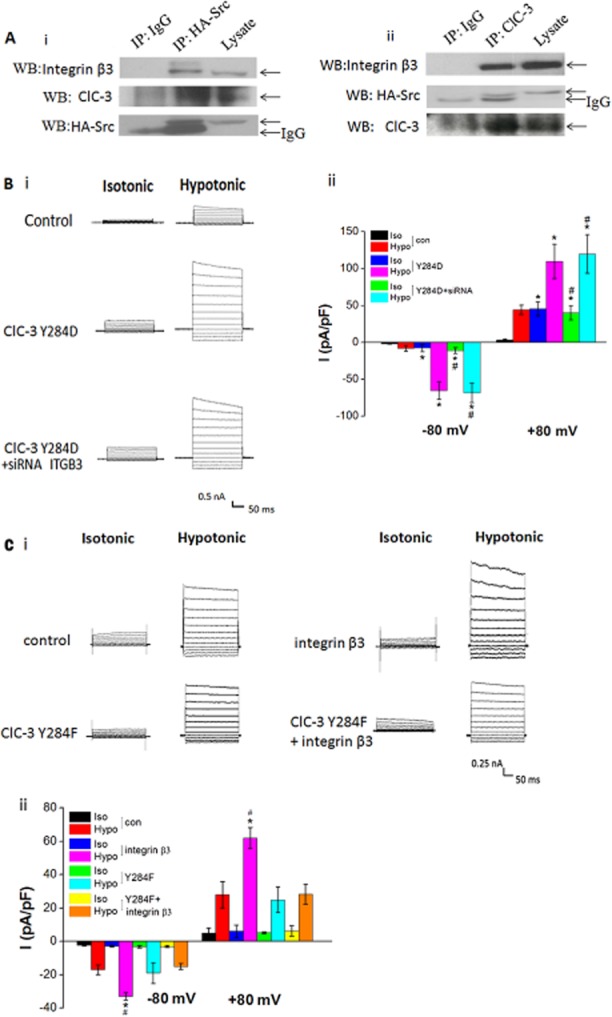
Effect of integrin β3 on ICl.vol in ClC-3 mutant (Y284D and Y284F) cells. (A) Immunoprecipitation of HA-Src, integrin β3 and ClC-3. (i) Immunoprecipitation of HA-Src coprecipitated with integrin β3 and ClC-3. Protein lysate from HA-SrcA BASMCs were immunoprecipated with HA-specific antibody or control IgG. The immunoprecipitations were blotted with anti-integrin β3, anti-ClC-3 or anti-HA-Src. (ii) Immunoprecipitation of ClC-3 coprecipitated with integrin β3 and HA-Src. Protein lysate from BASMCs were immunoprecipated with anti-ClC-3 antibody or control IgG and blotted with anti-integrin β3, anti-HA-Src, or anti-ClC-3 (n = 4). (B) Integrin β3 siRNA did not alter the ICl.vol in A10 cells stably overexpressing the ClC-3 Y284D mutant. (i) Representative traces of Cl− current. (ii) Mean current densities measured at −80 mv (downward bars) and+80 mv (upward bars; *P < 0.05 vs. corresponding control group, #P > 0.05 vs. Y284D group, n = 5). (C) Transfection of integrin β3 cDNA significantly increased the ICl.vol in A10 cells, but had no effect on the ICl.vol in ClC-3 Y284F mutant cells. (i) Representative traces of Cl− current. (ii) Mean current densities measured at −80 mV (downward bars) and +80 mV (upward bars) (*P < 0.05 vs. control group, #P > 0.05 vs. Y284F group, n = 5).
Next, we examined the role of integrin β3 in ICl.vol by using A10 cells stably expressing ClC-3 mutant proteins, Y284F or Y284D. The current densities of ICl.vol inY284D mutant cells were −11.1 ± 3.7 pA·pF−1 at −80 mV and 25.5 ± 7.3 pA·pF−1 at +80 mV in isotonic solution, which were significantly enhanced to −61.8 ± 8.8 pA·pF−1 at −80 mV and 111.4 ± 17.5 pA·pF−1 at +80 mV (n = 6, P < 0.05) by hypotonic solution. Knockdown of integrin β3 by siRNA did not alter these currents (Figure 4B). In contrast, in Y284F mutant cells hypotonic solution only induced a small ICl.vol, and overexpression of integrin β3 had little effect on the currents mediated by the Y284F mutant (Figure 4C). These results suggest that integrin β3 mediates ICl.vol through Src kinase-induced Tyr284 phosphorylation in the ClC-3.
Integrin β3 mediates Ang II-induced ICl.AngII through the Src/ClC-3 signalling pathway
It has been demonstrated that Ang II enhances integrin β3 expression (Brassard et al., 2006), which is consistent with our present results in vivo and in vitro (Figure 1C and see Supporting Information Figure S6). Previous studies found that Ang II activated outwardly rectifying Cl− currents (ICl.AngII) in rabbit cardiac myocytes (Ren et al., 2008) and Src TK played an important role in Ang II-initiated signalling transduction (Haendeler and Berk, 2000). We further investigated the effect of integrin β3/Src/ClC-3 signalling on ICl.AngII. In BASMCs Ang II (200 nM) activated an outwardly rectifying Cl− current (ICl.AngII) in isotonic solution, which was inhibited by hypertonic solution, and knockdown of integrin β3 significantly inhibited this ICl.AngII (Figure 5A).The ICl.AngII is dependent on Tyr284 phosphorylation of ClC-3 induced by Src kinase, as the current was almost completely inhibited by in the Y284F mutation but significantly potentiated in the Y284D mutation (Figure 5B, n = 6, P < 0.05). Interestingly, knockdown of integrin β3 did not altered the ICl.AngII mediated by the Y284D mutant (31.1 ± 3.3 pA·pF−1 at +80 mV), and overexpression of integrin β3 did not activate an ICl.AngII in Y284F (Figure 5B). These results reveal that the ICl.AngII is mediated through the integrin β3/Src/ClC-3 signalling pathway.
Figure 5.
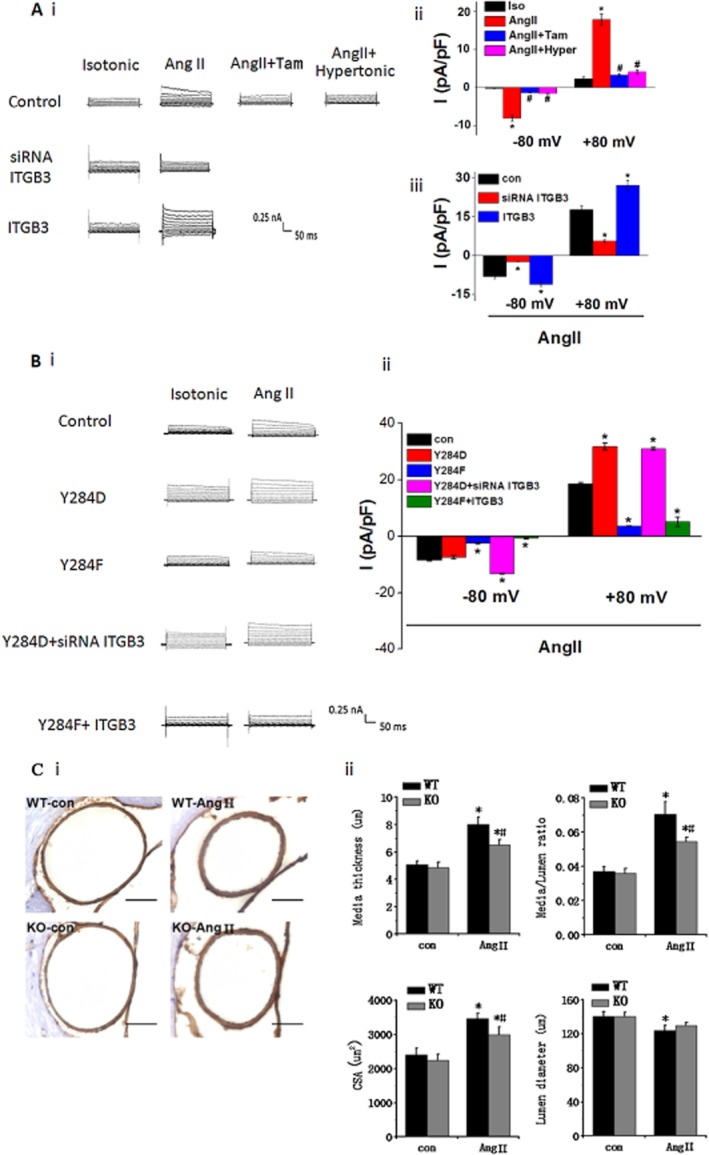
Involvement of integrin β3 in ICl.AngII and ClC-3 knockout attenuates cerebrovascular remodelling in Ang II-infused hypertensive mice. (A) In BASMCs, Ang II (200 nM) induced an outward rectifying current Cl− current (ICl.AngII) in isotonic solution, which was completely inhibited in hypertonic solution (Hyper) and by tamoxifen (Tam, 10 μm), indicating that this current was a volume-regulated Cl− current. ICl.AngII was also completely blocked by integrin β3 siRNA (siRNA ITGB3) and significantly enhanced by integrin β3 cDNA(ITGB3). (i) Representative traces of ICl.AngII. (ii) Mean current densities of control group with different treatment measured at −80 mV (downward bars) and +80 mV (upward bars). (iii) Mean current densities of angiotensin II-treated control, siRNA ITGB3 and ITGB3 groups at −80 mV (downward bars) and +80 Mv (upward bars; *P < 0.05 vs. control group, # P < 0.05 vs. Ang II group, n = 5). (B) ICl.AngII was regulated by Tyr284 phosphorylation in ClC-3. In ClC-3 Y284D and Y284F cells, ICl.AngII was enhanced and inhibited respectively. Integrin β3 cDNA transfection did not alter Y284F effect on ICl.AngII. Similarly, integrin β3 siRNA transfection did not reverse Y284D effect on ICl.AngII. (i) Representative traces of Cl− current. (ii) Mean current densities measured at −80 mV (downward bars) and +80 mV (upward bars) (*P < 0.05 vs. control group, n ≥ 6). (C) ClC-3 knockout (ClC-3−/−) ameliorated the morphological changes of mice cerebral basilar artery occurring during hypertension. (i) wt (ClC-3+/+) mice and ClC-3−/− mice were treated with or without Ang II. Vascular media smooth muscle was specifically stained as brown with α-actin antibody. Scale bars, 50 μm. Representative pictures are shown. (ii) Statistical analysis of media thickness, internal lumen diameter, media-to-lumen ratio and medial CSA (n = 6 per group, *P < 0.05, Ang II mice vs.control mice; #P < 0.05, wt-AngII mice vs. ClC-3−/−-AngII mice).
ClC-3 knockout attenuated cerebrovascular remodelling in Ang II hypertension
We further determined the functional role of the integrin β3/Src/ClC-3 signalling pathway in the development of cerebrovascular remodelling using ClC-3 knockout (ClC-3−/−) mice treated with Ang II. As indicated by smooth muscle α-actin immunocytochemical staining, ClC-3 knockout ameliorated the morphological changes in mice cerebral basilar artery that occur during hypertension (Figure 5C, i). Also Ang II treatment led to a significant increase in media thickness and decrease in the diameter of the internal lumen, resulting in an increased media-to-lumen ratio in basilar arteries from wt mice compared with those of control. Medial CSA was also significantly enhanced in AngII-treated wt mice. In contrast, cerebrovascular remodelling was significantly attenuated in the ClC-3−/− mice treated with AngII (Figure 5C, ii). The effects of ClC-3 knockdown on cerebrovascular remodelling were further confirmed by electron microscopy (see Supporting Information Figure S7).
Discussion and conclusions
Recent evidence obtained by us and others has demonstrated that the ClC-3 chloride channel plays an important role in the vascular remodelling that occurs during hypertension (Dai et al., 2005; Guan et al., 2006,2006; Zheng et al., 2013), by accelerating the proliferation of VSMCs (Wang et al., 2002; Tang et al., 2008) and preventing their apoptosis (Okada et al., 2006; Qian et al., 2011). We previously found that ClC-3 channel activity and protein expression were significantly increased in BASMCs from 2K2C hypertensive rats and these increases paralleled the severity of cerebrovascular remodelling (Shi et al., 2007). Moreover, simvastatin ameliorated hypertension-induced cerebrovascular remodelling by inhibiting the proliferation of cerebrovascular smooth muscle cell and suppressing ClC-3 chloride channel activity (Liu et al., 2010). Furthermore, ClC-3 knockout attenuated the cerebrovascular remodelling that occurs in DOCA-salt hypertensive mice (Zheng et al., 2013).
Although the functional roles of the ClC-3 chloride channel in VSMCs have been extensively characterized, the molecular signalling that is associated with ClC-3 channel activation has not been clarified. Recently, we found that Tyr284 phosphorylation of the ClC-3 protein targeted by Src kinase was required for ClC-3 channel activation; the ICl.vol was completely blocked in the non-phosphorylatable mutation Y284F, whereas this Cl− current mediated by ClC-3 was increased in the phosphomimetic mutation Y284D (Wang et al., 2013). Furthermore, there is accumulating evidence supporting the notion that Y284 phosphorylation of ClC-3 plays an important role in cell apoptosis (Dupere-Minier et al., 2004; Wang et al., 2013). However, the upstream and downstream components of this Src/ClC-3 signalling pathway remain to be identified. In this study, we demonstrated that integrin β3 acts as an upstream component of the Src/ClC-3 signalling pathway; integrin β3 siRNA and cDNA transfection inhibited and enhanced the ICl.vol mediated by ClC-3, respectively, but had no effect in ClC-3 Y284D or Y284F mutant cells.
Integrins are cell surface adhesion proteins that mediate a variety of physiological functions and pathological processes and consist of heterodimers composed of an α-chain and a β-chain. In mammals, 18 α- and 8 β-subunits have been characterized (Barczyk et al., 2010). We focused our study on β-subunits because of previous reports suggesting that activation of integrin β1 induced VRAC (Browe and Baumgarten, 2003). It should be noted that integrin β1 also mediated VRAC via Src in rabbit ventricular myocytes, indicating that our findings showing the involvement of a integrin/Src/ClC-3 signalling pathway may not be unique to VSMCs. The finding that different integrin β-subunits can mediate the Src/ClC-3 signalling pathway may be because the integrins expressed by different cell types vary greatly.
Hypertrophic stimuli, such as Ang II, and mechanical stress on VSMCs are important contributors to vascular remodelling during hypertension (Dzau and Gibbons, 1993; Lehoux and Tedgui, 1998). Integrin-mediated stretch-sensitive Cl− currents have been extensively studied (Browe and Baumgarten, 2003; 2006); however, it is unclear whether integrin-mediated signalling is involved in Ang II-activated Cl− currents. In the present investigation, we demonstrated that integrin β3/Src/ClC-3 signalling pathway mediated the volume-regulated Cl− current and Ang II-activated Cl− current, which resemble mechanical stress and hypertrophic stimuli respectively. To our knowledge, this is the first direct evidence that the integrin β3/Src/ClC-3 signalling pathway mediates the Cl− current induced by both hypertrophic stimuli (e.g. Ang II) and osmotic stimuli.
Furthermore, the present results obtained in hypertensive animal models in vivo suggest that integrin β3 mediated the cerebrovascular remodelling. The expressions of integrin β3 in the basilar artery were up-regulated in the different hypertensive animal models, which included an essential hypertensive (primary hypertensive) model of SHRSP, renovascular hypertensive (secondary hypertensive) model of 2K2C hypertensive rats and Ang II-infused hypertensive mice (Ang II-dependent hypertensive model). The increase in integrin β3 expression in the basilar artery from the 2K2C hypertensive rat was positively correlated with the increased BP, and with the hypertension-induced alteration in CSA. Notably, integrin β3 expression was increased 1 week after the operation, when there were no differences apparent between the basilar arteries of hypertensive mice and sham-operated controls. Furthermore, the in vitro results showed that knockdown of integrin β3 significantly inhibited Ang II-induced proliferation of the BASMCs. These results suggest that integrin β3 mediated the cerebrovascular remodelling during hypertension. We have noted the report that an up-regulation of αvβ3 integrin had a crucial role in the mesenteric artery of TGR (mRen2)27 (model of hypertension) during the eutrophic remodelling process (Heerkens et al., 2006). This report suggested that the αv subunit of αvβ3 integrin, but not the β3 subunit, played a key role in the eutrophic remodelling of resistance arteries. One possible explanation for this inconsistency is that vascular remodelling may be different among vessel types and models of hypertension. Consistent with our previous studies in 2K2C hypertensive rats (Shi et al., 2007; Liu et al., 2010; Wang et al., 2012), cerebral vessels from Ang II-infused hypertensive mice in the present study underwent the hypertrophic remodelling process, which is characterized by increases in blood vessel wall thickness, outer blood vessel wall diameter, CSA and a reduction in inner blood vessel wall diameter (Guan et al., 2006,2006; Shi et al., 2007; Liu et al., 2010). This is different from he αv-mediated eutrophic remodelling, which is characterized by a reduction in lumen diameter and unchanged CSA (Sonoyama et al., 2007). The signs of vascular remodelling are not uniform across vessel types or animal models, for example, pial arteries with smaller lumens and thicker walls were found in SHRSP (Chillon et al., 1996), L-NAME-induced hypertensive rats (Kitamoto et al., 2000), DOCA-salt hypertensive mice (Liu et al., 2010; Zheng et al., 2013); however, in 1-kindey 1-clip hypertensive rats, pial arterioles displayed thicker walls but normal lumens (Baumbach and Hajdu, 1993). It is noteworthy that these studies (Baumbach and Hajdu, 1993; Chillon et al., 1996; Kitamoto et al., 2000; Liu et al., 2010; Zheng et al., 2013) indicate that BP alone is not the sole stimulus for vascular remodelling. Interestingly, we recently demonstrated that in DOCA-salt hypertensive mice, propranolol, which reduced BP effectively, failed to prevent basilar artery from remodelling (Liu et al., 2010; Zheng et al., 2013). As integrin/Src/ClC-3 signalling is the common pathway in haemodynamic stress and hypertrophic stimuli-initiated pro-remodelling signalling, they may serve as more efficacious therapeutic candidates.
There is accumulating evidence supporting the important roles of ClC-3 in proliferation (Wang et al., 2002; Guan et al., 2006,2006; Tang et al., 2008), apoptosis (Guan et al., 2006,2006; Okada et al., 2006; Qian et al., 2011) and migration (Ganapathi et al., 2013) in VSMCs, making ClC-3 a novel therapeutic target against vascular remodelling. We recently found that knockout of ClC-3 in vivo attenuated the disorganization of VSMCs and extracellular matrix accumulation in DOCA-salt hypertension (Liu et al., 2010; Zheng et al., 2013). In addition to the influence of MMPs/ TIMP expression and TGF-b1/Smad3 signalling, the effects of ClC-3 on Akt (Tang et al., 2008), NF-κB (Miller et al., 2007) and redox (Miller et al., 2007; Matsuda et al., 2010) signalling are possible mechanisms linking ClC-3 to vascular remodelling.
Taken together, the results of the present study demonstrated that integrin β3/Src/ClC-3 signalling is involved in cerebrovascular remodelling in both a primary and secondary hypertension model. Mechanical strain and hypertrophic stimuli are important pathogenic factors in hypertensive vascular remodelling. We found that the integrin β3/Src/ClC-3 signalling pathway was involved both in hypo-osmotic stress (mimicking mechanical strain) and Ang II (main hypertrophic stimuli)-induced VRAC activation, triggering a series of mitogenic and anti-apoptotic signalling resulting in increased vascular cell proliferation and migration, and reduced apoptosis, finally leading to cerebrovascular remodelling (Figure 6).
Figure 6.
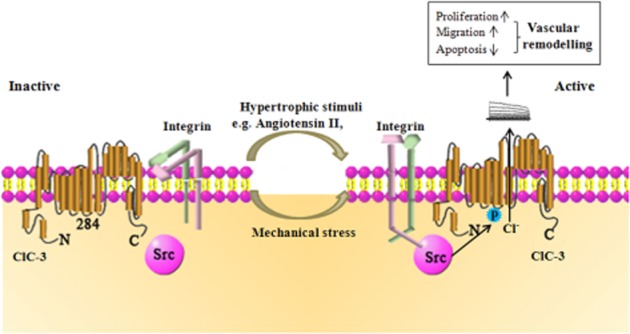
Integrin β3/Src/ClC-3 signalling pathway. Hypertrophic stimuli (such as Ang II), mechanical and osmotic stimuli could be sensed by integrin β3, leading to Src phosphorylation of ClC-3 at Tyr284, triggering activation of the ClC-3 chloride channel and efflux of Cl−. The ClC-3 chloride channel is associated with cerebrovascular remodelling, by promoting the proliferation and migration of VSMCs and preventing their apoptosis.
Acknowledgments
We thank Ding-Feng Su and Mark Ginsberg for their kindly help. This work was supported by the National Natural Science Foundation of China (Key grant, No.81230082, and No.81173055).
Glossary
- 2K2C
2-kidney 2-clip rats
- Ang II
angiotensin II
- BASMCs
basilar artery smooth muscle cells
- CSA
cross-sectional area
- DOCA
deoxycorticosterone acetate
- L-NAME
Nω-nitro-L-arginine methyl ester
- SHRSP
stroke-prone spontaneously hypertensive rats
- Tyr
tyrosine
- VRAC
volume-regulated chloride channel
- VSMCs
vascular smooth muscle cells
Conflict of interest
None.
Supporting information
Additional Supporting Information may be found in the online version of this article at the publisher's web-site:
http://dx.doi.org/10.1111/bph.12654
Figure S1 Integrin β1 and integrin β3 expression in BASMCs. Integrin β3 was detected while integrin β1 was hardly detected in BASMCs. Human umbilical vein endothelial cells (HUVECs) were used as positive control for integrin β1 expression. n = 8.
Figure S2 Blood pressure of hypertensive animals. (a) Blood pressure of 2K2C hypertensive group and sham group at different time point. PW, postoperative week. *P < 0.01 compared with sham in each group; #P < 0.01 compared with 1PW in each group, n = 6. (b) Blood pressure of SHRSP rats and WKY rat at 16 weeks, #P < 0.005, n ≥ 6. (c) Blood pressure of angiotensin II infusion induced hypertensive C57BL/6J mice, n = 5. Values are mean ± SEM.
Figure S3 (a) Integrin β3 (ITGB3) expression was positively correlated with the blood pressure, correlation coefficient was 0.8125 (n = 36 rats from six independent experiments, P < 0.001). (b) Integrin β3 expression was positively correlated with ClC-3 during hypertension (24 rats from three independent experiments, P < 0.005).
Figure S4 Changes in integrin β3 expression of basilar arteries in 2-kidney, 2-clip hypertensive rats and sham group rats (scale bars, 20 μm). Representative sections were shown, n = 6 per group.
Figure S5 The effect of integrin β3 siRNA (ITGB3 siRNA) transfection in BASMCs. BASMCs were transfected with 200 nM integrin β3 siRNA and assayed with reverse transcriptase-PCR (RT-PCR) 48 h later. *P < 0.05 versus negative group, n = 5. Values are mean ± SEM.
Figure S6 Densitometric analysis of integrin β3 expression induced by 1 nM, 10 nM, 100 nM and 1000 nM angiotensin II, the solvent of angiotensin II was dimethyl sulfoxide (DMSO, 0.1%; n = 5; *P < 0.05 vs. control).
Figure S7 Electron microscopy images of cerebral basilar arteries from wt and ClC-3−/− mice with or without angiotensin II implantation. Angiotensin II resulted in smooth muscle cell disarrangement (♦), intercellular space enlargement (→) and vacuolization (Δ) in wt mice; while in the basilar artery of ClC-3−/− mice, these ultrastructural changes did not occur. Black bar represents 1 μm. Six mice were studied in each group.
References
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, Peters JA, Harmar AJ, CGTP Collaborators The Concise Guide to PHARMACOLOGY 2013/14: Ion channels. Br J Pharmacol. 2013;170:1607–1651. doi: 10.1111/bph.12447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barczyk M, Carracedo S, Gullberg D. Integrins. Cell Tissue Res. 2010;339:269–280. doi: 10.1007/s00441-009-0834-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barfod ET, Moore AL, Melnick RF, Lidofsky SD. Src regulates distinct pathways for cell volume control through Vav and phospholipase Cγ. J Biol Chem. 2005;280:25548–25557. doi: 10.1074/jbc.M411717200. [DOI] [PubMed] [Google Scholar]
- Baumbach GL, Hajdu MA. Mechanics and composition of cerebral arterioles in renal and spontaneously hypertensive rats. Hypertension. 1993;21:816–826. doi: 10.1161/01.hyp.21.6.816. [DOI] [PubMed] [Google Scholar]
- Brassard P, Amiri F, Thibault G, Schiffrin EL. Role of angiotensin type-1 and angiotensin type-2 receptors in the expression of vascular integrins in angiotensin II–infused rats. Hypertension. 2006;47:122–127. doi: 10.1161/01.HYP.0000196272.79321.11. [DOI] [PubMed] [Google Scholar]
- Browe DM, Baumgarten CM. Stretch of β1 integrin activates an outwardly rectifying chloride current via FAK and Src in rabbit ventricular myocytes. J Gen Physiol. 2003;122:689–702. doi: 10.1085/jgp.200308899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Browe DM, Baumgarten CM. EGFR kinase regulates volume-sensitive chloride current elicited by integrin stretch via PI-3 K and NADPH oxidase in ventricular myocytes. J Gen Physiol. 2006;127:237–251. doi: 10.1085/jgp.200509366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao J, Davis M. The roles of integrins in mediating the effects of mechanical force and growth factors on blood vessels in hypertension. Curr Hypertens Rep. 2011;13:421–429. doi: 10.1007/s11906-011-0227-6. [DOI] [PubMed] [Google Scholar]
- Chillon J, Heistad DD, Baumbach GL. Effects of endothelin receptor inhibition on cerebral arterioles in hypertensive rats. Hypertension. 1996;27:794–798. doi: 10.1161/01.hyp.27.3.794. [DOI] [PubMed] [Google Scholar]
- Dahl S, Schliess F, Reissmann R, Görg B, Weiergr O, Kocalkova M, et al. Involvement of integrins in osmosensing and signaling toward autophagic proteolysis in rat liver. J Biol Chem. 2003;278:27088–27095. doi: 10.1074/jbc.M210699200. [DOI] [PubMed] [Google Scholar]
- Dai YP, Bongalon S, Hatton WJ, Hume JR, Yamboliev IA. ClC-3 chloride channel is upregulated by hypertrophy and inflammation in rat and canine pulmonary artery. Br J Pharmacol. 2005;145:5–14. doi: 10.1038/sj.bjp.0706135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daugherty A, Manning MW, Cassis LA. Angiotensin II promotes atherosclerotic lesions and aneurysms in apolipoprotein E-deficient mice. J Clin Invest. 2000;105:1605–1612. doi: 10.1172/JCI7818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Ciuceis C, Porteri E, Rizzoni D, Rizzardi N, Paiardi S, Boari GE, et al. Structural alterations of subcutaneous small-resistance arteries may predict major cardiovascular events in patients with hypertension. Am J Hypertens. 2007;20:846–852. doi: 10.1016/j.amjhyper.2007.03.016. [DOI] [PubMed] [Google Scholar]
- Du Y, Guan Y, Alp NJ, Channon KM, Chen AF. Endothelium-specific GTP cyclohydrolase I overexpression attenuates blood pressure progression in salt-sensitive low-renin hypertension. Circulation. 2008;117:1045–1054. doi: 10.1161/CIRCULATIONAHA.107.748236. [DOI] [PubMed] [Google Scholar]
- Duan D, Winter C, Cowley S, Hume JR, Horowitz B. Molecular identification of a volume-regulated chloride channel. Nature. 1997;390:417–421. doi: 10.1038/37151. [DOI] [PubMed] [Google Scholar]
- Dupere-Minier G, Hamelin C, Desharnais P, Bernier J. Apoptotic volume decrease, pH acidification and chloride channel activation during apoptosis requires CD45 expression in HPB-ALL T cells. Apoptosis. 2004;9:543–551. doi: 10.1023/B:APPT.0000038031.84705.84. [DOI] [PubMed] [Google Scholar]
- Dzau VJ, Gibbons GH. Introduction: vascular remodeling: mechanisms and implications. J Cardiovasc Pharmacol. 1993;21:S1–S5. [PubMed] [Google Scholar]
- Ganapathi SB, Wei S, Zaremba A, Lamb FS, Shears SB. Functional regulation of ClC-3 in the migration of vascular smooth muscle cells. Hypertension. 2013;61:174–179. doi: 10.1161/HYPERTENSIONAHA.112.194209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan YY, Wang GL, Zhou JG. The ClC-3 Cl-channel in cell volume regulation, proliferation and apoptosis in vascular smooth muscle cells. Trends Pharmacol Sci. 2006;27:290–296. doi: 10.1016/j.tips.2006.04.008. [DOI] [PubMed] [Google Scholar]
- Guan YY, Zhou JG, Zhang Z, Wang GL, Cai BX, Hong L, et al. Ginsenoside-Rd from panax notoginseng blocks Ca2+ influx through receptor-and store-operated Ca 2 + channels in vascular smooth muscle cells. Eur J Pharmacol. 2006;548:129–136. doi: 10.1016/j.ejphar.2006.08.001. [DOI] [PubMed] [Google Scholar]
- Haendeler J, Berk BC. Angiotensin II mediated signal transduction. Important role of tyrosine kinases. Regul Pept. 2000;95:1–7. doi: 10.1016/s0167-0115(00)00133-6. [DOI] [PubMed] [Google Scholar]
- Han M, Wen J, Zheng B, Liu Z, Chen Y. Blockade of integrin β3–FAK signaling pathway activated by osteopontin inhibits neointimal formation after balloon injury. Cardiovasc Pathol. 2007;16:283–290. doi: 10.1016/j.carpath.2007.04.002. [DOI] [PubMed] [Google Scholar]
- Häussinger D, Reinehr R. Osmotic regulation of bile acid transport, apoptosis and proliferation in rat liver. Cell Physiol Biochem. 2011;28:1089–1098. doi: 10.1159/000335845. [DOI] [PubMed] [Google Scholar]
- Heagerty AM, Aalkjær C, Bund SJ, Korsgaard N, Mulvany MJ. Small artery structure in hypertension. Dual processes of remodeling and growth. Hypertension. 1993;21:391–397. doi: 10.1161/01.hyp.21.4.391. [DOI] [PubMed] [Google Scholar]
- Heerkens EH, Shaw L, Ryding A, Brooker G, Mullins JJ, Austin C, et al. αV integrins are necessary for eutrophic inward remodeling of small arteries in hypertension. Hypertension. 2006;47:281–287. doi: 10.1161/01.HYP.0000198428.45132.02. [DOI] [PubMed] [Google Scholar]
- Hocher B, George I, Rebstock J, Bauch A, Schwarz A, Neumayer H, et al. Endothelin system–dependent cardiac remodeling in renovascular hypertension. Hypertension. 1999;33:816–822. doi: 10.1161/01.hyp.33.3.816. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG. Animal research: Reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitamoto S, Egashira K, Kataoka C, Koyanagi M, Katoh M, Shimokawa H, et al. Increased activity of nuclear factor-κB participates in cardiovascular remodeling induced by chronic inhibition of nitric oxide synthesis in rats. Circulation. 2000;102:806–812. doi: 10.1161/01.cir.102.7.806. [DOI] [PubMed] [Google Scholar]
- Lehoux S, Tedgui A. Signal transduction of mechanical stresses in the vascular wall. Hypertension. 1998;32:338–345. doi: 10.1161/01.hyp.32.2.338. [DOI] [PubMed] [Google Scholar]
- Liu YJ, Wang XG, Tang YB, Chen JH, Lv XF, Zhou JG, et al. Simvastatin ameliorates rat cerebrovascular remodeling during hypertension via inhibition of volume-regulated chloride channel. Hypertension. 2010;56:445–452. doi: 10.1161/HYPERTENSIONAHA.110.150102. [DOI] [PubMed] [Google Scholar]
- McGrath J, Drummond G, McLachlan E, Kilkenny C, Wainwright C. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuda JJ, Filali MS, Moreland JG, Miller FJ, Lamb FS. Activation of swelling-activated chloride current by tumor necrosis factor-α requires ClC-3-dependent endosomal reactive oxygen production. J Biol Chem. 2010;285:22864–22873. doi: 10.1074/jbc.M109.099838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mawatari K, Liu B, Craig Kent K. Activation of integrin receptors is required for growth factor-induced smooth muscle cell dysfunction. J Vasc Surg. 2000;31:375–381. doi: 10.1016/s0741-5214(00)90167-8. [DOI] [PubMed] [Google Scholar]
- Miller FJ, Filali M, Huss GJ, Stanic B, Chamseddine A, Barna TJ, et al. Cytokine activation of nuclear factor κB in vascular smooth muscle cells requires signaling endosomes containing Nox1 and ClC-3. Circ Res. 2007;101:663–671. doi: 10.1161/CIRCRESAHA.107.151076. [DOI] [PubMed] [Google Scholar]
- Moreau P, Takase H, Küng CF, van Rooijen M, Schaffner T, Lüscher TF. Structure and function of the rat basilar artery during chronic nitric oxide synthase inhibition. Stroke. 1995;26:1922–1929. doi: 10.1161/01.str.26.10.1922. [DOI] [PubMed] [Google Scholar]
- Nguyen Dinh Cat A, Touyz RM. Cell signaling of angiotensin II on vascular tone: novel mechanisms. Curr Hypertens Rep. 2011;13:122–128. doi: 10.1007/s11906-011-0187-x. [DOI] [PubMed] [Google Scholar]
- Okada Y, Shimizu T, Maeno E, Tanabe S, Wang X, Takahashi N. Volume-sensitive chloride channels involved in apoptotic volume decrease and cell death. J Membr Biol. 2006;209:21–29. doi: 10.1007/s00232-005-0836-6. [DOI] [PubMed] [Google Scholar]
- Panchatcharam M, Miriyala S, Yang F, Leitges M, Chrzanowska-Wodnicka M, Quilliam LA, et al. Enhanced proliferation and migration of vascular smooth muscle cells in response to vascular injury under hyperglycemic conditions is controlled by β3 integrin signaling. Int J Biochem Cell Biol. 2010;42:965–974. doi: 10.1016/j.biocel.2010.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedersen SF, Hoffmann EK, Mills JW. The cytoskeleton and cell volume regulation. Comp Biochem Physiol A Mol Integr Physiol. 2001;130:385–399. doi: 10.1016/s1095-6433(01)00429-9. [DOI] [PubMed] [Google Scholar]
- Qian J, Pang R, Zhu K, Liu D, Li Z, Deng C, et al. Static pressure promotes rat aortic smooth muscle cell proliferation via upregulation of volume-regulated chloride channel. Cell Physiol Biochem. 2009;24:461–470. doi: 10.1159/000257485. [DOI] [PubMed] [Google Scholar]
- Qian Y, Du YH, Tang YB, Lv XF, Liu J, Zhou JG, et al. ClC-3 chloride channel prevents apoptosis induced by hydrogen peroxide in basilar artery smooth muscle cells through mitochondria dependent pathway. Apoptosis. 2011;16:468–477. doi: 10.1007/s10495-011-0584-2. [DOI] [PubMed] [Google Scholar]
- Ren Z, Raucci FJ, Browe DM, Baumgarten CM. Regulation of swelling-activated Cl− current by angiotensin II signalling and NADPH oxidase in rabbit ventricle. Cardiovasc Res. 2008;77:73–80. doi: 10.1093/cvr/cvm031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruddy JM, Jones JA, Stroud RE, Mukherjee R, Spinale FG, Ikonomidis JS. Differential effects of mechanical and biological stimuli on matrix metalloproteinase promoter activation in the thoracic aorta. Circulation. 2009;120:S262–S268. doi: 10.1161/CIRCULATIONAHA.108.843581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato R, Koumi SI. Characterization of the stretch-activated chloride channel in isolated human atrial myocytes. J Membr Biol. 1998;163:67–76. doi: 10.1007/s002329900371. [DOI] [PubMed] [Google Scholar]
- Shi XL, Wang GL, Zhang Z, Liu YJ, Chen JH, Zhou JG, et al. Alteration of volume-regulated chloride movement in rat cerebrovascular smooth muscle cells during hypertension. Hypertension. 2007;49:1371–1377. doi: 10.1161/HYPERTENSIONAHA.106.084657. [DOI] [PubMed] [Google Scholar]
- Sonoyama K, Greenstein A, Price A, Khavandi K, Heagerty T. Review: vascular remodeling: implications for small artery function and target organ damage. Ther Adv Cardiovasc Dis. 2007;1:129–137. doi: 10.1177/1753944707086358. [DOI] [PubMed] [Google Scholar]
- Tang YB, Liu YJ, Zhou JG, Wang GL, Qiu QY, Guan YY. Silence of ClC-3 chloride channel inhibits cell proliferation and the cell cycle via G1/S phase arrest in rat basilar arterial smooth muscle cells. Cell Prolif. 2008;41:775–785. doi: 10.1111/j.1365-2184.2008.00551.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas SM, Brugge JS. Cellular functions regulated by Src family kinases. Annu Rev Cell Dev Biol. 1997;13:513–609. doi: 10.1146/annurev.cellbio.13.1.513. [DOI] [PubMed] [Google Scholar]
- Wang GL, Wang XR, Lin MJ, He H, Lan XJ, Guan YY. Deficiency in ClC-3 chloride channels prevents rat aortic smooth muscle cell proliferation. Circ Res. 2002;91:e28–e32. doi: 10.1161/01.res.0000042062.69653.e4. [DOI] [PubMed] [Google Scholar]
- Wang M, Yang H, Zheng L, Zhang Z, Tang Y, Wang G, et al. Downregulation of TMEM16A calcium-activated chloride channel contributes to cerebrovascular remodeling during hypertension by promoting basilar smooth muscle cell proliferation. Circulation. 2012;125:697–707. doi: 10.1161/CIRCULATIONAHA.111.041806. [DOI] [PubMed] [Google Scholar]
- Wang XG, Tao J, Ma MM, Tang YB, Zhou JG, Guan YY. Tyrosine 284 phosphorylation is required for ClC-3 chloride channel activation in vascular smooth muscle cells. Cardiovasc Res. 2013;98:469–478. doi: 10.1093/cvr/cvt063. [DOI] [PubMed] [Google Scholar]
- Ward MR, Pasterkamp G, Yeung AC, Borst C. Arterial remodeling mechanisms and clinical implications. Circulation. 2000;102:1186–1191. doi: 10.1161/01.cir.102.10.1186. [DOI] [PubMed] [Google Scholar]
- Zeng J, Zhang Y, Mo J, Su Z, Huang R, Kontos HA. Two-Kidney, two Clip renovascular hypertensive rats can be used as stroke-prone rats. Stroke. 1998;29:1708–1714. doi: 10.1161/01.str.29.8.1708. [DOI] [PubMed] [Google Scholar]
- Zheng LY, Li L, Ma MM, Liu Y, Wang GL, Tang YB, et al. Deficiency of volume-regulated ClC-3 chloride channel attenuates cerebrovascular remodeling in DOCA-salt hypertension. Cardiovasc Res. 2013;100:134–142. doi: 10.1093/cvr/cvt156. [DOI] [PubMed] [Google Scholar]
- Zhou JG, Ren JL, Qiu Q, He H, Guan YY. Regulation of intracellular Cl-concentration through volume-regulated ClC-3 chloride channels in A10 vascular smooth muscle cells. J Biol Chem. 2005;280:7301–7308. doi: 10.1074/jbc.M412813200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Integrin β1 and integrin β3 expression in BASMCs. Integrin β3 was detected while integrin β1 was hardly detected in BASMCs. Human umbilical vein endothelial cells (HUVECs) were used as positive control for integrin β1 expression. n = 8.
Figure S2 Blood pressure of hypertensive animals. (a) Blood pressure of 2K2C hypertensive group and sham group at different time point. PW, postoperative week. *P < 0.01 compared with sham in each group; #P < 0.01 compared with 1PW in each group, n = 6. (b) Blood pressure of SHRSP rats and WKY rat at 16 weeks, #P < 0.005, n ≥ 6. (c) Blood pressure of angiotensin II infusion induced hypertensive C57BL/6J mice, n = 5. Values are mean ± SEM.
Figure S3 (a) Integrin β3 (ITGB3) expression was positively correlated with the blood pressure, correlation coefficient was 0.8125 (n = 36 rats from six independent experiments, P < 0.001). (b) Integrin β3 expression was positively correlated with ClC-3 during hypertension (24 rats from three independent experiments, P < 0.005).
Figure S4 Changes in integrin β3 expression of basilar arteries in 2-kidney, 2-clip hypertensive rats and sham group rats (scale bars, 20 μm). Representative sections were shown, n = 6 per group.
Figure S5 The effect of integrin β3 siRNA (ITGB3 siRNA) transfection in BASMCs. BASMCs were transfected with 200 nM integrin β3 siRNA and assayed with reverse transcriptase-PCR (RT-PCR) 48 h later. *P < 0.05 versus negative group, n = 5. Values are mean ± SEM.
Figure S6 Densitometric analysis of integrin β3 expression induced by 1 nM, 10 nM, 100 nM and 1000 nM angiotensin II, the solvent of angiotensin II was dimethyl sulfoxide (DMSO, 0.1%; n = 5; *P < 0.05 vs. control).
Figure S7 Electron microscopy images of cerebral basilar arteries from wt and ClC-3−/− mice with or without angiotensin II implantation. Angiotensin II resulted in smooth muscle cell disarrangement (♦), intercellular space enlargement (→) and vacuolization (Δ) in wt mice; while in the basilar artery of ClC-3−/− mice, these ultrastructural changes did not occur. Black bar represents 1 μm. Six mice were studied in each group.