

Abstract
BACKGROUND AND PURPOSE
Sorafenib, a potent inhibitor that targets several kinases associated with tumourigenesis and cell survival, has been approved for clinical treatment as a single agent. However, combining sorafenib with other agents improves its anti-tumour efficacy in various preclinical tumour models. ABT-263, a second-generation BH3 mimic, binds to the anti-apoptotic family members Bcl-2, Bcl-xL and Bcl-w, and has been demonstrated to enhance TNFSF10 (TRAIL)-induced apoptosis in human hepatocarcinoma cells. Hence, we investigated the effects of ABT-263 treatment combined with sorafenib.
EXPERIMENTAL APPROACH
The effects of ABT-263 combined with sorafenib were investigated in vitro, on cell viability, clone formation and apoptosis, and the mechanism examined using western blot and flow cytometry. This combination was also evaluated in vivo, in a mouse xenograft model; tumour growth, volume and weights were measured and a TUNEL assay performed.
KEY RESULTS
ABT-263 enhanced sorafenib-induced apoptosis while sparing non-tumourigenic cells. Although ABT-263 plus sorafenib significantly stimulated intracellular reactive oxygen species production and subsequent mitochondrial depolarization, this was not sufficient to trigger cell apoptosis. ABT-263 plus sorafenib significantly decreased Akt activity, which was, at least partly, involved in its effect on apoptosis. Bax and p21 (CIP1/WAF1) were shown to play a critical role in ABT-263 plus sorafenib-induced apoptosis. Combining sorafenib with ABT-263 dramatically increased its efficacy in vivo.
CONCLUSION AND IMPLICATIONS
The anti-tumour activity of ABT-263 plus sorafenib may involve the induction of intrinsic cell apoptosis via inhibition of Akt, and reduced Bax and p21 expression. Our findings offer a novel effective therapeutic strategy for tumour treatment.
Keywords: ABT-263, sorafenib, combination therapy, cancer
Introduction
In human malignancies, the aberrant activation of certain kinases commonly occurs, and is associated with tumourigenesis, disease progression, resistance to chemotherapy and poor clinical outcome. The development of anti-cancer treatments targeting such cancer-specific kinase activities represents a promising therapeutic strategy. Sorafenib (Nexavar, BAY 43-9006) is an orally administered, small molecule multi-targeted kinase inhibitor displaying activity against multiple kinases (Liu et al., 2006; Nguyen et al., 2010). Recently, sorafenib has been approved by the U.S. Food and Drug Administration for clinical treatment in patients with advanced renal cell carcinoma and primary hepatocellular carcinoma (Wilhelm et al., 2006). Several clinical trials are currently ongoing against various malignancies, including leukaemia, prostate cancer, lung cancer, breast cancer and colon cancer (Singhal et al., 2010; Wei et al., 2010). The key anti-cancer effects of sorafenib appear to be mediated via the induction of apoptosis in a variety of tumour cells, as well as the inhibition of tumour cell proliferation and suppression of angiogenesis (Rahmani et al., 2005; Panka et al., 2006; Dasmahapatra et al., 2007; Meng et al., 2007; Plastaras et al., 2007; Rosato et al., 2007; Zhang et al., 2008). Previous studies have shown that sorafenib promotes apoptosis in cancer cells by inducing endoplasmic reticulum stress; stimulating the generation of reactive oxygen species (ROS); blocking the Raf/MEK/ERK pathway; and regulating the signal transduction of STAT3, NF-κB, Akt and AIF (Chen et al., 2010; Park et al., 2010; Ramakrishnan et al., 2010; Yang et al., 2010; Liu et al., 2011b). Alterations in the susceptibility of malignant cells to apoptosis and tumour recurrence due to acquired drug resistance, which are the hallmarks of cancer cells, have significantly limited the clinical applications of sorafenib as a targeted therapy.
ABT-263 (Navitoclax), a second-generation BH3 mimic, is a potent and orally bioavailable Bcl-2 family inhibitor (Shoemaker et al., 2008; Tse et al., 2008; Vogler et al., 2010). It binds to the anti-apoptotic family members, Bcl-2, Bcl-xL and Bcl-w, with high affinity, while it displays relatively lower affinity for Mcl-1 and Bcl2A1 (Shi et al., 2011; Tan et al., 2011; Yamaguchi et al., 2011). In preclinical models, ABT-263 has exhibited single-agent activity against a large panel of cancer cell lines by disrupting the interaction between these anti-apoptotic proteins and their pro-apoptotic counterparts, and subsequently evoking rapid apoptotic cell death via the intrinsic cell death cascade. Clinical trials involving lymphoid malignancies and small-cell lung cancer have indicated that ABT-263 is promising for some patients (Wilson et al., 2010; Tan et al., 2011). In addition to its single-agent activity, ABT-263 has also been shown to dramatically synergize with many cytotoxic agents to improve tumour regression and overall response rates. The combination of ABT-263 with rapamycin kills lymphoma cells, while the combined treatment with 2-deoxyglucose and Rituximab have been shown to eliminate other types of cancer cells (Ramanathan and Schreiber, 2009; Yamaguchi et al., 2011). We have also previously reported that ABT-263 sensitizes TNFSF10 (also known as TRAIL; see Alexander et al., 2013a)-resistant hepatocarcinoma cells by down-regulating the Bcl-2 family of anti-apoptotic proteins (Wang et al., 2012).
In this study, we investigated the efficacy and mechanism of the combined treatment of sorafenib and ABT-263 using both in vitro and in vivo models. Our results demonstrated that ABT-263 potently enhances sorafenib-induced apoptosis in human cancer cells. Inhibition of Akt, Bax and p21 (CIP1/WAF1) protein expression was shown to play a critical role in apoptosis induced by the dual-drug combination. These findings provide a novel therapeutic strategy and shed light on the potential anti-cancer mechanism of sorafenib and ABT-263 in both monotherapy and combined treatment.
Methods
Materials and antibodies
ABT-263 and sorafenib were purchased from Active Biochemicals Company (Hong Kong, China). The caspase-3 (#9665), caspase-9 (#9502), PARP (#9542), phospho-Akt (#4060), ERK (#4695), phospho-ERK (#9101), MEK (#9122), phospho-MEK (#9121), Mcl-1 (#5453), Bcl-2 (#2876), Bax (#2772), Bim (#2189), PUMA (#4976) and p21 (#2947) antibodies were purchased from Cell Signaling Technology (Beverly, MA, USA). The Akt (1076-2-AP), Bcl-xL (10783-1-AP) and Bid (10988-1-AP) antibodies were obtained from ProteinTech Group (Chicago, IL, USA). The caspase-8 (AC056), GAPDH (AG019) and horseradish peroxidase-conjugated secondary antibodies against mouse (A0216) and rabbit (A0208) IgG were purchased from Beyotime (Nantong, Jiangsu, China).
Cell lines and cell culture
The human colon cancer cell lines (HCT116, HCT116 Bax-/- and HCT116 p21-/-) were cultured in McCoy's 5A medium. The human hepatoma cell lines (BEL7402, Huh7, HepG2 and FHCC98), the human breast cancer epithelial cell line (MDA-MB-231), the human gastric cancer cell line (AGS), the human lung cancer cell line (A549) and the normal cell lines (L02, HFF and HEK293T) were cultured in DMEM. All cell culture media were supplemented with 10% FBS, penicillin 100 U mL−1 and 100 μg·mL−1 streptomycin at 37°C in a 5% CO2 incubator.
Plasmids and transient transfection
The constitutively active Akt plasmid (pUSE-CA-Akt), active MEK plasmid (pUSE-CA-MEK) and the empty vector (pUSE) were purchased from Upstate (Lake Placid, NY, USA). Cells were seeded in 24-well plates overnight and transfected for 36 h using FuGENE HD transfection reagent following the manufacturer's instructions (Roche, Indianapolis, IN, USA).
Cell viability and apoptosis assays
ABT-263 and sorafenib were dissolved in DMSO. Cell viability was determined using the trypan blue dye exclusion assay according to established protocols. For the apoptosis assays, the cells were harvested and washed with PBS and then fixed with 95% alcohol for 1 h in the dark at 4°C. The fixed cells were collected and washed twice with PBS, and then dyed with 3 μL 10 mg·mL−1 propidium iodide (PI) and 10 μL 1 mg·mL−1 RNase. The prepared cells were evaluated using the sub-G1 assay on a flow cytometer.
Clone formation assays
The cells were plated in six-well plates at 2000 cells per well. Twenty-four hours later, the drugs were added to the plates. After treatment, as indicated in the figure legends, fresh medium was applied to the plates. The cells were allowed to grow for 10 additional days before staining with crystal violet (Sigma, St Louis, MO, USA). All experiments were repeated at least three times, and similar results were obtained in each trial.
Cell extracts and Western blot analysis
After various treatments, both floating and adherent cells were harvested using trypsin and subsequently washed with cold PBS. The cells were then lysed with 1% SDS. The lysed samples were immediately heated to 100°C for 20 min and then centrifuged at 12 000× g for 15 min. The supernatant was collected, and the protein concentrations were determined using the Bicinchoninic Acid Protein Assay Kit (Pierce Rockford, IL, USA). Western blotting was then performed as previously described (Gong et al., 2012).
Assessment of ROS
The ROS levels were measured using a multi-mode microplate reader (Spectram Max M5, Molecular Devices, Sunnyvale, CA, USA) and a flow cytometer. Briefly, the BEL7402 cells were harvested, washed twice with PBS, resuspended in tubes with serum-free DMEM medium containing 10 nmol 5-(and-6)-carboxy-2′, 7′-dichlorodihydrofluorescein diacetate (Carboxy-H2DCFDA; Invitrogen, Carlsbad, CA, USA), and incubated at 37°C for 20 min. The cells were then washed twice with PBS and analysed via flow cytometry. The data were processed using the FCS Express V3 programme (DeNovo, Los Angeles, CA, USA). For the detection of Δψ, the cell pretreatments were performed as described for the ROS detection protocol, A 5 μL solution of 0.1 mg·mL−1 Rho123 (Sigma) (fluorescent probe) was added and the cells were incubated for 20 min at 37°C. The fluorescence was measured via flow cytometry (Gong et al., 2012).
Establishment of HCT116 p21-/- and MDA-MB-231 cells stably overexpressing p21
The p21 overexpression plasmids were transfected into the HCT116 and MDA-MB-231 cells according to the manufacturer's instructions on the lentivirus-based stable transfection system. The transfected cells were selected in medium containing 5 mg·mL−1 puromycin for 14 days, and the p21 expression levels were determined via immunoblotting analysis as described previously.
Tumour xenograft
Five-week-old male BALB/c nude mice were purchased from the Model Animal Research Center, MARC (Nanjing, Jiangsu, China). Animals were handled according to the Guidelines of the China Animal Welfare Legislation, as provided by the Committee on Ethics in the Care and Use of Laboratory Animals of Wuhan University. The experimental protocols were approved by the Experimental Animal Centre of Wuhan University.
The mice were housed in polycarbonate cages (5 per cage), provided with food and water ad libitum and maintained on a 12–12 h light-dark cycle at 22 ± 2°C and 55 ± 20% relative humidity. BALB/c mice were randomly assigned into three groups. Each mouse from group one was inoculated s.c. into the right oxter with 3 × 107 HCT116 cells in 0.2 mL PBS. Five days later, the mice bearing HCT116 tumours were randomized into four subgroups (n = 6), and each subgroup received one of the following daily oral treatments with 0.1% sodium carboxyl methylcellulose vehicle every other day for 20 days: ABT-263 (100 mg·kg−1), sorafenib (25 mg·kg−1), and ABT-263 (100 mg·kg−1) plus sorafenib (25 mg·kg−1). The second and third groups were inoculated s.c. with HCT116 p21-/- or HCT116 Bax-/- cells and the treatment just as group one. The tumour volumes and body weights were measured and recorded daily. The tumour volumes were calculated as length × width2 × ∏/6. All studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (Kilkenny et al., 2010; McGrath et al., 2010).
Statistical analysis
All the experiments were performed at least three times. All data are expressed as mean ± SD. Student's t test was used for all the statistical analyses, and the differences were considered significant if the P-value was less than 0.05.
Results
ABT-263 and sorafenib induced caspase-dependent apoptosis in cancer cells
To examine whether a cooperative effect exists between ABT-263 and sorafenib in tumour chemotherapy, we treated cancer cells with a series of concentrations of ABT-263 and sorafenib either alone or in combination. According to the results, a certain concentration of ABT-263 and sorafenib administered as single agent treatments failed to induce cell death, while synergistic anti-tumour activity was observed with a treatment of 0.4 μM ABT-263 in combination with 6 μM sorafenib in the human cancer cell lines Huh7, HepG2, BEL7402, HCT116, DLD1 and AGS (Figure 1A, Supporting Information Figs. S1a,b, S2). In contrast, the immortalized but non-malignant cell lines, HEK293T, L02 and HFF, displayed resistance to this combined treatment (Figure 1B). Furthermore, the analysis of long-term cell survival via colony formation assay revealed that the combination treatment of ABT-263 and sorafenib dramatically affected colony formation, while the effect on L02 cells was not significant (Figure 1C). These data suggest that the combination treatment of ABT-263 and sorafenib displayed a significant synergistic therapeutic effect on cancerous cells but not normal cells.
Figure 1.

The combination treatment of ABT-263 and sorafenib induces caspase-dependent apoptosis. The data represent the average of at least three independent experiments ± SD. **P < 0.01. (A) Huh7, HepG2, BEL7402, DLD1 and AGS cell lines were treated with 0.4 μM ABT-263 and 6 μM sorafenib either alone or in combination for 48 h, and the HCT116 cells were treated for 24 h, which were then assessed for cell viability using a trypan blue exclusion assay. (B) HEK293T, HFF and L02 cells were treated with 0.4 μM ABT-263 and 6 μM sorafenib either alone or in combination for 48 h. Cell viability was determined. (C) The cells were seeded in six-well plates and treated with ABT-263 and sorafenib, the HCT116 cells were treated for 24 h, and the BEL7402 and L02 cells were treated for 48 h. The attached cells were stained with crystal violet 10 days later. (D) FACS analysis of apoptosis following the treatment of ABT-263 (0.4 μM) and sorafenib (6 μM) either alone or in combination on the BEL7402, Huh7 and HepG2 cells for 48 h and the HCT116 cells for 24 h. The cells were dyed with 3 μL 10 mg·mL−1 PI and 10 μL 1 mg·mL−1 RNase, and the percentage of cells in sub-G1 determined. (E) The Huh7, BEL7402 and HCT116 cells were pretreated either with or without 50 μM z-VAD-fmk for 6 h, and then the trypan blue exclusion assay was performed to evaluate the cell viability after treatment with ABT-263 (0.4 μM) and sorafenib (6 μM) either alone or in combination. The percentage of cells dyed without trypan blue was determined.
To determine whether the reduction in cell survival induced by ABT-263 and sorafenib was associated with cancer cell apoptosis, Huh7, HepG2, BEL7402 and HCT116 cells were treated with ABT-263, sorafenib and a combination of the two drugs, and subsequently analysed via flow cytometry with PI. As shown in Figure 1D, Supporting Information Figs. S1c, S3, the combined treatment obviously leads to apoptosis in cancer cells. To evaluate the function of caspases in cell apoptosis, the cancer cells were pretreated with the pan-caspase inhibitor, z-VAD-fmk, for 1 h prior to the combined treatment. As shown in Figure 1E and Supporting Information Fig. S1d, the z-VAD-fmk pretreatment significantly prevented cell death and blocked both PARP cleavage and caspase activation in the Huh7, BEL7402 and HCT116 cells. Therefore, these data suggest that ABT-263 treatment combined with sorafenib induces caspase-dependent apoptosis in cancer cells.
ROS generation and mitochondrial depolarization are involved but not required in combination-induced apoptosis
Many studies showed that some chemotherapeutic agents activated intracellular ROS accumulation and subsequently induced apoptosis in certain types of cancer cells (Loor et al., 2010; Gong and Li, 2011). The excessive production of ROS often leads to mitochondrial depolarization. After that, the loss of mitochondrial membrane potential (ΔΨm) increases the permeability of the outer membrane, triggering intrinsic cell death (Bayir and Kagan, 2008; Vitiello et al., 2009; Pasdois et al., 2011). Therefore, we next investigated whether the combination-induced cell apoptosis was associated with ROS accumulation and mitochondrial depolarization. ROS accumulation was observed after cells were treated with ABT-263 and sorafenib either alone or in combination via flow cytometry using a specific ROS-detecting fluorescent dye, H2DCFDA (Figure 2A). To determine the role of ROS in combination treatment, ROS generation was abrogated by pretreating cancer cells with an ROS scavenger, N-acetyl-L-cysteine (NAC), 1 h prior to combination treatment (Supporting Information Fig. S4a). However, NAC does not rescue apoptosis (Figure 2B and Supporting Information Fig. S4b), and so did Tiron, another type of ROS scavenger (Figure 2C). In contrast, NAC and Tiron significantly enhanced cell apoptosis induced by ABT-263 alone or combined with sorafenib (Figure 2B, C, and Supporting Information Fig. S4b).
Figure 2.

Reactive oxygen species generation (ROS) and mitochondrial depolarization are involved but not required in ABT-263 and sorafenib combination-induced apoptosis. (A) BEL7402 cells were treated with ABT-263 and sorafenib either alone or in combination for 48 h and the intracellular ROS levels assessed. (B) BEL7402 cell viability was determined using the trypan blue exclusion assay in either the presence or absence of 15 mM NAC for 48 h. (C) BEL7402 cell viability was measured by trypan blue exclusion assay in either the presence or absence of 15 mM Tiron for 48 h. (D) To evaluate the time course of the mitochondrial membrane potential, BEL7402 cells were treated with ABT-263 and sorafenib either alone or in combination for 48 h followed by DCF fluorescence analysis. (E) BEL7402 cell viability was determined using the trypan blue exclusion assay in either the presence or absence of 2 μM CsA for 48 h.
On the other hand, ABT-263 treatment combined with sorafenib also dramatically decreased the Δψm of the mitochondrial membrane (Figure 2D). Cell pre-incubation with cyclosporine A (CsA), a mitochondrial membrane potential stabilizer, could prevent the loss of Δψm (Supporting Information Fig. S4c). However, CsA failed to rescue apoptotic cell death which was induced by the combination treatment (Figure 2E and Supporting Information Fig. S4d).
Therefore, these results above suggested that ROS generation and mitochondrial depolarization were accompanied by the ABT-263 and sorafenib combination treatment, but their functional role was not determined.
ABT-263 and sorafenib combination treatment-induced cell apoptosis involves Akt activation
The Ras/Raf/MEK/ERK cascade induces ERK translocation to the nucleus, the activation of a subset of transcription factors, and ultimately the regulation of cellular viability (Casar et al., 2009; Poulikakos et al., 2010). Sorafenib, which induces apoptosis as a single agent in a variety of tumour cells, is a potent inhibitor of the Ras/Raf/MEK/ERK pathway (Wilhelm et al., 2004; Fecteau et al., 2012). Therefore, we aimed to determine the function of ERK in combination treatment-induced cell apoptosis. As expected, sorafenib significantly down-regulated the phospho-ERK activation levels, thereby indicating the repression of ERK activity (Figure 3A). However, increasing ERK activity via the ectopic expression of MEK failed to block cell death induced by the ABT-263 and sorafenib combination treatment, thereby we guess ERK activity probably is not involved in combination treatment-induced cell apoptosis (Figure 3B).
Figure 3.
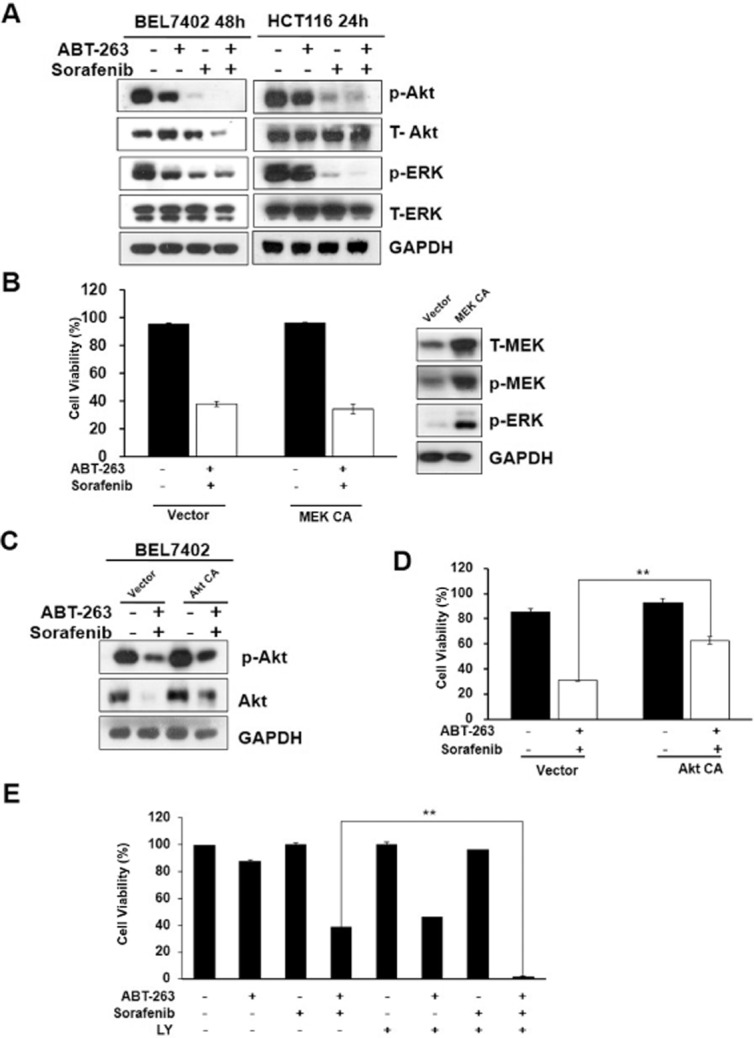
ABT-263 and sorafenib combination-induced cell apoptosis correlates with Akt activity. (A) BEL7402 and HCT116 cells were treated with ABT-263 and sorafenib either alone or in combination, and then analysed via Western blot to examine the protein activation levels of p-Akt, T-Akt, p-ERK and T-ERK. (B) HCT116 cells transfected with either vehicle or constitutively active MEK (MEK CA) were incubated with a combination of ABT-263 (0.2 μM) and sorafenib (6 μM), and the cells were then collected and analysed by use of the trypan blue exclusion assay. (C) BEL7402 cells transfected with either constitutively active Akt (CA-Akt) or vehicle were incubated with the combination of 0.4 μM ABT-263 and 6 μM sorafenib for 48 h, and protein lysates were then prepared from the cells and analysed by Western blot. (D) Cell viability was determined following the treatment of ABT-263 and sorafenib in the cells transfected expressing either CA-Akt or the vehicle plasmid. (E) After pretreatment with 25 μM LY294002 for 1 h, the BEL7402 cells were treated with ABT-263 and sorafenib for 24 h, and cell viability was then determined. **P < 0.01.
Akt is also a critical kinase that regulates a variety of biological processes, including cell survival, proliferation and differentiation (Bai et al., 2009). Many studies have shown that some clinical chemotherapeutic agents induce cancer cell apoptosis by regulating Akt kinase activity (Liu et al., 2011a). To examine the effects of Akt in combination treatment-induced cell apoptosis, the expression levels of phospho-Akt were examined to evaluate Akt activity via Western blot analysis. As shown in Figure 3A, ABT-263 combined with sorafenib significantly impeded Akt activity. However, expression of constitutively active Akt (Figure 3C) partially attenuated ABT-263 and sorafenib combination treatment-induced cell apoptosis (Figure 3D). In contrast, BEL7402 cells pretreated with LY294002, a PI3K inhibitor, prior to ABT-263 and sorafenib treatment displayed a synergistic chemotherapy effect, which further confirmed that Akt activity is at least partly involved in combination treatment-induced cell apoptosis (Figure 3E). Moreover, Western blot analysis of Akt level found that in drug-sensitive cell lines BEL7402, z-VAD failed to rescue the level of Akt activity at early time stage (20 h and 40 h), only at later time stage z-Vad can rescue the level of Akt activity. However, in drug-resistant cell lines p21-/- HCT116, z-VAD have no effects on the level of Akt activity (Supporting Information Fig. S5), which suggested that the change of Akt protein level is not due to caspase-mediated down-regulation. Therefore, these results suggest that cell apoptosis induced by the ABT-263 and sorafenib combination treatment is dependent on Akt but not ERK activity.
Expression of Bax and p21 plays a critical role in ABT-263 and sorafenib combination treatment-induced cell apoptosis
Bax is one of the vital pro-apoptotic Bcl-2-family proteins, which play a role in cell death regulation and are capable of regulating diverse cell death mechanisms, including apoptosis, necrosis and autophagy (Gautier et al., 2011). Bax deficiency causes blocking of cell death signalling and changes in chemosensitivity to drug-induced apoptosis in many cell types (Blatt et al., 2009). p21 (CIP1/WAF1), a potent cyclin-dependent kinase inhibitor, inhibits the activity of the cyclin-CDK complexes, and thus has functions as a regulator of cell cycle progression (Ding et al., 2008; Hoeferlin et al., 2011). Moreover, p21 has been identified as a senescent cell-derived inhibitor by inhibiting apoptosis during stress responses (Bhattacharya et al., 2007; Li et al., 2012). Therefore, we next investigated whether Bax and p21 expression are associated with the sensitivity to ABT-263 and sorafenib combination-induced apoptosis in cancer cells. We treated the wild type, Bax-/- and p21-/- HCT116 cell lines with ABT-263 and sorafenib for 24 h. The analysis of cell viability showed that the Bax-/- and p21-/- HCT116 cell lines exhibited significant resistance to the combination treatment (Figure 4A, C). The analysis of cell apoptosis and Western blot detection of PARP and caspase expression also indicated that the ABT-263 and sorafenib combination treatment did not induce apoptosis (Figure 4B, D, E), which is consistent with the cell viability results.
Figure 4.

Effect of Bax or p21 knockout on ABT-263 and sorafenib combination-induced apoptosis. (A) Cell viability was assessed in the HCT116 Bax-/- and HCT116 WT cells after the combination treatment of 0.2 μM ABT-263 and 6 μM sorafenib for 24 h. (B) Cell apoptosis was determined via FACS analysis by PI staining, and counting the percentage of cells in the sub-G1 phase. (C) Cell viability was determined with HCT116 p21-/- and HCT116 WT cells after the combination treatment of 0.2 μM ABT-263 and 6 μM sorafenib for 24 h. (D) Cell apoptosis was assessed via FACS. (E) Analysis of PARP, pro-caspase-3, caspase-8, caspsse-9 via Western blot. **P < 0.01.
To confirm these observations, Bax and p21 were overexpressed in the Bax-/- and p21-/- HCT116 cells, respectively, and the cells were then treated with both ABT-263 and sorafenib for 24 h. According to the results, the ectopic expression of Bax or p21 partially reversed the resistance of the knockout cells to the combination treatment (Figure 5A, B). To examine whether the relative expression levels of Bax and p21 determined the cell sensitivity to combination treatment, we analysed the Bax and p21 expression levels in various cells, including both sensitive and resistant cell lines. As shown in Figure 5C, the U251, A549, Hela and MDA-MB-231 cells were resistant to ABT-263 and sorafenib combination-induced apoptosis compared with the sensitive cells (Figure 1A). Western blot analysis indicated that the resistant cell lines U251, MDA-MB-231 and Hela expressed low levels of p21 compared with the sensitive cell lines (Figure 5D). We noted that the p21 protein levels varied greatly in the sensitive and resistant cells lines. To further verify this hypothesis, we ectopically expressed p21 in MDA-MB-231 cells, which express low levels of p21 and are resistant to the combination treatment, and subsequently treated the cells with both ABT-263 and sorafenib for 24 h. Consistent with the results observed for the HCT116 p21-/- cells, the overexpression of p21 enhanced the effects of combination treatment-induced cell apoptosis in the MDA-MB-231 cells (Figure 5E, F). Similar to the result of p21 knockout, knockdown p21 with p21-siRNA inhibit apoptosis of combo-treatment (Supporting Information Fig. S6a, b). Altogether, these findings strongly suggest that expression of Bax and p21 plays a key role in ABT-263 and sorafenib combination treatment-induced cell apoptosis.
Figure 5.

Bax and p21 expression play a critical role in ABT-263 and sorafenib combination treatment-induced cell apoptosis. (A) HCT116 Bax-/- cells transfected with pUSE-GFP/pUSE-GFP-Bax for 48 h were incubated with ABT-263 and sorafenib for 24 h. The protein lysates prepared from the cells were then analysed by Western blot, and cell viability was determined. (B) HCT116 p21-/- cells transfected with pHAGE/pHAGE-p21 using the lentivirus-based stable transfection system were incubated with ABT-263 and sorafenib for 24 h. The protein lysates were analysed by Western blot, and cell viability was determined. (C) The U251, A549, Hela and MDA-MB-231 cell lines were treated with 0.4 μM ABT-263 and 6 μM sorafenib either alone or in combination for 48 h, followed by assessment for cell viability using the trypan blue exclusion assay. (D) The expression levels of p21 and Bax in ABT-263 and sorafenib combination treated-sensitive cells (AGS, HCT116WT, Huh7 and BEL7402) compared with the resistant cells (MDA-MB-231, Hela and HCT116 p21-/-) were determined by Western blot. (E) MDA-MB-231 cells transfected with pHAGE/pHAGE-p21 using the lentivirus-based stable transfection system were incubated with 0.4 μM ABT-263 and 6 μM sorafenib for 48 h, and cell viability was then determined. (F) MDA-MB-231 cell protein lysates were analysed by Western blot using antibodies targeting PARP, caspase-9 and GAPDH. **P < 0.01.
The ABT-263 and sorafenib combined treatment exhibited synergistic anti-tumour activity in an in vivo xenograft model
To evaluate the synergistic anti-tumour effects and the safety of the ABT-263 and sorafenib combination treatment in vivo, we established HCT116 WT subcutaneous tumour xenograft models with athymic nude mice. Five days later, 24 mice were randomly divided into four groups and were intragastrically administered vehicle, ABT-263 (100 mg·kg−1 body weight), sorafenib (25 mg·kg−1 body weight) or the combination treatment (ABT-263 100 mg·kg−1 body weight and sorafenib 25 mg·kg−1 body weight) every other day for 20 days. Although ABT-263 and sorafenib administered alone inhibited tumour growth to a certain extent, the combination of ABT-263 and sorafenib was more effective than monotherapy at inhibiting tumour growth (Figure 6A). Consistent with the tumour volume results, the combination treatment also led to a lesser increase in tumour weight (Figure 6B). Notably, the combined treatment of ABT-263 and sorafenib at the dose treatment was well tolerated by all mice, as the animals did not display additional weight loss or other signs of acute or delayed toxicity compared with the monotherapy groups (Supporting Information Fig. S7). The TUNEL assay showed that the combined treatment induced a significant increase in the number of apoptotic cells in the tumour tissue (Figure 6C). Next, to examine the effect of Bax or p21 knockout on sensitivity to combination treatment in vivo, nude mice bearing either HCT116 Bax-/- or HCT116 p21-/- cell xenograft tumours were treated with either the vehicle or the combination treatment via gavage (ABT-263 100 mg·kg−1 body weight and sorafenib 25 mg·kg−1 body weight). We found that the HCT116 Bax-/- or HCT116 p21-/- cells xenograft tumours showed resistance to the combination treatment, which is consistent with the in vitro results (Figure 6D, E). Therefore, these data suggest that ABT-263 and sorafenib display significant synergistic anti-tumour activity by inducing cell apoptosis. Moreover, Bax and p21 protein expression plays a key role in the combination treatment in vivo.
Figure 6.
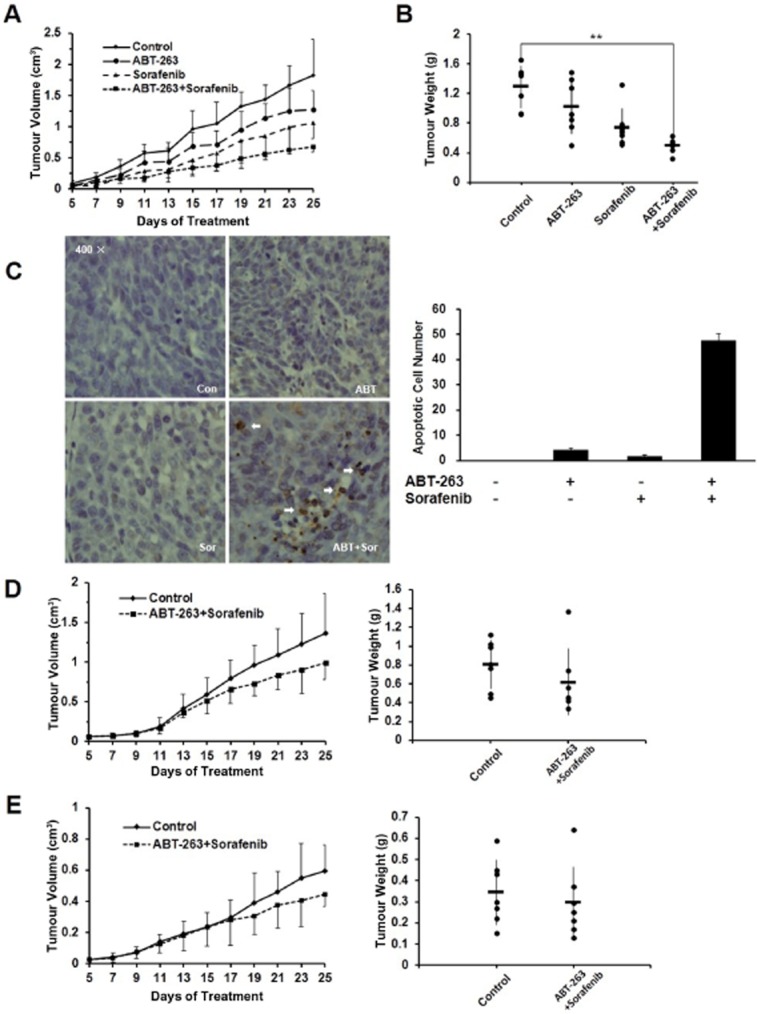
The combined treatment of ABT-263 and sorafenib demonstrated synergistic anti-tumour activity in an in vivo xenograft model. (A) HCT116 cells were inoculated into BALB/c mice (via s.c. injection) to establish a tumour model as indicated in the Materials and Methods section. Mice bearing tumours were randomly assigned into groups (six mice per group) and treated with vehicle, ABT-263 (100 mg·kg−1), sorafenib (25 mg·kg−1) either alone or as a combination of ABT-263 and sorafenib (ABT-263 100 mg·kg−1 and sorafenib 25 mg·kg−1) every other day. The tumour volume was then measured (the bars represented the means ± SD). (B) The tumours were dissected out and weighed after 20 days of treatment. (C) HCT116 transplanted tumours were dissected and analysed via TUNEL assay to examine the levels of apoptosis. The number of TUNEL-positive cells in five random (400×) fields was counted. (D) BALB/c mice (n = 6) were transplanted with HCT116 p21-/-cells and treated as above and then monitored for tumour development. (E) BALB/c mice (n = 7) were transplanted with HCT116 Bax-/-cells and treated as above and then monitored for tumour development. **P < 0.01.
Discussion
Sorafenib, originally developed as a specific Ras/Raf/MEK/ERK inhibitor, is a multi-kinase inhibitor with activity against the Ser/Thr kinase that is required to induce apoptosis and inhibit proliferation in a variety of tumour cells. Because the dose used in vivo is high, drug tolerance and toxic side effects are unavoidable. Due to its oral bioavailability, sorafenib displays a better clinical prospect in combination with other anti-tumour agents. ABT-263 is a second-generation BH3 mimic, which binds to Bcl-2, Bcl-xL and Bcl-w with high affinity and promotes cancer cell apoptosis with better physiochemical and pharmaceutical characteristics than the corresponding precursor, ABT-737 (Tse et al., 2008). In the present study, we demonstrated that ABT-263 enhances the anti-tumour effects of sorafenib in human cancer cells. The combination treatment both reduces the dosage requirement, and most importantly greatly improves the treatment efficacy compared with monotherapy. Mechanistically, the synergistic anti-tumour activity associated with the induction of apoptosis by ABT-263 and sorafenib primarily involved the intrinsic apoptotic pathway, as the observed cell death was manifested by intracellular ROS, mitochondrial depolarization, and the loss of mitochondrial membrane potential and the activation of caspase-9. Moreover, Akt activity and the expression of Bax and p21 play a critical role in ABT-263 and sorafenib-induced cancer cell apoptosis.
Activation of the apoptosis pathway is the most common mechanism for targeted chemotherapies that induce either cancer cell death or sensitivity to established cytotoxic agents or radiation therapy, which function via the activation of the intrinsic cell death pathway. ROS has been identified as a vital mediator of cell death and senescence in response to various stimuli, such as starvation, ionizing radiation and chemotherapeutic agents. Recently, we also demonstrated that a number of chemical anti-tumour agents induce apoptosis and autophagy in cancer cells primarily via the activation of intracellular ROS. In the present study, we observed that significant stimulation of ROS generation and mitochondrial depolarization were associated with the ABT-263 and sorafenib combination treatment, but their functional role was not determined, as both the ROS scavengers and the mitochondrial membrane potential stabilizer failed to protect against the drug combination-induced apoptosis. Therefore, ROS generation and mitochondrial depolarization were accompanied by the ABT-263 and sorafenib combination treatment, but their functional role was not determined. We speculated that intracellular ROS generation and the loss of ΔΨm are the result of cell death rather than the induction of apoptosis.
The Bcl-2 family, which includes the anti-apoptotic subfamily (Bcl-2, Bcl-xL, Bcl-w, Mcl-1 and A1), the pro-apoptotic subfamily (Bax, Bak and Bok) and the BH3-only proteins (Bik, Bad, Bid, Bim, Bmf, Hrk, Noxa and Puma), plays a central role in the regulation of cell death during chemotherapy (Adams and Cory, 2007; Youle and Strasser, 2008). Anti-apoptotic proteins protect cells from various cytotoxic conditions, while pro-apoptotic proteins promote cell apoptosis. BH3-only proteins, which have a conserved BH3 domain, selectively bind and regulate anti-apoptotic subfamily members to promote cell death (Vogler et al., 2009). However, the Bax-/- HCT116 cell line was highly resistant to the combination treatment. These observations demonstrate that the pro-apoptotic proteins, Bax and Bak, are crucial for inducing mitochondrial outer membrane permeabilization and the subsequent release of caspase-activating proteins and other cell death mediators, such as cytochrome c and SMAC. We speculate that this effect might explain why Bax-/- HCT116 cells displayed a strong resistance to the combination treatment. Additionally, the p21-/- HCT116 cells, p21 knockdown HCT116 cells and p21 knockdown Bel 7402 cells were also found to be resistant to the combination treatment of ABT-263 and sorafenib, for which the mechanism is unclear and will require further investigation.
Although sorafenib significantly repressed ERK activity via the inhibition of the Ras/Raf/MEK/ERK pathway, the up-regulation of ERK activity via the ectopic expression of MEK failed to prevent cell death (Figure 3B). However, the overexpression of Akt partially blocked cell death induced by ABT-263 and sorafenib (Figure 3C–E). Therefore, we believe that Akt activity and not ERK at least partly plays a role in the combination treatment. Akt is a crucial kinase of the cell signal network centre that significantly regulates gene expression and protein activity. In this study, Bax and p21 expression appeared to be closely associated with the level of cell sensitivity to combination treatment. We aimed to understand whether there was a relationship between Akt and either Bax or p21 expression. According to the experimental results, Bax and p21 protein expression did not correlate with Akt activity (data not shown). Therefore, Akt and Bax or p21 are involved in independent regulatory pathways in combination treatment-induced cell apoptosis.
In summary, we have presented a synergistic anti-tumour treatment, and our data suggest that the combination treatment of ABT-263 and sorafenib is safe and highly effective at both inducing cancer cell apoptosis in vitro and inhibiting tumour growth and progression in vivo. The potential molecular mechanisms involve Akt activity and Bax and p21 expression. Our findings provide the rationale for including ABT-263 and sorafenib in the therapeutic regimens for tumour patients, which may enhance the current clinical treatments involving sorafenib alone.
Acknowledgments
This work was supported by the National Basic Research Program of China (2014CB910600), the National Nature Science Foundation of China (81273540, 81072151), Distinguished Youth Foundation of Hubei Province of China (2012FFA019), and the Major Scientific and Technological Special Project for the ‘Significant Creation of New Drugs’ (2011ZX09102-001-32, 2011ZX09401-302-4). We thank Professor Bert Vogelstein (Howard Hughes Medical Institute) for the gift of these knockout cells line and Dr. Xiaodong Zhang (Wuhan University, China) for preparing these cells. We thank Dr. Shaoyong Chen (Harvard Medical School, USA) for critical reading and suggestions of this article.
Glossary
- ΔΨm
mitochondrial membrane potential
- CsA
cyclosporine A
- NAC
N-acetyl-L-cysteine
- PI
propidium iodide
- ROS
reactive oxygen species
Conflict of interest
Authors declare that they have not any conflict of interest.
Supporting information
Additional Supporting Information may be found in the online version of this article at the publisher's web-site:
http://dx.doi.org/10.1111/bph.12659
Figure S1 a,b. Cell viability for the combination of sorafenib and ABT-263 in various concentrations. c,d. Western blot to examine the protein of PARP and caspases.
{kind=link}
Figure S2 Cell viability of single drug effect.
{kind=link}
Figure S3 Primary sub-G1 histograms.
{kind=link}
Figure S4 a,c. Mitochondrail membrane potential for the combination of sorafenib and ABT-263 in presence of NAC or CsA. b,d. Western blot to examine the protein of PARP and caspases in presence of NAC or CsA.
{kind=link}
Figure S5 a. Western blot to detect the level of Akt activity in BEL7402 and HCT116 p21-/- cells treated with DMSO or z-VAD (50 μM) before combination treatment (ABT-263 0.4 μM, sorafenib 6 μM). b, Cell viability of BEL7402 and HCT116 p21-/- cells treated with DMSO of z-Vad (50 μM) before combination treatment (ABT-263 0.4 μM, sorafenib 6 μM).
{kind=link}
Figure S6 a. Western blot to detect the protein of PARP when cells transfected with p21 siRNA. b. Cell viability of cells transfected with p21 siRNA.
{kind=link}
Figure S7 a,b,c. The body weights of nude mice bearing established HVT116 tumour xenografts.
{kind=link}
References
- Adams JM, Cory S. The Bcl-2 apoptotic switch in cancer development and therapy. Oncogene. 2007;26:1324–1337. doi: 10.1038/sj.onc.1210220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, Peters JA, Harmar AJ, CGTP Collaborators The Concise Guide to PHARMACOLOGY 2013/14: Catalytic receptors. Br J Pharmacol. 2013a;170:1676–1705. doi: 10.1111/bph.12449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai D, Ueno L, Vogt PK. Akt-mediated regulation of NFkappaB and the essentialness of NFkappaB for the oncogenicity of PI3K and Akt. Int J Cancer. 2009;125:2863–2870. doi: 10.1002/ijc.24748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayir H, Kagan VE. Bench-to-bedside review: mitochondrial injury, oxidative stress and apoptosis – there is nothing more practical than a good theory. Crit Care. 2008;12:206. doi: 10.1186/cc6779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharya S, Guo H, Ray RM, Johnson LR. Basic helix-loop-helix protein E47-mediated p21Waf1/Cip1 gene expression regulates apoptosis of intestinal epithelial cells. Biochem J. 2007;407:243–254. doi: 10.1042/BJ20070293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blatt NB, Boitano AE, Lyssiotis CA, Opipari AW, Jr, Glick GD. Bz-423 superoxide signals B cell apoptosis via Mcl-1, Bak, and Bax. Biochem Pharmacol. 2009;78:966–973. doi: 10.1016/j.bcp.2009.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casar B, Arozarena I, Sanz-Moreno V, Pinto A, Agudo-Ibanez L, Marais R, et al. Ras subcellular localization defines extracellular signal-regulated kinase 1 and 2 substrate specificity through distinct utilization of scaffold proteins. Mol Cell Biol. 2009;29:1338–1353. doi: 10.1128/MCB.01359-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen KF, Tai WT, Liu TH, Huang HP, Lin YC, Shiau CW, et al. Sorafenib overcomes TRAIL resistance of hepatocellular carcinoma cells through the inhibition of STAT3. Clin Cancer Res. 2010;16:5189–5199. doi: 10.1158/1078-0432.CCR-09-3389. [DOI] [PubMed] [Google Scholar]
- Dasmahapatra G, Yerram N, Dai Y, Dent P, Grant S. Synergistic interactions between vorinostat and sorafenib in chronic myelogenous leukemia cells involve Mcl-1 and p21CIP1 down-regulation. Clin Cancer Res. 2007;13:4280–4290. doi: 10.1158/1078-0432.CCR-07-0835. [DOI] [PubMed] [Google Scholar]
- Ding H, Han C, Guo D, Wang D, Duan W, Chen CS, et al. Sensitivity to the non-COX inhibiting celecoxib derivative, OSU03012, is p21(WAF1/CIP1) dependent. Int J Cancer. 2008;123:2931–2938. doi: 10.1002/ijc.23895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fecteau JF, Bharati IS, O'Hayre M, Handel TM, Kipps TJ, Messmer D. Sorafenib-induced apoptosis of chronic lymphocytic leukemia cells is associated with downregulation of RAF and myeloid cell leukemia sequence 1 (Mcl-1) Mol Med. 2012;18:19–28. doi: 10.2119/molmed.2011.00164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautier F, Guillemin Y, Cartron PF, Gallenne T, Cauquil N, Le Diguarher T, et al. Bax activation by engagement with, then release from, the BH3 binding site of Bcl-xL. Mol Cell Biol. 2011;31:832–844. doi: 10.1128/MCB.00161-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong K, Li W. Shikonin, a Chinese plant-derived naphthoquinone, induces apoptosis in hepatocellular carcinoma cells through reactive oxygen species: a potential new treatment for hepatocellular carcinoma. Free Radic Biol Med. 2011;51:2259–2271. doi: 10.1016/j.freeradbiomed.2011.09.018. [DOI] [PubMed] [Google Scholar]
- Gong K, Chen C, Zhan Y, Chen Y, Huang Z, Li W. Autophagy-related gene 7 (ATG7) and reactive oxygen species/extracellular signal-regulated kinase regulate tetrandrine-induced autophagy in human hepatocellular carcinoma. J Biol Chem. 2012;287:35576–35588. doi: 10.1074/jbc.M112.370585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoeferlin LA, Oleinik NV, Krupenko NI, Krupenko SA. Activation of p21-dependent G1/G2 Arrest in the absence of DNA damage as an antiapoptotic response to metabolic stress. Genes Cancer. 2011;2:889–899. doi: 10.1177/1947601911432495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Wang J, Ye Z, Li JC. Oridonin up-regulates expression of P21 and induces autophagy and apoptosis in human prostate cancer cells. Int J Biol Sci. 2012;8:901–912. doi: 10.7150/ijbs.4554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C, Gong K, Mao X, Li W. Tetrandrine induces apoptosis by activating reactive oxygen species and repressing Akt activity in human hepatocellular carcinoma. Int J Cancer. 2011a;129:1519–1531. doi: 10.1002/ijc.25817. [DOI] [PubMed] [Google Scholar]
- Liu L, Cao Y, Chen C, Zhang X, McNabola A, Wilkie D, et al. Sorafenib blocks the RAF/MEK/ERK pathway, inhibits tumor angiogenesis, and induces tumor cell apoptosis in hepatocellular carcinoma model PLC/PRF/5. Cancer Res. 2006;66:11851–11858. doi: 10.1158/0008-5472.CAN-06-1377. [DOI] [PubMed] [Google Scholar]
- Liu Q, Mier JW, Panka DJ. Differential modulatory effects of GSK-3beta and HDM2 on sorafenib-induced AIF nuclear translocation (programmed necrosis) in melanoma. Mol Cancer. 2011b;10:115. doi: 10.1186/1476-4598-10-115. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG. Animal research: Reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loor G, Kondapalli J, Schriewer JM, Chandel NS, Vanden Hoek TL, Schumacker PT. Menadione triggers cell death through ROS-dependent mechanisms involving PARP activation without requiring apoptosis. Free Radic Biol Med. 2010;49:1925–1936. doi: 10.1016/j.freeradbiomed.2010.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath J, Drummond G, McLachlan E, Kilkenny C, Wainwright C. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng XW, Lee SH, Dai H, Loegering D, Yu C, Flatten K, et al. Mcl-1 as a buffer for proapoptotic Bcl-2 family members during TRAIL-induced apoptosis: a mechanistic basis for sorafenib (Bay 43-9006)-induced TRAIL sensitization. J Biol Chem. 2007;282:29831–29846. doi: 10.1074/jbc.M706110200. [DOI] [PubMed] [Google Scholar]
- Nguyen TK, Jordan N, Friedberg J, Fisher RI, Dent P, Grant S. Inhibition of MEK/ERK1/2 sensitizes lymphoma cells to sorafenib-induced apoptosis. Leuk Res. 2010;34:379–386. doi: 10.1016/j.leukres.2009.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panka DJ, Wang W, Atkins MB, Mier JW. The Raf inhibitor BAY 43-9006 (Sorafenib) induces caspase-independent apoptosis in melanoma cells. Cancer Res. 2006;66:1611–1619. doi: 10.1158/0008-5472.CAN-05-0808. [DOI] [PubMed] [Google Scholar]
- Park MA, Reinehr R, Haussinger D, Voelkel-Johnson C, Ogretmen B, Yacoub A, et al. Sorafenib activates CD95 and promotes autophagy and cell death via Src family kinases in gastrointestinal tumor cells. Mol Cancer Ther. 2010;9:2220–2231. doi: 10.1158/1535-7163.MCT-10-0274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasdois P, Parker JE, Griffiths EJ, Halestrap AP. The role of oxidized cytochrome c in regulating mitochondrial reactive oxygen species production and its perturbation in ischaemia. Biochem J. 2011;436:493–505. doi: 10.1042/BJ20101957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plastaras JP, Kim SH, Liu YY, Dicker DT, Dorsey JF, McDonough J, et al. Cell cycle dependent and schedule-dependent antitumor effects of sorafenib combined with radiation. Cancer Res. 2007;67:9443–9454. doi: 10.1158/0008-5472.CAN-07-1473. [DOI] [PubMed] [Google Scholar]
- Poulikakos PI, Zhang C, Bollag G, Shokat KM, Rosen N. RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild-type BRAF. Nature. 2010;464:427–430. doi: 10.1038/nature08902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahmani M, Davis EM, Bauer C, Dent P, Grant S. Apoptosis induced by the kinase inhibitor BAY 43-9006 in human leukemia cells involves down-regulation of Mcl-1 through inhibition of translation. J Biol Chem. 2005;280:35217–35227. doi: 10.1074/jbc.M506551200. [DOI] [PubMed] [Google Scholar]
- Ramakrishnan V, Timm M, Haug JL, Kimlinger TK, Wellik LE, Witzig TE, et al. Sorafenib, a dual Raf kinase/vascular endothelial growth factor receptor inhibitor has significant anti-myeloma activity and synergizes with common anti-myeloma drugs. Oncogene. 2010;29:1190–1202. doi: 10.1038/onc.2009.403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramanathan A, Schreiber SL. Direct control of mitochondrial function by mTOR. Proc Natl Acad Sci U S A. 2009;106:22229–22232. doi: 10.1073/pnas.0912074106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosato RR, Almenara JA, Coe S, Grant S. The multikinase inhibitor sorafenib potentiates TRAIL lethality in human leukemia cells in association with Mcl-1 and cFLIPL down-regulation. Cancer Res. 2007;67:9490–9500. doi: 10.1158/0008-5472.CAN-07-0598. [DOI] [PubMed] [Google Scholar]
- Shi J, Zhou Y, Huang HC, Mitchison TJ. Navitoclax (ABT-263) accelerates apoptosis during drug-induced mitotic arrest by antagonizing Bcl-xL. Cancer Res. 2011;71:4518–4526. doi: 10.1158/0008-5472.CAN-10-4336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoemaker AR, Mitten MJ, Adickes J, Ackler S, Refici M, Ferguson D, et al. Activity of the Bcl-2 family inhibitor ABT-263 in a panel of small cell lung cancer xenograft models. Clin Cancer Res. 2008;14:3268–3277. doi: 10.1158/1078-0432.CCR-07-4622. [DOI] [PubMed] [Google Scholar]
- Singhal SS, Sehrawat A, Sahu M, Singhal P, Vatsyayan R, Rao Lelsani PC, et al. Rlip76 transports sunitinib and sorafenib and mediates drug resistance in kidney cancer. Int J Cancer. 2010;126:1327–1338. doi: 10.1002/ijc.24767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan N, Malek M, Zha J, Yue P, Kassees R, Berry L, et al. Navitoclax enhances the efficacy of taxanes in non-small cell lung cancer models. Clin Cancer Res. 2011;17:1394–1404. doi: 10.1158/1078-0432.CCR-10-2353. [DOI] [PubMed] [Google Scholar]
- Tse C, Shoemaker AR, Adickes J, Anderson MG, Chen J, Jin S, et al. ABT-263: a potent and orally bioavailable Bcl-2 family inhibitor. Cancer Res. 2008;68:3421–3428. doi: 10.1158/0008-5472.CAN-07-5836. [DOI] [PubMed] [Google Scholar]
- Vitiello PF, Wu YC, Staversky RJ, O'Reilly MA. p21(Cip1) protects against oxidative stress by suppressing ER-dependent activation of mitochondrial death pathways. Free Radic Biol Med. 2009;46:33–41. doi: 10.1016/j.freeradbiomed.2008.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogler M, Dinsdale D, Dyer MJ, Cohen GM. Bcl-2 inhibitors: small molecules with a big impact on cancer therapy. Cell Death Differ. 2009;16:360–367. doi: 10.1038/cdd.2008.137. [DOI] [PubMed] [Google Scholar]
- Vogler M, Furdas SD, Jung M, Kuwana T, Dyer MJ, Cohen GM. Diminished sensitivity of chronic lymphocytic leukemia cells to ABT-737 and ABT-263 due to albumin binding in blood. Clin Cancer Res. 2010;16:4217–4225. doi: 10.1158/1078-0432.CCR-10-0777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G, Zhan Y, Wang H, Li W. ABT-263 sensitizes TRAIL-resistant hepatocarcinoma cells by downregulating the Bcl-2 family of anti-apoptotic protein. Cancer Chem Pharmacol. 2012;69:799–805. doi: 10.1007/s00280-011-1763-0. [DOI] [PubMed] [Google Scholar]
- Wei G, Wang M, Hyslop T, Wang Z, Carr BI. Vitamin K enhancement of sorafenib-mediated HCC cell growth inhibition in vitro and in vivo. Int J Cancer. 2010;127:2949–2958. doi: 10.1002/ijc.25498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilhelm S, Carter C, Lynch M, Lowinger T, Dumas J, Smith RA, et al. Discovery and development of sorafenib: a multikinase inhibitor for treating cancer. Nat Rev Drug Discov. 2006;5:835–844. doi: 10.1038/nrd2130. [DOI] [PubMed] [Google Scholar]
- Wilhelm SM, Carter C, Tang L, Wilkie D, McNabola A, Rong H, et al. BAY 43–9006 exhibits broad spectrum oral antitumor activity and targets the RAF/MEK/ERK pathway and receptor tyrosine kinases involved in tumor progression and angiogenesis. Cancer Res. 2004;64:7099–7109. doi: 10.1158/0008-5472.CAN-04-1443. [DOI] [PubMed] [Google Scholar]
- Wilson WH, O'Connor OA, Czuczman MS, LaCasce AS, Gerecitano JF, Leonard JP, et al. Navitoclax, a targeted high-affinity inhibitor of BCL-2, in lymphoid malignancies: a phase 1 dose-escalation study of safety, pharmacokinetics, pharmacodynamics, and antitumour activity. Lancet Oncol. 2010;11:1149–1159. doi: 10.1016/S1470-2045(10)70261-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi R, Janssen E, Perkins G, Ellisman M, Kitada S, Reed JC. Efficient elimination of cancer cells by deoxyglucose-ABT-263/737 combination therapy. PLoS ONE. 2011;6:e24102. doi: 10.1371/journal.pone.0024102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang F, Brown C, Buettner R, Hedvat M, Starr R, Scuto A, et al. Sorafenib induces growth arrest and apoptosis of human glioblastoma cells through the dephosphorylation of signal transducers and activators of transcription 3. Mol Cancer Ther. 2010;9:953–962. doi: 10.1158/1535-7163.MCT-09-0947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youle RJ, Strasser A. The BCL-2 protein family: opposing activities that mediate cell death. Nat Rev Mol Cell Biol. 2008;9:47–59. doi: 10.1038/nrm2308. [DOI] [PubMed] [Google Scholar]
- Zhang W, Konopleva M, Ruvolo VR, McQueen T, Evans RL, Bornmann WG, et al. Sorafenib induces apoptosis of AML cells via Bim-mediated activation of the intrinsic apoptotic pathway. Leukemia. 2008;22:808–818. doi: 10.1038/sj.leu.2405098. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 a,b. Cell viability for the combination of sorafenib and ABT-263 in various concentrations. c,d. Western blot to examine the protein of PARP and caspases.
Figure S2 Cell viability of single drug effect.
Figure S3 Primary sub-G1 histograms.
Figure S4 a,c. Mitochondrail membrane potential for the combination of sorafenib and ABT-263 in presence of NAC or CsA. b,d. Western blot to examine the protein of PARP and caspases in presence of NAC or CsA.
Figure S5 a. Western blot to detect the level of Akt activity in BEL7402 and HCT116 p21-/- cells treated with DMSO or z-VAD (50 μM) before combination treatment (ABT-263 0.4 μM, sorafenib 6 μM). b, Cell viability of BEL7402 and HCT116 p21-/- cells treated with DMSO of z-Vad (50 μM) before combination treatment (ABT-263 0.4 μM, sorafenib 6 μM).
Figure S6 a. Western blot to detect the protein of PARP when cells transfected with p21 siRNA. b. Cell viability of cells transfected with p21 siRNA.
Figure S7 a,b,c. The body weights of nude mice bearing established HVT116 tumour xenografts.