


Abstract
AbaSI, a member of the PvuRts1I-family of modification-dependent restriction endonucleases, cleaves deoxyribonucleic acid (DNA) containing 5-hydroxymethylctosine (5hmC) and glucosylated 5hmC (g5hmC), but not DNA containing unmodified cytosine. AbaSI has been used as a tool for mapping the genomic locations of 5hmC, an important epigenetic modification in the DNA of higher organisms. Here we report the crystal structures of AbaSI in the presence and absence of DNA. These structures provide considerable, although incomplete, insight into how this enzyme acts. AbaSI appears to be mainly a homodimer in solution, but interacts with DNA in our structures as a homotetramer. Each AbaSI subunit comprises an N-terminal, Vsr-like, cleavage domain containing a single catalytic site, and a C-terminal, SRA-like, 5hmC-binding domain. Two N-terminal helices mediate most of the homodimer interface. Dimerization brings together the two catalytic sites required for double-strand cleavage, and separates the 5hmC binding-domains by ∼70 Å, consistent with the known activity of AbaSI which cleaves DNA optimally between symmetrically modified cytosines ∼22 bp apart. The eukaryotic SET and RING-associated (SRA) domains bind to DNA containing 5-methylcytosine (5mC) in the hemi-methylated CpG sequence. They make contacts in both the major and minor DNA grooves, and flip the modified cytosine out of the helix into a conserved binding pocket. In contrast, the SRA-like domain of AbaSI, which has no sequence specificity, contacts only the minor DNA groove, and in our current structures the 5hmC remains intra-helical. A conserved, binding pocket is nevertheless present in this domain, suitable for accommodating 5hmC and g5hmC. We consider it likely, therefore, that base-flipping is part of the recognition and cleavage mechanism of AbaSI, but that our structures represent an earlier, pre-flipped stage, prior to actual recognition.
INTRODUCTION
In the deoxyribonucleic acid (DNA) of higher organisms, cytosine occurs in several chemical forms, including unmodified cytosine (C), 5-methylcytosine (5mC), 5-hydroxymethylcytosine (5hmC), 5-formylcytosine (5fC) and 5-carboxylcytosine (5caC) (1–7). These forms are genetically equivalent in terms of base-pairing and protein-coding, but they differ in how they interact with macromolecules and influence gene expression. There is much interest in the effects of these modifications in epigenetic regulation, in development and differentiation, in neuron function and in disease. In general, the modifications (or ‘marks’) are added to cytosine in situ, following its incorporation into DNA in the unmodified form. DNA methyltransferases convert certain cytosines to 5mC, usually within the sequence context CpG (8,9). And then ten-eleven translocation (Tet) dioxygenases convert a subset of these 5mC residues to 5hmC, 5fC and 5caC in consecutive, Fe(II)- and α-ketoglutarate-dependent oxidation reactions (10–13). The Tet dioxygenases are widely distributed across the eukaryotic tree of life (14), from mammals to the amoeboflagellate Naegleria gruberi (15).
To learn more about the functions of modified cytosines in the human genome, and about the mechanisms that control their genetic locations and levels, methods are needed to distinguish the modifications individually, and to map their positions accurately. Newly discovered ‘modification-dependent’ restriction endonucleases such as MspJI are helping in this regard (16). These enzymes recognize 5mC and 5hmC in certain sequence contexts and cleave the DNA wherever these occur, generating genomic fragments that can be sequenced and analyzed by bioinformatics (17,18).
In common with the chemical method of bisulfite conversion (19), enzymes of the MspJI-family cannot distinguish between 5mC and 5hmC. Enzymes from a different group, the PvuRts1I-family, can make this distinction, however, and these offer a promising way to map 5hmC specifically. PvuRts1I, from the bacterium Proteus vulgaris, was identified many years ago by its ability to restrict T-even bacteriophages containing 5hmC and g5hmC in their DNA (20,21), but it aroused little interest until the recent re-discovery of 5hmC in mammalian DNA (1,10). Since then, it has been purified and characterized (22,23) along with a number of homologs with similar, but subtly different, properties (24). One such homolog, AbaSI from Acinetobacter baumannii SDF, has been used successfully in conjunction with sequencing (Aba-seq) to map the locations of 5hmC in mouse embryonic stem cells (7).
AbaSI cleaves DNA containing g5hmC or 5hmC much more efficiently than DNA containing 5mC, by selectivity factors of 8000:500:1 (23). It has negligible activity on DNA containing only C. AbaSI cleaves with some variability 3′ to the modified cytosine, 11–13 nt away on the modified (‘top’) strand and 9–10 nt away on the complementary (‘bottom’) strand, producing fragments with short 3′-overhangs (23). AbaSI has no recognition sequence (‘context’) specificity, but optimal cleavage occurs when two (g)5hmC residues occur 21–23 bp apart on opposite DNA strands, whereupon cleavage takes place mid-way between them. Cleavage is less efficient if one of these two cytosines is unmodified, and much less efficient if the second cytosine is missing altogether (24). To understand this spatial requirement, and to learn more about the mechanism of modification-dependent recognition, we determined the crystal structures of AbaSI with substrate DNA, and without DNA. We report the structures, here, together with insights into the action of AbaSI gained from comparisons with the DNA co-crystal structures of the UHRF1 SRA domain, and the Vsr mismatch-repair endonuclease.
MATERIALS AND METHODS
Protein expression and purification
AbaSI from A. baumannii SDF, and originally designated ‘AbaSDFI’ (23), was expressed in Escherichia coli from a synthetic, codon-optimized, gene (Integrated DNA Technologies or IDT) and purified as previously described (23,24). A chitin-binding domain-intein tag was fused at its C-terminus for affinity purification purposes, and for crystallography, three cysteine residues were changed to serine to reduce oligomerization (described below). Typically, 6L cultures were grown at 30°C to late log phase, whereupon expression was induced by the addition of isopropyl β-d-1-thiogalactopyranoside (IPTG) to 0.2 mM, and overnight incubation at 16°C. Cells were harvested by centrifugation and lysed by French Press in 20 mM Tris-acetate (pH 8.0) and 500 mM potassium acetate (lysis buffer), followed by centrifugation at 18000 rpm. The cleared extract was loaded onto a chitin column [∼30 ml of chitin beads (NEB #S6651) were poured into a ∼80 ml gravity-flow column] pre-equilibrated with lysis buffer. The column was washed with 10+ column volumes of lysis buffer until a coomassie-stained blot revealed little further protein eluting from the column. To induce intein-mediated cleavage, 50 ml of the lysis buffer containing 30 mM dithiothreitol (DTT) was added to the column and incubated at 4°C overnight. Liberated AbaSI was then eluted from the column with lysis buffer containing 5 mM DTT until a blot revealed little further protein being recovered. At this stage, AbaSI was ∼90% pure.
Pooled protein was diluted 5-fold to ∼100 mM potassium acetate in 20 mM Tris-acetate (pH 8.0), 5 mM DTT and loaded onto tandem HiTrap Q/Heparin columns (GE Healthcare). Most of the AbaSI flowed through the Q column onto the Heparin column from which it was eluted as a single peak using a linear gradient of potassium acetate from 100 mM to 1 M. The position of the largest protein peak in a Superdex 200 column (GE Healthcare) appeared to indicate that it was mainly a dimer, with some higher molecular weight oligomers (Supplementary Figure S1a). This prompted us to create the variant ‘AbaSI-C3S’ by changing three cysteine residues at positions 2, 309 and 321, to serine. Cys2 (the first amino acid) and Cys321 (the last) are unique to AbaSI, whereas the equivalent of Cys309 in other family members is serine or threonine (Figure 1a). AbaSI-C3S, expressed and purified as the native protein, chromatographed as a single peak on the sizing column (Supplementary Figure S1b), was enzymatically active (Supplementary Figure S1c), and was the form used for crystallization (Supplementary Figure S1d).
Figure 1.
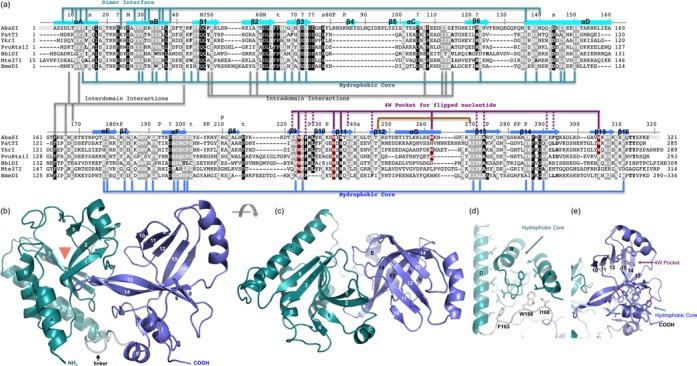
Monomeric structure of AbaSI. (a) Sequence alignment of AbaSI family members. The AbaSI residue numbering is shown above the sequence alignment. Amino acids highlighted are either invariant (white against black) among the seven proteins or similar (white against gray) as defined by the following groupings: V, L, I and M; F, Y and W; K and R, E and D; Q and N; E and Q; D and N; S and T and A, G and P. Helices are labeled αA-αG; strands are labeled β1–β16. Residues are indicated for forming the hydrophobic cores (see panels d and e), DNA phosphate binding (P), metal ion binding (M), structural turns (t) and surface exposed invariant residues (s). We note that residues 81–98 including strands β4 and β5 are unique to AbaSI, and helix αG is formed by the variable sequences. The first four N-terminal residues and the last three C-terminal residues (in gray) were not observed due to lack of continuous electron densities. (b and c) Two views of monomer structure of AbaSI, with N-terminal domain colored in turquoise, C-terminal domain colored in blue and the linker region in gray. The red triangle in panel b indicates a crevice formed between strands β2 and β3 where the putative cleavage active site is located (see Figure 2d). (d) Side chains from helices (αA, αB and αD) and strands (β1, β2, β3 and β6) (which are faded in the background) are buried in between forming a hydrophobic core of the N-terminal domain. (e) Side chains from helices (αE and αF) and strands β9, β13, β14 of the central curved sheet and strands β7 and β16 (which are faded in the background) are buried in between where they form a hydrophobic core of the C-terminal domain. In addition, four tryptophan residues of strand β9, β11, helix αG and strand β15 form a 4W pocket with additional contribution from hydrophobic residues from strands β9, β11, β12, β13 and β14 (see panel a).
Site-directed mutagenesis and activity assay
Alanine substitution mutagenesis was carried out by polymerase chain reaction (PCR) using vent polymerase (NEB #M0254) and pairs of synthetic mutagenic oligos (IDT). The PCR products were digested with DpnI to reduce template DNA carry-over, and transformed into E. coli strain T7 Express (NEB #C2566). All constructs were sequenced to verify that the desired mutations were present, with no additional changes.
Mutants were grown in 10 ml cultures and induced with IPTG, as described above. Crude cell-extracts were diluted 1, 10 and 100-fold in 250 mM potassium acetate, 10 mM Tris-acetate, pH 8.0, 0.2 mg/ml BSA (NEB Diluent E) and incubated with 200 ng of β-glucosylated phage T4 DNA in 50 mM potassium acetate, 20 mM Tris-acetate, 10 mM magnesium acetate, 1 mM DTT, pH 7.9 (NEB buffer 4) at room temperature (∼25ºC) for 20 min. The reaction products were then electrophoresed in 0.8% agarose gels. We note that the mutations may also affect protein expression level and/or stability, resulting in a similar observation of diminished enzyme activity in our assays using cell lysates. Under our assay conditions, the nonspecific nuclease activity of the extract from vector control (see Figure 2e, the last three lanes) is indistinguishable from that of the mutants with residual activity (<10%).
Figure 2.
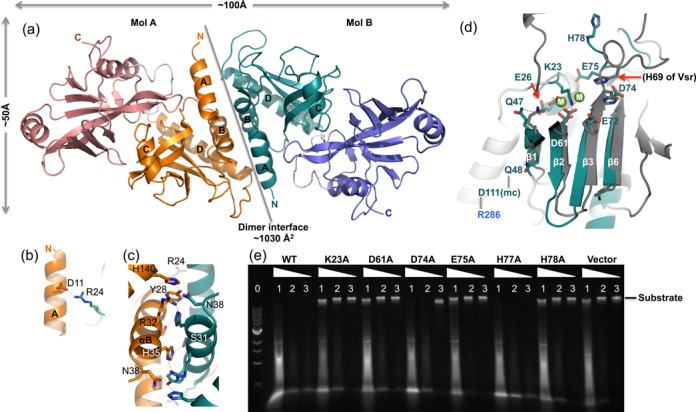
Dimeric structure of AbaSI. (a) Two AbaSI monomers (molecules A and B) form a dimer, mediated primarily by N-terminal helices (αA and αB). Labels ‘N’ and ‘C’ indicate amino and carboxyl termini of each molecule. (b and c) Examples of interactions involved in dimer interface. (d) A model of N-terminal Vsr-like endonuclease domain in complex with metal ions. Superimposition of the AbaSI N-terminal endonuclease domain (in turquoise) and the Vsr–DNA-Mg2+ complex (PDB: 1CW0) identified structural equivalent residues in AbaSI (in color) and in Vsr (in gray). The two Mg2+ ions (shown as green balls and labeled as ‘M’) are near the proposed catalytic residue Asp61 of strand β2. Other invariant charged residues among the AbaSI family near the modeled metal ions are Lys23, Glu26, Glu72, Asp74 and Glu75. (e) Activities of WT, mutants (K23A, D61A, D74A, E75A, H77A and H78A) and vector control. Lane 0: 1-kb DNA marker (NEB); lanes 1, 2 and 3: dilutions of 1-, 10- and 100-fold of crude cell-extracts.
Crystallography
We crystallized AbaSI-C3S in the absence of DNA, in the presence of substrate DNA and in the presence of product DNA (Supplementary Table S1). Crystallization was carried out by the sitting-drop vapor-diffusion method at 16°C, using equal amounts of protein (or protein–DNA mixtures) and well solutions. Crystals of protein alone (∼20 mg ml−1) could be grown with 2 M ammonium sulfate and 100 mM HEPES (pH 7.0).
For the protein in complex with substrate DNA, we started with a 28-bp double stranded oligonucleotide (oligo)—the minimum required length—containing one 5hmC five bases in from the 5′ end of the top strand and a second 5hmC (or C) 22-bp apart at position 28 on the opposite strand (Supplementary Figure S2a). This design was then lengthened one or two bp at a time to obtain oligos of 29-, 30- and 30-bp plus 5′-overhanging thymines (30 + 1), 32- and 32-bp plus 3′-overhanging thymines (32 + 1) (Supplementary Figure S2b). We used protein dimer:DNA ratios of 0.5:1, 1:1 and 2:1, with oligos of varying lengths. All combinations resulted in crystals in the same P21 space group, with varied diffraction limits. The best crystals contained ∼20 mg ml−1 protein, had a 1:1 ratio of dimeric protein to 32-bp DNA and grew in 21–24% (w/v) polyethylene glycol 3350, 200 mM ammonium tartrate, 100 mM BisTris (pH 6.0–6.4), 5 mM calcium acetate. Although calcium ions were present in the crystallization medium, they were not observed in the endonuclease catalytic sites in the structures, possibly due to chelation by the organic acids that were present.
For the protein in complex with product DNA, we used a 14-bp duplex oligo with a 4-nt, 3′-overhang, to mimic cleavage of the 32-bp oligo and dimer to DNA ratios of 0.5:1 or 1:1. At the time, a 4-base overhang seemed reasonable; now we suspect that oligos with a 2-base overhang, and slightly closer 5hmC bases, might be more informative. Experiments are in progress to address this. The crystals appeared under the conditions of 16–22% (w/v) polyethylene glycol 3000 or 3350, 100 mM HEPES (pH 7.2–7.8), with or without 170–220 mM ammonium tartrate or 2% Tacsimate (a mixture of weak organic acid salts including tartrate; Hampton Research). The crystallization conditions for the structure presented (Supplementary Table S1) were 20% (w/v) polyethylene glycol 3350, 200 mM ammonium tartrate and 100 mM HEPES (pH 7.4).
Selenomethionine (SeMet) was used for crystallographic phasing (25). Instead of Luria-Broth medium, AbaSI-C3S was expressed in E. coli BL21(DE3) utilizing M9 minimal medium (supplemented with glucose and the Difco yeast nitrogen base without amino acids and ammonium sulfate) where the l-amino acids Lys, Thr, Phe, Leu, Ile and Val were added immediately before L-SeMet addition and IPTG induction. A single anomalous dispersion (SAD) data set was collected from a crystal of selenomethionyl AbaSI-C3S (containing four methionines per molecule) in complex with 32-bp substrate DNA. The AutoSol module of the PHENIX software (26) identified a total of 16 selenium atoms. One set of four selenium atoms could be related to three other sets of four atoms, indicating four monomers in the asymmetric unit. The resulting electron density for α-helices, β-sheets and molecular envelopes could be visualized, but side chains and connecting loops could not be easily identified. A second Se-SAD dataset showed better traceable density that allowed a monomer to be completely traced. Molecular replacement using this monomer located the other three monomers and the DNA in the asymmetric unit.
All data sets were processed using the program HKL2000 (27). Phasing, map production, model refinement and molecular replacement were performed using PHENIX (28). Maps and models were visualized with COOT (29), which was also used for manual model manipulation during refinement rounds. Individual crystallographic thermal B-factors were refined only at the end stage of refinement process. In addition, rigid-body motion of domains and interdomain hinge motion, identified by the server of TLSMD (translation/libration/screw) (30,31), were also applied in the refinement. Molecular graphics were generated using PyMol (DeLano Scientific LLC).
RESULTS AND DISCUSSION
AbaSI forms a dimer
We determined the crystal structure of AbaSI on its own, with substrate DNA oligonucleotides (oligos), and with a product oligo. The crystallographic asymmetric unit contained one dimer in the absence of DNA and two dimers in the presence of DNA. The overall structures of the dimers were closely similar in all crystal forms, with pairwise root-mean-square deviations of ∼1.7 Å across 627 pairs of Cα atoms. We describe below the general structural features of AbaSI based on the complex with product DNA, which is representative of all of the structures, and was obtained at the highest resolution of 2.9 Å.
Monomeric AbaSI (37.7 kDa) consists of seven helices (αA-αG) and 16 strands (β1-β16) (Figure 1a–c). Sequence alignment of characterized members of this family shows that AbaSI has an 18-residue insertion between strand β3 and helix αC and five smaller insertions or deletions of four to eight residues, mostly in loops (Figure 1a). Sequence conservation is scattered throughout the protein, and includes residues involved in structural integrity, such as hydrophobic cores and inter- or intra-domain interactions (Figure 1d–e), as well as those with functional significance, such as DNA-binding, metal-ion coordination and catalysis.
Two monomers, A and B, interact with an interface of ∼1030 Å2 primarily through their two N-terminal helices, with additional contributions from the amino portion of helix αD (Figure 2a). These interactions include ion pairs between the side chains of Asp11 of helix αA and Arg24 (Figure 2b); hydrogen bonds (H-bonds) involving polar residues within and flanking helix αB (Tyr28, Ser31, Arg32, His35 and Asn38); and hydrophobic and aromatic residues of helix αA (Tyr22 and Ile14), helix αB (Tyr28 and Leu36) and helix αD (Leu136) (Figure 2c). Consistent with the dimerization observed in the crystal, purified AbaSI eluted mainly as what appeared to be a dimer during size-exclusion chromatography, with some tetramer and higher oligomers (Supplementary Figure S1a). The dimension along the long axis of the dimer is ∼100 Å (Figure 2a), sufficient to span ∼30 bp in B-form DNA. The recently reported monomeric structure of PvuRts1I (32) has the same dimeric interface, mediated by the crystallographic symmetry (Supplementary Figure S3).
The N-terminal Vsr-like endonuclease domain
The N-terminal dimerization helices αA and αB, together with αD, support and pack against one side of the four-stranded central β-sheet of the N-terminal domain. Curvature of these strands, a mix of anti-parallel (β1 and β2) and parallel (β3 and β6) topologies, results in a crevice between strands β2 and β3 where the putative catalytic site for DNA cleavage is located (Figure 1b, indicated by a triangle). The large majority of the conserved hydrophobic side chains intercalate with each other at the interface of the α helices and the β strands to form the hydrophobic core of the N-terminal domain (Figure 1d). Two small additional anti-parallel strands (β4 and β5) alongside strand β2 are unique to AbaSI due to the 18-residue insertion (Figure 1a and b).
VAST (vector alignment search tool) (33) and DALI (distance matric alignment) searches (34) against structures in the Protein Data Bank (PDB) showed that parts of the AbaSI N-terminal domain superimpose well on the very short patch repair DNA-nicking endonuclease, Vsr (35,36), on an intron homing endonuclease, I-Bth0305I (37), on the cleavage domain of Type IIG restriction endonuclease, BpuSI (38) and on several uncharacterized ‘restriction enzyme-like’ proteins (Supplementary Figure S4). Structurally similar elements include helices αB and αD, strands β2 and β3 containing the catalytic residues, and strand β6 (Supplementary Figure S4). Among these matching endonuclease structures, only Vsr has been crystallized with the essential catalytic co-factor, Mg2+ and with substrate DNA, which is cleaved (36).
Using the coordinates of the Vsr complex (PDB: 1CW0), we superimposed the proteins and positioned the Vsr DNA and Mg2+ ions over the N-terminal domain of AbaSI. The superimposition showed that the catalytic site of AbaSI is an unusual variant of the PD-D/EXK endonuclease superfamily (39–41) and perhaps also coordinates two Mg2+ ions (Figure 2d). The side chain of Asp61 in β2 (=Vsr Asp51), the main chain carbonyl of Val73 in β3 (=Vsr Thr63) and the side chain Glu75 in the loop after β3 are positioned to coordinate Mg2+ ions directly, as is customary in these catalytic sites. Mutation of Asp61 (D61A) and Glu75 (E75A) abolished activity (Figure 2e), as were also reported for the corresponding mutations (D57A and E71A) in PvuRts1I (32). In addition, the side chains of Glu72 and Asp74, conserved among the members of this enzyme family (Figure 1a) (23), might also participate in metal-ion coordination, but indirectly, via intermediate water molecules. Mutation of the latter residue (D74A) retained partial activity (Figure 2e).
Alternately, as occurs in BamHI (42), Asp74 or Glu75 might act as the general base in the catalytic reaction, assisting in the creation of the hydroxide nucleophile needed for in-line attack on the phosphorus atom, a hypothesized role usually assigned to the lysine (K) of the PD-D/EXK motif (43). In Vsr, His64 or His69 are positioned to act as the general base rather than lysine; His77 or His78 of AbaSI might act in this way, too. Mutation of His78 (H78A) eliminated activity whereas H77A has the usual WT activity (Figure 2e). Otherwise, Lys23, recruited from the loop preceding αB, is positioned to do this, instead. Lys23 also forms an ion bridge with Asp61 in the absence of metal ions (Figure 2d). Mutation of Lys23 (K23A) abolished activity (Figure 2e) as also did the corresponding lysine mutation (K17A) in PvuRts1I (32). Loss of activity in K23A and H78A mutants confirms the importance of these residues, both of which are highly conserved in members of this enzyme family.
Several other invariant residues are located near the catalytic site. Glu26 of helix αB (=Vsr Glu25) forms an H-bond with Gln47 of β1 (Vsr Gln42). Next to Gln47, Gln48 interacts with the main chain carbonyl oxygen of Asp111, which in turn forms an inter-domain interaction with Arg286 of the C-terminal domain (Figure 2d). His78 of AbaSI, located in the loop after strand β3, points away from the active site in the present model (Figure 2d). His69 of Vsr, located in the corresponding loop, is essential for Vsr endonuclease activity (35) and H-bonds with the cleaved phosphate group (36).
The DNA in the Vsr complex is significantly distorted (36). Phe67, Trp68, and His69 of Vsr penetrate the helix from the major groove and wedge apart adjacent base pairs by ∼60°. The equivalent residues of AbaSI, His77, His78, and Phe79, are also planar and might act in a similar way. Vsr-DNA superimposed on the AbaSI N-terminal domain fits well on one side of the catalytic site, but due to the distortion, poorly on the other. The loop preceding helix αD follows the major DNA groove, but the loop after strand β3 and the AbaSI-specific strands β4 and β5 do not; if the DNA were not distorted, these would occupy the minor groove. Vsr DNA superimposed on the N-terminal domain of one AbaSI subunit contacts the catalytic site of that subunit but not the catalytic site of other subunit in the homodimer. This might indicate that double-strand cleavage takes place in two steps by sequential strand-nicking reactions, or that conformational changes occur upon binding of DNA and/or metal ions that bring the components into proper register.
The C-terminal SRA-like DNA-binding domain
VAST and DALI searches also revealed that the C-terminal domain of AbaSI is structurally similar to the SET and RING-finger associated (SRA) domains of Arabidopsis SUVH5 (44), human and mouse UHRF1 (45–47), and the N-terminal DNA-binding domain of MspJI (48) (Supplementary Figure S5). In AbaSI, this domain contains eight β-strands (in the order 10, 11, 12, 15, 14, 13, 9 and 8) that together roughly form one twisted β-sheet resembling an arch (the ‘beta-arch’). Two long curved antiparallel strands, 15-residue β14 (residues 281–295) and 10-residue β13 (residues 268–277), are largely responsible for this conformation (Figure 1c and Supplementary Figure S5). Two short helices, αE and αF, pack on one side of this sheet against strands β8, β9, β13 and β14, with conserved hydrophobic residues in between (Figure 1b and e). One longer helix, αG, surmounts the sheet, and provides Trp262, one of four tryptophan residues that make up a unique ‘4W’ pocket (Figure 1e) within the beta-arch that accommodates (g)5hmC, we speculate, when this is flipped out of the helix (see below).
A rigid linker connects the two functional domains
A 10-residue linker (residues 161–170) connects the N-terminal Vsr-like domain to the C-terminal SRA-like domain. The linker contains three well-conserved hydrophobic residues (Phe163, Trp166 and Ile168) that pack against the hydrophobic core of the N-terminal domain (Figure 1d). Numerous additional inter-domain interactions account for ∼620 Å2 interface area. The extent and conservation of these interactions suggest that the overall AbaSI monomer has a rather stable structure that does not change greatly from what we observe in our crystal forms in the absence and presence of DNA.
DNA-protein interactions in the AbaSI co-crystals
For co-crystallization with AbaSI, substrate oligos of 28–32 bp were used (Supplementary Figure S2). All crystallized in the same P21 space group, with two AbaSI dimers and one DNA molecule in the crystallographic asymmetric unit. For product, a 14-bp oligo with complementary 4 nt, 3′-single-stranded ends was used. Two such molecules annealed via their ends in the product structure, forming a 32-bp duplex with one phosphodiester backbone break in each strand (Figure 3a and Supplementary Figure S6a). Regardless of whether substrate DNA or product DNA was used, the 32-bp duplexes stacked head-to-tail, with one neighboring DNA molecule at each end, forming a long helix, parallel to the crystal b-axis (Figure 4a). All four AbaSI subunits contribute to DNA backbone phosphate interactions, each dimer spanning ∼28 bp (Supplementary Figure S6b and c). The DNA in our co-crystals was aligned with the long axis of the AbaSI dimer, and the binding and catalytic domains were in the correct general locations for recognition and cleavage, but intimate contact with the DNA was completely absent.
Figure 3.
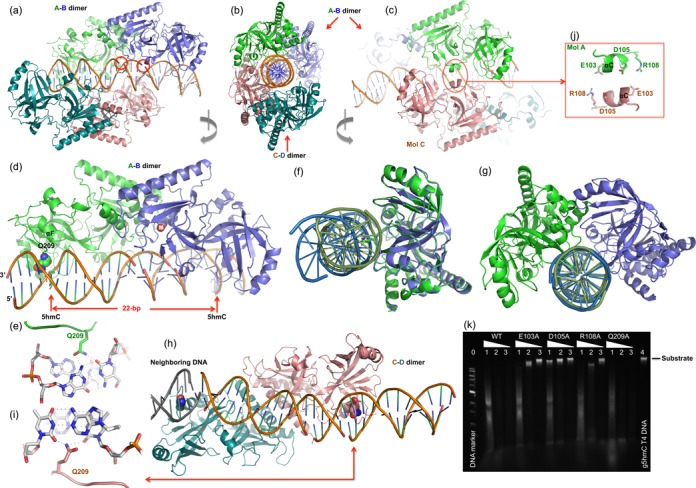
AbaSI-DNA interactions. (a, b and c) The DNA molecule is encircled by two pairs of AbaSI dimers, the A–B dimer and the C–D dimer, as indicated by colors. Three views are related via ∼90° rotations along a vertical axis as indicated. Two red circles in panels indicate the missing phosphate groups after annealing two product DNA molecules together by the 4-nt 3′ overhangs (Supplementary Figure S6a). (d) The A–B dimer approaches the DNA with Gln209 side chains (in space filling model) in the minor groove. (e) Gln209 of molecule A interacts with 5hmC:G pair from the minor groove. (f) Superimposition of molecules A (green) and B (blue) indicates relative rotation of the bound DNA molecules. (g) Superimposition of the corresponding DNA components indicates relative rotation between molecules A and B. (h) The C-D dimer, spanning two neighboring DNA molecules, has their Gln209 side chain (in space filling model) in the minor groove. (i) Gln209 of molecule D interacts with two thymines of neighboring base pairs (Supplementary Figure S6c). (j) The helix αC-mediated dimer–dimer interaction. (k) Activities of mutants of E103A, D105A, R108A and Q209A. Lane 0: 1-kb DNA marker (NEB); lanes 1, 2 and 3 for WT and mutants: dilutions of 1-, 10- and 100-fold; lane 4: substrate of β-glucosylated T4 DNA.
Figure 4.
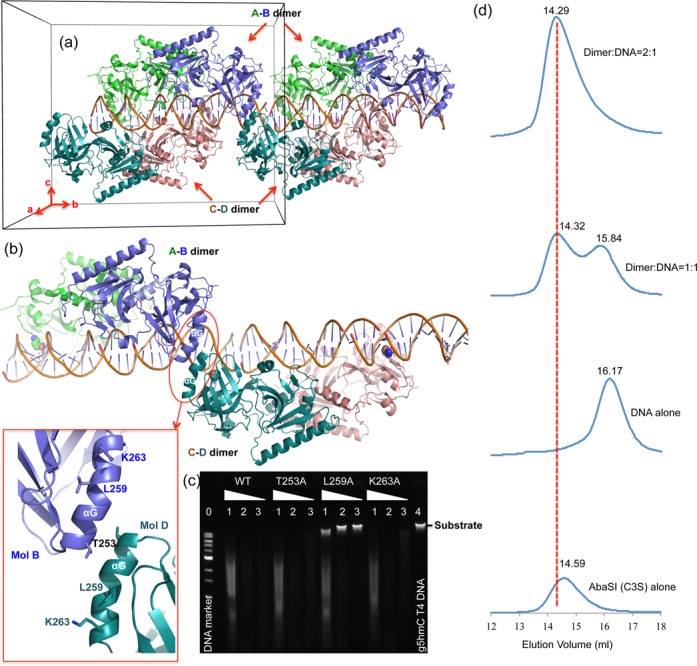
DNA-mediated dimer–dimer interaction. (a) The crystallographic asymmetric unit (indicated by a box) contains two dimers, A–B dimer and C–D dimer and one 32-bp DNA duplex. A second repeating unit along the b-axis is shown. (b) The helix αG-mediated dimer–dimer interaction in the crystal. (c) Activities of mutants of T253A, L259A and K263A. Lane 0: 1-kb DNA marker (NEB); lanes 1, 2 and 3 for WT and mutants: dilutions of 1-, 10- and 100-fold; lane 4: substrate of β-glucosylated T4 DNA. (d) Elution profiles of four consecutive runs on a Superdex 200 (10/300 GL) column (GE Healthcare) with 20 mM Tris acetate (pH 8.0), 150 mM potassium acetate, 5 mM DTT and 5 mM calcium acetate and containing (from bottom to top panels) protein alone (∼3 μM in 200 μl), DNA alone (∼3 μM in 200 μl; 29-bp duplex, see Supplementary Figure S2b), eqimolar AbaSI-C3S dimer and DNA duplex (∼3 μM in 200 μl), 2:1 molar ratio of AbaSI-C3S dimer (∼3 μM) to DNA duplex (∼1.5 μM in 400 μl). Peak heights reflected relative OD280 absorbance.
The only direct base contact we observed between AbaSI and the DNA is mediated by Gln209 in the minor groove (Figure 3d). Gln209 of molecule A of the A-B dimer forms two H-bonds (via the amide group) with the modified base pair, one with the O2 atom of 5hmC and the other with the 2-amino group of its partner guanine (Figure 3e). The Gln209 side chain also makes an additional H-bond with the O2 atom of the thymine 5′ to 5hmC. The phosphate-backbone contacts with molecule A are concentrated on four phosphate groups on each strand surrounding the 5hmC:G pair (Supplementary Figure S6b).
The side chain of the corresponding Gln209 of molecule B points toward the minor groove but is too far away to contact the second 5hmC 23 bp away at position 28. The corresponding phosphate contacts by molecule B are shifted 2–3 bp to the 3′ side of the second 5hmC as though the 5hmC residues were 2–3 bp too far apart (Supplementary Figure S6b). No major groove interactions with either 5hmC:G base pair is evident in the structures, suggesting that the modification status of the cytosine is not detected in the current crystal forms. Molecule A of the dimer interacts differently with the DNA than molecule B. Superimposing the protein components of molecules A and B, the corresponding bound DNA is misaligned, and must be rotated by ∼100° (Figure 3f), equivalent to 3 bp (360°/10.5 = 34°/bp), to coincide. Similarly, superimposing the DNA juxtaposed by molecules A and B requires that the latter be rotated by 100° in order to superimpose (Figure 3g). This difference suggests that the AbaSI dimer aligns most closely with modified cytosines that are only 19–20 bp apart. The DNA in the co-crystal structures is essentially straight. Bending at the center is needed for the DNA to contact the catalytic sites, some 15–20 Å away, and this might change the optimal spacing between the modified cytosines.
The second, C–D, dimer displays the same two glutamines in the minor groove of the DNA, separated by ∼22 bp in neighboring DNA molecules (Figure 3h and Supplementary Figure S6c). These do not juxtapose the modified cytosines, however. The side chain of Gln209 of molecule C interacts with the O2 atoms of adjacent thymine residues at positions 21 and 22 (Figure 3i), indicating that the Gln209-mediated interaction is not base-specific. Despite its singular interaction with DNA, Gln209 appears to be non-essential, as a Gln209-to-alanine (Q209A) mutant was found to display full wild-type activity (Figure 3k).
Dimer-dimer interactions
Dimers A–B and C–D have few direct contacts. These are confined to helix αC-mediated interaction between molecules A and C (Figure 3b and c) and helix αG-mediated interaction between molecule B and molecule D of the neighboring C–D dimer (Figure 4b). Two invariant residues, Asp105 and Arg108 (Figure 1a), are located in the helix αC and the following loop. Together with another acidic residue of helix αC, Glu103, which could form a potential ion bridge with Arg108, this charged surface appears to be important for catalysis as alanine-mutations of these three residues abolished (D105A), or severely reduced (E103A and R108A), activity (Figure 3k). Mutation of three surface residues of helix αG (T253A, L259A and K263A) indicated that only Leu259 is essential (Figure 4c). Neither the αC-mediated nor the αG-mediated interactions were observed in the absence of DNA, suggesting that they arise only upon DNA-binding. In solution, during gel-filtration chromatography, a 1:1 dimer:DNA mixture was found to elute as two peaks, one protein plus DNA, the other free DNA (Figure 4d). In contrast, a 2:1 dimer:DNA mixture eluted as a single peak of protein plus DNA. This suggests that the two-dimer plus one DNA complex seen in the crystal asymmetric unit (Figure 4a), although not specific, is consistent with the observation in solution under micromolar concentration of the complex.
Similarities with UHRF1 SRA–DNA complex
In the absence of a specific AbaSI–DNA recognition complex, we modeled 5hmC-containing DNA into the AbaSI C-terminal domain using the coordinates of the mouse UHRF1 (mUHRF1) SRA–DNA complex (PDB: 3FDE), in which the 5mC is extra-helical and flipped from the helix into a conserved binding pocket (47). The protein components were superimposed (Figure 5a) to position the DNA over the corresponding basic surface of AbaSI (Figure 5b and c) whereupon the flipped 5mC was found to occupy a cavity in the AbaSI C-terminal domain we term the ‘4W pocket’ (Figure 5d and e). In the UHRF1–SRA pocket, main chain atoms and the side chains of Asp474, Tyr471 and Tyr483 form the binding site for the methylated cytosine. Asp474 and Ala468 (main chain amide nitrogen, N), Gly470 (N) and Thr484 (main chain carbonyl oxygen, O), H-bond with the flipped base, compensating for the loss of the Watson–Crick H-bonds and the two tyrosine rings sandwich the base, compensating for the loss of aromatic base-pair stacking (Figure 5d and e). Comparable interactions are available for flipped 5(h)mC modeled into the AbaSI 4W pocket. The side chains of Asn236 (=Asp474), Glu247 (=Thr484 (O)), and the main chain of Arg227 (N) (=Ala468 (N)) are positioned to form H-bonds (Figure 5d), while Trp234 (=Tyr471) and perhaps Trp224 (=Tyr483) are positioned for aromatic stacking (Figure 5e). All of these amino acids are highly conserved among the members of this enzyme family (Figure 1a). Mutations to alanine of AbaSI residues that form the 4W pocket either abolished (W234A, R269A and W304A) or impaired (N236A and W224A) activity (Figure 5f), attesting to their importance. Equivalent mutations of two of these residues in PvuRts1I (W215A and E228) did the same (32).
Figure 5.
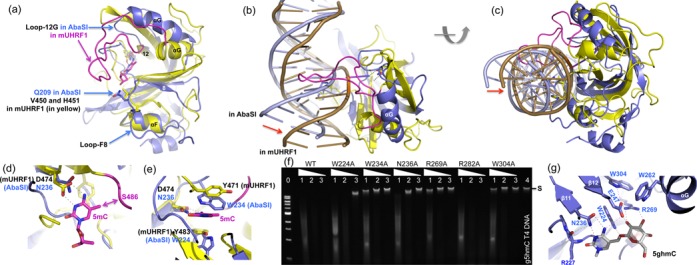
A model of AbaSI in complex with modified DNA. (a) Superimposition of the AbaSI C-terminal domain (in blue) with the SRA domain of mouse UHRF1 (in yellow; PDB: 3FDE). (b and c) Two 90° views of the DNA molecule observed in the current structure of AbaSI in comparison with the SRA-bound DNA, suggesting the AbaSI-bound DNA needs to move towards the 4W-binding pocket with a flipped cytosine. We note the long Loop-12G in mUHRF1 (colored in magenta) is in the DNA major groove. (d and e) The flipped 5mC nucleotide can be docked into the binding pocket of AbaSI (colored in blue). The SRA residues of mUHRF1 are in yellow and the long Loop-12G in magenta. (f) Activities of alanine mutants of residues that forms the 4W-pocket. Lane 0: 1-kb DNA marker (NEB); lanes 1, 2 and 3 for WT and mutants: dilutions of 1-, 10- and 100-fold; lane 4: substrate of β-glucosylated T4 DNA. For control, we included the phosphate-interacting Arg282 (Supplementary Figure S6b). (g) A model of glucosylated 5hmC in the 4W binding pocket of AbaSI.
Differences with UHRF1 SRA–DNA complex
Three interesting differences distinguish the AbaSI C-terminal domain from other SRA-domain proteins. The first concerns residue 236—asparagine in AbaSI (Asn236), but aspartate in UHRF1 (Asp474), MspJI (Asp103) and AspBHI (Asp71). The side chain of this residue accepts one H-bond from the 4-amino group of the flipped cytosine and donates one H-bond to its N3 ring atom, much as occurs during normal Watson–Crick base-pairing with guanine. Asparagine can donate to the N3 atom via its amide nitrogen (–NH2), but for aspartate to do so, its carboxylate group must be in the protonated state (–COOH). This is surprising since the pH (7–8) at which these enzymes operate is well above the pKa (3.9) of aspartate. The same is true for the conserved ‘motif V’ glutamate (ENV; pKa = 4.1) of the 5mC-methyltransferases (49–51), which likewise donates an H-bond to the flipped substrate cytosine and then protonates it preparatory to methyl transfer. The equivalent residue in the catalytic site of thymidylate synthase, whose substrate is dUMP, is also asparagine (52). For asparagine to H-bond with uracil rather than cytosine, its side chain must have the opposite orientation, rotated by 180° via the side chain χ2 torsion angle. If the Asn236 side chain can adopt both orientations, we anticipate that the 4W pocket of AbaSI might accommodate 5-hydroxymethyluracil (5hmU) and glucosyl-5hmU (base-J) in addition to modified cytosine. Preliminary data indicate that AbaSI is inactive on a 5hmU-containing DNA, however (not shown). This suggests that the Asn236 side chain cannot rotate, and indeed, close inspection reveals that its orientation is probably fixed by an H-bond with the main-chain oxygen of Arg227 (Figure 5g).
The second difference concerns the DNA-contact loops. Our model of the AbaSI C-terminal domain bound to the DNA specifically, derived from the mUHRF1 SRA–DNA complex, indicates that AbaSI contains an equivalent minor groove loop (Loop-F8), but lacks the long corresponding major groove loop (Loop-12G) used by mUHRF1 to recognize the modified cytosine and the CpG sequence-context in which it occurs (Figure 5a). AbaSI Loop-F8 (residues 201–211) contains Gln209, discussed previously. The corresponding minor groove loop of mUHRF1 contains Val451, which occupies the space left behind by the flipped 5mC and His450, which interacts with the 5′ base pair (47). The 24-residue major groove loop of mUHRF1 contains Arg496, which recognizes the orphan guanine via side chain H-bonds with the guanine O6 and N7 atoms, and Asn494, which recognizes the cytosine of the adjacent GC base pair via a main-chain H-bond (Figure 5b) (47). Consistent with the lack of sequence-specificity of AbaSI, its corresponding Loop-12G is only four residues long, making it too short to reach the DNA and recognize any sequence-context. In our model, Gln209 might make up for the lack of major groove H-bonds to the orphan guanine, when the (g)5hmC is flipped, by H-bonding with the guanine from the minor groove, instead. The two-loop mechanism used by mUHRF1 for substrate-recognition and base-flipping, in which the DNA is approached from opposite major and minor-groove directions, is also used by DNA 5mC-methyltransferases (53–55), DNA 5mC-dioxygenases (15,56), and DNA repair enzymes (57) including thymine DNA glycosylase which excises 5caC (58–60), an oxidation product of 5mC (12,13).
The third difference concerns the capacities of the binding-pockets. In mUHRF1, the methyl group of the flipped 5mC interacts with the Cα and Cβ atoms of Ser486 at the beginning of the Loop-12G (Figure 5d). There is no comparable interaction in AbaSI because Loop-12G of AbaSI is smaller and farther from the cytosine. As a result, the AbaSI pocket can accommodate cytosines with larger 5-modifications such as glucosylation. We modeled glucosylated hydroxymethylcytosine (g5hmC) into the 4W-pocket (Figure 5g) using the 5mC ring of the SRA–DNA complex as the foundation. Rotating three torsion angles between the cytosine ring and the glucosyl moiety allowed us to generate several possible conformations. Figure 5g shows one such conformation, in which the nitrogen of the indole ring of Trp224 interacts with the cytosine 5-methyl oxygen atom, and the guanidino group of Arg269 interacts with the glucose hydroxyl groups. In addition, Glu247, positioned alongside Asn236, could interact in several ways to stabilize the 4W pocket or the flipped nucleotide (Figure 5g).
A plausible model of AbaSI: coupling base recognition and DNA cleavage
A major difference between AbaSI and many other structurally characterized base-flipping enzymes is that AbaSI comprises two distinct domains—one for modified-base recognition, the other for DNA strand-cleavage. These likely communicate and cooperate in order to cleave DNA. The SRA-like, (g)5hmC-recognition domain comprises the C-terminus of AbaSI, and the Vsr-like endonuclease domain comprises the N-terminus. The order of these domains is the reverse of that in Type IIS restriction enzymes such as FokI (61), and in the modification-dependent restriction enzymes MspJI (48) and AspBHI (62). DNA-cleavage domains of restriction enzymes generally contain only one catalytic site, whereas two are required for duplex DNA cleavage. Usually, this shortfall is made up by dimerization (FokI) or tetramerization (MspJI and AspBHI) which juxtaposes pairs of catalytic sites in various ways that match the twist and the opposed polarities of the two DNA strands.
In the AbaSI–DNA complex reported here, the N-terminal domains are dimerized, but the two catalytic sites are too far from the DNA to cleave it, and too far apart to catalyze double-strand cleavage in unison. AbaSI is known to cleave with some variability, which might be a consequence of this geometric disparity. In PD-D/EXK catalytic sites, the principle Mg2+ ion is typically ∼3 Å from the target phosphorus atom, coordinated to one of its non-bridging oxygen atoms. Modeling Mg2+ ions from the Vsr–DNA complex into both catalytic sites of the AbaSI homodimer finds them to be ∼27 Å apart, a spacing appropriate therefore for hydrolyzing phosphates that are ∼21 Å apart. In B-form DNA, phosphates separated by a 2-nt, 3′-stagger, the principle substrates of AbaSI-hydrolysis, are on the order of 14 Å apart—∼7 Å closer, that is, than the catalytic sites in the dimer observed here (Figure 3d) [see, for example, the co-crystal structure of Eco29kI (63)]. Conformational changes like those seen in the Vsr complex (36), such as DNA bending and unwinding, might be needed, then, to bring the necessary elements together for catalysis. These conformational changes could accompany incorporation of Mg2+ ions into the active sites. In the absence of Mg2+ ions, AbaSI binds non-specifically to DNA containing different target modifications with approximately equal affinity (KD = 1.5–2 μM; Supplementary Figure S1e and f). It is possible that there is a temporal order for cleavage that proceeds by recognition of the target cytosine, flipping from DNA helix and capture by the 4W pocket, Mg2+-binding by the catalytic sites, and then double-strand DNA cleavage.
For optimal cleavage, AbaSI prefers two target cytosines symmetrically positioned around the cleavage site. One must be modified at ring position 5 by a hydroxymethyl (5hm) or glucosylated 5hm-group, but the other can be modified or unmodified (23,24). The requirement of the second cytosine appears not be absolute, however. In the ‘Aba-seq’ mapping of genomic 5hmC in mouse embryonic stem cells (7), the largest set of AbaSI cleavage sites (42.3%) have one CG and one CH (H = A/C/T) on opposite sides of the cleavage site, but the second largest set (27.3%) has one CG on one side and no cytosine at all on the other side. Asymmetric binding by the AbaSI dimer, with molecule A approaching one of the two 5hmC sites and molecule B is further away from the second 5hmC (Figure 3f and g), might account for this relaxed requirement for a second cytosine.
Diversity of restriction enzymes
Restriction enzymes have proven invaluable as laboratory tools for analyzing DNA molecules and rearranging them. They occur naturally in bacteria and archaea, and come in numerous different forms (64), from simple monomers [e.g. MspI (65)] and dimers [BamHI (66,67); PvuII (68)], to tetramers [NgoMIV (69); SfiI (70)], polymers [SgrAI (71)], and complex enzymes with allosteric regulatory domains [NaeI (72,73); EcoRII (74)]. The proteins can comprise one domain [HindIII (75)], two domains [FokI (61,76)], three [MmeI (77)] or more [TstI (78)]. Some cleave DNA exclusively one strand at a time [HinP1I (79,80)], others cleave both strands at once [EcoRI (81)] and some even multiple strands at once [BcgI (82)]. Most require one or two Mg2+ ions (83,84), but a few require no metal-ions [BfiI (85)], and others can use an array of different metal ions [HpyAV (86)]. Some barely distort DNA when they bind [BglII (87)], some distort it substantially [EcoRV (88)], and yet others distort it dramatically [PacI (89)]. All this variety reflects the amazing biochemical dexterity of microbes.
Most of the characterized restriction enzymes belong to the ‘Type II’ class and cleave unmodified DNA in which the bases are present in their ordinary, unaltered forms. Alongside these, we are learning, exists an alternative galaxy of enzymes with the opposite property of cleaving DNA only when it is modified. These ‘Type IV’ (90) restriction enzymes recognize DNA in which adenine or cytosine bases are altered by the addition of methyl groups, or small chemical derivatives, in the major DNA groove (48,62). AbaSI belongs to this latter group, about which little is yet known, including how diverse and numerous they are. Whereas Type II enzymes are used mainly for DNA cloning, Type IV enzymes are finding uses in epigenetic analysis. They cleave genomic DNA molecules into fragments that flank, or bracket, the sites of cytosine-modification, and that can be analyzed by sequencing and bioinformatics. Some successes in this regard have already been reported (7,17,18). The work described here contributes to our growing understanding of these new enzymes, and of their utility for investigating the epigenetic processes of higher organisms.
ACCESSION NUMBERS
The X-ray structures (coordinates and structure factor files) of AbaSI have been submitted to PDB under accession number 4PAR (protein-product DNA), 4PBA (protein-substrate DNA) and 4PBB (protein alone).
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
Acknowledgments
The authors thank Don Comb and Jim Ellard for enlightened support for the research presented here; Richard J. Roberts and Bill Jack for leadership and encouragement, and together with other members of the restriction enzymes and epigenetics research groups, for numerous thoughtful discussions. We thank John Buswell and his Organic Synthesis group for DNA oligonucleotides; and Hideharu Hashimoto for help with DNA-binding assays. X.C. is a Georgia Research Alliance Eminent Scholar.
Author contributions: J.R.H. performed crystallographic work and gel filtration chromatography, J.G.B. and A.Q. performed site-directed mutagenesis, R.M.G. and X.Z. performed initial purification and crystallization trials, X.Z. suggested the hydrodynamic experiment, S.G. duplicated experiments on mutant activities and tested AbaSI activity on 5hmU, G.G.W. performed structural analysis and assisted in preparing the manuscript, X.C., Y.Z. and Z.Z. organized and designed the scope of the study, and all were involved in analyzing data and preparing the manuscript.
FUNDING
National Institutes of Health (NIH) [GM049245-20 to X.C.]; NIH Small Business Innovation Research [GM096723 to Z.Z.]; Emory University School of Medicine, Department of Biochemistry, supports the use of Southeast Regional Collaborative Access Team (SER-CAT) 22-ID beamline at the Advanced Photon Source (APS), Argonne National Laboratory; Use of the APS was supported by the U.S. Department of Energy, Office of Science. Funding for open access charge: NIH.
Conflict of interest statement. AbaSI and others restriction enzymes mentioned in this article are products of New England Biolabs, Inc., a company that studies, purifies and sells restriction enzymes. The authors declare no competing interests.
REFERENCES
- 1.Kriaucionis S., Heintz N. The nuclear DNA base 5-hydroxymethylcytosine is present in Purkinje neurons and the brain. Science. 2009;324:929–930. doi: 10.1126/science.1169786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Globisch D., Munzel M., Muller M., Michalakis S., Wagner M., Koch S., Bruckl T., Biel M., Carell T. Tissue distribution of 5-hydroxymethylcytosine and search for active demethylation intermediates. PLoS One. 2010;5:e15367. doi: 10.1371/journal.pone.0015367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stroud H., Feng S., Morey Kinney S., Pradhan S., Jacobsen S.E. 5-Hydroxymethylcytosine is associated with enhancers and gene bodies in human embryonic stem cells. Genome Biol. 2011;12:R54. doi: 10.1186/gb-2011-12-6-r54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Booth M.J., Branco M.R., Ficz G., Oxley D., Krueger F., Reik W., Balasubramanian S. Quantitative sequencing of 5-methylcytosine and 5-hydroxymethylcytosine at single-base resolution. Science. 2012;336:934–937. doi: 10.1126/science.1220671. [DOI] [PubMed] [Google Scholar]
- 5.Yu M., Hon G.C., Szulwach K.E., Song C.X., Zhang L., Kim A., Li X., Dai Q., Shen Y., Park B., et al. Base-resolution analysis of 5-hydroxymethylcytosine in the mammalian genome. Cell. 2012;149:1368–1380. doi: 10.1016/j.cell.2012.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Raiber E.A., Beraldi D., Ficz G., Burgess H.E., Branco M.R., Murat P., Oxley D., Booth M.J., Reik W., Balasubramanian S. Genome-wide distribution of 5-formylcytosine in embryonic stem cells is associated with transcription and depends on thymine DNA glycosylase. Genome Biol. 2012;13:R69. doi: 10.1186/gb-2012-13-8-r69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sun Z., Terragni J., Borgaro J.G., Liu Y., Yu L., Guan S., Wang H., Sun D., Cheng X., Zhu Z., et al. High-resolution enzymatic mapping of genomic 5-hydroxymethylcytosine in mouse embryonic stem cells. Cell Rep. 2013;3:567–576. doi: 10.1016/j.celrep.2013.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bestor T., Laudano A., Mattaliano R., Ingram V. Cloning and sequencing of a cDNA encoding DNA methyltransferase of mouse cells. The carboxyl-terminal domain of the mammalian enzymes is related to bacterial restriction methyltransferases. J. Mol. Biol. 1988;203:971–983. doi: 10.1016/0022-2836(88)90122-2. [DOI] [PubMed] [Google Scholar]
- 9.Okano M., Xie S., Li E. Cloning and characterization of a family of novel mammalian DNA (cytosine-5) methyltransferases. Nat. Genet. 1998;19:219–220. doi: 10.1038/890. [DOI] [PubMed] [Google Scholar]
- 10.Tahiliani M., Koh K.P., Shen Y., Pastor W.A., Bandukwala H., Brudno Y., Agarwal S., Iyer L.M., Liu D.R., Aravind L., et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science. 2009;324:930–935. doi: 10.1126/science.1170116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ito S., D'Alessio A.C., Taranova O.V., Hong K., Sowers L.C., Zhang Y. Role of Tet proteins in 5mC to 5hmC conversion, ES-cell self-renewal and inner cell mass specification. Nature. 2010;466:1129–1133. doi: 10.1038/nature09303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ito S., Shen L., Dai Q., Wu S.C., Collins L.B., Swenberg J.A., He C., Zhang Y. Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science. 2011;333:1300–1303. doi: 10.1126/science.1210597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.He Y.F., Li B.Z., Li Z., Liu P., Wang Y., Tang Q., Ding J., Jia Y., Chen Z., Li L., et al. Tet-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA. Science. 2011;333:1303–1307. doi: 10.1126/science.1210944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Iyer L.M., Zhang D., Maxwell Burroughs A., Aravind L. Computational identification of novel biochemical systems involved in oxidation, glycosylation and other complex modifications of bases in DNA. Nucleic Acids Res. 2013;41:7635–7655. doi: 10.1093/nar/gkt573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hashimoto H., Pais J.E., Zhang X., Saleh L., Fu Z.Q., Dai N., Correa I.R., Jr., Zheng Y., Cheng X. Structure of a Naegleria Tet-like dioxygenase in complex with 5-methylcytosine DNA. Nature. 2014;506:391–395. doi: 10.1038/nature12905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zheng Y., Cohen-Karni D., Xu D., Chin H.G., Wilson G., Pradhan S., Roberts R.J. A unique family of Mrr-like modification-dependent restriction endonucleases. Nucleic Acids Res. 2010;38:5527–5534. doi: 10.1093/nar/gkq327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cohen-Karni D., Xu D., Apone L., Fomenkov A., Sun Z., Davis P.J., Kinney S.R., Yamada-Mabuchi M., Xu S.Y., Davis T., et al. The MspJI family of modification-dependent restriction endonucleases for epigenetic studies. Proc. Natl. Acad. Sci. U.S.A. 2011;108:11040–11045. doi: 10.1073/pnas.1018448108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Huang X., Lu H., Wang J.W., Xu L., Liu S., Sun J., Gao F. High-throughput sequencing of methylated cytosine enriched by modification-dependent restriction endonuclease MspJI. BMC Genet. 2013;14:56. doi: 10.1186/1471-2156-14-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Huang Y., Pastor W.A., Shen Y., Tahiliani M., Liu D.R., Rao A. The behaviour of 5-hydroxymethylcytosine in bisulfite sequencing. PLoS One. 2010;5:e8888. doi: 10.1371/journal.pone.0008888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ishaq M., Kaji A. Mechanism of T4 phage restriction by plasmid Rts1. Cleavage of T4 phage DNA by Rts1-specific enzyme. J. Biol. Chem. 1980;255:4040–4047. [PubMed] [Google Scholar]
- 21.Janosi L., Yonemitsu H., Hong H., Kaji A. Molecular cloning and expression of a novel hydroxymethylcytosine-specific restriction enzyme (PvuRts1I) modulated by glucosylation of DNA. J. Mol. Biol. 1994;242:45–61. doi: 10.1006/jmbi.1994.1556. [DOI] [PubMed] [Google Scholar]
- 22.Szwagierczak A., Brachmann A., Schmidt C.S., Bultmann S., Leonhardt H., Spada F. Characterization of PvuRts1I endonuclease as a tool to investigate genomic 5-hydroxymethylcytosine. Nucleic Acids Res. 2011;39:5149–5156. doi: 10.1093/nar/gkr118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang H., Guan S., Quimby A., Cohen-Karni D., Pradhan S., Wilson G., Roberts R.J., Zhu Z., Zheng Y. Comparative characterization of the PvuRts1I family of restriction enzymes and their application in mapping genomic 5-hydroxymethylcytosine. Nucleic Acids Res. 2011;39:9294–9305. doi: 10.1093/nar/gkr607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Borgaro J.G., Zhu Z. Characterization of the 5-hydroxymethylcytosine-specific DNA restriction endonucleases. Nucleic Acids Res. 2013;41:4198–4206. doi: 10.1093/nar/gkt102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hendrickson W.A., Horton J.R., LeMaster D.M. Selenomethionyl proteins produced for analysis by multiwavelength anomalous diffraction (MAD): a vehicle for direct determination of three-dimensional structure. EMBO J. 1990;9:1665–1672. doi: 10.1002/j.1460-2075.1990.tb08287.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Terwilliger T.C., Adams P.D., Read R.J., McCoy A.J., Moriarty N.W., Grosse-Kunstleve R.W., Afonine P.V., Zwart P.H., Hung L.W. Decision-making in structure solution using Bayesian estimates of map quality: the PHENIX AutoSol wizard. Acta Crystallogr. D Biol. Crystallogr. 2009;65:582–601. doi: 10.1107/S0907444909012098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Otwinowski Z., Borek D., Majewski W., Minor W. Multiparametric scaling of diffraction intensities. Acta Crystallogr. A. 2003;59:228–234. doi: 10.1107/s0108767303005488. [DOI] [PubMed] [Google Scholar]
- 28.Adams P.D., Afonine P.V., Bunkoczi G., Chen V.B., Davis I.W., Echols N., Headd J.J., Hung L.W., Kapral G.J., Grosse-Kunstleve R.W., et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Emsley P., Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 30.Painter J., Merritt E.A. Optimal description of a protein structure in terms of multiple groups undergoing TLS motion. Acta Crystallogr. D Biol. Crystallogr. 2006;62:439–450. doi: 10.1107/S0907444906005270. [DOI] [PubMed] [Google Scholar]
- 31.Painter J., Merritt E.A. A molecular viewer for the analysis of TLS rigid-body motion in macromolecules. Acta Crystallogr. D Biol. Crystallogr. 2005;61:465–471. doi: 10.1107/S0907444905001897. [DOI] [PubMed] [Google Scholar]
- 32.Kazrani A.A., Kowalska M., Czapinska H., Bochtler M. Crystal structure of the 5hmC specific endonuclease PvuRts1I. Nucleic Acids Res. 2014;42 doi: 10.1093/nar/gku186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gibrat J.F., Madej T., Bryant S.H. Surprising similarities in structure comparison. Curr. Opin. Struct. Biol. 1996;6:377–385. doi: 10.1016/s0959-440x(96)80058-3. [DOI] [PubMed] [Google Scholar]
- 34.Holm L., Rosenstrom P. Dali server: conservation mapping in 3D. Nucleic Acids Res. 2010;38:W545–W549. doi: 10.1093/nar/gkq366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tsutakawa S.E., Muto T., Kawate T., Jingami H., Kunishima N., Ariyoshi M., Kohda D., Nakagawa M., Morikawa K. Crystallographic and functional studies of very short patch repair endonuclease. Mol. Cell. 1999;3:621–628. doi: 10.1016/s1097-2765(00)80355-x. [DOI] [PubMed] [Google Scholar]
- 36.Tsutakawa S.E., Jingami H., Morikawa K. Recognition of a TG mismatch: the crystal structure of very short patch repair endonuclease in complex with a DNA duplex. Cell. 1999;99:615–623. doi: 10.1016/s0092-8674(00)81550-0. [DOI] [PubMed] [Google Scholar]
- 37.Taylor G.K., Heiter D.F., Pietrokovski S., Stoddard B.L. Activity, specificity and structure of I-Bth0305I: a representative of a new homing endonuclease family. Nucleic Acids Res. 2011;39:9705–9719. doi: 10.1093/nar/gkr669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shen B.W., Xu D., Chan S.H., Zheng Y., Zhu Z., Xu S.Y., Stoddard B.L. Characterization and crystal structure of the type IIG restriction endonuclease RM.BpuSI. Nucleic Acids Res. 2011;39:8223–8236. doi: 10.1093/nar/gkr543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bujnicki J.M., Rychlewski L. Grouping together highly diverged PD-(D/E)XK nucleases and identification of novel superfamily members using structure-guided alignment of sequence profiles. J. Mol. Microbiol. Biotechnol. 2001;3:69–72. [PubMed] [Google Scholar]
- 40.Laganeckas M., Margelevicius M., Venclovas C. Identification of new homologs of PD-(D/E)XK nucleases by support vector machines trained on data derived from profile-profile alignments. Nucleic Acids Res. 2011;39:1187–1196. doi: 10.1093/nar/gkq958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Steczkiewicz K., Muszewska A., Knizewski L., Rychlewski L., Ginalski K. Sequence, structure and functional diversity of PD-(D/E)XK phosphodiesterase superfamily. Nucleic Acids Res. 2012;40:7016–7045. doi: 10.1093/nar/gks382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Viadiu H., Aggarwal A.K. The role of metals in catalysis by the restriction endonuclease BamHI. Nat. Struct. Biol. 1998;5:910–916. doi: 10.1038/2352. [DOI] [PubMed] [Google Scholar]
- 43.Xie F., Briggs J.M., Dupureur C.M. Nucleophile activation in PD…(D/E)xK metallonucleases: an experimental and computational pK(a) study. J. Inorg. Biochem. 2010;104:665–672. doi: 10.1016/j.jinorgbio.2010.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rajakumara E., Law J.A., Simanshu D.K., Voigt P., Johnson L.M., Reinberg D., Patel D.J., Jacobsen S.E. A dual flip-out mechanism for 5mC recognition by the Arabidopsis SUVH5 SRA domain and its impact on DNA methylation and H3K9 dimethylation in vivo. Genes Dev. 2011;25:137–152. doi: 10.1101/gad.1980311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Arita K., Ariyoshi M., Tochio H., Nakamura Y., Shirakawa M. Recognition of hemi-methylated DNA by the SRA protein UHRF1 by a base-flipping mechanism. Nature. 2008;455:818–821. doi: 10.1038/nature07249. [DOI] [PubMed] [Google Scholar]
- 46.Avvakumov G.V., Walker J.R., Xue S., Li Y., Duan S., Bronner C., Arrowsmith C.H., Dhe-Paganon S. Structural basis for recognition of hemi-methylated DNA by the SRA domain of human UHRF1. Nature. 2008;455:822–825. doi: 10.1038/nature07273. [DOI] [PubMed] [Google Scholar]
- 47.Hashimoto H., Horton J.R., Zhang X., Bostick M., Jacobsen S.E., Cheng X. The SRA domain of UHRF1 flips 5-methylcytosine out of the DNA helix. Nature. 2008;455:826–829. doi: 10.1038/nature07280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Horton J.R., Mabuchi M.Y., Cohen-Karni D., Zhang X., Griggs R.M., Samaranayake M., Roberts R.J., Zheng Y., Cheng X. Structure and cleavage activity of the tetrameric MspJI DNA modification-dependent restriction endonuclease. Nucleic Acids Res. 2012;40:9763–9773. doi: 10.1093/nar/gks719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wu J.C., Santi D.V. Kinetic and catalytic mechanism of HhaI methyltransferase. J. Biol. Chem. 1987;262:4778–4786. [PubMed] [Google Scholar]
- 50.Baker D.J., Kan J.L., Smith S.S. Recognition of structural perturbations in DNA by human DNA(cytosine-5)methyltransferase. Gene. 1988;74:207–210. doi: 10.1016/0378-1119(88)90288-0. [DOI] [PubMed] [Google Scholar]
- 51.O'Gara M., Klimasauskas S., Roberts R.J., Cheng X. Enzymatic C5-cytosine methylation of DNA: mechanistic implications of new crystal structures for HhaI methyltransferase-DNA-AdoHcy complexes. J. Mol. Biol. 1996;261:634–645. doi: 10.1006/jmbi.1996.0489. [DOI] [PubMed] [Google Scholar]
- 52.Liu L., Santi D.V. Mutation of asparagine 229 to aspartate in thymidylate synthase converts the enzyme to a deoxycytidylate methylase. Biochemistry. 1992;31:5100–5104. doi: 10.1021/bi00137a002. [DOI] [PubMed] [Google Scholar]
- 53.Klimasauskas S., Kumar S., Roberts R.J., Cheng X. HhaI methyltransferase flips its target base out of the DNA helix. Cell. 1994;76:357–369. doi: 10.1016/0092-8674(94)90342-5. [DOI] [PubMed] [Google Scholar]
- 54.Reinisch K.M., Chen L., Verdine G.L., Lipscomb W.N. The crystal structure of HaeIII methyltransferase convalently complexed to DNA: an extrahelical cytosine and rearranged base pairing. Cell. 1995;82:143–153. doi: 10.1016/0092-8674(95)90060-8. [DOI] [PubMed] [Google Scholar]
- 55.Song J., Teplova M., Ishibe-Murakami S., Patel D.J. Structure-based mechanistic insights into DNMT1-mediated maintenance DNA methylation. Science. 2012;335:709–712. doi: 10.1126/science.1214453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hu L., Li Z., Cheng J., Rao Q., Gong W., Liu M., Shi Y.G., Zhu J., Wang P., Xu Y. Crystal structure of TET2-DNA complex: insight into TET-mediated 5mC oxidation. Cell. 2013;155:1545–1555. doi: 10.1016/j.cell.2013.11.020. [DOI] [PubMed] [Google Scholar]
- 57.Slupphaug G., Mol C.D., Kavli B., Arvai A.S., Krokan H.E., Tainer J.A. A nucleotide-flipping mechanism from the structure of human uracil-DNA glycosylase bound to DNA. Nature. 1996;384:87–92. doi: 10.1038/384087a0. [DOI] [PubMed] [Google Scholar]
- 58.Maiti A., Drohat A.C. Thymine DNA glycosylase can rapidly excise 5-formylcytosine and 5-carboxylcytosine: potential implications for active demethylation of CpG sites. J. Biol. Chem. 2011;286:35334–35338. doi: 10.1074/jbc.C111.284620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhang L., Lu X., Lu J., Liang H., Dai Q., Xu G.L., Luo C., Jiang H., He C. Thymine DNA glycosylase specifically recognizes 5-carboxylcytosine-modified DNA. Nat. Chem. Biol. 2012;8:328–330. doi: 10.1038/nchembio.914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hashimoto H., Hong S., Bhagwat A.S., Zhang X., Cheng X. Excision of 5-hydroxymethyluracil and 5-carboxylcytosine by the thymine DNA glycosylase domain: its structural basis and implications for active DNA demethylation. Nucleic Acids Res. 2012;40:10203–10214. doi: 10.1093/nar/gks845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wah D.A., Hirsch J.A., Dorner L.F., Schildkraut I., Aggarwal A.K. Structure of the multimodular endonuclease FokI bound to DNA. Nature. 1997;388:97–100. doi: 10.1038/40446. [DOI] [PubMed] [Google Scholar]
- 62.Horton J.R., Nugent R.L., Li A., Mabuchi M.Y., Fomenkov A., Cohen-Karni D., Griggs R.M., Zhang X., Wilson G.G., Zheng Y., et al. Structure and mutagenesis of the DNA modification-dependent restriction endonuclease AspBHI. Sci. Rep. 2014;4:4246. doi: 10.1038/srep04246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mak A.N., Lambert A.R., Stoddard B.L. Folding, DNA recognition, and function of GIY-YIG endonucleases: crystal structures of R.Eco29kI. Structure. 2010;18:1321–1331. doi: 10.1016/j.str.2010.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Pingoud A., Fuxreiter M., Pingoud V., Wende W. Type II restriction endonucleases: structure and mechanism. Cell. Mol. Life Sci. 2005;62:685–707. doi: 10.1007/s00018-004-4513-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Xu Q.S., Roberts R.J., Guo H.C. Two crystal forms of the restriction enzyme MspI-DNA complex show the same novel structure. Protein Sci. 2005;14:2590–2600. doi: 10.1110/ps.051565105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Newman M., Strzelecka T., Dorner L.F., Schildkraut I., Aggarwal A.K. Structure of restriction endonuclease BamHI and its relationship to EcoRI. Nature. 1994;368:660–664. doi: 10.1038/368660a0. [DOI] [PubMed] [Google Scholar]
- 67.Newman M., Strzelecka T., Dorner L.F., Schildkraut I., Aggarwal A.K. Structure of BamHI endonuclease bound to DNA: partial folding and unfolding on DNA binding. Science. 1995;269:656–663. doi: 10.1126/science.7624794. [DOI] [PubMed] [Google Scholar]
- 68.Cheng X., Balendiran K., Schildkraut I., Anderson J.E. Structure of PvuII endonuclease with cognate DNA. EMBO J. 1994;13:3927–3935. doi: 10.1002/j.1460-2075.1994.tb06708.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Deibert M., Grazulis S., Sasnauskas G., Siksnys V., Huber R. Structure of the tetrameric restriction endonuclease NgoMIV in complex with cleaved DNA. Nat. Struct. Biol. 2000;7:792–799. doi: 10.1038/79032. [DOI] [PubMed] [Google Scholar]
- 70.Bellamy S.R., Milsom S.E., Kovacheva Y.S., Sessions R.B., Halford S.E. A switch in the mechanism of communication between the two DNA-binding sites in the SfiI restriction endonuclease. J. Mol. Biol. 2007;373:1169–1183. doi: 10.1016/j.jmb.2007.08.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lyumkis D., Talley H., Stewart A., Shah S., Park C.K., Tama F., Potter C.S., Carragher B., Horton N.C. Allosteric regulation of DNA cleavage and sequence-specificity through run-on oligomerization. Structure. 2013;21:1848–1858. doi: 10.1016/j.str.2013.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Huai Q., Colandene J.D., Chen Y., Luo F., Zhao Y., Topal M.D., Ke H. Crystal structure of NaeI - an evolutionary bridge between DNA endonuclease and topoisomerase. EMBO J. 2000;19:3110–3118. doi: 10.1093/emboj/19.12.3110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Embleton M.L., Siksnys V., Halford S.E. DNA cleavage reactions by type II restriction enzymes that require two copies of their recognition sites. J. Mol. Biol. 2001;311:503–514. doi: 10.1006/jmbi.2001.4892. [DOI] [PubMed] [Google Scholar]
- 74.Tamulaitis G., Sasnauskas G., Mucke M., Siksnys V. Simultaneous binding of three recognition sites is necessary for a concerted plasmid DNA cleavage by EcoRII restriction endonuclease. J. Mol. Biol. 2006;358:406–419. doi: 10.1016/j.jmb.2006.02.024. [DOI] [PubMed] [Google Scholar]
- 75.Watanabe N., Takasaki Y., Sato C., Ando S., Tanaka I. Structures of restriction endonuclease HindIII in complex with its cognate DNA and divalent cations. Acta Crystallogr. D Biol. Crystallogr. 2009;65:1326–1333. doi: 10.1107/S0907444909041134. [DOI] [PubMed] [Google Scholar]
- 76.Vanamee E.S., Santagata S., Aggarwal A.K. FokI requires two specific DNA sites for cleavage. J. Mol. Biol. 2001;309:69–78. doi: 10.1006/jmbi.2001.4635. [DOI] [PubMed] [Google Scholar]
- 77.Morgan R.D., Bhatia T.K., Lovasco L., Davis T.B. MmeI: a minimal Type II restriction-modification system that only modifies one DNA strand for host protection. Nucleic Acids Res. 2008;36:6558–6570. doi: 10.1093/nar/gkn711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Smith R.M., Pernstich C., Halford S.E. TstI, a Type II restriction-modification protein with DNA recognition, cleavage and methylation functions in a single polypeptide. Nucleic Acids Res. 2014;42 doi: 10.1093/nar/gku187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Yang Z., Horton J.R., Maunus R., Wilson G.G., Roberts R.J., Cheng X. Structure of HinP1I endonuclease reveals a striking similarity to the monomeric restriction enzyme MspI. Nucleic Acids Res. 2005;33:1892–1901. doi: 10.1093/nar/gki337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Horton J.R., Zhang X., Maunus R., Yang Z., Wilson G.G., Roberts R.J., Cheng X. DNA nicking by HinP1I endonuclease: bending, base flipping and minor groove expansion. Nucleic Acids Res. 2006;34:939–948. doi: 10.1093/nar/gkj484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Rubin R.A., Modrich P. Substrate dependence of the mechanism of EcoRI endonuclease. Nucleic Acids Res. 1978;5:2991–2997. doi: 10.1093/nar/5.8.2991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Smith R.M., Marshall J.J., Jacklin A.J., Retter S.E., Halford S.E., Sobott F. Organization of the BcgI restriction-modification protein for the cleavage of eight phosphodiester bonds in DNA. Nucleic Acids Res. 2013;41:391–404. doi: 10.1093/nar/gks1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Pingoud V., Wende W., Friedhoff P., Reuter M., Alves J., Jeltsch A., Mones L., Fuxreiter M., Pingoud A. On the divalent metal ion dependence of DNA cleavage by restriction endonucleases of the EcoRI family. J. Mol. Biol. 2009;393:140–160. doi: 10.1016/j.jmb.2009.08.011. [DOI] [PubMed] [Google Scholar]
- 84.Dupureur C.M. One is enough: insights into the two-metal ion nuclease mechanism from global analysis and computational studies. Metallomics. 2010;2:609–620. doi: 10.1039/c0mt00013b. [DOI] [PubMed] [Google Scholar]
- 85.Lagunavicius A., Sasnauskas G., Halford S.E., Siksnys V. The metal-independent type IIs restriction enzyme BfiI is a dimer that binds two DNA sites but has only one catalytic centre. J. Mol. Biol. 2003;326:1051–1064. doi: 10.1016/s0022-2836(03)00020-2. [DOI] [PubMed] [Google Scholar]
- 86.Chan S.H., Opitz L., Higgins L., O'Loane D., Xu S.Y. Cofactor requirement of HpyAV restriction endonuclease. PLoS One. 2010;5:e9071. doi: 10.1371/journal.pone.0009071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Lukacs C.M., Kucera R., Schildkraut I., Aggarwal A.K. Understanding the immutability of restriction enzymes: crystal structure of BglII and its DNA substrate at 1.5 A resolution. Nat. Struct. Biol. 2000;7:134–140. doi: 10.1038/72405. [DOI] [PubMed] [Google Scholar]
- 88.Winkler F.K., Banner D.W., Oefner C., Tsernoglou D., Brown R.S., Heathman S.P., Bryan R.K., Martin P.D., Petratos K., Wilson K.S. The crystal structure of EcoRV endonuclease and of its complexes with cognate and non-cognate DNA fragments. EMBO J. 1993;12:1781–1795. doi: 10.2210/pdb4rve/pdb. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Shen B.W., Heiter D.F., Chan S.H., Wang H., Xu S.Y., Morgan R.D., Wilson G.G., Stoddard B.L. Unusual target site disruption by the rare-cutting HNH restriction endonuclease PacI. Structure. 2010;18:734–743. doi: 10.1016/j.str.2010.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Roberts R.J., Belfort M., Bestor T., Bhagwat A.S., Bickle T.A., Bitinaite J., Blumenthal R.M., Degtyarev S., Dryden D.T., Dybvig K., et al. A nomenclature for restriction enzymes, DNA methyltransferases, homing endonucleases and their genes. Nucleic Acids Res. 2003;31:1805–1812. doi: 10.1093/nar/gkg274. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.