


Abstract
While the structure of mature ribosomes is analyzed in atomic detail considerably less is known about their assembly process in living cells. This is mainly due to technical and conceptual hurdles. To analyze ribosome assembly in vivo, we designed and engineered an Escherichiacoli strain—using chromosomal gene knock-in techniques—that harbors large and small ribosomal subunits labeled with the fluorescent proteins EGFP and mCherry, respectively. A thorough characterization of this reporter strain revealed that its growth properties and translation apparatus were wild-type like. Alterations in the ratio of EGFP over mCherry fluorescence are supposed to indicate ribosome assembly defects. To provide proof of principle, subunit specific assembly defects were provoked and could be identified by both manual and fully automated fluorometric in vivo assays. This is to our knowledge the first methodology that directly detects ribosome assembly defects in vivo in a high-throughput compatible format. Screening of knock-out collections and small molecule libraries will allow identification of new ribosome assembly factors and possible inhibitors.
INTRODUCTION
Ribosomes belong to the most abundant and most important macromolecular entities in pro- and eukaryotic cells. Bacterial ribosomes consist of a small 30S and a larger 50S subunit, which together form the active 70S ribosome. While structural, functional and mechanistic aspects of the assembled ribosome are well explored (see for review (1,2)), less is known about the complex production process that yields ribosomal subunits and ribosomes. This process is referred to as ribosome subunit assembly. The task is to create two ribonucleoprotein particles out of 54 ribosomal proteins (r-proteins) and three ribosomal RNAs (5S, 16S and 23S rRNA) (3). The assembly of both ribosomal subunits follows a hierarchical order, where the binding of so-called early assembly r-proteins to rRNA precedes the integration of late assembly r-proteins. The assembly process is further coordinated by a pool of diverse r-protein and rRNA modifying enzymes (as well as rRNA chaperones and processing factors), which are in summary termed ribosome biogenesis factors (4,5). While ribosomal subunit assembly in vitro does not require any extra-ribosomal components (6,7) assembly in vivo strictly depends on ribosome biogenesis factors; there is no reason to assume that all of which are identified yet.
Ribosome assembly is also supposed to be an attractive target for new antimicrobial agents (8,9) for at least three reasons. (i) Inhibition of assembly by targeting rRNA or rRNA/r-protein interactions is supposed to slow the development of resistance due to the multiplicity of rRNA genes in most microorganisms (10). (ii) Due to significant differences in the assembly process of bacterial and mitochondrial ribosomes (11), less or no side effects on mitochondrial ribosomes are expected. (iii) Currently no specific inhibitors of ribosome assembly are available (12), since a suitable screening concept is lacking. This is mainly because of the difficulty to distinguish effects caused by inhibition of assembly from those caused by inhibition of translation (13,14).
A possible solution to this problem would be a screening method that focuses specifically on subunit assembly. Selective inhibition of assembly of one subunit should result in both impaired cell growth and an asymmetry in the number of functional ribosomal subunits (15), whereas inhibition of translation should not necessarily change the ratio between both ribosomal subunits. The method should be able to quantify the amount of both subunits in vivo, in order to identify subunit specific inhibitors by systematic screening approaches.
Ribosomal subunit assembly is known to be a fast process, taking place in the single-digit minute range in vivo (16). At the same time, the free cytosolic pool of ribosomal proteins is close to zero in most of the cases (17–19). It follows that the amounts of distinct ribosomal proteins from either subunit present in a cell can be taken as reference for the amount of the respective subunit in vivo.
Here, we report a method that can monitor the relative amounts of ribosomal subunits in vivo. The principle is that one protein of each ribosomal subunit is tagged with a fluorescent protein (FP). We have chosen a member of the GFP family for tagging the ribosomal protein L19 (L19-EGFP) and mCherry for S2 (S2-mCherry). The coding sequences of both FPs were chromosomally inserted in frame with the respective r-protein genes. Because subunit assembly is fast compared to fluorophore formation of FPs (20), it is granted that fluorescence does not appear before the r-protein-FP fusion protein becomes part of a ribosomal subunit. Since the two fluorophores are spectrally well distinguishable, each subunit is represented and quantifiable by a specific fluorescence signal. Reporter cells carrying both fusion proteins grew like wild-type cells indicating that ribosomes modified with both fusion proteins did not suffer from assembly defects and were as active. As proof of principle, assembly defects were generated for either subunit by depletion of the genes rpsQ (coding for S17) or rplC (L3). Polysome profiles revealed the expected assembly defects, which were detectable in vivo as well, using the reporter strain in a fluorescence-based assay. The fluorometric in vivo studies were corroborated by fluorescence microscopic imaging.
MATERIALS AND METHODS
Media, buffers, antibodies and antibiotics
LB medium (5 g yeast extract, 10 g trypton, 5 g NaCl/ l); M9 medium (12.8 g Na2HPO4.7H2O, 3 g KH2PO4, 0.5 g NaCl, 1 g NH4Cl, 0.4% glucose/ l, 2 mM MgSO4, 0.1 mM CaCl2); phosphate buffered saline (PBS) (137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 1.8 mM KH2PO4, pH 7.4); S2 and L19 specific antisera, raised in sheep were obtained from Dr. Nierhaus. Horseradish peroxidase (HRP)-conjugated rabbit anti-sheep secondary antibodies were from Jackson ImmunoResearch (CodeNo: 313–035–003; LotNo: 106383). HRP-substrate: for detection a mixture of 1 ml solution A + 100 μl solution B + 1 μl solution C was freshly prepared (solution A: 0.1 mM TRIS (pH8.6), 25 mg Luminol, 100 ml distilled H20; solution B: 11 mg p-hydroxycoumaric acid, in 10 ml DMSO; solution C: H2O2 (30%). The following antibiotics were used in concentrations as indicated: Ampicillin 100 μg/ml (Applichem-A0839,0100), kanamycin 50 μg/ml (Roth-T832.4), chloramphenicol 7 μg/ml (Sigma-C0378) and erythromycin 100 μg/ml (Sigma-E6367).
Plasmids and bacterial strains
rpsQ and rplC were amplified from genomic Escherichiacoli DNA using specific primers with SacI and XbaI restriction sites, respectively. Digested inserts were ligated with an opened pTRC99a vector (lacIq, trc promoter, bla-gene for ampicillin resistance) (21) using SacI/ XbaI restriction sites generating pTRC-rpsQ and pTRC-rplC, respectively. Plasmids were brought into DH5α-Z1 by chemical transformation for amplification and were isolated using Qiagen mini-prep kit.
MC4100 (F− [araD139]B/r Δ(argF-lac)169 lambda− e14- flhD5301 Δ(fruK-yeiR)725 (fruA25) relA1 rpsL150(strR) rbsR22 Δ(fimB-fimE)632(::IS1) deoC1); DY330 (W3110 Δ lacU169 gal490 λcI857 Δ (cro-bioA))(22), DH5α-Z1 (F endA1 hsdR17(rk mk+) supE44 thi-1 recA1 gyrA relA1 Δ (lacZYA-argF)U169 deoR Ф80 lacZΔ M15 LacR TetR and Spr) (23).
λ-red recombineering
Coding sequences of EGFP and mCherry (in combination with kanamycin resistance cassettes (kanR) derived from plasmid pKD4 (24)) with flanking homologous regions (40–50 nucleotides) for 3′prime genomic insertion in frame with rplS and rpsB, respectively, were amplified using Phusion DNA-Polymerase. Polymerase chain reaction (PCR) products of the expected size were purified and brought into competent DY330 cells via electroporation. Successful genomic integration was verified by colony PCR and DNA-sequencing. Genetic modifications were transferred to strains of interest using P1-phage transduction. Resistance cassettes were eliminated by transforming strains of interest with pCP20 (25) encoding FLP-recombinase from Saccharomyces cerevisiae, which eliminates kanR flanked by FRT-sites. Strains were cured from pCP20 by incubation at 42°C for 15 h.
Gene deletions of rpsQ and rplC were achieved similarly. First, strains of interest were transformed with pTRC-rpsQ or pTRC-rplC, respectively. The coding sequences of kanR with flanking homologous regions (40–50 nucleotides) upstream and downstream the gene of interest were generated by PCR using Phusion DNA-Polymerase. PCR products of the expected size were purified and brought into competent DY330 cells (harboring pTRC-rpsQ or pTRC-rplC) via electroporation. Single colonies growing on LB agar plates supplemented with kanamycin and IPTG were analyzed by colony PCR for replacement of the target gene. Gene deletions were transferred to strains of interest transformed with the appropriate complementation plasmid using P1-phage transduction. Pre-cultures of deletion strains were exclusively incubated in LB medium supplemented with ampicillin (100 μg/ml), kanamycin (50 μg/ml) and IPTG (1 mM).
In vivo fluorescence microscopy
E. coli cells of different strains were cultured either in LB or M9 medium, as indicated. Fifteen microliter of cell suspension was transferred to 0.5 ×0.5 cm agarose pads (0.5% agarose w/v in PBS) located on a slide and sealed with a cover slip. Samples were immediately analyzed by fluorescence microscopy using a Zeiss AX10 with 100× oil immersion objective (Plan Apochromat) and equipped with a Photometrics Cascade II 512 EMCCD camera. Images taken from identical specimens with different filter settings (red and green channels) were adjusted to the same dynamic range (using image-J software) and merges were built.
Cell growth analysis
Growth on solid media (LB agar): Stationary E. coli cells were diluted in LB medium to an initial cell density of OD600 = 0.025. 1:5 serial dilutions were prepared and transferred to LB agar plates with a plating stamp. Agar plates were incubated at 20, 30, 37 and 42°C until visible single colonies had formed.
Growth in liquid media: Stationary E. coli cells as indicated (cultured in LB medium) were diluted in LB medium to an initial cell density of OD600 = 0.05 (for incubation at 20°C) or 0.025 (for incubation at 37 and 42°C). Alternatively, stationary pre-cultures of the indicated strains were washed and diluted in M9 medium to an initial cell density of OD600 = 0.05. Cell suspensions were cultured in baffled flasks in a water bath incubator with a shaking frequency of 200 rpm until stationary phase was reached or a maximum of 10 h had passed. Cell density was determined using a photometer (Amersham ultrospec 3110 pro). Growth rates were calculated for periods of exponential growth.
SDS-PAGE and immunoblot analysis of purified ribosomes
Proteins from cell lysates (20 μg total protein) and purified ribosomes (15 pmol) from the indicated strains were resolved by a 13% SDS gel. Proteins were stained using Coomassie brilliant blue 250G.
For immunoblot analysis proteins from cell lysates (3 μg total protein) and purified ribosomes (3 pmol) were resolved by a 13% SDS gel and blotted to nitrocellulose membranes, which were decorated with S2 and L19 specific anti-sera (both 1:5000 in TBS +3% (w/v) milk powder) raised in sheep. An HRP-conjugated rabbit anti-sheep secondary antibody (1:10 000 in TBS + 3% (w/v) milk powder) was used in combination with HRP-substrate to allow immunodetection. Chemi-luminescence was monitored using LAS 3000 imager (Fuji Film). Additional bands appear upon incubation with S2 specific antisera (indicated by asterisks) due to chromophore hydrolysis of ds red derivatives (26).
Purification of ribosomes by sucrose cushion centrifugation
E. coli cells were cultured in LB medium at 37°C to cell densities as indicated, harvested by centrifugation, resuspended in buffer I (100 mM TRIS, 10 mM MgCl2, 100 mM NaCl, 15% sucrose, 100 μg/ml chloramphenicol, pH7.5), flash-frozen and stored at −80°C. Frozen cell pellets were resuspended in 3x volumes buffer II (10 mM MgCl2, 100 mM NaCl) and cells were lyzed using Fastprep-24. Three hundred microliter of cleared lysates was loaded on 700 μl 20% sucrose cushion (20 mM TRIS, 10 mM MgCl2, 100 mM KCl, 5 mM ß-mercaptoethanol, 20% sucrose (w/v), pH7.5) and centrifuged in an S140-AT rotor for 1:20 h at 65 krpm (220 000 average g). Pellets containing the ribosomes were resuspended in buffer III (10 mM TRIS, 12 mM MgCl2, 30 mM NaCl, 4 mM ß-mercaptoethanol, pH7.5) analyzed fluorometrically or subjected to sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE) and immunoblot analysis.
Sucrose gradient centrifugation and polysome analysis
Stationary pre-cultures of the individual strains were washed and diluted in M9 medium to OD600 = 0.05 and cultured at 37°C to cell densities as indicated. Chloramphenicol (250 μg/ml) was added 5 min before cells were harvested by centrifugation, flash-frozen and stored at −80°C. Frozen cell pellets were resuspended in buffer IV (10 mM TRIS, 10 mM MgCl2, 100 mM NH4Cl, 250 μg/ml chloramphenicol, pH7.5) and cells were lyzed using Fastprep-24. Cleared lysates (0.5 ml of a solution with A260 = 15 or 20) were loaded on 10–40% sucrose gradients (in buffer V: 10 mM TRIS, 10 mM MgCl2, 100 mM NH4Cl, 0.5 mM DTT, 1xCompleteTM (Roche, 05056489001), pH7.5) and centrifuged in a Sorvall TH-641 rotor for 2:40 h at 41 krpm. A254 profiles of sucrose centrifugates were obtained using a Teledyne Isco gradient reader. Fractions of the sucrose gradient were collected in 96-well plates (five drops per well) for further fluorometric analysis.
For testing MCrg with antibiotics, cells were washed and diluted in M9 medium to OD600 = 0.1 and grown to OD600 = 0.15 before chloramphenicol (7 μg/ml) or erythromycin (100 μg/ml) were added. Cells were cultured at 25°C. After 3 h of incubation cells were harvested and processed as described above with two exceptions: No Chloramphenicol was added 5 min before harvesting and 10–25% sucrose gradients were used for separation of ribosomal particles.
Agarose gel electrophoresis
Twenty microliter of sucrose gradient fractions was mixed with 6xDNA sample buffer (Thermo Scientific) and loaded on a 1% agarose gel. The RNA content was separated at 150 V for 1 h. The gel was stained in an ethidium bromide solution for 15 min and RNA bands were visualized at 302 nm on an UV-transilluminator (UVP).
Fluorometric analyses
Manual measurements using Fluorospectrometer (Jasco FP-6500): 1 ml aliquots of cell suspension of various strains was transferred to quartz cuvettes and EGFP- (excitation 480 nm/ emission 510 nm ± 5 nm band width) and mCherry-specific fluorescence intensities (excitation 580 nm/ emission 610 nm ± 5 nm band width) were determined. Fluorescence ratios were calculated by dividing EGFP by mCherry fluorescence intensities. Background-corrected ratios were normalized to the reporter strain (MCrg) and plotted in bar charts.
The fully automated in vivo assay was accomplished by a Freedom EVO® 200 robotic platform (Tecan) equipped with robotic manipulator arm, multi channel arm 384 (MCATM 384), carousel (LPT 220 EVOTM), monitored incubator option (MIO2TM) and microplate reader (Tecan F500).
Stationary cells of various strains were washed and diluted in M9 medium to a cell density of OD600 = 0.05. Eighty microliter aliquots of each strain was transferred to a 384-well plate in quadruplicates. Cells were incubated for 10 h at 37°C in a monitored incubator (MIO2TM) with 8.5 Hz shaking frequency. The samples were analyzed in the microplate reader in 1-h intervals for EGFP- and mCherry-specific fluorescence, using filter combinations 485/535 nm and 535/612 nm, respectively. Fluorescence ratios were calculated by dividing EGFP by mCherry fluorescence intensities. Background-corrected ratios were normalized to the reporter strain (MCrg) and plotted in bar charts. Cell densities were determined simultaneously by detecting the absorbance at 650 nm ± 5 nm (A650). Obtained values were plotted in a spread chart. Calculations and diagrams were made using Magellan 7 (Tecan) and Graph Pad Prism v6 (Graph Pad) software packages.
Sucrose fraction analysis using Infinite F500 (Tecan) fluorescence microplate reader: Sucrose gradient fractions collected in 96-well plates (five drops per well) were analyzed for EGFP- and mCherry-specific fluorescence using filter combinations 485/535 nm and 535/612 nm, respectively. Fluorescence intensities were normalized to a fraction containing 70S ribosomes (first, or second peak, as indicated).
RESULTS
Rationale
In order to generate a reporter strain able to indicate ribosomal subunit assembly defects, we selected ribosomal protein candidates from each subunit that are: (i) distant from functional sites, (ii) accessible to C-terminal tagging (27,28) with FPs, (iii) essential (29), (iv) late assembly proteins (17) and (v) subject of feedback regulation. The ribosomal proteins S2 and L19 fulfill all these criteria: Their surface exposed C-termini (Figure 1B) allow convenient tagging (with mCherry and EGFP). Due to their essentiality, growth will be severely hampered if the fusion proteins do not fully complement the wild-type protein's function. According to in vivo ribosome assembly maps (Supplementary Figure S1), both are late assembly proteins and consequently absent in premature ribosomal particles. In addition, feedback regulation by autogenous (30) and non-autogenous control (31), respectively, makes sure that they are not produced in excess. The two latter features are of equal importance as soon as subunit specific assembly defects appear, since in that case fluorescence emission is not only a relative measure for the abundance of a ribosomal subunit but rather a quality criterion reflecting the ‘intact’ portion of the respective subunit.
Figure 1.

Overview of constructed strains and 70S ribosome structure. (A) Given are the names of the constructed strains as used in this study (lab nomenclature in brackets), relevant genotype and fluorescent fusion proteins produced. pTRC-rpsQ, pTRC-rplC: complementation plasmids with copies of the chromosomally deleted genes. Endogenous genes are shown as gray arrows, genes encoding FPs as colored boxes. Genes to be deleted were replaced by kanamycin resistance cassettes (KanR). mCherry gene and protein portions are shown in red, EGFP accordingly in green. (B) Surface representation of an E. coli 70S ribosome crystal structure. The 16S rRNA is colored in light gray proteins of the small subunit in yellow. 23S and 5S rRNA are shown in dark gray, proteins of the large subunit in cyan. S2 is highlighted in red, L19 in green. Their surface exposed C-termini are shown in purple. The figure was generated with pymol, based on PDB files 3R8S and 4GD1 (28).
Physiological and biochemical characterization of the engineered strains
We used the technique of lambda red recombineering (22,24) to generate E. coli strains harboring modified genes coding for S2-mCherry (MCr) and L19-EGFP (MCg) fusion proteins (Figure 1A). Using phage transduction the final reporter strain MCrg was constructed, producing both S2-mCherry and L19-EGFP fusion proteins. When analyzed by fluorescence microscopy, this strain showed a normal morphology and notably a non-homogenous distribution of both red and green fluorescence (Figure 2A). This is in agreement with earlier observations demonstrating that ribosomes localize to cell poles and avoid the nucleoid region (32–34).
Figure 3.
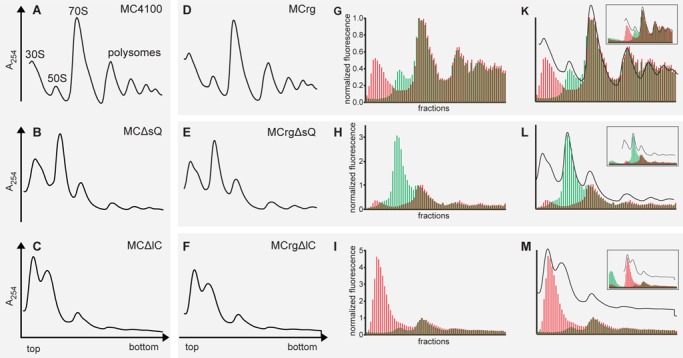
Polysome analysis and fluorescence detection of sucrose fractions. Cells were grown in M9 medium at 37°C to OD600 = 0.4 and harvested. Lysates were subjected to sucrose gradient centrifugation. Centrifugates were analyzed by A254 detection and fractionated. Polysome profiles derived from (A) MC4100, (B) MCΔsQ, (C) MCΔlC, (D) MCrg, (E) MCrgΔsQ, (F) MCrgΔlC. Sucrose gradient fractions of samples D–F were analyzed for EGFP- and mCherry-specific fluorescence and normalized results are given in bar charts for (G) MCrg, (H), MCrgΔsQ and (I) MCrgΔlC. Superposition of A254 profiles and corresponding fluorescence bar charts: (K) MCrg, (L) MCrgΔsQ, (M) MCrgΔlC. The inserts show fluorescence analysis of all available fractions from each sucrose gradient run. Red bars: normalized mCherry fluorescence; Green bars: normalized EGFP fluorescence. Fluorescence was normalized to 70S peak (‘monosome’) where subunits are supposed to be present in 1:1 ratio. However, the 70S peak contains the tail of the 50S peak, therefore normalization leads to underestimation of the EGFP signal intensity.
To exclude that tagging r-proteins with fluorescent moieties interferes with regular cell functions and growth, we analyzed the reporter strains in more detail. Spot tests revealed that growth of the genetically engineered strains did not differ from that of the wild-type strain at various temperatures (Figure 2B). To analyze potential differences in growth more precisely, wild-type and engineered strains were grown to stationary phase at different temperatures and growth rates were calculated (Figures 2C and D). It turned out that the growth rate of MCrg was <10% reduced compared to the wild-type strain at all temperatures tested.
Figure 2.

Physiological and biochemical characterization. (A) Fluorescence micrographs of MCrg cells. Cells were grown in LB medium at 37°C to mid-logarithmic phase and spotted onto a thin agarose pad. The agarose pad was sealed with a cover slip and subjected to microscopic analysis. Scale bar: 5 μm. (B) Growth comparison on solid medium: Cells of the indicated strains were spotted onto LB agar in a serial dilution and incubated at the given temperatures. For details see Materials and methods. (C) Growth comparison in liquid medium: Cells of the indicated strains were grown in LB medium at 37°C for 6 h. OD600 values were determined every hour and plotted against time. (D) Cells as indicated were grown at 20, 37 and 42°C to stationary phase. Growth rates were calculated and normalized values are given for each strain at each incubation temperature. N = 3. (E) Ribosomes from the indicated strains were isolated by sucrose cushion centrifugation and subjected to SDS-PAGE and western blot (WB) analysis. For immunodetection S2 and L19 specific antisera were used. Asterisks denote typical product of chromophore hydrolysis of ds red derivatives (26).
Next, the protein content of MCr-, MCg- and MCrg-derived ribosomes was analyzed by SDS-PAGE and immunoblotting (Figure 2E). While MCr and MCg ribosomes contained one fusion protein (migrating at 60 and 45 kDa, respectively), two fusion proteins were observed in MCrg ribosomes. Importantly, the fusion proteins seemed to be present in amounts similar to the wild-type proteins (see also Supplementary Figure S2).
Collectively, the data indicate that features such as growth behavior and functional competence of the ribosomes of the MCrg reporter strain are similar to those of the parental strain.
Generation of ribosome subunit specific assembly defects and in vitro analysis
To qualify MCrg as reporter strain for ribosome subunit assembly, it was necessary to provoke subunit specific assembly defects. The most obvious way to do so is to reduce expression levels of individual r-protein genes (15). Literature screening revealed two interesting candidates. A strain with a temperature sensitive point mutation in rpsQ (encoding S17) and an E. coli mutant lacking methylation of L3 (encoded by rplC) suffered from ribosome assembly defects (35,36). In addition, both proteins bind to rRNA and initiate (37) or at least stabilize (38) the assembly process of the subunit they associate with.
Therefore, we generated conditional gene knock-outs of rpsQ and rplC in MC4100 and MCrg strain backgrounds (Figure 1A). The resulting strains carried plasmids containing wild-type copies of the genes deleted from the chromosome. Consequently, these strains were able to grow on LB plates containing the inductor IPTG, while its absence prohibited cell growth (Supplementary Figure S3). However, the withdrawal of IPTG in liquid cultures should result in impaired growth and in subunit specific assembly defects (15) as soon as the number of intact ribosomes becomes limiting.
To this end, we grew MC4100, MCΔsQ and −ΔlC cells in the absence of IPTG to mid-logarithmic phase and examined the ribosomes by sucrose gradient centrifugation and polysome profile analysis (Figures 3A–C). Wild-type ribosomes showed the expected pattern consisting of 30S-, 50S-, 70S-, and polysome peaks (Figure 3A), whereas depletion of rpsQ (Figure 3B) led to reduced amounts of 70S ribosomes and polysomes and increased the 50S compared to the 30S peak. Likewise, depletion of rplC reduced the amount of 70S ribosomes but increased the number of 30S relative to 50S subunits (Figure 3C). This was expected because both absence of rpsQ and rplC should result in defective small and large ribosomal subunits, respectively. Consequently, the reduced number of functional subunits limited the amount of monosomes and polysomes.
Similarly, ribosome profiles were obtained from MCrg, MCrgΔsQ and MCrgΔlC cells (Figures 3D–F). Not surprisingly, their patterns were nearly identical to the MC4100 counterparts. Importantly, the fact that in these cases ribosomal subunits were labeled with FPs allowed fluorometric analysis of the sucrose gradient fractions (Figures 3G–I): Figure 3G shows the fluorescence analysis of the sucrose gradient-run profiled in Figure 3D. Red fluorescence represents the small, green fluorescence represents the large subunit forming a two-colored fluorescence equivalent of the polysome profile. The fact that Figure 3D and G was different readouts of the same sample allowed a superposition (Figure 3K), where A254 peaks and fluorescence peaks were in good agreement. The fluorescence readout of all sucrose fractions is given as insert, to demonstrate that there was no increased fluorescence in the low molecular weight fractions (region left of the 30S peak), indicating a low number of ribosomal precursors, which is in agreement with recent in vivo analysis (39).
Figure 3L provides three pieces of evidence. First of all, the green fluorescence signal matches the second A254 peak, indicating that the large subunit was fully intact. Secondly, there is a mismatch in intensity between the red fluorescence peak and the first A254 peak, meaning that a large portion of the small subunit—due to absence of rpsQ—was defective and consequently devoid of the late associating S2-mCherry. Finally, the insert shows that there was no accumulation of red fluorescence in the low molecular weight fractions, indicating proper autogenous control of S2-mCherry.
From Figure 3M it follows that the red fluorescence peak is in agreement with the first A254 peak, while the green fluorescence does not match the second A254 peak, which is in addition left shifted and overlaps the first. The interpretation is that the large subunit was defective, due to the absence of rplC and devoid of the late associating protein L19-EGFP. A closer investigation of the low molecular weight fractions in the insert uncovered a certain amount of green fluorescence, presumably due to incomplete feedback regulation of L19-EGFP. However, the portion of green compared to red fluorescence was clearly reduced.
In summary, selective assembly defects of the small and large ribosomal subunit could be provoked and were easily detectable by fluorescence analysis of sucrose gradient centrifugates, in particular when superimposed with the corresponding A254 profiles.
In vivo analysis of subunit specific assembly defects
The most important question was, whether subunit assembly defects would be detectable by fluorescence readout in vivo using our reporter strain? An assembly defect of the large subunit should reduce the amount of green fluorescence and consequently lower the normalized fluorescence emission ratio of EGFP/ mCherry, while an assembly defect of the small subunit in turn should increase the ratio.
First, we inoculated cultures with MCrg, MCrgΔsQ and MCrgΔlC cells in M9 medium and incubated them in Erlenmeyer flasks at 37°C for 9 h. Samples were taken every hour to determine cell densities and fluorescence intensities from which the normalized fluorescence ratios were calculated (Figure 4). The non-labeled strains MC4100, MCΔsQ and MCΔlC were cultivated in parallel to allow determination of background fluorescence, which was subtracted accordingly.
Figure 4.

Growth in flasks and manual fluorescence analysis of reporter strains. The strains MC4100, MCΔsQ, MCΔlC, MCrg, MCrgΔsQ and MCrgΔlC were cultured in Erlenmeyer flasks at 37°C in M9 medium for 9 h. In 1-h intervals samples were taken. (A) OD600 values were determined and (B) EGFP and mCherry fluorescence emission were detected and ratios were calculated for MCrg, MCrgΔsQ and MCrgΔlC. Fluorescence ratios of MCrg were normalized to 1. One exemplary growth curve is given and fluorescence ratios are means from three independent experiments; error bars show s.d. Growth in 384-well plates and fully automated fluorescence analysis of reporter strains. Aliquots of MC4100, MCΔsQ, MCΔlC, MCrg, MCrgΔsQ and MCrgΔlC cultures were transferred into 384- well plates in quadruplicates. Cells were grown in M9 medium at 37°C for 10 h. Samples were taken in 1-h intervals. (C) A650 values were determined and (D) EGFP and mCherry fluorescence emission were detected and ratios were calculated for MCrg, MCrgΔsQ and MCrgΔlC. Fluorescence ratios of MCrg were normalized to 1. A representative growth curve is shown and fluorescence ratios are mean values; n = 4, error bars show s.d.
After a lag-phase MCrg cells grew wild-type like, while MCrgΔsQ and MCrgΔlC cells showed impaired growth due to the individual assembly defects (Figure 4A). Normalized fluorescence ratios over time for MCrgΔlC lowered after 4 h and reached a minimum of 0.75 at 6 h of incubation, whereas the ratios of MCrgΔsQ rose after 6 h and reached a maximum of 2.4 at 9 h of incubation (Figure 4B).
To corroborate these findings, the same set of cells was transferred to 384-well plates and incubated at 37°C for 10 h in M9 medium. Using a robotic platform equipped with incubator, microplate reader and robotic arm allowed fully automated sample handling. Both A650 values and fluorescence intensities were measured (Figure 4) in 1-h intervals. While MCrg grew unperturbed, MCrgΔsQ and MCrgΔlC cells showed impaired growth and reached lower cell densities after 10 h (Figure 4C). The background-corrected and normalized fluorescence ratios of MCrgΔlC reached a minimum of 0.6 after 7 h, whereas the ratios of MCrgΔsQ increased instead reaching a maximum of 2.2 after 9 h (Figure 4D).
In addition, we analyzed MCrg, MCrgΔsQ and MCrgΔlC cells grown in M9 medium by in vivo fluorescence microscopy (Figure 5). MCrg cells were in mid-logarithmic growth phase, while MCrgΔsQ and MCrgΔlC had reached a cell density at which assembly defects had been recognized before. Microscopic investigation of MCrg revealed similar red and green fluorescence intensities, resulting in a yellow merge image. While MCrgΔsQ cells showed reduced red- and increased green fluorescence intensity, in MCrgΔlC cells red fluorescence was dominating. Hence, the results obtained by fluorometric analysis could be confirmed with an independent method.
Figure 5.
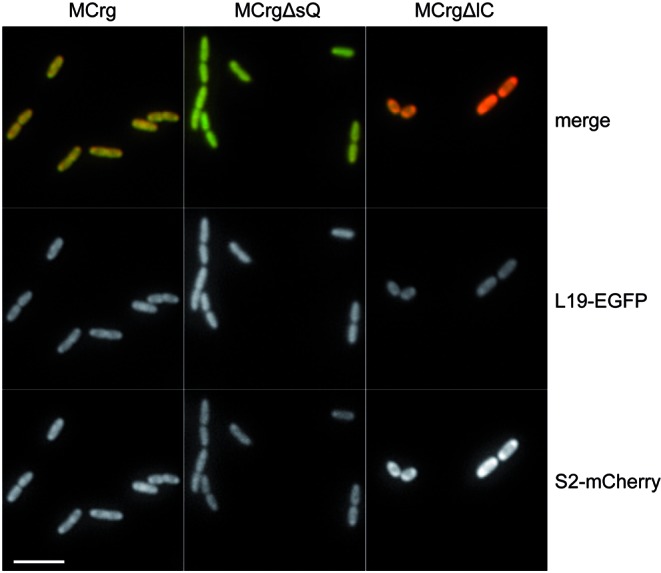
Fluorescence microscopic comparison of MCrg, MCrgΔsQ and MCrgΔlC. MCrg and MCrgΔsQ cells were grown in M9 medium to OD600 = 0.4, whereas MCrgΔlC cells were grown to OD600 = 0.12. Representative fluorescence images were taken. Lower row: red channel (S2-mCherry); middle row: green channel (L19-EGFP); the upper row is an overlay of both channels (merge). Scale bar: 5 μm.
Probing MCrg with chloramphenicol and erythromycin
It is known since a long time that application of translation inhibitors such as chloramphenicol (40,41) and erythromycin (42,43) provoke assembly defects of the large ribosomal subunit in E. coli. Albeit, it has been shown only recently that the two antibiotics cause assembly defects of both ribosomal subunits (14,44). The underlying mechanisms are discussed controversially (42–44). While our reporter strain cannot contribute to the clarification of mechanistic questions it should nevertheless be able to indicate, whether treatment of cells with chloramphenicol or erythromycin influences assembly of both ribosomal subunits equally or not.
To this end MCrg cells were cultured in M9 medium for 7 h in the absence or presence of chloramphenicol (7 μg/ml) or erythromycin (100 μg/ml). Treatment with either one led to impaired cell growth, as expected (Figure 6A). Fluorometric analysis revealed that the normalized fluorescence ratio in the presence of erythromycin decreased continuously within 7 h by 15%, while both chloramphenicol treated and non-treated cells did not show any significant change in the fluorescence ratio (Figure 6B). This indicates that the antibiotic erythromycin—in contrast to chloramphenicol—should disturb assembly of the large subunit stronger than that of the small subunit.
Figure 6.
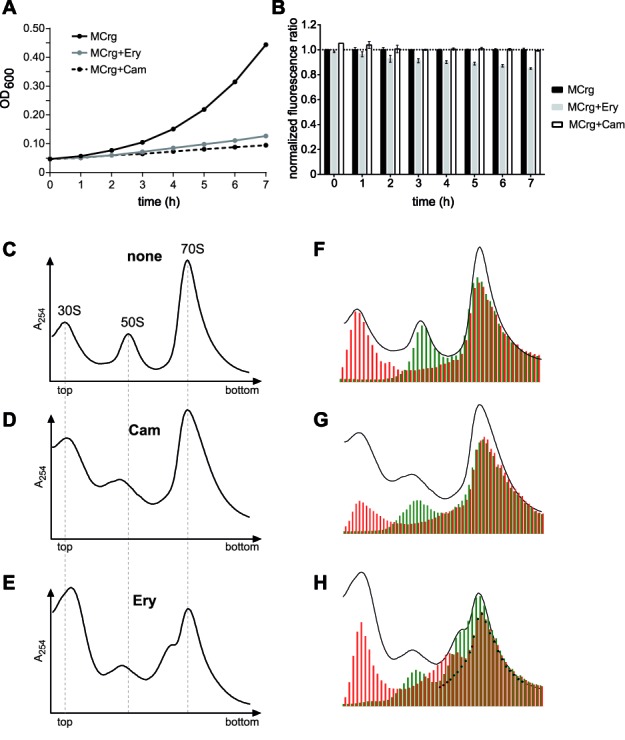
Testing MCrg with inhibitors of translation. Whole-cell analyses: MCrg cells were cultured in Erlenmeyer flasks at 25°C in M9 medium for 7 h in the absence and presence of antibiotics, as indicated. Samples were taken every hour. (A) OD600 values were determined and (B) EGFP and mCherry fluorescence emission were detected and ratios were calculated. Fluorescence ratios of MCrg were normalized to 1. Exemplary growth curves are given and fluorescence ratios are means from three independent experiments; error bars show s.d. Analyses of isolated ribosomal particles: Sucrose density gradient (10–25%) centrifugation profiles from (C) control cells with no antibiotic (none), (D) chloramphenicol (Cam) and (E) erythromycin (Ery) treated cells. Sucrose gradient fractions from (C), (D) and (E) were analyzed for fluorescence by a microplate reader. A254 profiles and fluorescence bar charts were superimposed for (F) control cells with no antibiotic (none), (G) chloramphenicol (Cam) and (H) erythromycin (Ery) treated cells. Cells in presence and absence of antibiotics were cultured in M9 medium at 25°C for 3 h before subsequent polysome analysis. Red bars: normalized mCherry fluorescence; Green bars: normalized EGFP fluorescence. Fluorescence was normalized to the first polysome peak (‘disome’) where subunits are present in 1:1 ratio.
This hypothesis was tested by analyzing ribosome profiles obtained from MCrg cells that grew in the presence of the antibiotics or without. Compared to untreated control cells (Figure 6C) the presence of chloramphenicol slightly reduced the 50S peak (Figure 6D), while erythromycin caused a somewhat stronger reduction of the 50S peak (Figure 6E). Fluorometric analyses of the sucrose gradient fractions supported the A254 profiles and exhibited surprising additional information.
In profiles derived from untreated cells A254 peaks and fluorescence peaks were in good agreement (Figure 6F).
The A254 profile derived from chloramphenicol treated cells (Figure 6G) showed a reduced and left shifted 50S peak but did not exhibit an obvious difference in the pattern of EGFP versus mCherry fluorescence. This indicated that the assembly process of small and large ribosomal subunits seemed to be perturbed equally, which is in agreement with the fluorescence ratios determined from whole cells (Figure 6B).
Erythromycin treatment resulted in an A254 profile with an apparently slightly stronger reduction of the 50S peak, which was confirmed by fluorescence readout (Figure 6H). Fluorescence analysis in addition delivered details of the unexpected composition of the 70S peak. Apart from a regular 70S ‘core’ (black dots) it seemed to be composed of exclusively red or green fluorescent components. However, it turned out that the green fluorescence particularly in the 50S peak and in the total profile was reduced compared to the profile derived from the control strain (Figure 6F). Again this is in agreement with the cell-based analysis where a 15% reduction of the ratio was obtained after 7 h of incubation in the presence of erythromycin. Detailed analyses of ribosome profiles obtained after antibiotic treatment covering the entire fractions and a quantitation of 16S and 23S rRNA levels are given in the supplementary material (Supplementary Figure S4).
Testing MCrg with the two antibiotics chloramphenicol and erythromycin demonstrated that the reporter strain reacts with a moderate change in the fluorescence ratio when treated with erythromycin. This indicates small relative changes between the intact portion of large and small ribosomal subunits, caused by erythromycin as shown by fluorescence-based analysis of the corresponding sucrose gradient fractions.
Taken together our in vivo studies demonstrated that subunit specific assembly defects were detectable in real time by both fluorometer- or fluorescence microscope-based readouts. Depletion of rpsQ led to an increased fluorescence ratio (EGFP/mCherry) indicating an assembly defect of the small subunit beginning after 6–7 h, whereas depletion of rplC resulted in decreased fluorescence ratios indicating an earlier assembly defect of the large subunit detectable after 5–6 h.
In addition, treatment of the reporter strain with the translation inhibitor erythromycin resulted in a moderate change of the EGFP/mCherry fluorescence ratio, reflecting changes in the ratio between intact large and small ribosomal subunits.
This justifies the use of MCrg as reporter strain detecting imbalances in the amounts of functional ribosomal subunits.
DISCUSSION
We report the design, construction and validation of the reporter strain MCrg harboring EGFP-labeled large- and mCherry labeled small ribosomal subunits that has growth features similar to the parental strain. We have provided proof of principle that this strain reveals assembly defects of ribosomal subunits by fluorescence-based readouts both in vitro and in vivo.
To our best knowledge this is the first time that two ribosomal proteins were labeled with FPs simultaneously, using chromosomal gene knock-in techniques. While English et al. (33) used r-proteins and RelA tagged with (photo-activatable) FPs to study mechanistic aspects of the stringent response, Bakshi et al. (32) used fusions of S2 and RNA polymerase with FPs to address questions of transcriptional and translational coupling. In our case, a thorough characterization of physiological and biochemical properties of our reporter strain demonstrated that there are no substantial limitations of the translation apparatus resulting from two ribosomal proteins fused to different FPs (Figure 2).
One major criterion for the selection of ribosomal protein candidates for fluorescence labeling was that their insertion into the subunit structure had to be late (Supplementary Figure S1). Consequently, the observed fluorescence will be a measure of intact ribosomal subunits. S2 and L19 fulfilled this criterion as shown by in vitro analysis (Figure 3).
To test our system for its response to ribosome assembly defects, we used conditional deletions of rpsQ and rplC, because so far no specific inhibitors of ribosome assembly are available. Ribosome profiles derived from the reporter strain MCrg and its derivatives MCrgΔsQ and MCrgΔlC demonstrated that fluorescence labeling of S2 and L19 allowed to observe and analyze assembly defects fast and accurately avoiding radio-isotope labeling. Sucrose gradient analysis of MCrg derived ribosomes therefore turned out to be a strong diagnostic in vitro tool to study ribosome assembly defects.
The reporter strain is similarly attractive for in vivo analyses of ribosome assembly defects. Its advantage is that it is self-calibrated in a way that one fluorescence serves as reference-value for the other. Therefore, the critical parameter is the ratio of green to red fluorescence (EGFP/mCherry) allowing investigation of samples irrespective of their cell count. From analysis of the unperturbed reporter strain over time a signature line EGFP/mCherry can be derived. Values above this critical ratio indicate assembly defects of the small ribosomal subunit, while values below suggest assembly defects of the large subunit.
However, the in vivo analysis requires an additional feature, the ribosomal reporter proteins must be under the regime of negative feedback regulation. This means that the relative concentration of the reporter protein must not exceed the amount of intact ribosomal subunit precursors they are supposed to associate with. The best guarantee for such a behavior is the so-called autogenous control. It is dedicated to a number of rRNA binding r-proteins (S1, S2, S4, S7, S8, S15, S20, L1, L4, L7/L12, L10 and L20 (45)) and refers to their ability to interact with their own mRNA and thereby blocking its translation.
When analyzing the small subunit specific assembly defect (using MCrgΔsQ) in vivo the fluorescence ratio increased by 100–150% compared to MCrg indicating both a decrease of functional small subunits and an accurate feedback regulation of S2-mCherry (Figure 3L). In vivo analysis of the large subunit assembly defect (using MCrgΔlC) resulted in decreased fluorescence ratios by 30–40% only, which was due to a decrease of functional large subunit but was compromised by insufficient feedback regulation of L19-EGFP (Figure 3M, insert). Importantly, it turned out that missing feedback regulation of L19-EGFP was less dramatic when MCrgΔlC was grown in M9 medium maybe due to more economic use of resources in minimal medium. L19 is supposed to be regulated by non-autogenous mechanisms possibly acting at transcriptional or translational levels (31). Therefore, its regulation could require additional proteins (adapters, proteases, transcription factors, etc.) and consequently protein translation, which is extremely reduced in MCrgΔlC due to lack of functional ribosomes. Nevertheless, it is difficult to find a better candidate, since late assembly proteins of the large subunit are not subject to autogenous control. Consequently, L19 might be not optimal but the best choice presently available. Finally, MCrgΔlC was able to indicate the genetically provoked assembly defect of the large subunit repeatedly and reliably.
To test how MCrg reacts upon treatment with classical antibiotics that inhibit translation, the reporter strain was grown in the presence of chloramphenicol or erythromycin. Chloramphenicol binds to the peptidyl transferase center (PTC) and inhibits fixation of the CCA-aminoacyl end of an aminoacyl-tRNA at the A-site region of the PTC (46), while the macrolide erythromycin binds in the upper region of the ribosomal exit tunnel and hinders protein synthesis by blocking a full occupation of the ribosomal tunnel (47). There is evidence that both antibiotics cause assembly defects of both ribosomal subunits (13,39) but the exact molecular mechanism is still under debate. Our analysis revealed three important findings.
MCrg should allow the identification of primary assembly inhibitors, in particular when considering cell growth. The two antibiotics in the concentrations used here (according to Siibak et al. (13)) led to strong growth impairment but little or no change in fluorescence ratios (Figure 6A and B). The opposite holds true for the two knock-out scenarios we tested that directly caused assembly defects and thereby mimic effects of primary assembly inhibitors (Figure 4). However, the example case of erythromycin treatment shows that in the search of assembly inhibitors, stringent criteria concerning cell growth and EGFP/mCherry ratio must be applied. The screening of small molecule libraries revealed that the growth of the reporter strain should exceed 30% and the EGFP/ mCherry ratios should be either clearly below 80% (for 50S assembly inhibitors) or above 150% (for 30S assembly inhibitors), compared with the untreated control (our unpublished results).
While the presence of both antibiotics leads to formation of defective ribosomal subunits, as indicated by A254 profiles and rRNA quantitation (Supplementary Figure S4B and C), according to our study erythromycin has a measurable stronger negative effect on the assembly of the large ribosomal subunit (Figure 6).
Even though chloramphenicol and erythromycin cause defective assembly intermediates of both subunits, no such particles were detected when analyzing the sucrose gradient fractions fluorometrically. This confirms our assumption that S2-mCherry and L19-EGFP are only present in fully assembled subunits.
Fluorometric analysis of sucrose fractions derived from MCrg cells after erythromycin treatment surprisingly revealed a heterogeneous composition of the 70S peak. We assumed that it contains a regular 70S ‘core’ that is depicted with black dots (Figure 6 H) and a broadened base (fractions 50–56 (Supplementary Figure S4C)). The base region of the 70S peak is obviously build by a portion of mCherry labeled particles, while an overshoot of EGFP-labeled particles contributes to the rest of the left 70S flank. One explanation for this ensemble could be the presence of assembled ribosomes with one regular and one defective—and therefore non-fluorescent—subunit. It is known that 22 of the 54 E. coli r-protein genes can be individually deleted (48). It follows that cells lacking certain r-proteins are viable and therefore capable of translation with incomplete ribosomal subunits (49,50). Well-documented cases are gene deletions of rpsO (S15) and rpsT (S20) (51,52). It might be possible that ribosomes lacking two or even more r-proteins are translationally active. Alternatively, it could be the case that proteins within incompletely assembled subunits are not stably integrated and fall of during sample preparation (i.e. sucrose gradient ultracentrifugation). This means the lower molecular weight 70S region (Supplementary Figure S4C, fractions 46–56) with dominating mCherry signal could consist of intact 30S and defective 50S particles. The higher molecular weight 70S region (fractions 56–66) with dominating EGFP signal could contain 50S and defective 30S particles. However, for an in-depth evaluation of this unexpected phenomenon detailed analyses of the material within the 70S region would be required, using techniques such as quantitative mass spectrometry or cryo-electron microscopy.
Collectively, the methodology described here appears to be suitable to screen knock-out collections and small molecule libraries to identify factors involved in ribosomal subunit assembly. This should pave the way for a first identification of assembly inhibitors and thus for the development of new classes of antimicrobial agents.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
Acknowledgments
We thank L. Sironi for excellent technical assistance in fluorescence microscopy. We would like to thank K. H. Nierhaus for critical reading of the manuscript and helpful suggestions. We are grateful to W. Boos and members of the Deuerling lab for proof reading of the manuscript and critical discussions.
FUNDING
Fellowship of the Zukunftskolleg from the University of Konstanz (to R.N.); German Science Foundation [DE 783/3–1, SFB 969/A01 to E.D.]; Human Frontier in Science Program [RGP0025/2012 to E.D.]. Source of Open Access funding: University of Konstanz.
Conflict of interest statement. A patent application is pending.
REFERENCES
- 1.Wilson D.N., Doudna Cate J.H. The structure and function of the eukaryotic ribosome. Cold Spring Harb. Perspect. Biol. 2012;4 doi: 10.1101/cshperspect.a011536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Frank J., Gonzalez R.L., Jr Structure and dynamics of a processive Brownian motor: the translating ribosome. Annu. Rev. Biochem. 2010;79:381–412. doi: 10.1146/annurev-biochem-060408-173330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.de Narvaez C.C., Schaup H.W. In vivo transcriptionally coupled assembly of Escherichia coli ribosomal subunits. J. Mol. Biol. 1979;134:1–22. doi: 10.1016/0022-2836(79)90411-x. [DOI] [PubMed] [Google Scholar]
- 4.Kaczanowska M., Ryden-Aulin M. Ribosome biogenesis and the translation process in Escherichia coli. Microbiol. Mol. Biol. Rev. 2007;71:477–494. doi: 10.1128/MMBR.00013-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shajani Z., Sykes M.T., Williamson J.R. Assembly of bacterial ribosomes. Annu. Rev. Biochem. 2011;80:501–526. doi: 10.1146/annurev-biochem-062608-160432. [DOI] [PubMed] [Google Scholar]
- 6.Traub P., Nomura M. Structure and function of E. coli ribosomes. V. Reconstitution of functionally active 30S ribosomal particles from RNA and proteins. Proc. Natl Acad. Sci. U.S.A. 1968;59:777–784. doi: 10.1073/pnas.59.3.777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nierhaus K.H., Dohme F. Total reconstitution of functionally active 50S ribosomal subunits from Escherichia coli. Proc. Natl Acad. Sci. U.S.A. 1974;71:4713–4717. doi: 10.1073/pnas.71.12.4713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Champney W.S. Bacterial ribosomal subunit assembly is an antibiotic target. Curr. Top. Med. Chem. 2003;3:929–947. doi: 10.2174/1568026033452186. [DOI] [PubMed] [Google Scholar]
- 9.Comartin D.J., Brown E.D. Non-ribosomal factors in ribosome subunit assembly are emerging targets for new antibacterial drugs. Curr. Opin. Pharmacol. 2006;6:453–458. doi: 10.1016/j.coph.2006.05.005. [DOI] [PubMed] [Google Scholar]
- 10.Tenson T., Mankin A. Antibiotics and the ribosome. Mol. Microbiol. 2006;59:1664–1677. doi: 10.1111/j.1365-2958.2006.05063.x. [DOI] [PubMed] [Google Scholar]
- 11.Bogenhagen D.F., Martin D.W., Koller A. Initial steps in RNA processing and ribosome assembly occur at mitochondrial DNA nucleoids. Cell Metab. 2014;19:618–629. doi: 10.1016/j.cmet.2014.03.013. [DOI] [PubMed] [Google Scholar]
- 12.Bharat A., Blanchard J.E., Brown E.D. A high-throughput screen of the GTPase activity of Escherichia coli EngA to find an inhibitor of bacterial ribosome biogenesis. J. Biomol. Screen. 2013;18:830–836. doi: 10.1177/1087057113486001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Maguire B.A. Inhibition of bacterial ribosome assembly: a suitable drug target. Microbiol. Mol. Biol. Rev. 2009;73:22–35. doi: 10.1128/MMBR.00030-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Siibak T., Peil L., Donhofer A., Tats A., Remm M., Wilson D.N., Tenson T., Remme J. Antibiotic-induced ribosomal assembly defects result from changes in the synthesis of ribosomal proteins. Mol. Microbiol. 2011;80:54–67. doi: 10.1111/j.1365-2958.2011.07555.x. [DOI] [PubMed] [Google Scholar]
- 15.Maguire B.A., Wild D.G. The roles of proteins L28 and L33 in the assembly and function of Escherichia coli ribosomes in vivo. Mol. Microbiol. 1997;23:237–245. doi: 10.1046/j.1365-2958.1997.2131578.x. [DOI] [PubMed] [Google Scholar]
- 16.Lindahl L. Intermediates and time kinetics of the in vivo assembly of Escherichia coli ribosomes. J. Mol. Biol. 1975;92:15–37. doi: 10.1016/0022-2836(75)90089-3. [DOI] [PubMed] [Google Scholar]
- 17.Chen S.S., Williamson J.R. Characterization of the ribosome biogenesis landscape in E. coli using quantitative mass spectrometry. J. Mol. Biol. 2013;425:767–779. doi: 10.1016/j.jmb.2012.11.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Marvaldi J., Pichon J., Delaage M., Marchis-Mouren G. Individual ribosomal protein pool size and turnover rate in Escherichia coli. J. Mol. Biol. 1974;84:83–96. doi: 10.1016/0022-2836(74)90213-7. [DOI] [PubMed] [Google Scholar]
- 19.Ulbrich B., Nierhaus K.H. Pools of ribosomal proteins in Escherichia coli. Studies on the exchange of proteins between pools and ribosomes. Eur. J. Biochem. 1975;57:49–54. doi: 10.1111/j.1432-1033.1975.tb02275.x. [DOI] [PubMed] [Google Scholar]
- 20.Lentini R., Forlin M., Martini L., Del Bianco C., Spencer A.C., Torino D., Mansy S.S. Fluorescent proteins and in vitro genetic organization for cell-free synthetic biology. ACS Synth. Biol. 2013;2 doi: 10.1021/sb400003y. [DOI] [PubMed] [Google Scholar]
- 21.Amann E., Ochs B., Abel K.J. Tightly regulated tac promoter vectors useful for the expression of unfused and fused proteins in Escherichia coli. Gene. 1988;69:301–315. doi: 10.1016/0378-1119(88)90440-4. [DOI] [PubMed] [Google Scholar]
- 22.Yu D., Ellis H.M., Lee E.C., Jenkins N.A., Copeland N.G., Court D.L. An efficient recombination system for chromosome engineering in Escherichia coli. Proc. Natl Acad. Sci. U.S.A. 2000;97:5978–5983. doi: 10.1073/pnas.100127597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lutz R., Bujard H. Independent and tight regulation of transcriptional units in Escherichia coli via the LacR/O, the TetR/O and AraC/I1-I2 regulatory elements. Nucleic Acids Res. 1997;25:1203–1210. doi: 10.1093/nar/25.6.1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Datsenko K.A., Wanner B.L. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl Acad. Sci. U.S.A. 2000;97:6640–6645. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cherepanov P.P., Wackernagel W. Gene disruption in Escherichia coli: TcR and KmR cassettes with the option of Flp-catalyzed excision of the antibiotic-resistance determinant. Gene. 1995;158:9–14. doi: 10.1016/0378-1119(95)00193-a. [DOI] [PubMed] [Google Scholar]
- 26.Gross L.A., Baird G.S., Hoffman R.C., Baldridge K.K., Tsien R.Y. The structure of the chromophore within DsRed, a red fluorescent protein from coral. Proc. Natl Acad. Sci. U.S.A. 2000;97:11990–11995. doi: 10.1073/pnas.97.22.11990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schuwirth B.S., Borovinskaya M.A., Hau C.W., Zhang W., Vila-Sanjurjo A., Holton J.M., Cate J.H. Structures of the bacterial ribosome at 3.5 A resolution. Science. 2005;310:827–834. doi: 10.1126/science.1117230. [DOI] [PubMed] [Google Scholar]
- 28.Dunkle J.A., Wang L., Feldman M.B., Pulk A., Chen V.B., Kapral G.J., Noeske J., Richardson J.S., Blanchard S.C., Cate J.H. Structures of the bacterial ribosome in classical and hybrid states of tRNA binding. Science. 2011;332:981–984. doi: 10.1126/science.1202692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bubunenko M., Baker T., Court D.L. Essentiality of ribosomal and transcription antitermination proteins analyzed by systematic gene replacement in Escherichia coli. J. Bacteriol. 2007;189:2844–2853. doi: 10.1128/JB.01713-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Aseev L.V., Levandovskaya A.A., Tchufistova L.S., Scaptsova N.V., Boni I.V. A new regulatory circuit in ribosomal protein operons: S2-mediated control of the rpsB-tsf expression in vivo. RNA. 2008;14:1882–1894. doi: 10.1261/rna.1099108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wikstrom P.M., Bystrom A.S., Bjork G.R. Non-autogenous control of ribosomal protein synthesis from the trmD operon in Escherichia coli. J. Mol. Biol. 1988;203:141–152. doi: 10.1016/0022-2836(88)90098-8. [DOI] [PubMed] [Google Scholar]
- 32.Bakshi S., Siryaporn A., Goulian M., Weisshaar J.C. Superresolution imaging of ribosomes and RNA polymerase in live Escherichia coli cells. Mol. Microbiol. 2012;85:21–38. doi: 10.1111/j.1365-2958.2012.08081.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.English B.P., Hauryliuk V., Sanamrad A., Tankov S., Dekker N.H., Elf J. Single-molecule investigations of the stringent response machinery in living bacterial cells. Proc. Natl Acad. Sci. U.S.A. 2011;108:E365–373. doi: 10.1073/pnas.1102255108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mascarenhas J., Weber M.H., Graumann P.L. Specific polar localization of ribosomes in Bacillus subtilis depends on active transcription. EMBO Rep. 2001;2:685–689. doi: 10.1093/embo-reports/kve160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Herzog A., Yaguchi M., Cabezon T., Corchuelo M.C., Petre J., Bollen A. A missense mutation in the gene coding for ribosomal protein S17 (rpsQ) leading to ribosomal assembly defectivity in Escherichia coli. Mol. Gen. Genet. 1979;171:15–22. doi: 10.1007/BF00274010. [DOI] [PubMed] [Google Scholar]
- 36.Lhoest J., Colson C. Cold-sensitive ribosome assembly in an Escherichia coli mutant lacking a single methyl group in ribosomal protein L3. Eur. J. Biochem. 1981;121:33–37. doi: 10.1111/j.1432-1033.1981.tb06425.x. [DOI] [PubMed] [Google Scholar]
- 37.Nowotny V., Nierhaus K.H. Initiator proteins for the assembly of the 50S subunit from Escherichia coli ribosomes. Proc. Natl Acad. Sci. U.S.A. 1982;79:7238–7242. doi: 10.1073/pnas.79.23.7238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ramaswamy P., Woodson S.A. Global stabilization of rRNA structure by ribosomal proteins S4, S17, and S20. J. Mol. Biol. 2009;392:666–677. doi: 10.1016/j.jmb.2009.07.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chen S.S., Sperling E., Silverman J.M., Davis J.H., Williamson J.R. Measuring the dynamics of E. coli ribosome biogenesis using pulse-labeling and quantitative mass spectrometry. Mol. Biosyst. 2012;8:3325–3334. doi: 10.1039/c2mb25310k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dodd J., Kolb J.M., Nomura M. Lack of complete cooperativity of ribosome assembly in vitro and its possible relevance to in vivo ribosome assembly and the regulation of ribosomal gene expression. Biochimie. 1991;73:757–767. doi: 10.1016/0300-9084(91)90055-6. [DOI] [PubMed] [Google Scholar]
- 41.Osawa S., Otaka E., Itoh T., Fukui T. Biosynthesis of 50 s ribosomal subunit in Escherichia coli. J. Mol. Biol. 1969;40:321–351. doi: 10.1016/0022-2836(69)90158-2. [DOI] [PubMed] [Google Scholar]
- 42.Chittum H.S., Champney W.S. Erythromycin inhibits the assembly of the large ribosomal subunit in growing Escherichia coli cells. Curr. Microbiol. 1995;30:273–279. doi: 10.1007/BF00295501. [DOI] [PubMed] [Google Scholar]
- 43.Usary J., Champney W.S. Erythromycin inhibition of 50S ribosomal subunit formation in Escherichia coli cells. Mol. Microbiol. 2001;40:951–962. doi: 10.1046/j.1365-2958.2001.02438.x. [DOI] [PubMed] [Google Scholar]
- 44.Siibak T., Peil L., Xiong L., Mankin A., Remme J., Tenson T. Erythromycin- and chloramphenicol-induced ribosomal assembly defects are secondary effects of protein synthesis inhibition. Antimicrob. Agents Chemother. 2009;53:563–571. doi: 10.1128/AAC.00870-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fu Y., Deiorio-Haggar K., Anthony J., Meyer M.M. Most RNAs regulating ribosomal protein biosynthesis in Escherichia coli are narrowly distributed to Gammaproteobacteria. Nucleic Acids Res. 2013;41:3491–3503. doi: 10.1093/nar/gkt055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ulbrich B., Mertens G., Nierhaus K.H. Cooperative binding of 3’-fragments of transfer ribonucleic acid to the peptidyltransferase center of Escherichia coli ribosomes. Arch. Biochem. Biophys. 1978;190:149–154. doi: 10.1016/0003-9861(78)90262-x. [DOI] [PubMed] [Google Scholar]
- 47.Tenson T., Lovmar M., Ehrenberg M. The mechanism of action of macrolides, lincosamides and streptogramin B reveals the nascent peptide exit path in the ribosome. J. Mol. Biol. 2003;330:1005–1014. doi: 10.1016/s0022-2836(03)00662-4. [DOI] [PubMed] [Google Scholar]
- 48.Shoji S., Dambacher C.M., Shajani Z., Williamson J.R., Schultz P.G. Systematic chromosomal deletion of bacterial ribosomal protein genes. J. Mol. Biol. 2011;413:751–761. doi: 10.1016/j.jmb.2011.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dabbs E.R. Mutants lacking individual ribosomal proteins as a tool to investigate ribosomal properties. Biochimie. 1991;73:639–645. doi: 10.1016/0300-9084(91)90043-z. [DOI] [PubMed] [Google Scholar]
- 50.Akanuma G., Nanamiya H., Natori Y., Yano K., Suzuki S., Omata S., Ishizuka M., Sekine Y., Kawamura F. Inactivation of ribosomal protein genes in Bacillus subtilis reveals importance of each ribosomal protein for cell proliferation and cell differentiation. J. Bacteriol. 2012;194:6282–6291. doi: 10.1128/JB.01544-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bubunenko M., Korepanov A., Court D.L., Jagannathan I., Dickinson D., Chaudhuri B.R., Garber M.B., Culver G.M. 30S ribosomal subunits can be assembled in vivo without primary binding ribosomal protein S15. RNA. 2006;12:1229–1239. doi: 10.1261/rna.2262106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tobin C., Mandava C.S., Ehrenberg M., Andersson D.I., Sanyal S. Ribosomes lacking protein S20 are defective in mRNA binding and subunit association. J Mol. Biol. 2010;397:767–776. doi: 10.1016/j.jmb.2010.02.004. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.