

It has been nearly 10 years since the identification and cloning of the cystic fibrosis (CF) gene was announced (1). Despite rapid advances in our understanding of the molecular determinants of the disease, CF remains one of the most common lethal inherited autosomal recessive disorders in the Caucasian population worldwide. Seventy percent of patients carry at least one copy of the most common mutation — ΔF508 — but over 800 unique mutations have been described (http://www.genet.sickkids.on.ca/cftr/). Based on the molecular outcome, genotypes can be assigned to one of five classes of mutations. It is hoped that by designing novel therapeutic molecules whose activity is specialized to correct specific mRNA or protein defects in CF transmembrane conductance regulator (CFTR), the morbidity and mortality of CF could be dramatically improved. Several prototype therapies directed at restoration of CFTR function by targeting the underlying molecular defect are already in Phase I clinical trials. These designer drugs require that genotype be determined to guide the choice of therapy.
Spectrum of defects in CFTR.
The CFTR protein is a 1480 amino acid, glycosylated membrane glycoprotein that functions as a cAMP-regulated chloride channel in exocrine glands and secretory epithelia. The nucleotide sequence predicts a structure with two sets of six transmembrane domains (TMD), two nucleotide binding domains (NBDs), and a regulatory (R) domain (1). The NBDs are involved in the gating of the CFTR chloride channel and portions of the TMDs form the pore. The R domain is intracellular and also is involved in gating the channel (2) by a mechanism dependent on its phosphorylation state (3). CFTR is required for normal activity of the outwardly rectifying chloride channel (ORCC), perhaps via CFTR-mediated ATP transport (4). CFTR is also critical to control of the epithelial sodium channel (EnaC) (5). Disruption of CFTR function alters the salt and water content of luminal secretions and impairs airway mucosal defense (6).
Most of the reported mutations in CFTR are point mutations involving one or a few nucleotides. They are distributed as follows: missense (40%), nonsense (or termination, 18%), splice-site (18%), frameshift (22%), and other (promoter, in-frame deletions, etc., 2%). Missense and nonsense mutations change a single amino acid or introduce a premature termination site. A splice-site mutation can destroy one or more exons or introduce a novel exon, but because this mutation is leaky, it can permit low levels of the normally spliced mRNA. Frameshift mutations often disrupt the synthesis of the mRNA. Nevertheless, the most useful way to look at genotype in CF is by classification according to the molecular fate.
Class I mutations lead to defects in the synthesis of stable CFTR mRNA transcripts resulting in absence of the CFTR protein. About half of all mutations in CFTR (encompassing premature termination, exon skipping, aberrant mRNA splicing, and frameshifts) are thought to fall into this class and result in complete loss of CFTR protein/function.
Class II mutations, including ΔF508, complete protein translation but produce an abnormal protein that fails to escape the endoplasmic reticulum. Little or no CFTR reaches the plasma membrane, and the absence of all surface CFTR results in a severe phenotype. It is being increasingly recognized that mutations in unrelated genes can create defective proteins, which fail to traffic properly through the cell. Classically, missense mutations creating an abnormal protein were thought to be relatively benign or less consequential than nonsense mutations (null) or large deletions. This is no longer strictly the case because examples from CF and other inherited disorders demonstrate that a synthesized protein that fails to mature along the normal biosynthetic pathway often becomes quite destructive (7).
Class III mutations disrupt activation and regulation of CFTR at the plasma membrane. Thus biosynthesis, trafficking, and processing are undisturbed, but the channel may be defective with respect to ATP binding and hydrolysis, or phosphorylation. Mutations, such as G551D, tend to be associated with a severe phenotype.
Class IV mutations affect chloride conductance or channel gating and thus result in reduced chloride current. As might be expected, mutations in this class, such as R117H or P574H, are thought to confer a milder phenotype.
Class V mutations reduce the level of normal CFTR protein by alterations in the promoter or by altering splicing. Currently it is thought that a reduction in mRNA to less than 10% of normal results in disease in CF. Examples of Class V mutations include 3849 + 10kb C→T, A455E, and 5T.
No classification scheme works perfectly, and the genetics are often complicated. A single missense mutation can be associated with features from two different classes. For example, the ΔF508 CFTR is misprocessed (Class II) but also has a reduced channel open time (Class IV) (8). Many patients have two different mutations in CFTR, a situation termed compound heterozygosity. It is thought that presence of a Class IV or Class V mutation on one allele can ameliorate the consequences of a severe mutation on the other allele, such as ΔF508. Much rarer is the occurrence of two different mutations in a single gene on the same allele. The best example of this is the thymidine tract in intron 8, where 5T and 7T modify disease expression. The Class IV mutation, R117H, occurs in cis with either the 5T or 7T tract variant in intron 8. The 5T allele confers a pancreatic sufficient CF phenotype, whereas the 7T allele has been associated with male infertility caused by congenital bilateral absence of the vas deferens (CBAVD). Interestingly, women with the R117H-7T allele tend to be asymptomatic.
New approaches to Class I defects in CFTR.
Mutations leading to premature stop codons produce truncated mRNA transcripts that are inherently unstable and fail to undergo translation into proteins (Fig. 1). The efficiency of translation termination tends to vary as a function of the surrounding sequence. Certain aminoglycosides, such as G418 and gentamicin, appear to suppress termination and restore readthrough to full-length CFTR (9). This has been demonstrated for W1282X and R553X in vitro. Given that W1282X mRNA is unusually stable in certain patients (10), topical and inhaled gentamicin is under study in the United States and Israel (11, 12). Preliminary data from these small clinical trials are encouraging. For example, investigators in Israel, using measurements of nasal epithelial chloride transport, have been able to demonstrate modest responses to isoproterenol in CF patients homozygous for stop codon mutations placed on gentamicin nasal drops three times daily for two weeks (12). Similar measurements are being made in the United States for patients on intravenous gentamicin (E. Sorscher, personal communication).
Figure 1.
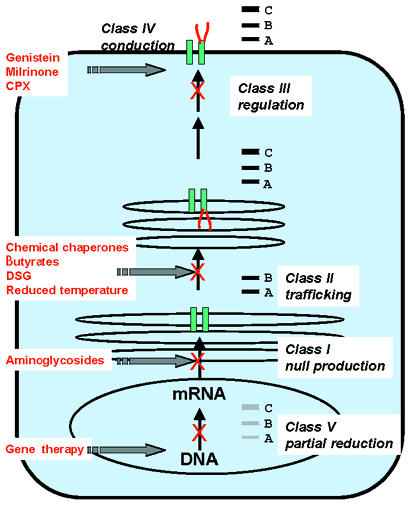
The five general classifications of CFTR mutations are shown within the context of a single epithelial cell. The corresponding protein electrophoresis pattern for each class is depicted in the box to the right of each mutation class. A, B, and C refer to the typical mobilities of nascent CFTR (A, 130 kDa), core glycosylated CFTR (B, 150 kDa), and fully glycosylated CFTR (C, 180 kDa). Null refers to the absence of CFTR protein. CFTR, cystic fibrosis transmembrane conductance regulator.
Class II trafficking defects in CFTR can be corrected in vitro and in vivo.
The most common CFTR mutation, ΔF508, is synthesized, inserted into the endoplasmic reticulum, but then fails to progress to the Golgi apparatus and the cell surface (Fig. 1, band B). Failure to pass through the Golgi can be recognized by failure to become fully glycosylated and attain a mobility of 170–180 kDa on an SDS-PAGE gel (Fig. 1, band C). The deletion of the phenylalanine at position 508 is hypothesized to cause “misfolding” of the nascent chain, such that molecular chaperones target it for early proteolysis. Similar “misfolding” and impairment of protein trafficking is seen for mutations in the α1-antitrypsin gene, Tay-Sachs, familial hypercholesterolemia, and for accumulation of insoluble protein aggregates in Alzheimer's and prion-associated disorders. Evidence for misfolding of CFTR includes a reduced stability of ΔF508 at the cell membrane (13), slightly altered circular dichroism spectra of a peptide containing this site (14), restoration of processing by incubation of cells at temperatures between 25°C and 29°C (15, 16), and restoration of processing by chemical chaperones (17). It is important to keep in mind, however, that wild-type CFTR is targeted for early proteolysis 75% of the time. Only about 25% of normal CFTR trafficks through the Golgi to the cell surface (15, 18, 19). CFTR is marked for destruction by ubiquitination and proteolysis by proteosome-dependent and -independent pathways (20). It was initially hypothesized that by blocking these pathways, a greater amount of mature CFTR could be produced. This proved not to be the case when it was shown that interference with proteolysis by lactacystin or ALLN does not force CFTR through the normal pathway (20). Thus, therapeutic interventions designed to restore normal trafficking should be targeted proximal to ubiquitination and proteolysis in the endoplasmic reticulum.
Wild-type CFTR “folding” and progression to a protease-resistant state is assisted by ER– resident chaperone proteins. Hsp70 (21) and calnexin (22), but not BiP and Grp94, appear to associate with CFTR transiently. Prolongation of the complex of Hsp70 and ΔF508 CFTR has been observed, suggesting that Hsp70 may recognize CFTR for early removal. Hsc70, a member of the Hsp70 family, is constitutively expressed, and has also been implicated as a signal for removal to the proteolytic pathway (23). Because ΔF508 retains some chloride channel activity (24, 25) modulation of chaperone associations or addition of a chemical chaperone are reasonable approaches.
Deoxyspergualin (DSG) is a stable synthetic analog of the natural product spergualin (Fig. 2) and is known to compete effectively for binding with Hsp70 and Hsp90. DSG has potent immunosuppressive activity mediated in part through interactions with Hsc70 and Hsp90. One research group has extensively studied DSG in CF cells expressing the ΔF508 mutation. The researchers report appearance of functional cAMP-stimulated CFTR chloride channel activity at the cell surface in both immortalized human CF airway and biliary epithelial cells, although confirmation of CFTR trafficking by immunoprecipitation techniques could not be demonstrated. The effect was observed at reasonable concentrations from 10 to 100 μg/ml for at least 24 hours (26). These cells were also responsive to 5 mM sodium butyrate used as a positive control.
Figure 2.

Chemical structures of compounds in Phase I testing in cystic fibrosis.
The butyrate compounds (Fig. 2) are low-molecular-weight fatty acids that restore ΔF508 to the cell surface at 37°C (27). Treatment of cell cultures with sodium butyrate or sodium 4-phenylbutyrate at non-toxic concentrations increases normal CFTR processing and leads to cAMP-mediated chloride conduction at the cell surface (28). Butyrate has a short half-life in vivo and must be administered by intravenous drip. It has been demonstrated to induce fetal hemoglobin (29, 30) and favorably induce differentiation in a variety of solid tumors (31). Phenylbutyrate is an oral pro-drug of phenylacetate that is also capable of inducing fetal hemoglobin, acting as an adjunct chemotherapeutic agent, and inducing CFTR expression and trafficking. It does not appear to affect CFTR single channel properties (28). Preliminary data implicate phenylbutyrate-mediated reduction in Hsc70 to explain the shift of mutant CFTR onto the trafficking pathway (32). Interestingly, both DSG and the butyrates may regulate interactions of CFTR with ER chaperones. In a randomized, double-blind, placebo-controlled trial in CF patients homozygous for ΔF508, one week of phenylbutyrate at 20 gm/day was associated with induction of chloride transport based on nasal potential difference measurements (33). Importantly, no safety issues arose in the CF trial, suggesting that whatever effects occurred on chaperone activity, widespread disruption of the quality control mechanism for folding or degrading proteins was not detectable. Buphenyl (4-phenylbutyrate) is a newly approved drug for the rare inherited urea cycle disorders where it functions to provide an alternative vehicle for waste nitrogen excretion by conjugation with glutamine, and it is under study in the hemoglobinopathies, adrenoleukodystrophy (34), and in cancer trials. Dose escalation and pharmacokinetic studies are continuing in adults with CF.
A number of laboratories have confirmed that growth of ΔF508 CFTR-expressing cells at reduced temperature dramatically corrects the trafficking defect in CFTR and restores cAMP-mediated chloride transport at the plasma membrane. This effect can be mimicked by chemicals such as glycerol, dimethylsulfoxide (DMSO), trimethylamine-N-oxide (TMAO), and deuterated water (35). Glycerol is active at a relatively high concentration (1.25 M) making in vivo application impractical. Nevertheless, screening for low-molecular-weight compounds that can restore trafficking may lead to identification of a useful molecule. Such a compound might also be effective for other genetic disorders that have a trafficking defect.
Certain tissues and organs, upon exposure to hyper-osmotic stress, are able to accumulate low-molecular-weight “cellular osmolytes” to protect themselves. Carbohydrates such as glycerol or myoinositol, free amino acids such as glycine or alanine, and methylamines such as betaine or TMAO are either transported in or synthesized by the tissue (35). As urea accumulates in the renal medulla, TMAO is produced to reduce urea-induced protein denaturation. It may be possible to adapt these principles locally within the lung, liver, or intestine.
Both trafficking and chloride channel properties appear, at least in vivo, to be favorably altered by CPX (8-cyclopentyl-1,3-dipropylxanthine) (Fig. 2) (36, 37), an adenosine receptor antagonist that directly binds to ΔF508 CFTR in a region that structurally resembles the adenosine receptor. Chloride transport is stimulated in vitro in a variety of cells expressing the ΔF508 mutation. A Phase I dose escalation and safety trial was recently completed, and a Phase II trial is in progress. Another potent analog, 1,3-diallyl-8-cyclohexylxanthine, is undergoing laboratory assessment and looks very promising (38).
Genistein (Fig. 2), a member of the naturally occurring flavonoid class, has been extensively studied for its ability to activate CFTR through a highly debated mechanism (39). Genistein is a tyrosine kinase inhibitor; however, not all tyrosine kinase inhibitors have activity on CFTR. Genistein does not increase cAMP levels (40) and more recent data implicate direct binding to the NBD of phosphorylated CFTR (41). Genistein and related flavonoids are ubiquitous in the human diet in plant foods such as soybeans and legumes. Their pharmacology and toxicology have been intensely investigated. It is known that flavonoid intake from a normal diet can result in a blood concentration of 1–2 μM in humans (42). Illek and Fischer tested the ability of perfusion of the flavonoids, quercetin (Fig. 2), genistein, kaempferol, and apigenin, to augment the nasal potential difference response to isoproterenol in normal volunteers (43). On average, 30 μM concentrations of flavonoids stimulated nasal PD at about 30% of levels seen with 10 μM isoproterenol.
Genistein and other flavonoid compounds effectively activate chloride conduction through the ΔF508 CFTR (44), although they do not appear to favorably restore trafficking. One group has demonstrated that pretreatment of CF cells with 4-phenylbutyrate or low temperature to induce ΔF508 trafficking to the plasma membrane allowed genistein to activate chloride transport (45). Results from a pilot clinical trial of topical genistein in CF were recently presented (46). Genistein was applied by perfusion to the nasal epithelia alone or in combination with isoproterenol. Genistein alone or in combination with isoproterenol induced a modest hyperpolarization in the nasal potential difference response to low chloride in normal volunteers but was not active in CF patients homozygous for ΔF508, perhaps reflecting inactivity with respect to the trafficking defect. On the other hand, genistein alone induced a small chloride response in CF patients carrying one copy of the G551D mutation. G551D is a Class III mutation that is synthesized, resides in the plasma membrane but is not activated with isoproterenol. The ability of genistein to activate G551D in the intestine of the G551D CF mouse has also been observed (47).
Class III regulation or activation mutations are difficult to repair.
CFTR mutants in this class have reduced responses to regulatory molecules (Fig. 1). G551D is a common mutation associated with pancreatic insufficiency and the severe phenotype. G551D CFTR does not conduct chloride in response to elevated cAMP but is resident in the plasma membrane. Genistein and related compounds are the first chemicals ever shown to stimulate G551D chloride conductance in vitro. Other mutations in the nucleotide binding domains can interfere with nucleotide binding and hydrolysis, and disrupt CFTR function to a variable degree. These mutants sustain a reduced response to ATP-examples include S1255P, G551S, G1244E, and G1349D. It may be possible to find molecules that alter the interaction of the NBDs with ATP and restore activity to these mutants.
Class IV conduction mutations are associated with a mild phenotype.
Class IV mutants such as R117H, G314E, R334W, and R347P are associated with normal PKA-dependent phosphorylation and ATP binding. These CFTR proteins manifest a reduction in chloride conductance or abnormal channel gating (see Fig. 1). The mutations occur within the regions of the TMDs that form the chloride pore. R347P affects the rate of chloride flow, whereas R117H and P574H reduce the channel open time. The latter mutation is sometimes placed in Class II because of a reduced efficiency of processing.
Class IV mutations such as R117H, R334W, R347P, A455E, and P574H are associated with a pancreatic sufficient phenotype or late onset pancreatic insufficiency. Attempts have been made to estimate severity of disease based on mRNA transcript level (48); however, strict correlations between mRNA transcript level and a functional CFTR protein at the cell surface cannot be made. Nevertheless, the clinical phenotype seen with Class IV mutations suggests that treatments aimed at promoting CFTR trafficking and activating or increasing chloride conductance would tend to support a milder course.
Milrinone (Fig. 2), a Class III phosphodiesterase inhibitor, is an approved drug that sustains elevated cAMP and should theoretically improve chloride conduction through CFTR. Milrinone itself is used for short-term intravenous therapy of congestive heart failure as a positive inotrope and vasodilator, and thus it is unlikely to be practical in CF. However, demonstration of activity would spark further development of related pharmaceuticals. Milrinone (together with forskolin to initiate elevated cAMP) favorably restores cAMP-mediated chloride secretion to nasal epithelia of the ΔF508 murine model, but in preliminary in vivo studies it did not alter nasal potential difference measurements of patients carrying the ΔF508 mutation (49). The murine nasal epithelium appears to process and traffic the ΔF508 CFTR more efficiently than human tissue, and this is a likely explanation for the different responses of the two species. A second murine model, the R117H mouse, expresses a mutant which is found in the cell surface in humans. Encouraging responses to milrinone and forskolin in the nasal epithelia in this model suggest further studies with human carriers of R117H would be useful (49).
Class V mutations generate a small amount of normal CFTR.
Mutations in the CFTR promoter, in introns that regulate mRNA splicing, or that cause inefficient levels of protein to be produced are grouped together in Class V (see Fig. 1). Class V mutations typically are associated with a pancreatic sufficient phenotype or even the absence of classic CF (as seen in males with CBAVD). When splicing efficiency is reduced, variable levels of normal CFTR can escape to provide some function, as is seen with 3849 + 10kb C→T. This mutation creates a partially active splice site in intron 19 that leads to insertion of a new 84-bp exon with a stop codon. Patients with this mutation are diagnosed later, have pancreatic sufficiency, better nutritional status, variable male fertility, and can have normal or slightly elevated sweat chlorides. Along the same theme, the 5T tract in intron 8 disrupts exon 9 in CFTR and similarly reduces the amount of normal CFTR. Therapies that would maximally activate the normal CFTR already present in these patients might confer clinical benefit.
Therapies directed at alternative chloride channels.
Organs that are less affected by mutations in CFTR, such as the kidney, express abundant levels of alternative, non-CFTR chloride channels. Because glands and secretory epithelia that are affected or diseased in CF express one or more of these “alternative” chloride channel conductances, a number of groups have chosen to develop agonists for these channels. The calcium-activated chloride channels are highly expressed in the murine airway and appear to compensate for absent CFTR and prevent lung disease in the CFTR-null mouse. On the other hand, the murine gastrointestinal tract lacks this calcium-activated chloride conductance, and the CFTR-null mouse develops serious intestinal complications. The human airways express a calcium-activated chloride channel that can be stimulated by topical application of nucleotides that bind to purinergic receptors to increase calcium or directly via a calcium ionophore. A Phase I trial of a UTP analog by aerosol delivery is under way in normal volunteers and patients with CF (50). Similarly, studies of peptide drugs (e.g., duramycin) that increase intracellular calcium have begun Phase I testing (51).
Summary
The diagnosis of inherited disorders is increasingly more complex. It is no longer sufficient to make a clinical diagnosis. We must determine the precise sequence abnormalities in order to select the appropriate therapeutic approach. Whereas currently approved pharmaceuticals target the symptoms of CF, progress must now be made in repairing the nucleotide abnormality or intervening to assist a defective protein. This will also be true in other disorders. Monogenic inherited diseases affecting widely different gene families, tissues, or enzymatic pathways reveal common themes in their disruption of cellular processes. It is increasingly more likely that progress made with one orphan disease may favorably influence therapeutic development for another, as is currently being recognized for diseases that result from protein folding or trafficking abnormalities.
It has been harder to foresee that drugs developed for one purpose, such as the butyrates for excretion of waste nitrogen, also have profound effects on gene regulation and alternative enzymatic pathways in diseases such as the hemoglobinopathies, cystic fibrosis, adrenoleukodystrophy, and cancer. CF is a complex systemic inherited disorder with a wide variety of severity. The relative abundance of new compounds directed at the Class II trafficking mutation ΔF508 is encouraging and likely to benefit the majority of patients with CF. Because a single mutation can affect both protein function and location, combination therapies may be needed. Multiple approaches will almost certainly be required to control this disease.
Acknowledgments
The author thanks M. Heffler for handling the manuscript, and M. Boyle, W. Guggino, and B.J. Rosenstein for editorial comments. P.L. Zeitlin has received support from the National Heart, Lung, and Blood Institute and the Cystic Fibrosis Foundation for work described in this manuscript. A licensing agreement exists between The Johns Hopkins University School of Medicine, Ucyclyd Pharma Inc., and P.L. Zeitlin. The terms of this arrangement are being managed by The Johns Hopkins University School of Medicine in accordance with its conflict-of-interest policies.
References
- 1.Riordan JR, et al. Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA [erratum published Science.245:1437] Science. 1989;245:1066–1073. doi: 10.1126/science.2475911. [DOI] [PubMed] [Google Scholar]
- 2.Rich DP, et al. Effect of deleting the R domain on CFTR-generated chloride channels. Science. 1991;253:205–207. doi: 10.1126/science.1712985. [DOI] [PubMed] [Google Scholar]
- 3.Ma J, et al. Phosphorylation-dependent block of cystic fibrosis transmembrane conductance regulator chloride channel by exogenous R domain protein. J Biol Chem. 1996;271:7351–7356. doi: 10.1074/jbc.271.13.7351. [DOI] [PubMed] [Google Scholar]
- 4.Schwiebert EM, et al. CFTR regulates outwardly rectifying chloride channels through an autocrine mechanism involving ATP. Cell. 1995;81:1063–1073. doi: 10.1016/s0092-8674(05)80011-x. [DOI] [PubMed] [Google Scholar]
- 5.Ismailov II, et al. Regulation of epithelial sodium channels by the cystic fibrosis transmembrane conductance regulator. J Biol Chem. 1996;271:4725–4732. doi: 10.1074/jbc.271.9.4725. [DOI] [PubMed] [Google Scholar]
- 6.Smith JJ, Travis SM, Greenberg EP, Welsh MJ. Cystic fibrosis airway epithelia fail to kill bacteria because of abnormal airway surface fluid [erratum published Cell.87(2)] Cell. 1996;85:229–236. doi: 10.1016/s0092-8674(00)81099-5. [DOI] [PubMed] [Google Scholar]
- 7.Ashkenas J, Byers PH. The final stage of gene expression: chaperones and the regulation of protein fate. Am J Hum Genet. 1997;61:267–272. doi: 10.1086/514865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dalemans W, et al. Altered chloride ion channel kinetics associated with the delta F508 cystic fibrosis mutation [see comments] Nature. 1991;354:526–528. doi: 10.1038/354526a0. [DOI] [PubMed] [Google Scholar]
- 9.Howard M, Frizzell RA, Bedwell DM. Aminoglycoside antibiotics restore CFTR function by overcoming premature stop mutations [see comments] Nat Med. 1996;2:467–469. doi: 10.1038/nm0496-467. [DOI] [PubMed] [Google Scholar]
- 10.Shoshani T, et al. Similar levels of mRNA from the W1282X and the delta F508 cystic fibrosis alleles, in nasal epithelial cells. J Clin Invest. 1994;93:1502–1507. doi: 10.1172/JCI117128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bedwell DM, et al. Aminoglycoside repair of premature translation termination mutations. Pediatr Pulmonol. 1998;17(Suppl.):168. (Abstr.) [Google Scholar]
- 12.Wilschanski M, et al. Topical gentamicin rectifies abnormal nasal potential difference measurements in CF patients homozygous for STOP mutations. Pediatr Pulmonol. 1998;17(Suppl.):253. (Abstr.) [Google Scholar]
- 13.Denning GM, et al. Processing of mutant cystic fibrosis transmembrane conductance regulator is temperature-sensitive [see comments] Nature. 1992;358:761–764. doi: 10.1038/358761a0. [DOI] [PubMed] [Google Scholar]
- 14.Ko YH, Thomas PJ, Delannoy MR, Pedersen PL. The cystic fibrosis transmembrane conductance regulator. Overexpression, purification, and characterization of wild type and delta F508 mutant forms of the first nucleotide binding fold in fusion with the maltose-binding protein. J Biol Chem. 1993;268:24330–24338. [PubMed] [Google Scholar]
- 15.Brown CR, Hong-Brown LQ, Welch WJ. Correcting temperature-sensitive protein folding defects. J Clin Invest. 1997;99:1432–1444. doi: 10.1172/JCI119302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Egan ME, Schweibert EM, Guggino WB. Differential expression of ORCC and CFTR induced by low temperature in CF airway epithelial cells. Am J Physiol. 1995;268:C243–C251. doi: 10.1152/ajpcell.1995.268.1.C243. [DOI] [PubMed] [Google Scholar]
- 17.Sato S, Ward CL, Krouse ME, Wine JJ, Kopito RR. Glycerol reverses the misfolding phenotype of the most common cystic fibrosis mutation. J Biol Chem. 1996;271:635–638. doi: 10.1074/jbc.271.2.635. [DOI] [PubMed] [Google Scholar]
- 18.Jensen TJ, et al. Multiple proteolytic systems, including the proteasome, contribute to CFTR processing. Cell. 1995;83:129–135. doi: 10.1016/0092-8674(95)90241-4. [DOI] [PubMed] [Google Scholar]
- 19.Ward CL, Kopito RR. Intracellular turnover of cystic fibrosis transmembrane conductance regulator. Inefficient processing and rapid degradation of wild-type and mutant proteins. J Biol Chem. 1994;269:25710–25718. [PubMed] [Google Scholar]
- 20.Ward CL, Omura S, Kopito RR. Degradation of CFTR by the ubiquitin-proteasome pathway. Cell. 1995;83:121–127. doi: 10.1016/0092-8674(95)90240-6. [DOI] [PubMed] [Google Scholar]
- 21.Yang Y, Janich S, Cohn JA, Wilson JM. The common variant of cystic fibrosis transmembrane conductance regulator is recognized by hsp70 and degraded in a pre-Golgi nonlysosomal compartment. Proc Natl Acad Sci USA. 1993;90:9480–9484. doi: 10.1073/pnas.90.20.9480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pind S, Riordan JR, Williams DB. Participation of the endoplasmic reticulum chaperone calnexin (p88, IP90) in the biogenesis of the cystic fibrosis transmembrane conductance regulator. J Biol Chem. 1994;269:12784–12788. [PubMed] [Google Scholar]
- 23.Strickland E, Qu BH, Millen L, Thomas PJ. The molecular chaperone hsc70 assists the in vitro folding of the N- terminal nucleotide-binding domain of the cystic fibrosis transmembrane conductance regulator [In Process Citation] J Biol Chem. 1997;272:25421–25424. doi: 10.1074/jbc.272.41.25421. [DOI] [PubMed] [Google Scholar]
- 24.Pasyk EA, Foskett JK. Mutant (delta F508) cystic fibrosis transmembrane conductance regulator Cl– channel is functional when retained in endoplasmic reticulum of mammalian cells. J Biol Chem. 1995;270:12347–12350. doi: 10.1074/jbc.270.21.12347. [DOI] [PubMed] [Google Scholar]
- 25.Haws CM, et al. Delta F508-CFTR channels: kinetics, activation by forskolin, and potentiation by xanthines. Am J Physiol. 1996;270:C1554–C1555. doi: 10.1152/ajpcell.1996.270.5.C1544. [DOI] [PubMed] [Google Scholar]
- 26.Jiang C, et al. Partial restoration of cAMP-stimulated CFTR chloride channel activity in DeltaF508 cells by deoxyspergualin. Am J Physiol. 1998;275:C171–C178. doi: 10.1152/ajpcell.1998.275.1.C171. [DOI] [PubMed] [Google Scholar]
- 27.Cheng SH, et al. Functional activation of the cystic fibrosis trafficking mutant delta F508-CFTR by overexpression. Am J Physiol. 1995;268:L615–L624. doi: 10.1152/ajplung.1995.268.4.L615. [DOI] [PubMed] [Google Scholar]
- 28.Rubenstein RC, Egan ME, Zeitlin PL. In vitro pharmacologic restoration of CFTR-mediated chloride transport with sodium 4-phenylbutyrate in cystic fibrosis epithelial cells containing delta F508-CFTR. J Clin Invest. 1997;100:2457–2465. doi: 10.1172/JCI119788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Constantoulakis P, Knitter G, Stamatoyannopoulos G. On the induction of fetal hemoglobin by butyrates: in vivo and in vitro studies with sodium butyrate and comparison of combination treatments with 5-AzaC and AraC. Blood. 1989;74:1963–1971. [PubMed] [Google Scholar]
- 30.Perrine SP, et al. Butyrate derivatives. New agents for stimulating fetal globin production in the beta-globin disorders. Am J Pediatr Hematol Oncol. 1994;16:67–71. [PubMed] [Google Scholar]
- 31.Carducci M, et al. Phenylbutyrate (PB) for refractory solid tumors: a phase I clinical and pharmacological evaluation. Proc Am Assoc Cancer Res. 1998;39:506. [Google Scholar]
- 32.Rubenstein RC, Zeitlin PL. Sodium 4-phenylbutyrate promotes DF508-CFTR trafficking by down-regulation of HSC70. Pediatr Pulmonol. 1998;17(Suppl.):211. (Abstr.) [Google Scholar]
- 33.Rubenstein RC, Zeitlin PL. A pilot clinical trial of sodium 4-phenylbutyrate (Buphenyl) in delta F508-homozygous cystic fibrosis patients: evidence of restoration of nasal epithelial CFTR function. Am J Resp Crit Care Med. 1998;157:484–490. doi: 10.1164/ajrccm.157.2.9706088. [DOI] [PubMed] [Google Scholar]
- 34.Kemp S, et al. Gene redundancy and pharmacological gene therapy: implications for X-linked adrenoleukodystrophy [see comments] Nat Med. 1998;4:1261–1268. doi: 10.1038/3242. [DOI] [PubMed] [Google Scholar]
- 35.Brown CR, Hong-Brown LQ, Welch WJ. Strategies for correcting the delta F508 CFTR protein-folding defect. J Bioenerg Biomembr. 1997;29:491–502. doi: 10.1023/a:1022491124939. [DOI] [PubMed] [Google Scholar]
- 36.Cohen BE, et al. 8-cyclopentyl-1,3-dipropylxanthine and other xanthines differentially bind to the wild-type and delta F508 first nucleotide binding fold (NBF-1) domains of the cystic fibrosis transmembrane conductance regulator. Biochemistry. 1997;36:6455–6461. doi: 10.1021/bi970150v. [DOI] [PubMed] [Google Scholar]
- 37.Guay-Broder C, et al. A1 receptor antagonist 8-cyclopentyl-1,3-dipropylxanthine selectively activates chloride efflux from human epithelial and mouse fibroblast cell lines expressing the cystic fibrosis transmembrane regulator delta F508 mutation. Biochemistry. 1995;34:9079–9087. doi: 10.1021/bi00028a017. [DOI] [PubMed] [Google Scholar]
- 38.Arispe N, Ma J, Jacobson KA, Pollard HB. Direct activation of cystic fibrosis transmembrane conductance regulator channels by 8-Cyclopentyl-1,3-dipropylxanthine (CPX) and 1,3-Diallyl-8-cyclohexylzanthine (DAX) J Biol Chem. 1997;273:5727–5734. doi: 10.1074/jbc.273.10.5727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Illek B, Fischer H, Machen TE. II. Regulation of CFTR by small molecules including HCO-3. Am J Physiol. 1998;275:G1221–G1226. doi: 10.1152/ajpgi.1998.275.6.G1221. [DOI] [PubMed] [Google Scholar]
- 40.He Z, Raman S, Guo Y, Reenstra W. Cystic fibrosis transmembrane conductance regulator activation by cAMP-independent mechanisms. Am J Physiol. 1998;275:C958–C966. doi: 10.1152/ajpcell.1998.275.4.C958. [DOI] [PubMed] [Google Scholar]
- 41.Weinreich F, Wood PG, Riordan JR, Nagel G. Direct action of genistein on CFTR. Pflugers Arch. 1997;434:484–491. doi: 10.1007/s004240050424. [DOI] [PubMed] [Google Scholar]
- 42.Janssen K, et al. Effects of the flavonoids quercetin and apigenin on hemostasis in healthy volunteers: results from an in vitro and a dietary supplement study. Am J Clin Nutr. 1998;67:255–262. doi: 10.1093/ajcn/67.2.255. [DOI] [PubMed] [Google Scholar]
- 43.Illek B, Fischer H. Flavonoids stimulate Cl conductance of human airway epithelium in vitro and in vivo. Am J Physiol. 1998;275:L90–L910. doi: 10.1152/ajplung.1998.275.5.L902. [DOI] [PubMed] [Google Scholar]
- 44.Hwang TC, Wang F, Yang IC, Reenstra WW. Genistein potentiates wild-type and delta F508-CFTR channel activity. Am J Physiol. 1997;273:C988–C998. doi: 10.1152/ajpcell.1997.273.3.C988. [DOI] [PubMed] [Google Scholar]
- 45.Fischer H, Illek B. Effects of genistein on disease-causing mutant CFTRs. Pediatr Pulmonol. 1998;17(Suppl.):239. (Abstr.) [Google Scholar]
- 46.Gondor M, et al. Genistein stimulates chloride secretion in normal volunteers and CF patients with a G551D mutation. Pediatr Pulmonol. 1998;17(Suppl.):253. (Abstr.) [Google Scholar]
- 47.Smith SN, et al. Effect of IBMX and alkaline phosphatase inhibitors on Cl– secretion in G551D cystic fibrosis mutant mice. Am J Physiol. 1998;274:C492–C499. doi: 10.1152/ajpcell.1998.274.2.C492. [DOI] [PubMed] [Google Scholar]
- 48.Hamosh A, et al. Severe deficiency of cystic fibrosis transmembrane conductance regulator messenger RNA carrying nonsense mutations R553X and W1316X in respiratory epithelial cells of patients with cystic fibrosis. J Clin Invest. 1991;88:1880–1885. doi: 10.1172/JCI115510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Drumm ML, Steagall WK, Kelley TJ, Milgram LJ, Konstan MW. Phosphodiesterase inhibitor-stimulated chloride secretion from gut and respiratory epithelia. Pediatr Pulmonol. 1998;17(Suppl.):168–169. [Google Scholar]
- 50.Knowles MR, Clarke LL, Boucher RC. Extracellular ATP and UTP induce chloride secretion in nasal epithelia of cystic fibrosis patients and normal subjects in vivo. Chest. 1992;101:60s–63s. doi: 10.1378/chest.101.3_supplement.60s. [DOI] [PubMed] [Google Scholar]
- 51.Zeitlin PL, Guggino WB, Molina L. Activation by the nonadecapeptide 2622U90 (Duramycin) of chloride secretion in healthy volunteers and patients with cystic fibrosis. Pediatr Pulmonol. 1998;17(Suppl.):277. [Google Scholar]