


Abstract
Phosphorothioate (PS) antisense oligonucleotides (ASOs) have been successfully developed as drugs to reduce the expression of disease-causing genes. PS-ASOs can be designed to induce degradation of complementary RNAs via the RNase H pathway and much is understood about that process. However, interactions of PS-ASOs with other cellular proteins are not well characterized. Here we report that in cells transfected with PS-ASOs, the chaperonin T-complex 1 (TCP1) proteins interact with PS-ASOs and enhance antisense activity. The TCP1-β subunit co-localizes with PS-ASOs in distinct nuclear structures, termed phosphorothioate bodies or PS-bodies. Upon Ras-related nuclear protein (RAN) depletion, cytoplasmic PS-body-like structures were observed and nuclear concentrations of PS-ASOs were reduced, suggesting that TCP1-β can interact with PS-ASOs in the cytoplasm and that the nuclear import of PS-ASOs is at least partially through the RAN-mediated pathway. Upon free uptake, PS-ASOs co-localize with TCP1 proteins in cytoplasmic foci related to endosomes/lysosomes. Together, our results indicate that the TCP1 complex binds oligonucleotides with TCP1-β subunit being a nuclear PS-body component and suggest that the TCP1 complex may facilitate PS-ASO uptake and/or release from the endocytosis pathway.
INTRODUCTION
Recent progress in sequencing techniques greatly advances personal genomic information and helps pinpoint genetic changes involved in diseases (1). To rapidly and fully exploit this wealth of genetic information that provides mechanistic insights into diseases, antisense technology provides an efficient approach to rationally design drugs to target disease-causing genes at the ribonucleic acid (RNA) level, with high specificity provided by base pairing between the antisense molecules and the target RNAs (2). Regulation of gene expression by antisense techniques can be achieved through different mechanisms, such as RNA interference (RNAi) mediated by the RNA-induced silencing complex (RISC) pathway, antisense oligonucleotides (ASOs) that modulate messenger RNA (mRNA) translation or pre-mRNA splicing and, importantly, ASO-directed RNA cleavage through the ribonuclease (RNase) H1 pathway (2,3). The only antisense drugs currently marketed are RNaseH-dependent ASOs; for example, Mipomersen targets apolipoprotein B mRNA and is approved for treatment of patients with homozygous familial hypercholesterolemia (4).
Second-generation RNase H-dependent ASO drugs are ∼20 nucleotides in size and are constructed as ‘gapmer’ with 10 deoxynucleotides flanked at both ends with five ribonucleotides (5-10-5 gapmer). To improve stability and potency, these ASOs have a phosphorothioate (PS) backbone and the flanking ribonucleotides are additionally modified with 2′-O-methyloxyethyl (MOE). PS-ASOs base-pair with the target RNAs and trigger sequence-specific cleavage of the RNA by RNase H1, which is present in both cytoplasm and nucleus (2,3,5). Inside cells, the potency of PS-ASOs is affected by many factors, including RNA structure and/or protein binding of target RNAs, the subcellular localization of the PS-ASOs, the nature of proteins that bind to the PS-ASOs and the recruitment of RNase H1 to the ASO/RNA duplex. Understanding of ASO–protein interactions may enable the design of more effective ASOs. ASOs modified with PSs bind more proteins than ASOs with a phosphodiester backbone (PO) (3). Differences in protein binding may result in different pharmacological profiles and alter subcellular distribution (6,7). Although some proteins that interact with PS-ASOs, such as albumin, nucleolin, hnRNP A1 and hnRNP C, have been previously identified (8,9), the effects of these proteins on ASO activity and subcellular localization have not been studied.
PS-ASOs distribute differently within cells depending on the delivery approaches used. In the case of free uptake, that is in the absence of delivery agent, PS-ASOs predominately localize in the cytoplasm, mainly in punctate structures and/or perinuclear structures corresponding to endosomes or lysosomes (10,11). Upon transfection, PS-ASOs can be quickly released from the cationic vesicles and appear diffusely in the cytoplasm, in the perinuclear structures and in some punctate dot structures (10,12,13). Transfected PS-ASOs can be quickly distributed to and mainly accumulate in the nucleus, and the nuclear localization positively correlates with the antisense activity of PS-ASOs (13). PS-ASOs undergo nucleocytoplasmic shuttling and the nuclear import of PS-ASOs may be mediated by an active import pathway (14,15). After intravenous administration to rats, ASOs accumulate in the nuclei and cytoplasm of liver cells (16). Thus the localization of ASOs in vivo can be predicted by the results of transfection experiments in cultured cells.
In the nucleus, fluorescently labeled PS-ASOs can exist in a diffuse form and in distinct nuclear structures, especially in multiple dot-like structures, termed phosphorothioate-bodies, or PS-bodies (15). PS-bodies do not co-localize with other known nuclear bodies, such as splicing speckles, promyelocytic leukemia bodies or centromeres. PS-bodies are formed in a dose-dependent-yet-sequence-independent manner when PS-ASOs are transfected or microinjected into cells (15). PS-body formation requires neither antisense activity nor active transcription (15). However, formation of PS-bodies requires PS modifications. The PS-bodies are round, 0.15–0.3 μm in diameter structures that are stable and undergo noticeable reorganization only during mitosis (15). PS-bodies form only in living cells, suggesting that cellular processes are involved.
To better understand the mechanisms of ASO action, we sought to identify cellular proteins that associate with PS-ASOs, with a hope to identify the protein components of PS-bodies. Using an affinity selection approach to isolate intracellular PS-ASO binding proteins, we identified and characterized around 50 cellular proteins that associate with PS-ASOs and found that many proteins can affect the subcellular localization and/or the antisense activity of ASOs (our unpublished data). As the first of several reports summarizing this work, in this report we show that T-complex 1 (TCP1, also known as CCT or TRiC) proteins bind to PS-ASOs. TCP1 is an ∼1-mDa protein complex belonging to the group II chaperone family. The TCP1 complex has a double-ring structure with central cavities where protein folding takes place (17). Each ring is composed of eight different subunits, α, β, γ, δ, ϵ, ζ, η and θ, which are ∼60 kDa each and share ∼30% sequence identity (18). TCP1 has been estimated to be involved in the folding of ∼10% of cellular proteins (19), including actin, tubulin, cyclin E1 and histone deacetylase (20). Like other chaperonins, the TCP1 complex requires magnesium ions for adenosine triphosphate (ATP) binding and hydrolysis (21). TCP1 proteins have been shown to be involved in multiple cellular processes, such as cell cycle progression and cytoskeletal organization (22).
Previous studies have demonstrated that the interactions between TCP1 and its major substrates, actin and tubulin, are sequence specific and electrostatic in nature (22). The interactions involve charged and polar residues in both the chaperonin and the substrate (20). In addition, actin, tubulin, huntingtin and Von Hippel–Lindau tumor suppressor are all recognized by distinct subsets of the TCP1 subunits via specific motifs (23,24), suggesting that the TCP1 complex can contact substrates through different mechanisms and that different subunits can have distinct roles. Despite the diversity of the substrate proteins, it is not clear if TCP1 proteins can interact with deoxyribonucleic acid (DNA) or RNA.
In the current study, TCP1 complex proteins were co-isolated with PS-ASOs, in a PS-dependent manner. Importantly, we found that TCP1 proteins affected the antisense activity of PS-ASOs. Only the TCP1-β subunit co-localized with PS-ASOs in the ASO-induced nuclear PS-bodies. Upon inhibition of expression of Ras-related nuclear protein (RAN), PS-body-like structures were observed in the cytoplasm. Thus, our study identified the first PS-body protein component and the first cellular ASO binding protein that enhances PS-ASO activity.
MATERIALS AND METHODS
Oligonucleotides, siRNAs and antibodies used are described in supplementary data
Cell culture and transfection
HeLa cells were cultured in Dulbecco's modified Eagle's medium supplemented with 10% Fetal Bovine Serium (FBS), 0.1-μg/ml streptomycin and 100 units/ml penicillin. Cells were seeded at ∼50% confluency and grown for 16 h before transfection. siRNA was transfected into cells using 5-μg/ml Lipofectamine RNAiMax (Life Technologies), based on manufacturer's procedure, at 3–5-nM final siRNA concentration. For ASO activity assays, HeLa cells were treated or not treated with siRNAs for 24 h and were then re-seeded in 6-well dishes at ∼75% confluency. After 4 h, cells were transfected with ASOs using 4-μg/ml Lipofectamine 2000 in Opti-MEM medium (Life Technologies). After 4 h, total RNA was isolated for quantitative real-time PCR (qRT-PCR) or whole cell extract was prepared for western analysis. For immunofluorescence staining, cells treated or not treated with siRNAs for 8 h or 30 h were reseeded in glass bottom dishes at ∼70% confluence. After overnight incubation, cells were transfected with fluorescently labeled ASOs for 4–6 h.
Affinity selection of ASO binding proteins
Neutravidin beads (50 μl) were incubated with 50 μl of 200-μM biotinylated ASO ISIS386652 at 4°C for 2 h in W-100 buffer [50-mM Tris–HCl (pH 7.5), 100-mM KCl, 5-mM ethylenediaminetetraacetic acid (EDTA), 0.1% NP-40, 0.05% sodium dodecyl sulphate (SDS)]. Beads were then incubated for 30 min in block buffer [10-mg/ml bovine serum albumin (BSA), 1.2-mg/ml glycogen and 0.2-mg/ml transfer RNA in W-100]. After three washes with W-100, ASO-coated beads were incubated at 4°C for 3 h with 300-μg HeLa cell extract prepared in buffer A [25-mM Tris-HCl pH 8.0, 5-mM MgCl2, 150-mM KCl, 10% glycerol, 0.5-mM Phenylmethanesulfonylfluoride (PMSF), 5-mM β-mercaptoethanol and one tablet of Protease Inhibitor Cocktail/50 ml (Roche)]. After three washes in tube with 500-μl W-300 buffer [50-mM Tris-HCl (pH 7.5), 300-mM KCl, 5-mM EDTA, 0.1% NP-40, 0.05% SDS], beads were transferred to a 1-ml column and washed seven times with W-300. Bound proteins were eluted by incubation with 100 μl of 50-μM ASO 116847 in W-100 at room temperature for 30 min. The eluted proteins were precipitated, separated on 4–12% polyacrylamide gel electrophoresis (PAGE) and visualized by silver staining. Interested protein bands were excised and identified by mass spectrometry (Alphalyse, CA, USA). To analyze the effects of PS backbone or 2′-modifications on protein binding, affinity selection was performed essentially as described above, except that 120-μl or 240-μl beads were pre-coated with 120 μl or 240 μl of 200-μM ASO 586183. After binding and washing, beads in W-100 buffer were equally separated into four or eight aliquots depending on the volume of beads and transferred to 1-ml columns. After removal of W-100 by centrifugation, proteins were eluted with 100 μl of 50-μM ASOs. Eluted proteins were analyzed by western analyses.
Western blot analysis
Equal portion of ASO binding proteins isolated by affinity selection or an equal amount of protein from whole cell extract (∼20 μg) was separated by 4–12% SDS-PAGE and transferred to a membrane. Blocking and detection of proteins with enhanced chemiluminescence (ECL) was performed as described previously (25).
Subcellular fractionation
Untreated HeLa cells or cells treated with indicated concentrations of PS-ASOs for indicated times were harvested, washed with phosphate buffered saline (PBS) and pelleted. Cytoplasmic and nuclear fractions were prepared using Qproteome Nuclear Protein kit (Qiagen), based on the manufacturer's instructions. Cytoplasmic and nuclear proteins were separated by 4–12% SDS-PAGE and analyzed by western.
RNA preparation and qRT-PCR
Total RNA was prepared from HeLa cells using RNeasy mini kit (Qiagen). qRT-PCR using TaqMan primer probe sets was performed as described previously (25).
Immunofluorescence staining
Cells were grown in glass bottom culture dishes (MatTek) for 16 h and were transfected or not transfected with fluorescently labeled ASOs for 4–6 h. For free uptake, cells were incubated with 1000-nM fluorescent-labeled ASOs for 16–24 h. Immunofluorescence staining was performed as described (26), with minor revisions. Briefly, cells were fixed with 4% formaldehyde for 30 min at room temperature and were permeabilized for 5 min using 0.15% Triton X-100 in PBS. Following three washes with 1 × PBS, cells were treated with blocking buffer (1-mg/ml BSA in 1 × PBS) at room temperature for 30 min and were incubated with primary antibodies (1:100) at room temperature for 2 h. After three washes with wash buffer (0.1% NP-40 in 1 × PBS), cells were incubated with secondary antibodies (1:200) in blocking buffer at room temperature for 1 h and washed three times with wash buffer. For double staining, two antibodies were used together. Images were taken using a confocal microscope (Olympus) and were analyzed with Fluoview Ver. 2.0b Viewer (Olympus).
RESULTS
TCP1 complex proteins co-isolate with PS-ASOs
To identify intracellular ASO binding proteins, we developed a two-step affinity selection approach. Proteins were first co-isolated from HeLa cell extracts using a biotinylated 5-10-5 gapmer PS-ASO with ribonucleotides modified with 2′-MOE (PS/MOE-ASOs, ISIS386652). To eliminate non-specific binding, the isolated proteins were eluted by competition using the same PS-ASO without biotin (Figure 1A). Co-isolated proteins were separated on an SDS-PAGE gel and visualized by silver staining (Figure 1B). The protein bands of interest were excised and identified by mass spectrometry. Six out of the eight subunits of the TCP1 complex, α, β, ϵ, δ, γ and θ, were identified, along with several other proteins, including LRPPRC, nucleolin and the Ku70/Ku80 complex. The binding of TCP1 proteins to PS-ASOs appeared to be independent of the ASO sequence and the biotin position, as determined by affinity selection using different ASOs, followed by western analysis (Figure 1C). TCP1-β and TCP1-ϵ, together with Ku70, were co-isolated with each of the six 20-mer PS-ASOs of different sequences or biotin positions (5′- or 3′-linked).
Figure 1.
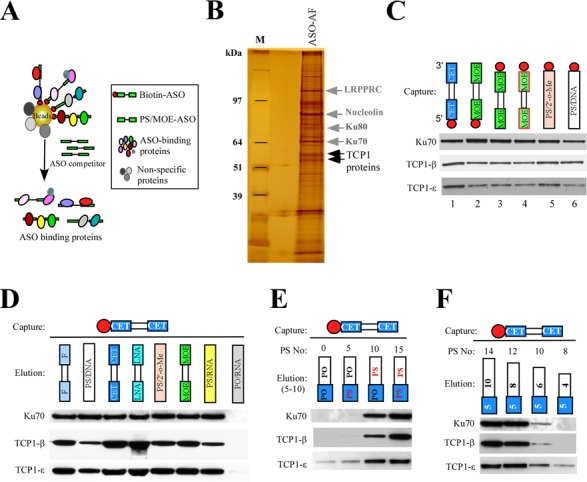
TCP1 proteins can be co-isolated with PS-ASOs. (A) Schematic representation of a two-step affinity selection approach used to isolate PS-ASO binding proteins. (B) Proteins co-isolated with ASOs were separated by SDS-PAGE, and an image of the silver staining gel is shown. The identified protein bands are indicated. M: protein size marker. ASO-AF: proteins isolated by affinity selection using ASOs. (C) Affinity selection was performed using indicated PS-ASOs. Co-selected TCP1 proteins were analyzed by western. Ku70 protein was detected and served as a loading control. The biotinylated ASOs used in lanes 1–6 were 58 6183, 38 6652, 55 7442, 36 7070, XL169 and XL168, respectively. The circle indicates a biotin. (D) 2′-modifications influence the binding of TCP1 proteins. ASO binding proteins were isolated using a biotinylated PS/cEt-ASO (58 6183) and eluted by competition using ASOs with the same sequence, but different 2′-modifications, or a PO-ASO. The eluted proteins were analyzed by western. (E) The number of PS-modified nucleotides is important for TCP1 binding. The affinity-selected proteins were eluted using 15-mer ASOs with different numbers of PS-modified nucleotides, and eluted proteins were analyzed by western. (F) Binding of TCP1 proteins requires a minimum number of PS modifications. Affinity selection was performed as in panel (D), but proteins were eluted with PS/cEt -ASOs of different lengths. The numbers in the white and gray boxes indicate the lengths of the PS-DNA portion and the cEt-modified portion of the ASOs.
To determine whether different 2′-modifications of PS-ASOs affect the affinity of TCP1 proteins for PS-ASOs, affinity selection was performed using a biotinylated 5-10-5 gapmer PS-ASO modified at the ribonucleotides with constrained ethyl (cEt, ISIS586183). Bound proteins were eluted by competition using PS-ASOs of the same sequence but different 2′ moieties. As shown in Figure 1D, western analyses indicated that although all PS-ASOs eluted TCP1 proteins, 2′-modifications influenced binding, especially of TCP1-β, which exhibited stronger binding to ASOs modified with cEt, locked nucleic acid (LNA) or 2′-fluro (F) than others, such as 2′-MOE, 2′-O-methyl (Me) or unmodified ribonucleotides. As a control, the binding of Ku70 protein was less affected by 2′-modifications than was binding of TCP1-β, as we observed in multiple different experiments (data not shown). Thus, 2′-modifications have different effects on binding of proteins like TCP1-β and TCP1-ϵ that share significant homology. Ku70, TCP1-β and TCP1-ϵ were not significantly eluted using an unmodified phosphodiester (PO) RNA oligonucleotide, and the failure of PO-ASOs to elute these proteins was not due to degradation of the relatively unstable PO-ASOs (Supplementary Figure S1A). These results suggest that the observed binding requires PS moieties. A minimum of 10 PS substitutions was required for significant binding of TCP1 proteins, as determined using different ASOs of the same length, sequence and 2′-compositions (Figure 1E). This is further confirmed by a similar competition elution experiment using PS/cEt-ASOs of various lengths (Figure 1F). Consistently, the two different TCP1 subunits exhibited moderately different binding properties, as TCP1-ϵ was less affected than TCP1-β by reductions in the number of PS backbone modifications. Similarly, Ku70 binding was also affected by the number of PS modifications. In addition, it appears that TCP1 and RNase H1 are not present in the same ASO/protein complex, as TCP1 proteins were not co-immunoprecipitated with RNase H1 from the ASO–protein pools pre-enriched using the two-step affinity selection approach (Supplementary Figure S1B). Together, these results show that (i) PS-ASOs can bind TCP1 proteins in a sequence-independent manner, (ii) significant protein binding requires 10 or more PS-modified nucleotides, (iii) 2′-modifications affect the affinity of PS-ASOs for TCP1-β and (iv) TCP1-β interacts differently with PS-ASOs than does the closely related protein TCP1-ϵ.
TCP1-β subunit co-localizes with PS-ASOs in nuclear PS-bodies
To determine whether PS-ASOs co-localize with TCP1 proteins in cells, HeLa cells were transfected for 4 h with a Cy3-labeled PS-ASO (ISIS446654) at 60 nM, a concentration much higher than required to knock down the mRNA target (less than 10 nM; data not shown), in order to monitor the potential co-localization. Cells were fixed and stained for various TCP1 subunits, and the localization of ASOs and TCP1 proteins was visualized by confocal microscopy. Consistent with previous studies, transfected PS-ASOs accumulated in the nucleus in a diffuse pattern and as distinct structures, namely PS-ASO-induced PS-bodies (15) (Figure 2A). No significant co-localization was observed between TCP1-α, ϵ, η or θ subunits and PS-ASOs, even using three different antibodies specific for the same subunit, i.e. TCP1-α (Supplementary Figure S2A; data not shown). However, TCP1-β was co-localized with PS-ASOs in distinct nuclear foci (Figure 2A, lower panel). In mock-transfected control cells, TCP1-β was evenly distributed in the cytoplasm and nuclear staining was also observed (Figure 2A, upper panel). Surprisingly, upon ASO transfection, TCP1-β was observed in the nucleus as aggregated, round-shaped structures that exactly co-localized with PS-ASOs in the PS-bodies (15). We note that not all ASO-containing nuclear structures co-localized with TCP1-β (marked with a blue arrow), since PS-ASOs were also observed in ASO-induced filaments and nuclear paraspeckles that differ from PS-bodies (our unpublished data).
Figure 2.
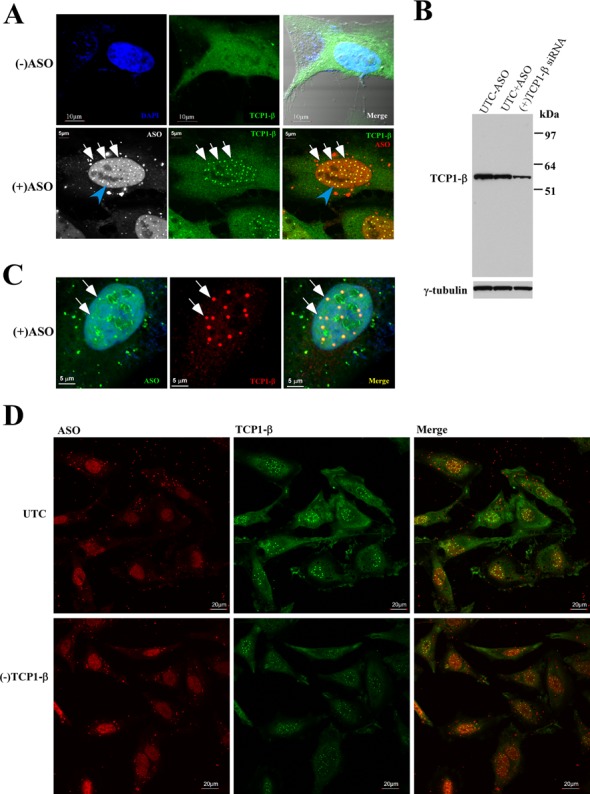
TCP1-β localizes in nuclear PS-bodies upon PS-ASO transfection. (A) Immunofluorescence staining of TCP1-β in control HeLa cells [(−)ASO] or HeLa cells transfected with 60-nM Cy3-labeled PS-ASO ISIS446654. The co-localization of TCP1-β (FITC) and PS-ASO in nuclear PS-bodies is indicated with white arrows. The scale bars: upper panels, 10 μm; lower panels, 5 μm. (B) Western analysis of TCP1-β protein in control cells transfected with (UTC+ASO) or without (UTC−ASO) 60-nM ASO ISIS116847 for 24 h, or in cells transfected with 5-nM TCP1-β siRNA for 24 h. TCP1-β protein was detected using antibody ab92756 (Abcam). γ-tubulin served as a loading control. (C) The PS-body localization of TCP1-β was not due to channel crosstalk. HeLa cells were transfected with 60 nM of FITC-labeled PS-ASO ISIS256903 and stained for TCP1-β (AF647) as described in panel (A). Scale bars, 5 μm. (D) Reduction in levels of TCP1-β did not completely block the formation of PS-bodies. HeLa cells treated with TCP1-β siRNA [(−)TCP1-β] for 24 h were transfected with 60-nM ISIS446654 for 5 h and stained for TCP1-β protein (FITC). UTC: untreated control cells. Scale bars: 20 μm.
The aggregated TCP1-β localization pattern was unlikely to stem from non-specific staining by the antibody since the antibody detected a single protein band on western blot that was sensitive to TCP1-β siRNA treatment (Figure 2B). Note that the TCP1-β protein level did not change in PS-ASO-transfected cells, relative to mock-transfected control cells, suggesting that the accumulation of TCP1-β in nuclear PS-bodies was not due to enhanced expression of the protein. In addition, a similar staining pattern upon ASO transfection was also detected using a different TCP1-β antibody (Supplementary Figure S2B). Finally, the PS-body localization of TCP1-β does not appear to be a result of channel crosstalk, since a different fluorescein isothiocyanate (FITC)-labeled PS-ASO also co-localized with TCP1-β stained with a secondary antibody conjugated with AF647 (Figure 2C). Together, these results indicate that TCP1-β protein co-localizes with PS-ASOs in nuclear PS-bodies. TCP1-β can thus serve as a PS-body marker protein.
A previous study using fluorescently labeled PS-ASOs showed that PS-body formation was dependent on ASO concentration but not sequence (15). Similar properties of PS-bodies were also observed with TCP1-β staining in cells transfected with non-labeled PS-ASO ISIS116847 at different concentrations (Supplementary Figure S3A) and with 60-nM PS-ASOs of different sequences (Supplementary Figure S3B). Finally, similar TCP1-β localization in ASO-induced PS-bodies was also observed in different human and mouse cell lines transfected with a variety of PS-ASOs (data not shown), suggesting that TCP1-β/PS-body localization is not unique to certain cell types or species.
Next, we analyzed whether TCP1-β protein is required for PS-body formation. Control cells and cells depleted of TCP1-β with siRNA treatment (Figure 2B) were transfected with 50-nM PS-ASO (ISIS446654) for 4 h and stained for TCP1-β. Consistent with the western results, the overall fluorescent signal of TCP1-β was significantly reduced in TCP1-β-depleted cells as compared with that in control cells (Figure 2D). Interestingly, an ∼70% reduction of TCP1-β protein did not completely block the formation of nuclear PS-bodies, as evidenced by the nuclear TCP1-β staining pattern. PS-body formation was not completely abolished even when TCP1-β protein level was reduced by more than 85% in another experiment (data not shown). However, the TCP1-β-stained PS-bodies appeared smaller in TCP1-β-depleted cells than that in control cells (Figure 2D), suggesting that TCP1-β protein is at least involved in higher order aggregation of PS-bodies, if not required for seeding the formation of PS-bodies. Note that the nuclear accumulation of PS-ASOs was not significantly affected by TCP1-β depletion, suggesting that the reduced size of PS-bodies is not due to reduced levels of nuclear ASOs. Taken together, our results show that TCP1-β is a PS-body component, the first protein component identified thus far for these nuclear bodies.
TCP1 proteins enhance the antisense activity of PS-ASOs
To determine whether binding of TCP1 proteins impacts the antisense activity of PS-ASOs designed to direct RNase H1-mediated cleavage of complementary RNAs, HeLa cells were treated with individual siRNAs targeting three different TCP1 subunits. Since siRNAs can cause unexpected effects (25), results of treatment with three additional control siRNAs were also evaluated. Two control siRNAs target LRPPRC and KHSRP, PS-ASO binding proteins that we recently identified and the third is an inactivate siRNA targeting U16 snoRNA (27). To reduce potential secondary effects resulting from depletion of the TCP1 proteins, siRNA treatment was performed for 24 h, followed by transfection of 2′-MOE gapmer PS-ASOs targeting NCL1 mRNA or Malat1 nuclear RNA for 4 h. The effects of reduction of individual TCP1 proteins on ASO activity were determined by analyzing reduction of the RNA targets of the PS-ASOs.
As expected, mRNA levels of TCP1-α, β and ϵ were specifically reduced by their corresponding siRNAs, but not by the other siRNAs, as analyzed by qRT-PCR assay (Figure 3A). Consistently, the protein levels were also significantly reduced by specific siRNAs (Figure 3B). The siRNA-treated cells were subsequently transfected with PS-ASOs targeting NCL1 mRNA (Figure 3C) or Malat1 RNA (Figure 3D). Levels of NCL1 or Malat1 RNAs were significantly reduced in a dose-dependent manner by the corresponding ASOs, however, less reduction of the ASO-targeted RNAs was observed in cells depleted of TCP1 proteins, as compared with those in control cells (Figure 3C and D). This experiment was repeated two more times and similar results were obtained (Supplementary Figure S4). In addition, reduced ASO activity was also observed for three other tested PS-ASOs targeting PTEN, Drosha or NEAT1 RNAs (data not shown), suggesting that the effects of reducing TCP1 proteins on ASO activity are not restricted to certain ASO sequences or target RNAs. Reduced ASO activity appears not to be due to siRNA off-target effects, since reduction of TCP1-β using two different siRNAs led to similar effects (Supplementary Figure S5A and B). Importantly, treatment in the same way with control siRNAs targeting LRPPRC, KHSRP or U16 RNAs had no significant effects on ASO activity (Supplementary Figure S5C and D). The effects of siRNA-mediated reduction of TCP1 subunits on ASO activity appears not to stem from multiple transfection steps, since similar results were obtained when TCP1-β was reduced using lentiviral shRNA (Supplementary Figure S5E–H). These results indicate that the observed effects resulted from depletion of the TCP1 proteins. In addition, the reduced ASO activity seems not to stem from a disrupted cytoskeleton network that may be caused by TCP1 depletion (22), since, under the experimental conditions we used, the steady state protein levels of β-actin, α-tubulin and γ-tubulin were not affected (Figure 3B) and the actin and tubulin network appeared to be normal (Supplementary Figure S5I and J). Together, our data indicate that TCP1 proteins can enhance PS-ASO activity in cells.
Figure 3.
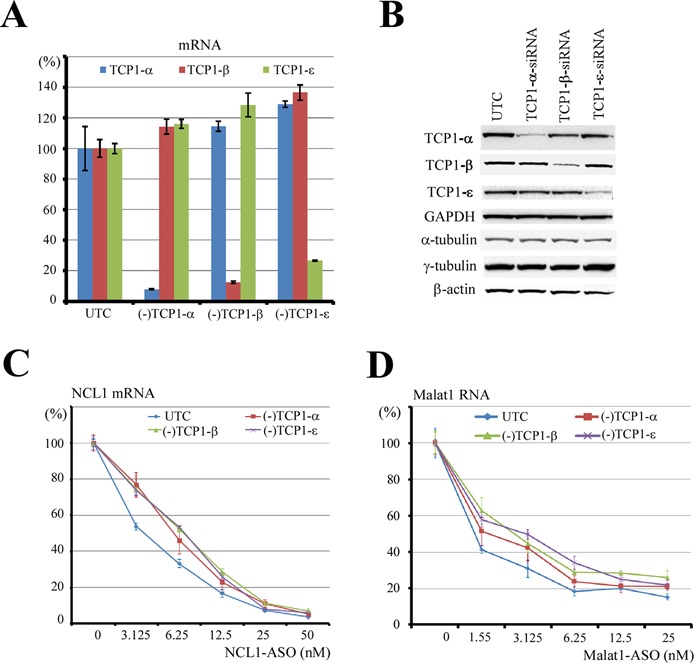
Reduction of TCP1 proteins can decrease the antisense activity of PS-ASOs. (A) qRT-PCR analyses for the mRNA levels of three different TCP1 subunits in cells treated with siRNAs targeting the TCP1 subunits. UTC: mock-transfected control cells. (B) Western analyses for the protein levels of three TCP1 subunits in siRNA-treated cells. ACTB, α-tubulin and γ-tubulin were also detected. GAPDH served as a loading control. (C) The antisense activity of a PS-ASO targeting NCL1 mRNA was reduced in cells depleted of TCP1 subunits, as determined by qRT-PCR for the levels of NCL1 mRNA. (D) qRT-PCR for the level of Malat1 RNA in test cells transfected with a PS-ASO targeting Malat1 RNA, as in panel (C). In all panels, the error bars indicate standard deviation from three experiments.
PS-body localization of TCP1-β requires at least 10 PS-modified nucleotides in ASOs
A previous study showed that PS-body formation depends on the presence of the PS modification and is sequence independent (15). To determine whether different 2′-modifications of PS-ASOs affect TCP1-β-stained PS-body formation, HeLa cells were transfected for 4 h with 50-nM PS-ASOs with different 2′-modifications, including 5-10-5 PS-ASOs with MOE, cEt or LNA 2′-modifications, and 20-mer PS-DNA and PS-RNA oligonucleotides. PS-bodies formed in the presence of each of these PS-ASOs (Figure 4A), suggesting that 2′-modifications do not have significant effects on PS-body formation, although the affinities of TCP1-β to these different PS-ASOs varied moderately (Figure 1D). Consistent with the previous study (15), a PO-RNA fully modified with 2′-O-methyl failed to form nuclear PS-bodies (Figure 4A).
Figure 4.
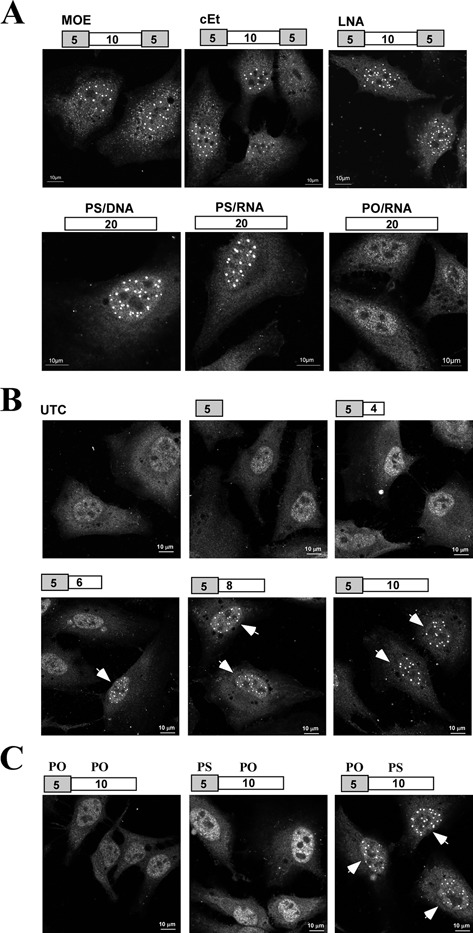
PS-body formation depends on PS backbone but not 2′ ribose modification. (A) HeLa cells were transfected for 4 h with 60-nM 20-mer PS-ASOs with different 2′-modifications, a 20-mer PS-RNA ASO or a 20-mer 2′-O-methylated PO-RNA ASO. The gray boxes represent 2′-modified portion of the gapmer ASOs. The numbers in the boxes indicate the number of nucleotides in the portion of ASOs. Cells were fixed and stained for TCP1-β protein (AF647) and representative cells are shown. (B) HeLa cells were transfected with 60-nM PS-ASOs with different lengths, as indicated above the panels. The gray and white boxes indicate the cEt-modified portion and the PS-DNA portion of the ASOs, respectively. Cells were stained for TCP1-β (AF647), as in panel (A). The nuclear PS-bodies are indicated by arrows. (C) HeLa cells were transfected for 4 h with 60-nM 15-mer ASOs with different numbers of PS nucleotides, as indicated above the panels. The nuclei containing PS-bodies are indicated by arrows.
As the PS modification is required for PS-body formation (15), we investigated the relationship between the number of PS-modified nucleotides and the formation TCP1-β/PS-bodies by transfection of HeLa cells with 50-nM PS/cEt-ASOs of different lengths (Figure 4B). Significant PS-body formation was detected in cells transfected with 11-mer or longer PS-ASOs based on TCP1-β staining. To further determine that PS-body formation is related to the number of PS-modified nucleotides but not the length of ASOs, HeLa cells were next transfected with 15-mer ASOs containing 0, 5 or 10 PS-modified nucleotides and stained for TCP1-β. In these cases, significant PS-body formation was only observed with the 15-mer ASO containing 10 PS-modified nucleotides (Figure 4C), indicating that the number of PS-modified nucleotides, but not the entire length of the ASOs, is important for PS-body formation. We note that the ability of these ASOs to form PS-bodies strongly correlated with their ability to bind TCP1-β protein (Figure 1E and F), implying that the interaction between PS-ASOs and TCP1-β protein may play a significant role in PS-body formation or higher order assembly. Together, these results indicate that binding to TCP1-β and PS-body formation requires at least 10 PS-modified nucleotides, whereas the nature of 2′-modifications has no significant effect on PS-body assembly.
The nuclear level of TCP1-β protein increases moderately upon PS-ASO transfection
The TCP1 complex is known to localize in the cytoplasm (28). However, TCP1 proteins were observed in the nucleus in control cells by immunofluorescence staining (Supplementary Figure S2), and, upon ASO transfection, TCP1-β was found to accumulate in nuclear PS-bodies (Figure 2A). These observations suggest that TCP1-β may be present in the nucleus where it can form PS-bodies with ASOs or that TCP1-β is imported to the nucleus upon ASO transfection, or both. To investigate these possibilities, HeLa cells were transfected with 60-nM PS-ASOs (ISIS116847) for 5 h, and cytoplasmic and nuclear proteins were separated and subjected to western analyses. As shown in Figure 5A, both TCP1-β and TCP1-ϵ proteins were detected in the nuclear fraction in mock-transfected control cells, indicating that under normal conditions some TCP1 proteins are present in the nucleus, albeit at very low levels. This nuclear localization appeared not to stem from cytoplasmic contamination during fractionation, since an abundant cytoplasmic control protein, GAPDH, was not detected in the nuclear fraction. Ku70 and hnRNP K, which are mainly present in the nucleus but are also found in the cytoplasm (29,30), were used as loading controls. Upon PS-ASO transfection, the nuclear level of TCP1-β, but not TCP1-ϵ, was moderately increased (Figure 5A). The increase in levels of nuclear TCP1-β protein was ASO dose-dependent, as transfection of PS-ASOs at 40 nM, but not 20 nM, caused a detectable increase of nuclear TCP1-β as compared with the level in control cells (Figure 5B). In addition, the increase in the nuclear TCP1-β upon transfection with PS-ASOs was time dependent. Six hours after transfection, the nuclear TCP1-β level was ∼3.3-fold that of control cells. At earlier time points only a 1.6-fold was observed (Figure 5C). A slight increase in the nuclear TCP1-ϵ subunit was also observed 6 h after transfection, yet to a lesser extent than TCP1-β, suggesting that the nuclear import of other TCP1 subunits, although not present in PS-bodies, may also be increased upon ASO transfection.
Figure 5.
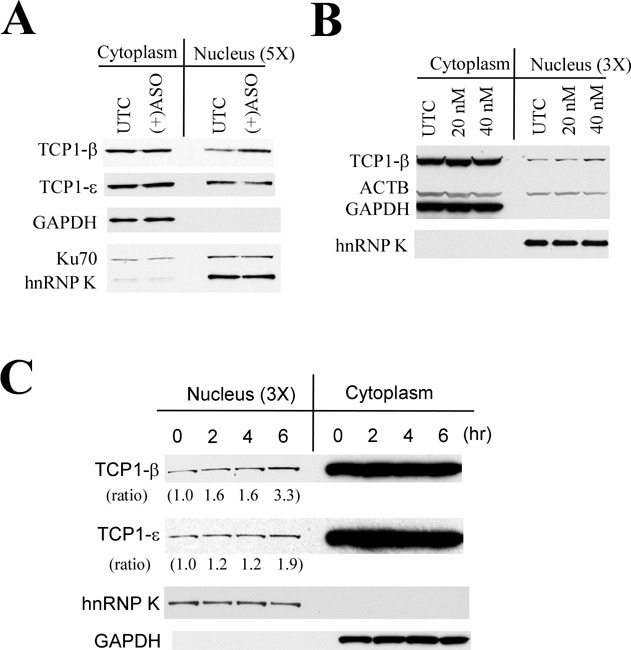
The nuclear level of TCP1-β protein can be increased upon PS-ASO transfection. (A) HeLa cells were transfected with 60-nM PS-ASOs ISIS116847 for 5 h, and cytoplasmic and nuclear fractions were prepared. Since the nuclear level of TCP1 proteins is much lower than the cytoplasmic level (data not shown), the cytoplasmic and nuclear fractions were loaded in a 1:5 ratio (5×). Proteins were separated on SDS-PAGE and subjected to western analyses. Cytoplasmic protein GAPDH and nuclear protein hnRNP K were detected as controls for the quality of fractionation. Ku70 protein is present in both nucleus and cytoplasm and served as a loading control. (B) HeLa cells were transfected for 5 h with 0 (UTC), 20 or 40-nM PS-ASO ISIS116847. Subcellular fractionation and western analyses were performed as in panel (A). ACTB served as a loading control. (C) HeLa cells transfected with 60-nM PS-ASO ISIS116847 for different times were fractionated, and proteins were subjected to western analyses, as in panel (A). The relative levels of TCP1-β and TCP1-ϵ measured using ImageJ and normalized to hnRNP K are indicated below the lanes.
The increase in nuclear TCP1-β protein appeared to lag the PS-body formation. PS-bodies, though small in size and only observed in a small portion of cells (∼15%), were detected around 2 h after transfection, even in cells lacking high levels of diffuse nuclear ASOs (Supplementary Figure S6). Four hours after transfection, PS-bodies, as well as strong nuclear accumulation of PS-ASOs, were observed in the majority of cells (Supplementary Figure S6; data not shown). Thus, it appears that PS-body formation can occur in the absence of or before strong nuclear accumulation of PS-ASOs. However, over time as the nuclear ASO concentration increases, more and larger nuclear PS-bodies were observed. These observations suggest that PS-ASOs may form PS-bodies with TCP1-β proteins that are present in the nucleus or that ASO-TCP1-β can be co-imported to the nucleus.
Reduction of RAN can cause the formation of PS-body-like structure in the cytoplasm
Previous studies have shown that PS-ASOs can shuttle between the nucleus and cytoplasm, and the shuttling process shares characteristics of active transport (14). To determine whether nuclear PS-body formation and/or TCP1-β localization in the PS-bodies are mediated by an active transport pathway, levels of the small GTPase RAN, which is required for cytoplasmic/nuclear transport of proteins and RNAs (31), were reduced in HeLa cells by siRNA treatment (Figure 6A). Untreated or siRNA-treated cells were then transfected with fluorescently labeled PS-ASOs for 5 h. As shown in representative live cell images in Figure 6B, reduction of RAN led to lower levels of nuclear accumulation of PS-ASOs than observed in control cells. This result suggests that nuclear import of PS-ASOs is at least partially mediated by the RAN pathway. Despite reduced levels of nuclear PS-ASOs, depletion of RAN protein did not significantly block the formation of nuclear PS-bodies (Supplementary Figure S7). Interestingly, in a subpopulation of RAN-depleted cells, TCP1-β formed aggregates in the cytoplasm that co-localized with PS-ASOs (Figure 6C), we refer to these as PS-body-like structures. In some cells exhibiting multiple cytoplasmic PS-body-like structures few or no nuclear PS-bodies were observed (data not shown; Figure 6C, lower panel). Similar cytoplasmic PS-body-like structures were not observed in control cells (exemplified in Figure 6C, upper panel), suggesting that upon RAN reduction, PS-ASOs can interact with TCP1-β in the cytoplasm to form PS-body-like structures. Like nuclear PS-bodies, the cytoplasmic PS-body-like structures appeared to contain only TCP1-β, not other TCP1 subunits (Figure 6D). These results indicate that the interaction of PS-ASOs with TCP1-β can take place in the cytoplasm and suggest that the RAN pathway is at least partially involved in nuclear import of PS-ASOs and perhaps the import of TCP1-β/ASO complex as well.
Figure 6.
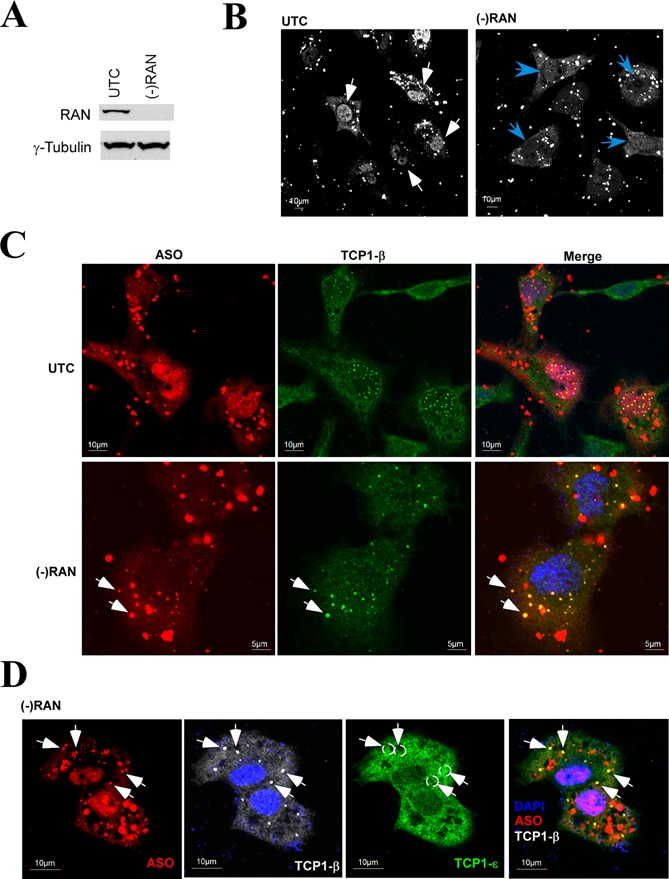
Reduction of RAN can lead to the formation of cytoplasmic PS-body-like structures. (A) Western analysis for RAN protein in cells treated or not with siRNA targeting RAN. Levels of γ-tubulin were detected and served as a loading control. (B) Reduction of RAN reduces the level of nuclear ASOs. Control HeLa cells (UTC) or cells treated with RAN siRNA [(−)RAN] were transfected with 50-nM Cy3-labeled ISIS446654 for 4 h, washed and images were taken of live cells. The PS-ASOs were pseudo-colored to white. The white arrows indicate the nuclear accumulation of PS-ASOs in control cells, whereas blue arrows indicate cells with reduced nuclear levels of PS-ASOs upon RAN reduction. Scale bar: 10 μm. (C) Reduction of RAN can cause formation of PS-body-like structures in the cytoplasm. Cells as used in panel (B) were fixed and stained for TCP1-β. White arrows indicate the cytoplasmic PS-body-like structures that contain PS-ASOs and TCP1-β protein. Scale bars: upper panels, 10 μm; lower panels, 5 μm. (D) TCP1-ϵ subunit does not localize in the cytoplasmic PS-body-like structures. RAN-depleted cells were transfected with PS-ASOs and co-stained for TCP1-β and TCP1-ϵ subunits. The PS-body-like structures are indicated with arrows, whereas the corresponding positions are marked with dashed circles in the TCP1-ϵ panel. Scale bar: 10 μm.
The TCP1 complex can co-localize with PS-ASOs in cytoplasmic organelles related to endocytosis pathways upon free uptake
Previous studies have demonstrated that PS-ASOs predominantly localize in the cytoplasm when cells were treated with PS-ASOs in the absence of transfection reagents (free uptake) (11,13). To determine whether TCP1 proteins co-localize with PS-ASOs after free uptake, HeLa cells were incubated with FITC-labeled PS-ASOs (ISIS256903) at 1-μM final concentration for 16 h and were then stained for TCP1-β and TCP1-ϵ subunits (Figure 7A). Upon ASO incubation, both TCP1-β and TCP1-ϵ subunits appeared in distinct cytoplasmic structures that contrasted with the even distribution pattern in control cells (Figure 2A). Importantly, these TCP1-containing structures co-localized with PS-ASOs (Figure 7A, right panel). This co-localization was not due to channel crosstalk, since no TCP1 signals were detected for a nearby ASO locus (Figure 7A, right panel, marked with a gray arrow) and labeling with different dyes resulted in similar co-localization patterns (data not shown).
Figure 7.
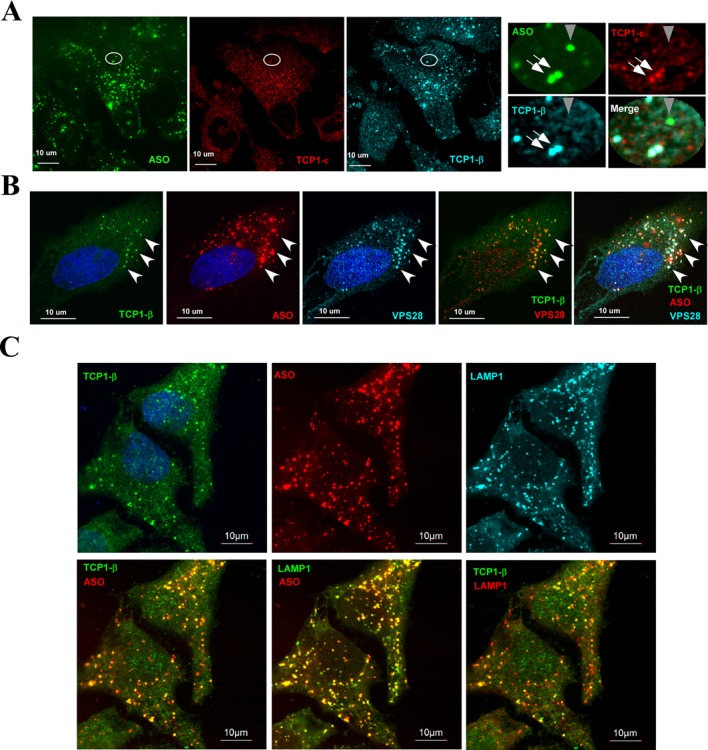
TCP1 proteins can co-localize with PS-ASOs in endosome/lysosome-related structures upon free uptake. (A) TCP1-β and TCP1-ϵ can co-localize with PS-ASOs in cytoplasmic foci upon free uptake. HeLa cells incubated for 16 h with 1-μM FITC-labeled PS-ASOs ISIS256903 without transfection reagent were stained for TCP1-β (AF647) and TCP1-ϵ (AF555) subunits. The right panels represent a zoomed area as indicated by a circle. The co-localized TCP1 proteins and PS-ASO are indicated by white arrows, whereas a PS-ASO locus that does not co-localize with TCP1 proteins is marked with a gray arrowhead. (B) The cytoplasmic PS-ASO/TCP1 foci can co-localize with ESCRT-1 protein VPS28. HeLa cells were incubated with 1-μM Cy3-labeled PS-ASO ISIS446654 for 24 h, fixed and co-stained for TCP1-β (FITC) and VPS28 (AF647) proteins. The TCP1-β/PS-ASO/VPS28 co-localized foci are exemplified by arrowheads. The co-localization of TCP1-β and VPS28 is shown in a merge panel. (C) Immunofluorescence staining for TCP1-β (FITC) and lysosomal protein LAMP1 (AF647) in HeLa cells incubated with 1.5-μM ASO ISIS446654 (Cy3) for 24 h, as in panel (B). The TCP1-β/PS-ASO, LAMP1/PS-ASO or TCP1-β/LAMP1 co-localizations are shown by two-channel merging. Scale bars, 10 μm. The nuclei were stained by DAPI (blue).
It has been shown that PS-ASOs can enter cells through endocytosis and accumulate in endosome-related structures in the cytoplasm (11,13). To determine whether the cytoplasmic TCP1/PS-ASO structures are related to endosome, lysosome or multivesicular bodies (MVBs), HeLa cells were incubated with 1-μM Cy3-labeld PS-ASO (ISIS446654) for 24 h and co-stained for TCP1-β and VPS28, a protein component of the ESCRT-I complex that is required for the biogenesis of MVBs (32). Indeed, significant co-localization of TCP1-β, PS-ASO and VPS28 was observed, as exemplified by arrows (Figure 7B). This can be clearly seen from the merged channels between TCP1-β and VPS28. Similarly, TCP1-β and PS-ASOs also significantly co-localized with LAMP1, a lysosome marker protein, as shown for merged images between different channels (Figure 7C). TCP1-β loci exactly co-localized with ASOs (Figure 7C, lower left panel) and LAMP1 (lower right panel), as indicated by the merged yellow dot structures. Co-localization between TCP1-β, ASO and LAMP1 was also confirmed by a 3D analysis (Supplementary Figure S8). These results suggest that TCP1 proteins can be enriched in endosome/lysosome-related organelles upon ASO free uptake. Note that most of the VPS28- or LAMP1-stained foci contained PS-ASOs, whereas not all such endosome/lysosome-related structures contained TCP1-β, indicating that TCP1 proteins are not always present in these structures.
As described above, upon PS-ASO transfection, reduction of RAN can cause the formation of cytoplasmic PS-body-like structures that contain TCP1-β and PS-ASOs (Figure 6). These PS-body-like structures do not appear to be related to the cytoplasmic structures that form upon free uptake of PS-ASOs, since PS-body-like structures, demarked by the structures containing PS-ASOs and TCP1-β upon RAN depletion, did not co-localize with Lamp1 protein (Supplementary Figure S9). The nuclear PS-bodies (white arrow) and cytoplasmic PS-body-like structures (blue arrow) did not overlap with LAMP1 (yellow dots in the merge panel), whereas some PS-ASOs present in lysosomes (white dots in the merge panel) also did not co-localize with TCP1-β protein (gray arrow). Together, these results indicate that the TCP1 complex can co-localize with PS-ASOs in cytoplasmic endosome/lysosome-related structures upon PS-ASO free uptake and that PS-body-like loci are different from endosome/lysosome-related structures.
DISCUSSION
In this study, we found that PS-ASOs can interact with the members of the TCP1 complex. The TCP1/PS-ASO interaction requires at least 10 PS-modified nucleotides, whereas the 2′-modification, despite influential, is not essential for TCP1 protein binding. Although it is known that TCP1 proteins can bind ATP, the binding of TCP1 complex to PS-ASOs apparently utilizes a mechanism different from ATP binding, since PO-ASOs do not bind TCP1 proteins and the binding of PS-ASOs requires multiple PS backbone modification. Unlike other chaperones that recognize substrates through hydrophobic interactions, TCP1 proteins recognize and interact with the substrate proteins in a specific manner involving charged and polar residues in both the chaperone and the substrates (20). Clearly, the interactions with ASOs involve charge recognition, but the greater hydrophobicity of PS-ASOs as compared with PO-ASOs is also critical. Further, ASOs with more hydrophobic 2′-modifications, such as 2′-LNA, cEt and F, may bind more tightly to TCP1-β and other TCP1 proteins than do ASOs with less hydrophobic modifications such as 2′-MOE. In future studies, it will be important to more precisely map the protein domains involved in the PS-ASO binding and to better characterize the influence of specific ASO modifications on binding.
The interaction between TCP1 proteins and PS-ASOs affected the potency of PS-ASOs. Depletion of three different subunits of TCP1 complex individually inhibited ASO-directed target reduction (Figure 3), suggesting that these proteins enhance the antisense activity of PS-ASOs. The effects were modest, however, suggesting that many other cellular proteins also impact ASO efficacy. This view is supported by the fact that PS-ASOs, as short as 20-mer, bind many intracellular proteins (our unpublished data). Thus, it is possible that each protein binds a portion of, but not all, PS-ASOs, leading to moderate effect on ASO activity upon reduction of a particular ASO binding protein. How TCP1 proteins enhance PS-ASO activity in directing RNase H1 cleavage is currently unknown. Since TCP1 and RNase H1 are not present in the same ASO/protein complex, it is unlikely that TCP1 proteins directly recruit RNase H1 to ASOs or ASO-RNase H1 complex to mRNA targets. However, it is possible that the binding of TCP1 proteins to PS-ASOs prevents the binding of other proteins that would reduce the ASO activity. TCP1 proteins may be involved in nuclear import of PS-ASOs or serve to retain ASOs in the nuclear PS-bodies to ensure high levels of nuclear PS-ASOs. In addition, these proteins may also play a role in ASO uptake and/or in exchange between endocytosis-related organelles and cytosol environment. Finally, we cannot rule out the possibility that TCP1 proteins may help to melt the intra-molecular structures of PS-ASOs to increase their base-pairing potential with target RNAs.
It was previously reported that TCP1-β subunit is exclusively found in the cytoplasm, whereas the TCP1-ϵ subunit is also detected in the nucleus by immunohistochemistry analyses (33). However, both TCP1-β and TCP1-ϵ subunits were found in the nuclear fraction in our study, though at low levels (Figure 5). These observations suggest that TCP1 proteins may also function in the nucleus. Upon PS-ASO transfection, the nuclear level of TCP1-β protein moderately increased, in a dose- and time-dependent manner, presumably accompanying the nuclear import of PS-ASOs. This is not surprising, since nuclear localization of chaperone proteins has been well documented for heat shock proteins, which can translocate to the nucleus upon stress or at certain stages in the growth of cells (34,35).
Upon transfection or microinjection, PS-ASOs quickly accumulate in the nucleus and at high concentrations form PS-bodies in a sequence-independent manner (15). PS-bodies may be built around PS-ASOs together with other cellular components, as PS-ASOs can interact with many nuclear proteins, especially RNA binding proteins [(8) and our unpublished data]. Our results indicate that TCP1-β is a key PS-body component as this protein interacts with PS-ASOs and co-localizes with PS-ASOs in nuclear PS-bodies and in cytoplasmic PS-body-like structures. In addition, the ability of PS-ASOs with different PS-modified oligonucleotides to interact with TCP1-β protein correlates with the ability of these ASOs to form PS-bodies. Currently it is not clear if TCP1-β is essential for PS-body formation, since significant reduction of TCP1-β protein by siRNA treatment did not completely block the formation of PS-bodies, although the number and size of PS-bodies were reduced (Figure 2D; data not shown). It is likely that this protein is required for PS-body formation, but we failed to observe strong defects solely due to incomplete protein depletion.
It is possible that PS-bodies contain other proteins in addition to TCP1-β, since PS-ASOs can interact with many proteins including nuclear RNA/DNA binding proteins, such as NCL1 (8). Several tested RNA binding proteins, including NCL1 and hnRNP A1 that associate with PS-ASOs, do not co-localize in the PS-bodies (8,9,15), suggesting that PS-bodies are not formed randomly with any associated proteins solely based on high affinities. No PS-body localization was found for several other tested nuclear proteins, including NPM1, HMGB1, PC4/SUB1 and La/SSB, which we found to interact with PS-ASOs (data not shown). In addition, attempts to isolate PS-bodies using immunoprecipitation with the TCP1-β antibody from nuclear fractions of PS-ASO-transfected cells also failed, likely due to the large size of PS-bodies and/or the tight structure that reduces the accessibility of antibodies (data not shown). Identification of other PS-body components that may regulate PS-body formation and PS-ASO transport/activity thus awaits further investigation. It is possible that PS-bodies may contain other TCP1 subunits, since PS-ASOs can interact with multiple TCP1 subunit proteins. However, no PS-body localization was found for any of these tested TCP1 subunits, even using multiple different antibodies. Although we cannot completely rule out the possibility that other TCP1 subunits are present in PS-bodies, our current data do not support this view. In addition, TCP1-ϵ was not found in cytoplasmic PS-body-like structures upon RAN reduction. These observations suggest that TCP1-β subunit may have unique properties that favor the binding of PS-ASOs and the formation of aggregated structures. These findings also imply that individual TCP1 subunits may have distinct roles, as supported by previous studies showing that different substrate proteins may be recognized and contacted by different subsets of TCP1 subunits (23,24). In addition, it has been shown that TCP1-α and TCP1-β, but not other TCP1 subunits, were up-regulated in tumor tissues (36) and that over-expression of TCP1-α (CCT1), and to a lesser degree, TCP1-θ (CCT4), caused morphological changes of polyglutamine aggregate (24), further suggesting different functions for individual TCP1 subunits.
PS-body formation appears to be an early event after PS-ASO transfection that occurs prior to accumulation of diffuse nuclear PS-ASOs, suggesting that PS-bodies are not a result of self-aggregation of high levels of diffuse ASOs. As no increase in cytotoxic effects of ASOs was found in cells depleted of TCP1 proteins (data not shown), it is unlikely that PS-bodies serve as depots for excess amount of PS-ASOs that prevent cytotoxicity. Although PS-bodies appear to form with low levels of nuclear TCP1-β protein, it is currently not clear how PS-body formation is initiated. One possibility is that PS-ASOs interact with TCP1-β in the cytoplasm and the ASO–protein complex is imported to the nucleus. This view is supported by previous findings that the nuclear import of PS-ASOs is carrier-mediated (14). In addition, we showed that PS-ASOs and TCP1-β can form cytoplasmic PS-body-like structures upon reduction of RAN. Therefore, PS-ASOs do interact with TCP1-β in the cytoplasm. However, we cannot rule out the possibility that imported PS-ASOs interact in the nucleus with pre-existing TCP1-β protein to form PS-bodies.
Interestingly, we found that both TCP1-β and TCP1-ϵ can co-localize with PS-ASOs in cytoplasmic structures related to endosome/lysosomes upon free uptake (Figure 7 and Supplementary Figure S8). Although TCP1 proteins have been reported to be present in endosome/lysosome-related organelles (37), these proteins do not appear as punctuated structures in control cells without PS-ASO treatment, suggesting that accumulation of TCP1 proteins in these endocytosis-related structures is induced by PS-ASO uptake. Since depletion of TCP1 proteins can reduce the antisense activity of PS-ASOs, which requires escape of ASOs from these membrane enclosed structures, it is therefore possible that TCP1 proteins may facilitate the cellular uptake of PS-ASOs from extracellular environments and/or the release of PS-ASOs from endosomes or lysosomes. This view is supported by a recent study showing that TCP1 complex is required for efficient delivery of anthrax toxin into the cytosol of host cells (38), likely by facilitating the action of crossing plasma membrane by mediating conformational changes of the protein complex. A similar situation may also apply to PS-ASOs, as PS-ASOs bind multiple proteins and structural or compositional changes of ASO/protein complex may be involved in ASO translocation across membranes, which can be facilitated by TCP1 chaperone proteins. In addition, hsc70, a protein that can form a stable complex with TCP1 (39), is involved in the release of clathrin from clathrin-coated vesicles (40), implying that TCP1 complex may also be able to play a role in ASO release from the endocytosis-related structures. Further investigation of the underlying mechanisms of how TCP1 proteins improve ASO drug potency will be important for better drug design.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
ACKNOWLEDGMENTS
We wish to thank Frank Bennett for critical reading of this manuscript and Walt Lima, Timothy Vickers and Hongjiang Wu for stimulating discussions.
FUNDING
ISIS Pharmaceuticals. Funding for open access charge: ISIS Pharmaceuticals.
Conflict of interest statement. None declared.
REFERENCES
- 1.Rehm H.L. Disease-targeted sequencing: a cornerstone in the clinic. Nat. Rev. Genet. 2013;14:295–300. doi: 10.1038/nrg3463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Crooke S.T., Vickers T., Lima W., Wu H.-J. Mechanisms of antisense drug action, an introduction. In: Crooke ST, editor. Antisense Drug Technology—Principles, Strategies, and Application. Boca Raton, FL: CRC Press; 2006. pp. 3–46. [Google Scholar]
- 3.Bennett C.F. Pharmacological properties of 2'-O-methyoxyethyl-modified oligonucleotides. In: Crooke ST, editor. Antisense Drug Technology—Principles, Strategies, and Applications. Boca Raton, FL: CRC Press; 2006. pp. 273–304. [Google Scholar]
- 4.Toth P.P. Emerging LDL therapies: mipomersen-antisense oligonucleotide therapy in the management of hypercholesterolemia. J. Clin. Lipidol. 2013;7:S6–S10. doi: 10.1016/j.jacl.2013.02.004. [DOI] [PubMed] [Google Scholar]
- 5.Dias N., Stein C.A. Antisense oligonucleotides: basic concepts and mechanisms. Mol. Cancer Ther. 2002;1:347–355. [PubMed] [Google Scholar]
- 6.Geary R.S. Antisense oligonucleotide pharmacokinetics and metabolism. Expert Opin. Drug Metab. Toxicol. 2009;5:381–391. doi: 10.1517/17425250902877680. [DOI] [PubMed] [Google Scholar]
- 7.Bennett C.F., Swayze E.E. RNA targeting therapeutics: molecular mechanisms of antisense oligonucleotides as a therapeutic platform. Annu. Rev. Pharmacol. Toxicol. 2010;50:259–293. doi: 10.1146/annurev.pharmtox.010909.105654. [DOI] [PubMed] [Google Scholar]
- 8.Weidner D.A., Valdez B.C., Henning D., Greenberg S., Busch H. Phosphorothioate oligonucleotides bind in a non sequence-specific manner to the nucleolar protein C23/nucleolin. FEBS Lett. 1995;366:146–150. doi: 10.1016/0014-5793(95)00517-d. [DOI] [PubMed] [Google Scholar]
- 9.Abdul-Manan N., Williams K.R. hnRNP A1 binds promiscuously to oligoribonucleotides: utilization of random and homo-oligonucleotides to discriminate sequence from base-specific binding. Nucleic Acids Res. 1996;24:4063–4070. doi: 10.1093/nar/24.20.4063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Marcusson E.G., Bhat B., Manoharan M., Bennett C.F., Dean N.M. Phosphorothioate oligodeoxyribonucleotides dissociate from cationic lipids before entering the nucleus. Nucleic Acids Res. 1998;26:2016–2023. doi: 10.1093/nar/26.8.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Koller E., Vincent T.M., Chappell A., De S., Manoharan M., Bennett C.F. Mechanisms of single-stranded phosphorothioate modified antisense oligonucleotide accumulation in hepatocytes. Nucleic Acids Res. 2011;39:4795–4807. doi: 10.1093/nar/gkr089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zelphati O., Szoka F.C., Jr Mechanism of oligonucleotide release from cationic liposomes. Proc. Natl. Acad. Sci. U.S.A. 1996;93:11493–11498. doi: 10.1073/pnas.93.21.11493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bennett C.F., Chiang M.Y., Chan H., Shoemaker J.E., Mirabelli C.K. Cationic lipids enhance cellular uptake and activity of phosphorothioate antisense oligonucleotides. Mol. Pharmacol. 1992;41:1023–1033. [PubMed] [Google Scholar]
- 14.Lorenz P., Misteli T., Baker B.F., Bennett C.F., Spector D.L. Nucleocytoplasmic shuttling: a novel in vivo property of antisense phosphorothioate oligodeoxynucleotides. Nucleic Acids Res. 2000;28:582–592. doi: 10.1093/nar/28.2.582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lorenz P., Baker B.F., Bennett C.F., Spector D.L. Phosphorothioate antisense oligonucleotides induce the formation of nuclear bodies. Mol. Biol. Cell. 1998;9:1007–1023. doi: 10.1091/mbc.9.5.1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Graham M.J., Crooke S.T., Monteith D.K., Cooper S.R., Lemonidis K.M., Stecker K.K., Martin M.J., Crooke R.M. In vivo distribution and metabolism of a phosphorothioate oligonucleotide within rat liver after intravenous administration. J. Pharmacol. Exp. Ther. 1998;286:447–458. [PubMed] [Google Scholar]
- 17.Cong Y., Baker M.L., Jakana J., Woolford D., Miller E.J., Reissmann S., Kumar R.N., Redding-Johanson A.M., Batth T.S., Mukhopadhyay A., et al. 4.0-A resolution cryo-EM structure of the mammalian chaperonin TRiC/CCT reveals its unique subunit arrangement. Proc. Natl. Acad. Sci. U.S.A. 2010;107:4967–4972. doi: 10.1073/pnas.0913774107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kubota H., Hynes G., Willison K. The chaperonin containing t-complex polypeptide 1 (TCP-1). Multisubunit machinery assisting in protein folding and assembly in the eukaryotic cytosol. Eur. J. Biochem. 1995;230:3–16. doi: 10.1111/j.1432-1033.1995.tb20527.x. [DOI] [PubMed] [Google Scholar]
- 19.Thulasiraman V., Yang C.F., Frydman J. In vivo newly translated polypeptides are sequestered in a protected folding environment. EMBO J. 1999;18:85–95. doi: 10.1093/emboj/18.1.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gomez-Puertas P., Martin-Benito J., Carrascosa J.L., Willison K.R., Valpuesta J.M. The substrate recognition mechanisms in chaperonins. J. Mol. Recognit. 2004;17:85–94. doi: 10.1002/jmr.654. [DOI] [PubMed] [Google Scholar]
- 21.Frydman J., Nimmesgern E., Erdjument-Bromage H., Wall J.S., Tempst P., Hartl F.U. Function in protein folding of TRiC, a cytosolic ring complex containing TCP-1 and structurally related subunits. EMBO J. 1992;11:4767–4778. doi: 10.1002/j.1460-2075.1992.tb05582.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Brackley K.I., Grantham J. Activities of the chaperonin containing TCP-1 (CCT): implications for cell cycle progression and cytoskeletal organisation. Cell Stress Chaperones. 2009;14:23–31. doi: 10.1007/s12192-008-0057-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Spiess C., Miller E.J., McClellan A.J., Frydman J. Identification of the TRiC/CCT substrate binding sites uncovers the function of subunit diversity in eukaryotic chaperonins. Mol. Cell. 2006;24:25–37. doi: 10.1016/j.molcel.2006.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tam S., Geller R., Spiess C., Frydman J. The chaperonin TRiC controls polyglutamine aggregation and toxicity through subunit-specific interactions. Nat. Cell Biol. 2006;8:1155–1162. doi: 10.1038/ncb1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liang X.H., Hart C.E., Crooke S.T. Transfection of siRNAs can alter miRNA levels and trigger non-specific protein degradation in mammalian cells. Biochim. Biophys. Acta. 2013;1829:455–468. doi: 10.1016/j.bbagrm.2013.01.011. [DOI] [PubMed] [Google Scholar]
- 26.Liang X.H., Crooke S.T. Depletion of key protein components of the RISC pathway impairs pre-ribosomal RNA processing. Nucleic Acids Res. 2011;39:4875–4889. doi: 10.1093/nar/gkr076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liang X.H., Vickers T.A., Guo S., Crooke S.T. Efficient and specific knockdown of small non-coding RNAs in mammalian cells and in mice. Nucleic Acids Res. 2011;39:1–17. doi: 10.1093/nar/gkq1121. e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Valpuesta J.M., Martin-Benito J., Gomez-Puertas P., Carrascosa J.L., Willison K.R. Structure and function of a protein folding machine: the eukaryotic cytosolic chaperonin CCT. FEBS Lett. 2002;529:11–16. doi: 10.1016/s0014-5793(02)03180-0. [DOI] [PubMed] [Google Scholar]
- 29.Cohen H.Y., Lavu S., Bitterman K.J., Hekking B., Imahiyerobo T.A., Miller C., Frye R., Ploegh H., Kessler B.M., Sinclair D.A. Acetylation of the C terminus of Ku70 by CBP and PCAF controls Bax-mediated apoptosis. Mol. Cell. 2004;13:627–638. doi: 10.1016/s1097-2765(04)00094-2. [DOI] [PubMed] [Google Scholar]
- 30.Habelhah H., Shah K., Huang L., Ostareck-Lederer A., Burlingame A.L., Shokat K.M., Hentze M.W., Ronai Z. ERK phosphorylation drives cytoplasmic accumulation of hnRNP-K and inhibition of mRNA translation. Nat. Cell Biol. 2001;3:325–330. doi: 10.1038/35060131. [DOI] [PubMed] [Google Scholar]
- 31.Freitas N., Cunha C. Mechanisms and signals for the nuclear import of proteins. Curr. Genomics. 2009;10:550–557. doi: 10.2174/138920209789503941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Henne W.M., Buchkovich N.J., Emr S.D. The ESCRT pathway. Dev. Cell. 2011;21:77–91. doi: 10.1016/j.devcel.2011.05.015. [DOI] [PubMed] [Google Scholar]
- 33.Coghlin C., Carpenter B., Dundas S.R., Lawrie L.C., Telfer C., Murray G.I. Characterization and over-expression of chaperonin t-complex proteins in colorectal cancer. J. Pathol. 2006;210:351–357. doi: 10.1002/path.2056. [DOI] [PubMed] [Google Scholar]
- 34.Velazquez J.M., Lindquist S. hsp70: nuclear concentration during environmental stress and cytoplasmic storage during recovery. Cell. 1984;36:655–662. doi: 10.1016/0092-8674(84)90345-3. [DOI] [PubMed] [Google Scholar]
- 35.Mehlen P., Arrigo A.P. The serum-induced phosphorylation of mammalian hsp27 correlates with changes in its intracellular localization and levels of oligomerization. Eur. J. Biochem. 1994;221:327–334. doi: 10.1111/j.1432-1033.1994.tb18744.x. [DOI] [PubMed] [Google Scholar]
- 36.Hamelin C., Cornut E., Poirier F., Pons S., Beaulieu C., Charrier J.P., Haidous H., Cotte E., Lambert C., Piard F., et al. Identification and verification of heat shock protein 60 as a potential serum marker for colorectal cancer. FEBS J. 2011;278:4845–4859. doi: 10.1111/j.1742-4658.2011.08385.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hu Z.Z., Valencia J.C., Huang H., Chi A., Shabanowitz J., Hearing V.J., Appella E., Wu C. Comparative bioinformatics analyses and profiling of lysosome-related organelle proteomes. Int. J. Mass Spectrom. 2007;259:147–160. doi: 10.1016/j.ijms.2006.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Slater L.H., Hett E.C., Clatworthy A.E., Mark K.G., Hung D.T. CCT chaperonin complex is required for efficient delivery of anthrax toxin into the cytosol of host cells. Proc. Natl. Acad. Sci. U.S.A. 2013;110:9932–9937. doi: 10.1073/pnas.1302257110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cuellar J., Martin-Benito J., Scheres S.H., Sousa R., Moro F., Lopez-Vinas E., Gomez-Puertas P., Muga A., Carrascosa J.L., Valpuesta J.M. The structure of CCT-Hsc70 NBD suggests a mechanism for Hsp70 delivery of substrates to the chaperonin. Nat. Struct. Mol. Biol. 2008;15:858–864. doi: 10.1038/nsmb.1464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Eisenberg E., Greene L.E. Multiple roles of auxilin and hsc70 in clathrin-mediated endocytosis. Traffic. 2007;8:640–646. doi: 10.1111/j.1600-0854.2007.00568.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.