

Abstract
BACKGROUND AND PURPOSE
The novel macrocyclic peptide cyclo[Phe-D-Pro-Phe-D-Trp] ([D-Trp]CJ-15,208) exhibits κ opioid (KOP) receptor antagonist activity in both in vitro and in vivo assays. The four alanine analogues of this peptide were synthesized and characterized both in vitro and in vivo to assess the contribution of different amino acid residues to the activity of [D-Trp]CJ-15,208.
EXPERIMENTAL APPROACH
The peptides were synthesized by a combination of solid phase peptide synthesis and cyclization in solution. The analogues were evaluated in vitro in receptor binding and functional assays, and in vivo with mice using a tail-withdrawal assay for antinociceptive and opioid antagonist activity. Mice demonstrating extinction of cocaine conditioned-place preference (CPP) were pretreated with selected analogues to evaluate prevention of stress or cocaine-induced reinstatement of CPP.
KEY RESULTS
The alanine analogues displayed pharmacological profiles in vivo distinctly different from [D-Trp]CJ-15,208. While the analogues exhibited varying opioid receptor affinities and κ and μ opioid receptor antagonist activity in vitro, they produced potent opioid receptor-mediated antinociception (ED50 = 0.28–4.19 nmol, i.c.v.) in vivo. Three of the analogues also displayed KOP receptor antagonist activity in vivo. Pretreatment with an analogue exhibiting both KOP receptor agonist and antagonist activity in vivo prevented both cocaine- and stress-induced reinstatement of cocaine-seeking behaviour in the CPP assay in a time-dependent manner.
CONCLUSIONS AND IMPLICATIONS
These unusual macrocyclic peptides exhibit in vivo opioid activity profiles different from the parent compound and represent novel compounds for potential development as therapeutics for drug abuse and possibly as analgesics.
Keywords: CJ-15,208; macrocyclic peptide; κ opioid receptor antagonist; analgesic; cocaine reinstatement
Introduction
Evidence suggests that κ opioid (KOP) receptor antagonists may have therapeutic potential in the treatment of drug abuse (Aldrich and McLaughlin, 2009). For example, pretreatment with the KOP receptor-selective small-molecule antagonists nor-binaltorphimine (nor-BNI) and ((3R)-7-hydroxy-N-((S)-1-[[(3R,4R)-4-(3-hydroxyphenyl)-3,4-dimethyl-1-piperidinyl]methyl]-2-methylpropyl)-1,2,3,4-tetrahydro-3-isoquinolinecarboxamide) (JDTic) has been shown to prevent the stress-induced reinstatement of extinguished cocaine-seeking behaviour (Beardsley et al., 2005; Redila and Chavkin, 2008), and heroin-dependent patients treated for 12 weeks with a ‘functional KOP receptor antagonist’ (buprenorphine plus naltrexone) showed significantly improved drug abstinence relative to patients treated only with naltrexone (Rothman et al., 2000; Gerra et al., 2006). However, the prototypical KOP receptor-selective non-peptide antagonists nor-BNI, 5′-guanidinylnaltrindole (GNTI) and JDTic exhibit exceptionally long activity, antagonizing KOP receptors for weeks after a single dose (Metcalf and Coop, 2005; Aldrich and McLaughlin, 2009). This profile could potentially complicate their use as pharmacological tools and as possible therapeutic agents, spurring the search for shorter acting KOP receptor-selective antagonists (Brugel et al., 2010; Runyon et al., 2010; Grimwood et al., 2011; Peters et al., 2011; Frankowski et al., 2012).
In addition to dynorphin A derivatives (Bennett et al., 2002; Patkar et al., 2005), we are examining novel peptides unrelated to the endogenous opioid peptides as KOP receptor antagonists. Of particular interest was the novel tetrapeptide CJ-15,208 (cyclo[Phe-D-Pro-Phe-Trp]) that was reported to exhibit KOP receptor antagonist activity in vitro (Saito et al., 2002). The relatively low molecular weight (577 Da) and macrocyclic structure of CJ-15,208 suggested it would be a promising lead candidate for potential development. Macrocyclic tetrapeptides are stable to proteolytic degradation (Delaforge et al., 1997), so it was expected that these peptides would exhibit activity in vivo after systemic administration.
Because the stereochemistry of the Trp residue in CJ-15,208 was not determined when this natural product was isolated, we synthesized both tryptophan isomers of this macrocyclic peptide (Kulkarni et al., 2009; Ross et al., 2010), and found that the peptide containing L-Trp was the natural product based on its optical rotation. The D-Trp containing peptide (Figure 1A) also preferentially bound to KOP receptors and exhibited antagonist activity at these receptors, both in vitro (Dolle et al., 2009; Ross et al., 2010; 2012) and in vivo (Ross et al., 2012; Eans et al., 2013). Therefore, we are exploring the structure–activity relationships of both Trp isomers of CJ-15,208. To determine which amino acid side chains were important for the observed opioid activity, each amino acid in CJ-15,208 was initially replaced by alanine (Aldrich et al., 2011). Unexpected differences in their opioid activity in vivo were found for the alanine analogues and CJ-15,208 compared with the results obtained in vitro. Therefore, we also examined the alanine-substituted analogues of cyclo[Phe-D-Pro-Phe-D-Trp] ([D-Trp]CJ-15,208; Figure 1B) both in vitro and in vivo to determine the contribution of each residue to this parent peptide's opioid receptor interactions and its opioid activity profile in vivo.
Figure 1.
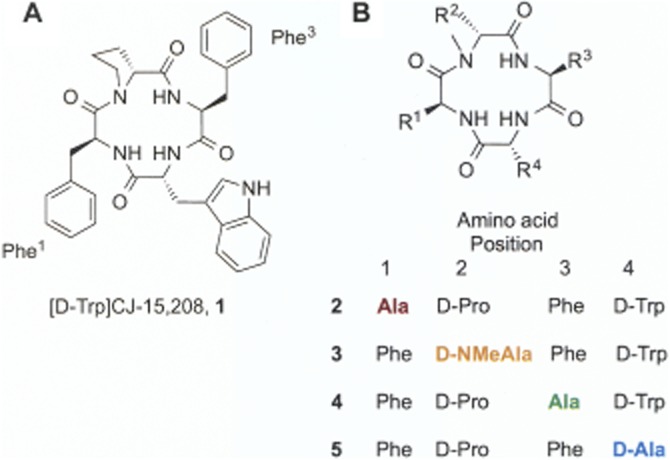
Structures of (A) [D-Trp]CJ-15,208 (1) and (B) alanine analogues 2–5. The residues are numbered 1–4, arbitrarily starting with the Phe C-terminal to the D-Trp residue.
Statement on drug and receptor nomenclature
All drug and molecular target terms conform to parameters specified in the British Journal of Pharmacology's Guide to Receptors and Channels (Alexander et al., 2013).
Methods
Materials
The sources of reagents, amino acids, 2-chlorotrityl chloride resin and solvents are the same as reported previously (Ross et al., 2010; Aldrich et al., 2011). Amino acids are the L-isomer unless otherwise specified and abbreviations for amino acids follow the IUPAC-IUB Joint Commission of Biochemical Nomenclature [Eur J Biochem (1984) 138: 9–37]. All chemicals other than analogues of [D-Trp]CJ-15,208 were obtained from Sigma-Aldrich (St. Louis, MO, USA). [D-Trp]CJ-15,208 and synthesized analogues (Figure 1) were dissolved daily prior to administration initially in dimethyl sulfoxide (DMSO), and sufficient warm (40°C) sterile saline (0.9% ) then added so that the final vehicle for in vivo administration consisted of one part DMSO and one part sterile saline.
Peptide synthesis and purification
The linear peptide precursors (based on the parent sequence H-D-Trp-Phe-D-Pro-Phe-OH with substitution of D-Ala, Ala, D-NMeAla and Ala in positions 1–4, respectively) were synthesized on a 2-chlorotrityl choride resin by Fmoc solid phase synthesis, and the peptides cleaved from the resin with 1% trifluoroacetic acid in dichloromethane as described previously (Ross et al., 2010; Aldrich et al., 2011). The crude linear peptides were used in the cyclization reactions without further purification. The peptides were initially cyclized using the previously reported procedure (Ross et al., 2010; Aldrich et al., 2011). This procedure was subsequently modified as follows to increase the yields of the cyclic peptides (Senadheera et al., 2011): the crude linear peptide (1 equiv, 0.62 mM in 20 mL N,N-dimethylformamide, DMF) was added dropwise at a rate of 1.4 mL·h−1 (using a KD Scientific single infusion syringe pump, Lab Source Inc., Romeoville, IL, USA) to a dilute solution of HATU [2-(1H-7-azabenzotriazol-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate, 1.5 equiv, 0.938 mM] and N,N-diisopropylethylamine (8 equiv, 5 mM) in DMF. After 15 h additional HATU (1.5 equiv) was then added to the reaction in one portion, and additional linear peptide (1 equiv, 0.62 mM in 20 mL DMF) was added dropwise at a rate of 1.2 mL·h−1 as described above. The reaction was then stirred for 12 h at room temperature, followed by an additional 20–24 h at 37°C. The solvent was evaporated under reduced pressure, and the crude cyclic tetrapeptides isolated as previously described (Ross et al., 2010; Aldrich et al., 2011).
Initially, the peptides were purified by reversed-phase HPLC as described previously (Ross et al., 2010; Aldrich et al., 2011). Larger quantities of peptides for in vivo evaluation were purified by silica gel chromatography using a step gradient of 60–90% EtOAc in hexane (with EtOAc increased in 10% increments), followed by 0–3% MeOH in EtOAc (with MeOH increased in 1% increments). For the more polar analogues 2 and 4, the gradient used was 0–3% MeOH in EtOAc. The purified peptides were dissolved in aqueous acetonitrile (water : MeCN, 4:1) and then lyophilized to give the peptides as white solids. The yields of the Ala analogues after purification were 45–55%.
The purified peptides were analysed by electrospray ionization mass spectrometry, thin-layer chromatography and analytical HPLC (see Supporting Information Appendix S1). All peptides were >99% pure in both HPLC systems.
In vitro pharmacological analysis
Radioligand binding assays
Opioid receptor affinities were determined in radioligand binding assays as previously described (Arttamangkul et al., 1997; Aldrich et al., 2011; Ross et al., 2012) with membranes from Chinese hamster ovary (CHO) cells stably expressing rat KOP, rat μ opioid (MOP) or mouse δ opioid (DOP) receptors using the radioligands [3H]diprenorphine, [3H]-[D-Ala2,NMePhe4,glyol5]enkephalin ([3H]DAMGO) and [3H]cyclo[D-Pen2,D-Pen5]enkephalin ([3H]DPDPE) respectively. IC50 values were determined by non-linear regression analysis using Prism software (GraphPad Software Co., La Jolla, CA, USA). Ki values were calculated from the IC50 values by the Cheng and Prusoff equation (Cheng and Prusoff, 1973) using KD values of 0.45, 0.49 and 1.76 nM for [3H]diprenorphine, [3H]DAMGO and [3H]DPDPE respectively. These results are presented as the mean ± SEM from at least three separate experiments each performed in triplicate.
GTPγS assays
The binding of [35S]GTPγS to membranes from CHO cells stably expressing KOP or MOP receptors was assayed as described previously (Siebenaller and Murray, 1999; Aldrich et al., 2011; Ross et al., 2012). Efficacy was determined relative to the reference full agonists dynorphin (Dyn) A-(1–13) amide for KOP receptors and DAMGO for MOP receptors. The antagonist profiles of the peptides at KOP and MOP receptors were determined by measuring the EC50 values of Dyn A-(1–13)amide and DAMGO, respectively, in the absence and presence of four different concentrations (0.1 nM−10 μM) of the peptide, performed in triplicate. The pA2 values were determined by Schild analysis (Schild, 1947), and the results are reported as KB values ± SEM from at least three experiments except where noted.
In vivo pharmacological evaluation
Animals and drug administration
All animal care and experimental procedures complied with the National Institute of Health Guide for the Care and Use of Laboratory Animals and were approved by the Institutional Animal Care and Use Committee at the Torrey Pines Institute for Molecular Studies. All results of animal testing are reported in accordance with the ARRIVE guidelines (Kilkenny et al., 2010; McGrath et al., 2010). A total of 852 animals were used in the experiments described here.
Adult male wild-type C57BL/6J mice weighing 20–25 g were obtained from Jackson Laboratory (Bar Harbor, ME, USA). MOP receptor gene-disrupted (MOP KO) and KOP receptor gene-disrupted (KOP KO) mice were obtained from colonies established at the Torrey Pines Institute for Molecular Studies from homozygous breeding pairs of mice backcrossed to C57BL/6J inbred mice for at least 12 generations and obtained from the Jackson Laboratory. All mice were kept on a 12 h light–dark cycle and food pellets and distilled water were available ad libitum.
Antinociceptive testing
The 55°C warm-water tail-withdrawal assay was performed in mice as previously described (McLaughlin et al., 1999), with the latency of the mouse to withdraw its tail from the water taken as the end point (a cut-off time of 15 s was used in this assay). Peptides were administered by i.c.v. injection as described previously (Aldrich et al., 2011). Antinociception was calculated according to the following formula: % antinociception = 100× (test latency − control latency)/(15 − control latency). Tail withdrawal data points are the means of 8–12 mice, unless otherwise indicated, with SEM shown by error bars. Tail withdrawal latencies in KO mice were determined in six to eight mice.
To determine the opioid receptor involvement in the agonist activity of macrocyclic peptides 2–5, mice were pretreated with a single dose of β-funaltrexamine (β-FNA, 5 mg·kg−1, s.c.) or nor-BNI (10 mg·kg−1, i.p.) 23.5 h in advance of administration of a dose of a macrocyclic peptide. Additional mice were pretreated with a single dose of naltrindole (20 mg·kg−1, i.p.) 15 min prior to administration of the macrocyclic tetrapeptide.
To determine antagonist activity, mice were pretreated with the macrocyclic tetrapeptide 140 min prior to the administration of the MOP receptor-preferring agonist morphine (10 mg·kg−1, i.p.), the KOP receptor-selective agonist U50,488 (10 mg·kg−1, i.p.) or the DOP receptor-selective agonist SNC-80 ((+)-4-[(αR)-α-((2S,5R)-4-allyl-2,5-dimethyl-1-piperazinyl)-3-methoxybenzyl]-N,N-diethylbenzamide, 100 nmol, i.c.v.); at this time the antinociceptive activity of the macrocyclic peptides had dissipated. Antinociception produced by these established agonists was then measured 40 min after their administration. Additionally, to determine the duration of KOP receptor antagonist activity, other mice were pretreated 2.3–23.3 h prior to administration of U50,488 as described above.
Conditioned-place preference (CPP) evaluation
Male C57BL/6J mice (weighing 20–25 g at the beginning of the experiment) were subjected to an unbiased and counterbalanced cocaine CPP paradigm, where the compartment in which the animal receives vehicle or drug is randomly assigned regardless of initial preference, using similar timing as detailed previously (Eans et al., 2013). The time the animals spent in the two outer and middle compartments was measured for 30 min in boxes outfitted with infrared beams (San Diego Instruments, San Diego, CA, USA); data are reported as the difference in time spent in the cocaine-minus the vehicle-paired compartment.
Mice were subjected to 4 days of place conditioning to 0.9% saline and cocaine (10 mg·kg−1, s.c. in 0.9% saline) as previously described (Aldrich et al., 2009; Ross et al., 2012; Eans et al., 2013). The mice were then evaluated for their place preference for 30 min twice weekly over a 3 week period until extinction was established (see Figure 8).
Figure 8.
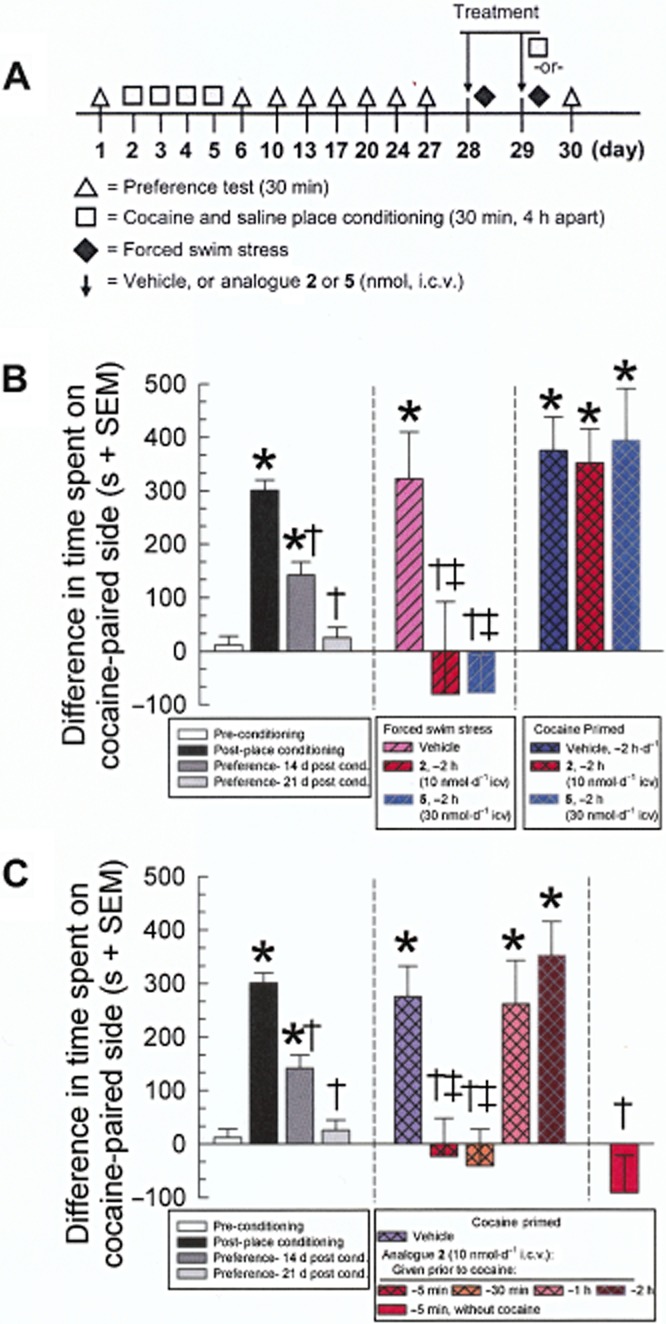
Time-dependent prevention of reinstatement of extinguished cocaine–CPP following pretreatment with analogues 2 and 5. (A) Schematic showing timing of the extinction, treatment and reinstatement protocol. Vehicle (50% DMSO) or analogues 2 or 5 (10 or 30 nmol, i.c.v., respectively) was administered on days 28 and 29, 2 h prior to initial exposure to forced swim stress; for cocaine place conditioning the mice were treated on days 28 and 29 with vehicle or peptide, followed by cocaine place conditioning 5 min to 2 h after the injection (square) on day 29. (B) Mice exhibited significant preference for the cocaine (10 mg kg−1, s.c. daily for 4 days)-paired compartment, with extinction occurring over the next 3 weeks (left bars). Mice were then exposed to forced swim stress (centre bars) or an additional round of cocaine place conditioning (right bars), resulting in the reinstatement of place preference in vehicle-treated mice. Pretreatment for 2 h with either alanine analogue 2 or 5 prevented stress-induced, but not cocaine-primed, reinstatement of cocaine-seeking behaviour in the CPP assay. (C) Analogue 2 also prevented cocaine-primed reinstatement of cocaine CPP when administered 5 or 30 min, but not 1 or 2 h, prior to cocaine. Pretreatment with analogue 2 without cocaine (rightmost bar) did not induce reinstatement by itself. Data shown are mean (± SEM) difference in time spent on the drug-paired side. *P < 0.05, significantly different from preconditioning place preference response (leftmost bar); †P < 0.05, significantly different from post-CPP response (leftmost solid black bar); ‡P < 0.05, significantly different from vehicle-treated, stress-induced or cocaine-primed reinstatement of place preference response; anova followed by Tukey's post hoc test.
Following demonstration of extinction, groups of mice (10–24) were exposed to either forced swim stress or an additional cycle of cocaine place conditioning (see Figure 8A) as described previously (Carey et al., 2007; Aldrich et al., 2009; Ross et al., 2012). Mice were pretreated with vehicle, macrocyclic peptide 2 (10 nmol, i.c.v.) or peptide 5 (30 nmol, i.c.v.) on days 28 and 29 2 h prior to a forced swimming session on each day, performed as previously described (Aldrich et al., 2009; Ross et al., 2012; Eans et al., 2013). Additional mice were treated for 2 days with vehicle or macrocyclic peptide 5 min, 30 min, 1 h or 2 h prior to an additional session of cocaine conditioning on day 29 (Figure 8A). Mice were tested for place preference on the day following completion of the stress exposure or cocaine place conditioning (day 30, see Figure 8A).
Data analysis
All data are presented as mean ± SEM. Graded dose–response curves were constructed, and potencies are reported as the effective dose producing 50% antinociception (ED50). All dose–response lines were analysed by non-linear regression, and ED50 values and 95% confidence intervals determined using individual data points with the Prism 6.02 software package (GraphPad). Data for antinociception experiments were evaluated with anova using Tukey's or Dunnett's multiple comparison post hoc tests, as appropriate. Analyses were used to compare baseline and post-treatment tail-withdrawal latencies and to determine statistical significance for all tail-withdrawal data. Statistical significance of differences between ED50 values was determined by evaluation of the ED50 value shift via non-linear regression modeling with Prism 6.02. One-way anova was performed on all in vivo receptor agonist and antagonist selectivity data. Data for CPP experiments were analysed by multivariate anova with the main effect of CPP phase (e.g. postconditioning, week of preference test, reinstatement) and the interaction of drug pretreatment (macrocyclic peptide or vehicle) and reinstatement condition (stress or cocaine exposure). Significant (P < 0.05) effects were further analysed using Tukey's honestly significant difference (HSD) post hoc test.
Results
Synthesis
The alanine analogues of [D-Trp]CJ-15,208 (Figure 1) were synthesized by a combination of solid phase synthesis of the linear precursors followed by cyclization in solution (Kulkarni et al., 2009; Ross et al., 2010). Modifications were made to the cyclization reaction, namely increasing the reaction temperature and decreasing the rate of addition of the linear peptide to the reaction, to improve the yields of the macrocyclic peptides (Senadheera et al., 2011; Aldrich et al., 2013). Initially, the peptides were purified by reversed-phase HPLC; subsequent purifications were performed by flash chromatography on silica gel, which permitted the facile purification of larger quantities of the macrocyclic peptides for in vivo pharmacological evaluation.
In vitro pharmacological evaluation
The affinities of the alanine analogues of [D-Trp]CJ-15,208 for KOP receptors varied substantially (Table 1). Analogue 2, in which Phe1 was replaced by Ala (see Figure 1B for residue numbering), exhibited the highest KOP receptor affinity, sevenfold higher than the parent macrocyclic peptide 1. In contrast, analogue 5, in which D-Trp4 was replaced by D-Ala, exhibited very low KOP receptor affinity (Ki = 1.7 μM), suggesting that the D-Trp4 residue was important for binding to these receptors. Analogues 3 and 4 exhibited intermediate affinities for KOP receptors, 3.5- and 7.7-fold lower than that of analogue 1. Similar results were reported by Dolle et al. (2009) for analogues 2 and 5, but the KOP receptor affinity of analogue 4 found here (167 nM) is substantially higher than that reported by Dolle et al. (2009) (Ki = 1300 nM). (Note that analogue 3 has not been previously reported.)
Table 1.
Opioid receptor affinities of the alanine analogues of [D-Trp]CJ-15,208
| Peptide | Ki (nM ± SEM) | Selectivity | ||
|---|---|---|---|---|
| KOP receptor | MOP receptor | DOP receptor | KOP/MOP/DOP | |
| 2, Ala1 | 3.07 ± 0.30 | 27.3 ± 2.7 | 8330 ± 1220 | 1/6.7/2620 |
| 3, D-NMeAla2 | 76.9 ± 18.2 | 257 ± 14 | 5690 ± 610 | 1/3.3/74 |
| 4, Ala3 | 167 ± 15 | 299 ± 114 | >10 000 | 1/1.8/>59 |
| 5, D-Ala4 | 1720 ± 420 | 775 ± 71 | >10 000 | 2.2/1/>13 |
| 1 | 21.8 ± 4.8 | 259 ± 29 | 4190 ± 858 | 1/12/192 |
CHO membranes expressing cloned KOP, MOP or DOP receptors were incubated with different concentrations of each macrocyclic tetrapeptide in the presence of [3H]diprenorphine, [3H]DAMGO or [3H]DPDPE, for KIL KOP, MOP and DOP receptors, respectively. Data are the mean Ki values ± SEM from at least three experiments.
MOP receptor affinity was generally less sensitive to alanine substitution in the macrocyclic tetrapeptide than was affinity for the KOP receptor (Table 1), although there was some variation among the analogues. Substitution of Phe1 by Ala to give 2 substantially increased MOP receptor affinity, while substitution of D-Trp4 by D-Ala to give 5 decreased affinity threefold. Replacement of D-Pro2 by D-NMeAla or of Phe3 by Ala had little effect on MOP receptor affinity. Analogous to the results for the KOP receptor, the MOP receptor affinities found for analogues 2 and 5 are similar to those reported previously by Dolle et al. (2009), although those authors reported substantially lower affinity (Ki > 2 μM) for peptide 4. None of the analogues showed appreciable affinity for DOP receptors in the binding assay.
As observed with the parent peptide, none of the Ala analogues exhibited appreciable agonist activity in the [35S]GTPγS assay at KOP and MOR receptors. All of the peptides antagonized the stimulation of [35S]GTPγS binding by Dyn A-(1–13)NH2 at KOP receptors with varying potencies (Table 2). Analogue 2 exhibited potent antagonist activity at KOP receptors (KB = 0.8 nM), consistent with the report by Dolle et al. (2009). The potencies of the other analogues as KOP receptor antagonists in this assay generally paralleled their receptor affinities. The potencies of the analogues as MOP receptor antagonists also generally correlated with their respective MOP receptor affinities, except for the D-Ala4 analogue 5 that exhibited weaker antagonist activity than might be expected based on its affinity.
Table 2.
Opioid antagonist activity of the alanine analogues in vitro in the [35S]GTPγS assaya
| Peptide | Antagonism: KB (nM ± SEM) | |
|---|---|---|
| KOP receptor | MOP receptor | |
| 2, Ala1 | 0.81 ± 0.58 | 8.08 ± 2.44 |
| 3, D-NMeAla2 | 103 ± 38 | 321 ± 186 |
| 4, Ala3 | 397 ± 118 | 1470 ± 160b |
| 5, D-Ala4 | 922 ± 309 | 1100 ± 480 |
| 1 | 20.2 ± 7.9 | – c |
CHO membranes expressing cloned KOP or MOP receptors were incubated with Dyn A-(1–13) amide or DAMGO concentrations, respectively, either alone or in the presence of each macrocyclic tetrapeptide, as described in Methods. Data presented are the mean KB values ± SEM from at least three experiments, except where noted, which were derived from Schild regression analysis.
The analogues exhibited negligible efficacy at KOP and MOP receptors; <10% relative to Dyn A-(1–13) amide and DAMGO at KOP and MOP receptors, respectively.
n = 2.
No antagonist activity observed at concentrations up to 300 nM (Ross et al., 2012).
In vivo pharmacological evaluation
Antinociceptive activity
The analogues were initially evaluated for their antinociceptive activity in the 55°C warm-water tail-withdrawal assay in C57BL/6J mice following i.c.v. administration. The parent peptide 1 exhibited minimal antinociceptive activity at doses up to 10 nmol (Ross et al., 2012), with moderate antinociceptive activity (40%) detected only at 30 nmol, i.c.v. (Figure 2A). In contrast, all of the alanine analogues exhibited potent antinociceptive activity, which was unexpected given the lack of efficacy observed for the analogues in vitro in the GTPγS assay. Significant antinociceptive activity was detected for 80–100 min after administration of each analogue at their highest respective dose (Figure 2B). The D-Ala analogue 5 produced potent antinociception [with an ED50 (and 95% confidence interval) of 0.28 (0.15–0.55) nmol, i.c.v.], which was particularly surprising given its low affinity for opioid receptors. The Ala1 (2) and Ala3 (4) analogues exhibited similar antinociceptive potencies in this in vivo assay, with ED50 values of 1.99 (0.36–13.2) and 1.04 (0.45–2.47) nmol, i.c.v. respectively. The D-NMeAla analogue (3) was the least potent of the alanine analogues [ED50 = 4.19 (1.40–11.9) nmol, i.c.v.]. Although the potencies differed, all four analogues produced antinociception comparable with that produced by morphine [ED50 value = 2.35 (1.13–5.03) nmol, i.c.v.].
Figure 2.
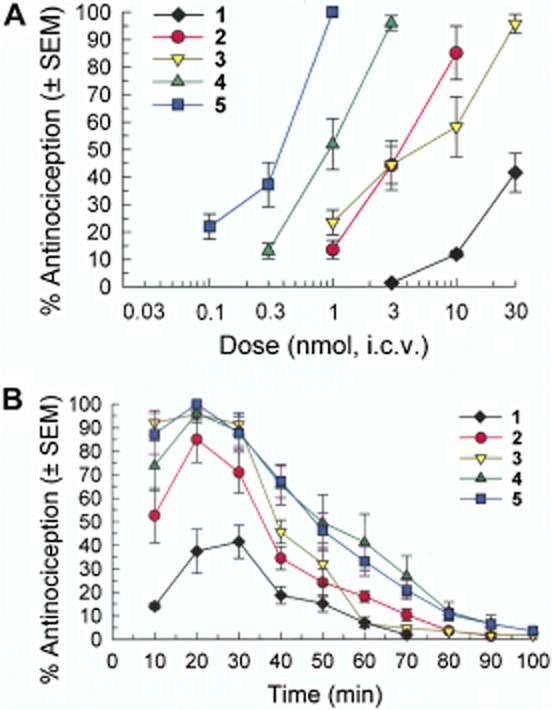
Antinociceptive activity of [D-Trp]CJ-15,208 (1) and the alanine analogues 2–5 in vivo following i.c.v. administration in the 55°C warm-water tail-withdrawal assay in C57BL/6J mice. Data shown are mean % antinociception ± SEM. (A) Dose–response curves at peak response, which was 30 min for 1 or 20 min for analogues 2–5. (B) Time course for antinociceptive activity for 1 (30 nmol), 2 (10 nmol), 3 (30 nmol), 4 (3 nmol) and 5 (1 nmol).
To determine the involvement of MOP, KOP and DOP receptors in the observed antinociceptive activity, mice were pretreated with the selective receptor antagonists β-FNA, nor-BNI and naltrindole, respectively, prior to administration of a macrocyclic tetrapeptide (Figure 3). Antinociception produced by analogue 2 was significantly antagonized only by nor-BNI pretreatment [F(3,27) = 7.26; P = 0.001; one-way anova with Tukey's HSD]. In contrast, the antinociception of analogues 3 and 4 were significantly reduced by pretreatment with selective antagonists for all three opioid receptors [F(3,28) = 100.0; P < 0.0001 and F(3,27) = 62.7; P < 0.0001 respectively]. The antinociception induced by the D-Ala4 analogue 5 was significantly antagonized by either β-FNA or nor-BNI pretreatment [F(3,28) = 41.0; P < 0.0001], but naltrindole pretreatment was without significant effect.
Figure 3.
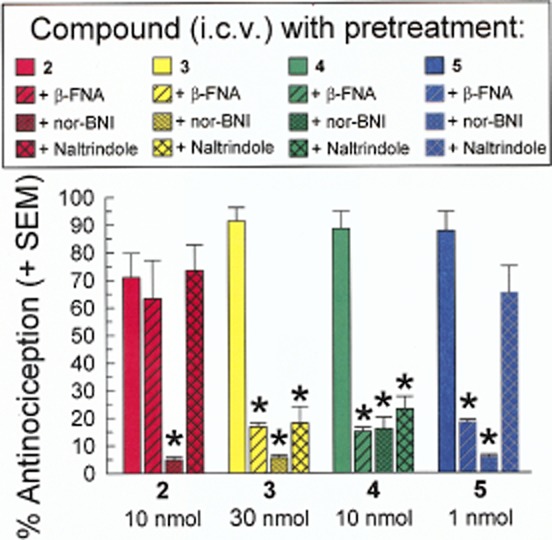
Opioid receptor selectivity of the antinociceptive activity produced by alanine analogues of [D-Trp]CJ-15,208 in the mouse 55°C warm-water tail-withdrawal assay in C57BL/6J mice. The antinociceptive activity of the analogues was determined at the indicated doses after i.c.v. administration alone (solid bars), 24 h after administration of β-FNA or nor-BNI, and 15 min after pretreatment with naltrindole. Tail withdrawal latencies were measured 30 min after analogue administration. Data shown are mean % antinociception ± SEM.*P < 0.05, significantly different from response of matching administered analogue alone; one-way anova followed by Tukey's post hoc test.
The opioid receptor mediation of antinociception induced by the Ala1 analogue 2 was further assessed in MOP KO, KOP KO mice and wild-type (WT) mice pretreated with naltrindole (Figure 4). The antinociceptive activity of this peptide in MOP KO mice [ED50 = 5.47 (2.02–14.9) nmol, i.c.v.] or WT mice pretreated with naltrindole [ED50 = 5.47 (2.62–8.49) nmol, i.c.v.] was not statistically different from the response observed in naïve WT mice [F(1,65) = 2.07; P = 0.155; non-linear regression modeling], suggesting little if any contribution of MOP or DOP receptors to the antinociceptive activity of analogue 2. However, the antinociceptive dose–response curve for analogue 2 was shifted substantially to the right in KOP KO mice, with significant antinociception observed only at doses of 30 nmol or higher (Figure 4). These data confirmed significant mediation by KOP receptors of the observed antinociception.
Figure 4.
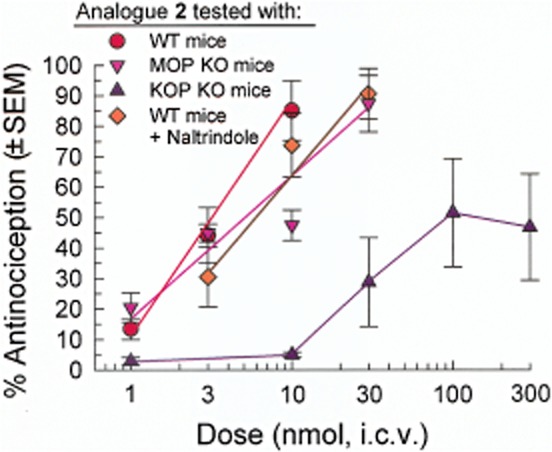
Further analysis of opioid receptor mediation of the antinociceptive activity of analogue 2. The antinociceptive dose–response of analogue 2 in the mouse 55°C warm-water tail-withdrawal assay was determined in MOP KO mice, KOP KO mice and C57BL/6J WT mice pretreated 15 min with naltrindole (20 mg·kg−1, i.p.) prior to administration of 2. Tail withdrawal latencies were measured 20 min after analogue administration, except in KOP KO mice where the tail withdrawal latencies were measured 50 min after analogue administration. Data shown are mean % antinociception ± SEM.
Antagonist activity
The Ala analogues of [D-Trp]CJ-15,208 were evaluated for their ability to antagonize the KOP receptor-selective agonist U50,488 (10 mg·kg−1, i.p.) (Figure 5). Consistent with the action of the parent compound 1 (Ross et al., 2012), the Ala1 (2) and D-Ala4 (5) analogues significantly antagonized U50,488 in a dose-dependent manner following a 3 h pretreatment [F(4,39) = 13.6; P < 0.0001 and F(4,39) = 42.0; P < 0.0001, respectively; Figure 5]. The Ala3 analogue 4 proved less potent, antagonizing U50,488 antinociception only after pretreatment with a higher dose [30 nmol, i.c.v.; F(3,32) = 17.3; P < 0.0001], while the D-NMePhe2 analogue 3 did not antagonize U50,488 at doses up to 30 nmol, i.c.v. [F(4,39) = 0.19; P = 0.94]. The duration of the KOP receptor antagonist activity of the three active alanine analogues differed (Figure 6). The duration of the antagonist activity produced by the Ala1 analogue (2, 1 nmol, i.c.v.) was similar to that of the parent macrocyclic peptide 1 (3 nmol, i.c.v.) (Ross et al., 2012), with significant KOP receptor antagonism detected for 6–8 h [F(4,39) = 12.8; P < 0.0001; Figure 6]. In contrast, the antagonist activity of analogue 5 (30 nmol, i.c.v.) was substantially reduced by 4 h, and had completely dissipated by 8 h [F(3,32) = 27.2; P < 0.0001]. The antagonist activity of the Ala3 analogue 4 dissipated even more rapidly, with significant KOP receptor antagonism detected after a 3 h pretreatment [F(3,32) = 18.1; P < 0.0001] that was gone by 4 h (P > 0.05, not significant; Figure 6).
Figure 5.
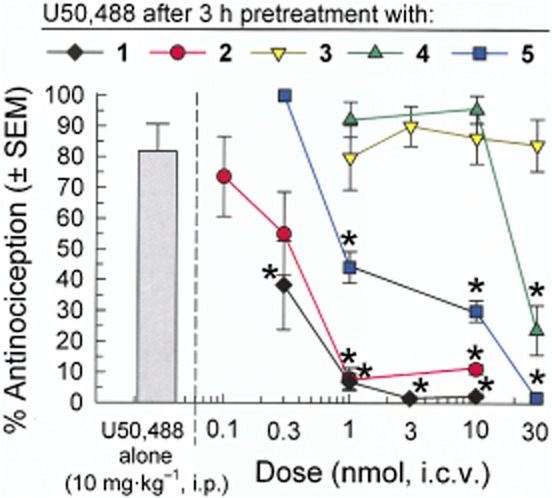
Dose-dependent antagonism of U50,488-induced antinociception by pretreatment with the alanine analogues 2, 4 and 5 compared with [D-Trp]CJ-15,208 [1; from Ross et al. (2012)] in the mouse 55°C warm-water tail-withdrawal assay. Mice were pretreated i.c.v. with the [D-Trp]CJ-15,208 or one of the analogues 2–5, 140 min prior to administration of the KOP receptor-selective agonist U50,488, and antinociceptive activity determined 40 min later. Alanine analogue 3 did not antagonize U50,488-induced antinociception at doses up to 30 nmol. Data shown are mean % antinociception ± SEM. *P < 0.05, significantly different from response of U50,488 administered alone; one-way anova followed by Tukey's post hoc test.
Figure 6.
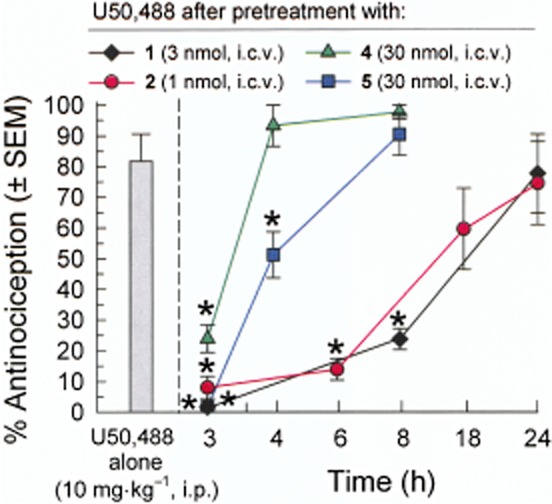
Duration of macrocyclic tetrapeptide-mediated antagonism of U50,488-induced antinociception in the mouse 55°C warm-water tail-withdrawal assay. Mice were pretreated i.c.v. with [D-Trp]CJ-15,208 [1, 3 nmol; from Ross et al. (2012)] or one of the analogues displaying KOP receptor antagonism (2, 1 nmol; 4 or 5, 30 nmol each), and the antinociception induced by U50,488 was determined 3–24 h later. Tail withdrawal latencies were determined 40 min after U50,488 administration. Data shown are mean % antinociception ± SEM. *P < 0.05, significantly different from response of U50,488 administered alone; one-way anova followed by Tukey's post hoc test.
The receptor selectivity of the analogues' antagonist activity was determined by further examining the peptides' ability to antagonize the MOP receptor-preferring agonist morphine (10 mg·kg−1, i.p.) and the DOP receptor-selective agonist SNC-80 (100 nmol, i.c.v., Figure 7). At the same doses tested for KOP receptor antagonism, none of the alanine analogues significantly antagonized morphine [F(5,45) = 1.54; P = 0.20]. Although a globally significant effect was detected [F(5,46) = 2.68; P = 0.03], none of the individual macrocyclic tetrapeptides significantly antagonized SNC-80-induced antinociception (P > 0.05, Tukey's post hoc test).
Figure 7.
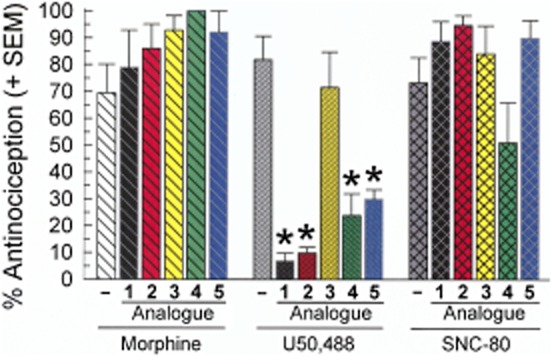
Opioid receptor selectivity of antagonism by the alanine analogues of [D-Trp]CJ-15,208 in the mouse 55°C warm-water tail-withdrawal assay. Antinociception induced by morphine (left group) or SNC-80 (right group) was not significantly decreased by a 3 h pretreatment with 1 [3 nmol; from Ross et al. (2012)], 2 (10 nmol), 3 (30 nmol), 4 (30 nmol) or 5 (10 nmol), in contrast to the antinociceptive effect of U50,488 (centre group), which was significantly antagonized by pretreatment with 1, 2, 4 and 5. Tail withdrawal latencies were determined 40 min after selective agonist administration. Data shown are mean % antinociception ± SEM. *P < 0.05, significantly different from response of U50,488 administered alone; one-way anova followed by Tukey's post hoc test.
Prevention of stress-induced reinstatement of cocaine CPP
We evaluated the ability of the Ala1 (2) and D-Ala4 (5) analogues to prevent reinstatement of cocaine CPP (Figure 8). Following 4 days of cocaine place conditioning, mice demonstrated significant CPP [F(3,827) = 50.6; P < 0.0001] and subsequent extinction 3 weeks after conditioning (Figure 8B). Mice were then pretreated daily for two days with either analogue 2 (10 nmol, i.c.v.) or analogue 5 (30 nmol, i.c.v.) and subjected to forced swimming or an additional round of cocaine conditioning (see schematic, Figure 8A). Mice pretreated with vehicle demonstrated significant reinstatement of cocaine CPP after exposure to forced swimming [F(5,693) = 35.1; P < 0.0001] or cocaine [F(5,703) = 40.6; P < 0.0001]. Both alanine analogues 2 and 5 prevented stress-induced reinstatement of cocaine CPP (P < 0.005; Figure 8B, central bars), but not cocaine-induced reinstatement (P > 0.05; Figure 8B, rightmost bars), when administered 2 h prior to the forced swim or administration of cocaine, a time point coinciding with their KOP receptor antagonist activity. These results are consistent with the action of other KOP receptor-selective antagonists (Carey et al., 2007; Aldrich et al., 2009), including the parent peptide 1 (Ross et al., 2012). Because the Ala1 analogue 2 demonstrated short-duration KOP receptor agonism (<2 h), additional mice were treated with analogue 2 for 5, 30 or 60 min prior to cocaine exposure (Figure 8C). Mice pretreated with analogue 2 for shorter time periods (5 or 30 min) prior to the additional cycle of cocaine place conditioning demonstrated significant prevention of cocaine-induced reinstatement [F(8,772) = 25.6; P < 0.0001] (Figure 8C). Pretreatment of mice demonstrating extinction of cocaine CPP with the Ala1 analogue 5 min before place preference testing, but without the additional cocaine conditioning, did not result in a significant change from the extinction response (Figure 8C, rightmost bar).
Discussion and conclusions
Overall, the in vitro pharmacological results for the alanine-substituted analogues of [D-Trp]CJ-15,208 were strikingly similar to those observed for the corresponding substitutions in the natural product CJ-15,208 containing L-Trp (Aldrich et al., 2011). In general, alanine substitution had a greater impact on binding affinities for KOP than for MOP receptors in both macrocyclic tetrapeptides. Substitution of the Trp residue in both peptides substantially decreased binding affinity for both KOP and MOP receptors, suggesting the importance of the indole moiety for receptor binding, while substitution of Phe1 with Ala increased affinity for both KOP and MOP receptors. However, in our studies, the substitution of Phe3 with Ala appeared to be better tolerated in [D-Trp]CJ-15,208 than in the L-Trp isomer (Aldrich et al., 2011).
As observed with the alanine analogues of CJ-15,208 (Aldrich et al., 2011), the alanine analogues of the D-Trp isomer exhibited in vivo pharmacological profiles that were unexpected based on their opioid receptor affinities and activity in the [35S]GTPγS assay in vitro. Consistent with the results for the alanine analogues of CJ-15,208, agonist activity was not detected in vitro in the GTPγS assay at KOP or MOP receptors for any of the alanine analogues of 1. While the parent peptide 1 exhibited significant antinociceptive activity in the 55°C warm-water tail-withdrawal assay only at an elevated dose (30 nmol, i.c.v.), alanine substitution for any of the residues increased antinociceptive potency in this assay following i.c.v. administration. Interestingly, the relative potencies of the alanine analogues of 1 in the in vivo assay were almost completely reversed compared with their relative affinities for KOP receptors, with the D-Ala4 analogue 5 exhibiting the most potent antinociceptive activity in vivo despite its micromolar affinity for these receptors. Removal of the indole in analogue 5 resulted in a profile more like the natural product CJ-15,208, with mixed KOP/ MOP receptor agonism and KOP receptor antagonist activity.
For most of the analogues, multiple opioid receptors appear to contribute to their antinociceptive activity. For all the alanine analogues pretreatment with nor-BNI significantly reduced the antinociceptive activity of the analogues, suggesting KOP receptor mediation of their antinociception. This is in contrast to the alanine analogues of CJ-15,208 where the antinociceptive activity was mediated predominantly by MOP receptors (Aldrich et al., 2011). For all of the analogues except analogue 2, pretreatment with the MOP receptor antagonist β-FNA significantly decreased antinociceptive activity, suggesting that these receptors also contributed to their antinociception. Similar to the results for KOP receptors, the relative antinociceptive potencies of analogues 3, 4 and 5 in vivo did not correlate with their affinities for MOP receptors. Naltrindole also significantly decreased the antinociception produced by analogues 3 and 4, suggesting DOP receptors contributed to their observed antinociceptive activity. This was unexpected given their very low affinity for DOP receptors. Together, these results suggest that all of the amino acid side chains contribute to minimizing the agonist (antinociceptive) activity in vivo of the parent macrocyclic peptide 1.
The KOP receptor mediation of the antinociceptive activity of the Ala1 analogue 2 was confirmed in the KOP KO mice, which showed a significant rightward shift in the dose–response curve compared with WT mice (Figure 4). The lowered maximal response in the KOP KO mice suggests analogue 2 also produces partial agonism through the other opioid receptors, but only at higher doses. The antinociceptive potency of analogue 2 was not significantly different in MOP KO mice compared with WT mice, further supporting the minimal contribution of MOP receptors to the antinociception of analogue 2 in spite of its relatively high affinity for this receptor (Ki = 27 nM).
Each of the alanine analogues of [D-Trp]CJ-15,208 except the D-NMeAla2 analogue 3 antagonized the antinociceptive effect of U50,488 in vivo, suggesting that the D-Pro2 residue is important for KOP receptor antagonist activity in the parent peptide. The lack of such antagonist activity of analogue 3 in vivo was unexpected given its KOP receptor antagonist activity in the GTPγS assay. While the potency of analogue 3 as a KOP receptor antagonist in vitro was modest, it proved more potent than analogues 4 and 5, which did exhibit KOP receptor antagonism in vivo. These results contrast with those for the alanine analogues of CJ-15,208 where all of the analogues exhibited KOP receptor antagonism in vivo (Aldrich et al., 2011). Consistent with other peptide KOP receptor antagonists (Aldrich et al., 2009; 2011; Ross et al., 2012), the duration of the antagonist activity of analogues 2, 4 and 5 was relatively short (<18 h). These results contrast with the established non-peptide KOP receptor-selective antagonists nor-BNI, GNTI and JDTic and their exceptionally long duration of antagonist activity (weeks after a single dose) (Metcalf and Coop, 2005). Notably, the very short KOP receptor antagonism of analogue 4 (<4 h) suggests it could serve as a useful pharmacological tool to study KOP receptor-mediated physiological and pharmacological activities.
While these analogues produce antinociception and, except for analogue 3, antagonist activity that is clearly mediated through opioid receptors, the differences between the in vitro and in vivo activity profiles suggest that these compounds produce their opioid activity through more complex mechanisms than utilized by typical opioid receptor ligands. As discussed above, similar differences were noted for the alanine analogues of the natural product CJ-15,208 (Aldrich et al., 2011), and have also been found for other novel antinociceptive compounds that are structurally unrelated to these macrocyclic peptides (Reilley et al., 2010). There are a number of unconventional mechanisms that could potentially account for the differences observed between the in vitro and in vivo assays, including activation of different signalling pathways, modulation of opioid receptors by other proteins in vivo (including other receptors) and modulation of endogenous opioid peptide levels (Szeto et al., 2003). We are very interested in understanding the mechanism(s) behind the complex in vivo activity profiles of these compounds that could offer important new insights into both opioid function and approaches to drug discovery. These investigations, however, will involve extensive additional studies that are beyond the scope of this report describing the basic characterization of these unusual opioid compounds. Nevertheless, the observed differences highlight the important contributions that in vivo studies can make to understanding the pharmacological activity profiles of novel ligands.
Because of their KOP receptor antagonist activity, alanine analogues 2 and 5 were examined for their ability to prevent both stress- and cocaine-induced reinstatement of cocaine CPP. Consistent with the action of other KOP receptor-selective antagonists (Carey et al., 2007; Aldrich et al., 2009; Ross et al., 2012), pretreatment with either peptide at a time point when they exhibited KOP receptor antagonist activity (2 h) prevented stress-induced, but not cocaine-primed, reinstatement of cocaine CPP. When evaluated for its ability to prevent cocaine-primed reinstatement following shorter pretreatment times, the Ala1 analogue 2 prevented reinstatement with a time course consistent with its antinociceptive activity. Evaluation in KOP KO mice (Figure 4) verified that at this dose (10 nmol) used in the CPP studies the agonist activity of 2 was solely due to activation of KOP receptors. The ability of analogue 2 to prevent both stress- and cocaine-induced reinstatement of extinguished cocaine-seeking behaviour is an unusual property. This sets this peptide apart from typical single-action KOP receptor ligands; compounds that produce only KOP receptor antagonism do not prevent cocaine-primed reinstatement (Beardsley et al., 2005; Carey et al., 2007; Aldrich et al., 2009), and repeated treatment with KOP receptor-selective agonists may reinstate cocaine-seeking behaviour (Redila and Chavkin, 2008). The results with analogue 2 are consistent with those found for the mixed agonist/ KOP receptor antagonist CJ-15,208, which also prevented cocaine-induced reinstatement following a short pretreatment time (Aldrich et al., 2013). With both KOP receptor agonist and antagonist activities, these macrocyclic peptides can counteract both stress- and drug-induced triggers of reinstatement in a time-dependent manner, suggesting compounds with broader therapeutic value in maintaining abstinence in cocaine abusers.
In conclusion, different in vivo opioid profiles were identified in this series of alanine analogues. The Ala1 analogue 2 appears to produce both its agonist and antagonist activity predominantly through KOP receptors, whereas the Ala4 analogue 5 exhibits a profile of mixed KOP/MOP receptor agonism with KOP receptor antagonist activity similar to the natural product CJ-15,208. In contrast to the other macrocyclic peptides examined, the D-NMeAla2 analogue 3 exhibited only antinociceptive activity without KOP receptor antagonism in vivo, suggesting the importance of the D-Pro2 residue to such antagonist activity in [D-Trp]CJ-15,208. Taken together, these novel ligands with distinct opioid activity profiles in vivo represent intriguing compounds for further study. The successful prevention by analogue 2 of both cocaine- and stress-induced reinstatement of an extinguished cocaine–CPP response supports the development of these novel macrocyclic tetrapeptides for potential therapeutic application as treatments for drug abuse treatments.
Acknowledgments
This research was supported by grants from the National Institute on Drug Abuse R01 DA018832, R01 DA023924 and R01 DA032928, and funds from the State of Florida, Executive Office of the Governor's Office of Tourism, Trade, and Economic Development.
Glossary
- CHO
Chinese hamster ovary
- CJ-15,208
cyclo[Phe-D-Pro-Phe-Trp]
- CPP
conditioned-place preference
- DAMGO
[D-Ala2,NMePhe4,glyol5]enkephalin
- DMF
N,N-dimethylformamide
- DMSO
dimethyl sulfoxide
- DOP receptor
δ opioid receptor
- DPDPE
cyclo[D-Pen2,D-Pen5]enkephalin (Pen = penicillamine)
- [D-Trp]CJ-15,208
cyclo[Phe-D-Pro-Phe-D-Trp]
- Dyn
dynorphin
- β-FNA
β-funaltrexamine
- GNTI
5′-guanidinylnaltrindole
- JDTic
(3R)-7-hydroxy-N-((1S)-1-[[(3R,4R)-4-(3-hydroxyphenyl)-3,4-dimethyl-1-piperidinyl]methyl]-2-methylpropyl)-1,2,3,4-tetrahydro-3-isoquinolinecarboxamide
- KOP receptor
κ opioid receptor
- KOP KO
KOP receptor gene-disrupted mice
- MOP receptor
μ opioid receptor
- MOP KO
MOP receptor gene-disrupted mice
- nor-BNI
nor-binaltorphimine
- SNC-80
(+)-4-[(αR)-α-((2S,5R)-4-allyl-2,5-dimethyl-1-piperazinyl)-3-methoxybenzyl]-N,N-diethylbenzamide
- WT
wild type
- U50,488
(±)-trans-3,4-dichloro-N-methyl-N-[2-(1-pyrrolidinyl)cyclohexyl] benzeneacetamide
Conflict of interest
The authors state no conflict of interest.
Supporting information
Additional Supporting Information may be found in the online version of this article at the publisher's web-site:
http://dx.doi.org/10.1111/bph.12664
Appendix S1 Analytical data for compounds 2-5.
References
- Aldrich JV, McLaughlin JP. Peptide kappa opioid receptor ligands: potential for drug development. AAPS J. 2009;11:312–322. doi: 10.1208/s12248-009-9105-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aldrich JV, Patkar KA, McLaughlin JP. Zyklophin, a systemically active selective kappa opioid receptor peptide antagonist with short duration of action. Proc Natl Acad Sci U S A. 2009;106:18396–18401. doi: 10.1073/pnas.0910180106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aldrich JV, Kulkarni SS, Senadheera SN, Ross NC, Reilley KJ, Eans SO, et al. Unexpected opioid activity profiles of analogs of the novel peptide kappa opioid receptor ligand CJ-15,208. ChemMedChem. 2011;6:1739–1745. doi: 10.1002/cmdc.201100113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aldrich JV, Senadheera SN, Ross NC, Ganno ML, Eans SO, McLaughlin JP. The macrocyclic peptide natural product CJ-15,208 is orally active and prevents reinstatement of extinguished cocaine-seeking behavior. J Nat Prod. 2013;76:433–438. doi: 10.1021/np300697k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The concise guide to pharmacology 2013/14: G protein-coupled receptors. Br J Pharmacol. 2013;170:1459–1581. doi: 10.1111/bph.12445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arttamangkul S, Ishmael JE, Murray TF, Grandy DK, DeLander GE, Kieffer BL, et al. Synthesis and opioid activity of conformationally constrained dynorphin A analogues. 2. Conformational constraint in the ‘address’ sequence. J Med Chem. 1997;40:1211–1218. doi: 10.1021/jm960753p. [DOI] [PubMed] [Google Scholar]
- Beardsley PM, Howard JL, Shelton KL, Carroll FI. Differential effects of the novel kappa opioid receptor antagonist, JDTic, on reinstatement of cocaine-seeking induced by footshock stressors vs cocaine primes and its antidepressant-like effects in rats. Psychopharmacology (Berl) 2005;183:118–126. doi: 10.1007/s00213-005-0167-4. [DOI] [PubMed] [Google Scholar]
- Bennett MA, Murray TF, Aldrich JV. Identification of arodyn, a novel acetylated dynorphin A-(1–11) analogue, as a κ opioid receptor antagonist. J Med Chem. 2002;45:5617–5619. doi: 10.1021/jm025575g. [DOI] [PubMed] [Google Scholar]
- Brugel TA, Smith RW, Balestra M, Becker C, Daniels T, Hoerter TN, et al. Discovery of 8-azabicyclo[3.2.1]octan-3-yloxy-benzamides as selective antagonists of the kappa opioid receptor. Part 1. Bioorg Med Chem Lett. 2010;20:5847–5852. doi: 10.1016/j.bmcl.2010.07.113. [DOI] [PubMed] [Google Scholar]
- Carey AN, Borozny K, Aldrich JV, McLaughlin JP. Reinstatement of cocaine place-conditioning prevented by the peptide kappa-opioid receptor antagonist arodyn. Eur J Pharmacol. 2007;569:84–89. doi: 10.1016/j.ejphar.2007.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng YC, Prusoff WH. Relationship between the inhibition constant (Ki) and the concentration of inhibitor which causes 50 percent inhibition (IC50) of an enzymatic reaction. Biochem Pharmacol. 1973;22:3099–3108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- Delaforge M, Andre F, Jaouen M, Dolgos H, Benech H, Gomis JM, et al. Metabolism of tentoxin by hepatic cytochrome P-450 3A isozymes. Eur J Biochem. 1997;250:150–157. doi: 10.1111/j.1432-1033.1997.00150.x. [DOI] [PubMed] [Google Scholar]
- Dolle RE, Michaut M, Martinez-Teipel B, Seida PR, Ajello CW, Muller AL, et al. Nascent structure–activity relationship study of a diastereomeric series of kappa opioid receptor antagonists derived from CJ-15,208. Bioorg Med Chem Lett. 2009;19:3647–3650. doi: 10.1016/j.bmcl.2009.04.105. [DOI] [PubMed] [Google Scholar]
- Eans SO, Ganno ML, Reilley KJ, Patkar KA, Senadheera SN, Aldrich JV, et al. The macrocyclic tetrapeptide [D-Trp]CJ-15,208 produces short acting κ opioid receptor antagonism in the CNS after oral administration. Br J Pharmacol. 2013;169:426–436. doi: 10.1111/bph.12132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frankowski KJ, Hedrick MP, Gosalia P, Li K, Shi S, Whipple D, et al. Discovery of small molecule kappa opioid receptor agonist and antagonist chemotypes through a HTS and hit refinement strategy. ACS Chem Neurosci. 2012;3:221–236. doi: 10.1021/cn200128x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerra G, Fantoma A, Zaimovic A. Naltrexone and buprenorphine combination in the treatment of opioid dependence. J Psychopharmacol. 2006;20:806–814. doi: 10.1177/0269881106060835. [DOI] [PubMed] [Google Scholar]
- Grimwood S, Lu Y, Schmidt AW, Vanase-Frawley MA, Sawant-Basak A, Miller E, et al. Pharmacological characterization of 2-methyl-N-((2′-(pyrrolidin-1-ylsulfonyl)biphenyl-4-yl)methyl)propan-1-amine (PF-04455242), a high-affinity antagonist selective for kappa opioid receptors. J Pharmacol Exp Ther. 2011;339:555–566. doi: 10.1124/jpet.111.185108. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG. Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulkarni SS, Ross NC, McLaughlin JP, Aldrich JV. Synthesis of cyclic tetrapeptide CJ 15,208: a novel kappa opioid receptor antagonist. Adv Exp Med Biol. 2009;611:269–270. doi: 10.1007/978-0-387-73657-0_121. [DOI] [PubMed] [Google Scholar]
- McGrath JC, Drummond GB, McLachlan EM, Kilkenny C, Wainwright CL. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaughlin JP, Hill KP, Jiang Q, Sebastian A, Archer S, Bidlack JM. Nitrocinnamoyl and chlorocinnamoyl derivatives of dihydrocodeinone: in vivo and in vitro characterization of mu-selective agonist and antagonist activity. J Pharmacol Exp Ther. 1999;289:304–311. [PubMed] [Google Scholar]
- Metcalf MD, Coop A. Kappa opioid antagonists: past successes and future prospects. AAPS J. 2005;7:E704–E722. doi: 10.1208/aapsj070371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patkar KA, Yan X, Murray TF, Aldrich JV. [Nα-BenzylTyr1,cyclo(D-Asp5,Dap8)]-dynorphin A-(1–11)NH2 cyclized in the ‘address’ domain is a novel kappa-opioid receptor antagonist. J Med Chem. 2005;48:4500–4503. doi: 10.1021/jm050105i. [DOI] [PubMed] [Google Scholar]
- Peters MF, Zacco A, Gordon J, Maciag CM, Litwin LC, Thompson C, et al. Identification of short-acting kappa-opioid receptor antagonists with anxiolytic-like activity. Eur J Pharmacol. 2011;661:27–34. doi: 10.1016/j.ejphar.2011.04.017. [DOI] [PubMed] [Google Scholar]
- Redila VA, Chavkin C. Stress-induced reinstatement of cocaine seeking is mediated by the kappa opioid system. Psychopharmacology (Berl) 2008;200:59–70. doi: 10.1007/s00213-008-1122-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reilley KJ, Giulianotti M, Dooley CT, Nefzi A, McLaughlin JP, Houghten RA. Identification of two novel, potent, low-liability antinociceptive compounds from the direct in vivo screening of a large mixture-based combinatorial library. AAPS J. 2010;12:318–329. doi: 10.1208/s12248-010-9191-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross NC, Kulkarni SS, McLaughlin JP, Aldrich JV. Synthesis of CJ-15,208, a novel κ-opioid receptor antagonist. Tetrahedron Lett. 2010;51:5020–5023. doi: 10.1016/j.tetlet.2010.07.086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross NC, Reilley KJ, Murray TF, Aldrich JV, McLaughlin JP. Novel opioid cyclic tetrapeptides: Trp isomers of CJ-15,208 exhibit distinct opioid receptor agonism and short-acting kappa opioid receptor antagonism. Br J Pharmacol. 2012;165:1097–1108. doi: 10.1111/j.1476-5381.2011.01544.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothman RB, Gorelick DA, Heishman SJ, Eichmiller PR, Hill BH, Norbeck J, et al. An open-label study of a functional opioid κ antagonist in the treatment of opioid dependence. J Subst Abuse Treat. 2000;18:277–281. doi: 10.1016/s0740-5472(99)00074-4. [DOI] [PubMed] [Google Scholar]
- Runyon SP, Brieaddy LE, Mascarella SW, Thomas JB, Navarro HA, Howard JL, et al. Analogues of (3R)-7-hydroxy-N-[(1S)-1-{[(3R,4R)-4-(3-hydroxyphenyl)-3,4-dimethyl-1-piperidinyl]methyl}-2-methylpropyl)-1,2,3,4-tetrahydro-3-isoquinolinecarboxamide (JDTic). Synthesis and in vitro and in vivo opioid receptor antagonist activity. J Med Chem. 2010;53:5290–5301. doi: 10.1021/jm1004978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito T, Hirai H, Kim Y-J, Kojima Y, Matsunaga Y, Nishida H, et al. CJ-15,208, a novel kappa opioid receptor antagonist, from a fungus, Ctenomyces serratus ATCC15502. J Antibiot. 2002;55:847–854. doi: 10.7164/antibiotics.55.847. [DOI] [PubMed] [Google Scholar]
- Schild HO. pA, a new scale for the measurement of drug antagonism. Br J Pharmacol Chemother. 1947;2:189–206. doi: 10.1111/j.1476-5381.1947.tb00336.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senadheera SN, Kulkarni SS, McLaughlin JP, Aldrich JV. Improved synthesis of CJ-15,208 isomers and their pharmacological activity at opioid receptors. In: Lebl M, editor. Peptides: Building Bridges. San Diego, CA: American Peptide Society; 2011. pp. 346–347. [Google Scholar]
- Siebenaller JF, Murray TF. Hydrostatic pressure alters the time course of GTP[S] binding to G proteins in brain membranes from two congeneric marine fishes. Biol Bull. 1999;197:388–394. doi: 10.2307/1542793. [DOI] [PubMed] [Google Scholar]
- Szeto HH, Soong Y, Wu D, Qian X, Zhao GM. Endogenous opioid peptides contribute to antinociceptive potency of intrathecal [Dmt1]DALDA. J Pharmacol Exp Ther. 2003;305:696–702. doi: 10.1124/jpet.102.048561. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1 Analytical data for compounds 2-5.