

Highlights
-
•
We generated SMP30/SOD1-double knockout (DKO) mice for oxidative stress research.
-
•
SMP30/SOD1-DKO mice showed low levels of ascorbic acid and premature death.
-
•
SMP30/SOD1-DKO mice exhibited high levels of oxidative stress and liver injury.
-
•
SMP30/SOD1-DKO mice manifest hepatic steatosis due to decreased levels of Apolipoprotein B.
Abbreviations: AA, l-ascorbic acid; ApoB, Apolipoprotein B; AST, aspartate aminotransferase; DHA, dehydroascorbic acid; DHE, dihydroethidium; DKO, double knockout; EDTA, ethylenediaminetetraacetic acid; FFA, free fatty acid; Grp78, glucose-regulated protein 78 kDa; KO, knockout; MTP, microsomal triglyceride transfer protein; NAFLD, non-alcoholic fatty liver disease; NASH, non-alcoholic steatohepatitis; PL, phospholipid; PPARα, peroxisome proliferator-activated receptor-α; qPCR, quantitative real-time polymerase chain reaction; SDS, sodium dodecyl sulfate; SMP30, senescence marker protein-30; SOD, superoxide dismutase; SREBP, sterol regulatory element binding protein; T-cho, total cholesterol; TBARS, thiobarbituric acid reactive substances; TG, triglyceride; VLDL, very low-density lipoprotein
Keywords: Ascorbic acid, Non-alcoholic fatty liver disease, Reactive oxygen species, SMP30, SOD1
Abstract
Superoxide dismutase 1 (SOD1) is an antioxidant enzyme that converts superoxide anion radicals into hydrogen peroxide and molecular oxygen. The senescence marker protein-30 (SMP30) is a gluconolactonase that functions as an antioxidant protein in mammals due to its involvement in ascorbic acid (AA) biosynthesis. SMP30 also participates in Ca2+ efflux by activating the calmodulin-dependent Ca2+-pump. To reveal the role of oxidative stress in lipid metabolism defects occurring in non-alcoholic fatty liver disease pathogenesis, we generated SMP30/SOD1-double knockout (SMP30/SOD1-DKO) mice and investigated their survival curves, plasma and hepatic lipid profiles, amounts of hepatic oxidative stress, and hepatic protein levels expressed by genes related to lipid metabolism. While SMP30/SOD1-DKO pups had no growth retardation by 14 days of age, they did have low plasma and hepatic AA levels. Thereafter, 39% and 53% of male and female pups died by 15–24 and 89 days of age, respectively. Compared to wild type, SMP30-KO and SOD1-KO mice, by 14 days SMP30/SOD1-DKO mice exhibited: (1) higher plasma levels of triglyceride and aspartate aminotransferase; (2) severe accumulation of hepatic triglyceride and total cholesterol; (3) higher levels of superoxide anion radicals and thiobarbituric acid reactive substances in livers; and (4) decreased mRNA and protein levels of Apolipoprotein B (ApoB) in livers – ApoB is an essential component of VLDL secretion. These results suggest that high levels of oxidative stress due to concomitant deficiency of SMP30 and/or AA, and SOD1 cause abnormal plasma lipid metabolism, hepatic lipid accumulation and premature death resulting from impaired VLDL secretion.
1. Introduction
Non-alcoholic fatty liver disease (NAFLD) represents a spectrum of liver disease ranging from steatosis to non-alcoholic steatohepatitis (NASH), and cirrhosis [1]. In NASH, intralobular inflammation and hepatocellular ballooning are present in addition to steatosis, and are often accompanied by progressive fibrosis. Several population studies showed that NAFLD is closely associated with obesity, insulin resistance and metabolic syndrome, diabetes, and dyslipidemia. However, the pathogenesis of NAFLD/NASH has not been completely elucidated. One popular hypothesis is the “two-hit theory” wherein the “first hit” is insulin resistance that causes hepatic fat accumulation and the “second hit” induces hepatocyte injury and inflammation [2]. Although the mechanisms that cause hepatocyte injury in fatty liver are not fully understood, oxidative stress is a commonly proposed mechanism for hepatocellular injury.
Many antioxidant enzymes and molecules protect mammalian cells from oxidative stress caused by reactive oxygen species such as superoxide anion radicals, hydrogen peroxide, hydroxyl radicals, and singlet oxygen, as well as reactive nitrogen species such as peroxynitrite [3]. Superoxide dismutase (SOD) is an antioxidant enzyme that converts superoxide anion radicals into hydrogen peroxide and molecular oxygen. SOD consists of three isozymes: copper/zinc SOD (Cu, Zn-SOD, SOD1) that localizes in the cytosol, nucleus, and mitochondrial intermembrane space, manganese SOD (Mn-SOD, SOD2), which is localized in the mitochondrial matrix, and extracellular Cu, Zn-SOD (EC-SOD, SOD3). In a previous study we reported that SOD1-knockout (KO) mice showed age-related macular degeneration [4], retinal degeneration [5], skin atrophy [6], osteoporosis [7], deterioration of Alzheimer’s disease [8], and luteal [9] and lacrimal degeneration [10]. Other groups showed SOD1-KO phenotypes that included age-related cataracts [11], hepatocellular carcinoma [12], muscle atrophy [13], and hemolytic anemia [14].
The liver plays a key role in lipid metabolism including uptake and de novo synthesis of free fatty acids (FFA) followed by conversion of FFA into triglycerides (TG) by esterification [15]. TG are secreted into the circulation as Apolipoprotein B (ApoB)-containing very low-density lipoprotein (VLDL) or stored in hepatocytes as TG vacuoles. FFA that is not esterified into TG is metabolized by β-oxidation in the liver. VLDL transports not only endogenously synthesized lipids, in particular TG, but also some cholesterol and cholesteryl esters to peripheral tissues. ApoB100 is absolutely required for VLDL assembly and secretion. The quality of VLDL is strictly controlled in multiple steps from production to secretion by ApoB100 degradation [16]. Regarding lipid metabolism in the liver, we showed that SOD1-KO mice exhibited increased lipid peroxidation and hepatic TG accumulation due to decreases in VLDL secretion from the liver caused by oxidative degradation of ApoB. This result suggests that superoxide anion radicals may be involved in abnormal lipid metabolism in the mouse liver [17].
We originally identified the 34 kDa senescence marker protein-30 (SMP30) in rat liver and also showed that SMP30 levels decrease with age [18,19]. SMP30 participates in Ca2+ efflux by promoting calmodulin-dependent Ca2+ pump activity in HepG2 cells, which confers resistance to cell injury caused by high intracellular Ca2+ concentrations [20]. We reported that SMP30-KO mice livers are highly susceptible to tumor necrosis factor-α- and Fas-induced apoptosis, suggesting that SMP30 has a protective role in cell injury [21]. Furthermore, SMP30-KO mice hepatocytes exhibited increased hepatic TG accumulation, total cholesterol (T-cho), and phospholipid (PL), as well as abnormally enlarged mitochondria [22]. Previously, we identified SMP30 as a gluconolactonase that catalyzes lactonization of l-gulonic acid in the penultimate step of mammalian l-ascorbic acid (AA) biosynthesis [23,24]. AA is a free radical scavenger that acts as an electron donor and cofactor in reactions catalyzed by dopamine-β-hydroxylase and collagen prolyl- and lysyl- hydroxylase [25,26]. In guinea pigs and primates including humans, the capacity to synthesize AA has been lost due to many mutations in the l-gulono-γ-lactone oxidase gene, whereas most vertebrates can synthesize AA in vivo. We reported that AA-depleted SMP30-KO mice showed ultraviolet radiation type B-induced cataracts [27], skin atrophy [28], impaired catecholamine synthesis [29], age-related hearing loss [30], increased superoxide anion radical generation in the brain [31], pulmonary emphysema [32], and abnormal insulin release from pancreatic islets [33]. We also reported that SMP30 deficiency independent of AA worsens glucose tolerance by impairing acute insulin secretion [34], causes coronary artery spasms that are triggered by chronic thiol oxidation [35], impairs myocardium-induced dilation of coronary arterioles [36] and affects astrocyte activation, which exacerbates Parkinson’s pathology [37]. In addition, SMP30-KO mice on a Leprdb/db background exhibit increases in small dense-LDL and severe fatty liver accompanied by numerous inflammatory cells and increased oxidative stress, suggesting that SMP30 is closely associated with NAFLD pathogenesis [38].
To address the pathological role of reactive oxygen species in lipid metabolism involved in NAFLD/NASH pathogenesis, we generated SMP30/SOD1-double knockout (SMP30/SOD1-DKO) mice, and investigated oxidative stress and lipid metabolism in the livers of these mice. In this study, we show that SMP30/SOD1-DKO mice died after 15 days without obvious growth retardation, and at 14 days of age exhibited low AA levels, high levels of plasma TG and aspartate aminotransferase (AST), and lipid accumulation in response to impaired VLDL secretion, which is caused by decreased ApoB gene expression and ApoB protein degradation that occurs in the presence of high levels of superoxide anion radicals and/or lipid peroxide.
2. Results
2.1. Smp30Y/−Sod1−/− male and Smp30−/−Sod1−/− female mice died after day 15 of the neonatal period
Smp30Y/−Sod1−/− male and Smp30−/−Sod1−/− female mice were born healthy with expected Mendelian frequencies. Out of 243 pups examined in the mating pair of Smp30Y/−Sod1+/− male and Smp30−/−Sod1+/− female mice, there were 70 (28.8%) Smp30Y/−Sod1+/+ and Smp30−/−Sod1+/+, 116 (47.7%) Smp30Y/−Sod1+/− and Smp30−/−Sod1+/−, 57 (23.5%) Smp30Y/−Sod1−/− mice and Smp30−/−Sod1−/− mice. For the control mice, out of 102 pups examined in the mating pair of Smp30Y/+Sod1+/− male and Smp30+/+Sod1+/− female mice, there were 24 (23.5%) Smp30Y/+Sod1+/+ and Smp30+/+Sod1+/+, 55 (53.9%) Smp30Y/+Sod1+/− and Smp30+/+Sod1+/−, 23 (22.5%) Smp30Y/+Sod1−/− and Smp30+/+Sod1−/− mice. From survival analysis of the male mice, Smp30Y/−Sod1−/− mice died starting at 15 days of age, and eventually 39% of the mice died by 24 days of age, although mice in the other five experimental groups survived (p < 0.05 versus Smp30Y/+Sod1+/+, Smp30Y/+Sod1+/−, and Smp30Y/+Sod1−/−, p < 0.01 versus Smp30Y/−Sod1+/− using a log-rank test) (Fig. 1A). Similarly, Smp30−/−Sod1−/− female mice began to die at 15 days of age, and 53% of these mice eventually died up until 89 days of age, although the other five experimental groups had hardly any deaths (p < 0.001 versus Smp30+/+Sod1+/+, Smp30+/+Sod1+/−, and Smp30+/+Sod1−/−, p < 0.05 versus Smp30−/−Sod1+/+ and Smp30−/−Sod1+/− by using log-rank test). At 14 days of age, the appearance and size of the Smp30Y/−Sod1−/− male and Smp30−/−Sod1−/− female mice were similar to the other five experimental groups (Fig. 1B). Smp30Y/−Sod1−/− male mice gained weight and the body weight change was not significantly different from the other five experimental groups at 14 days of age (Fig. 1C). While the body weight of Smp30−/−Sod1−/− female mice also increased, the body weight changes were significantly (16%) lower than that of Smp30+/+Sod1+/− animals at 14 days of age (p < 0.05).
Fig. 1.
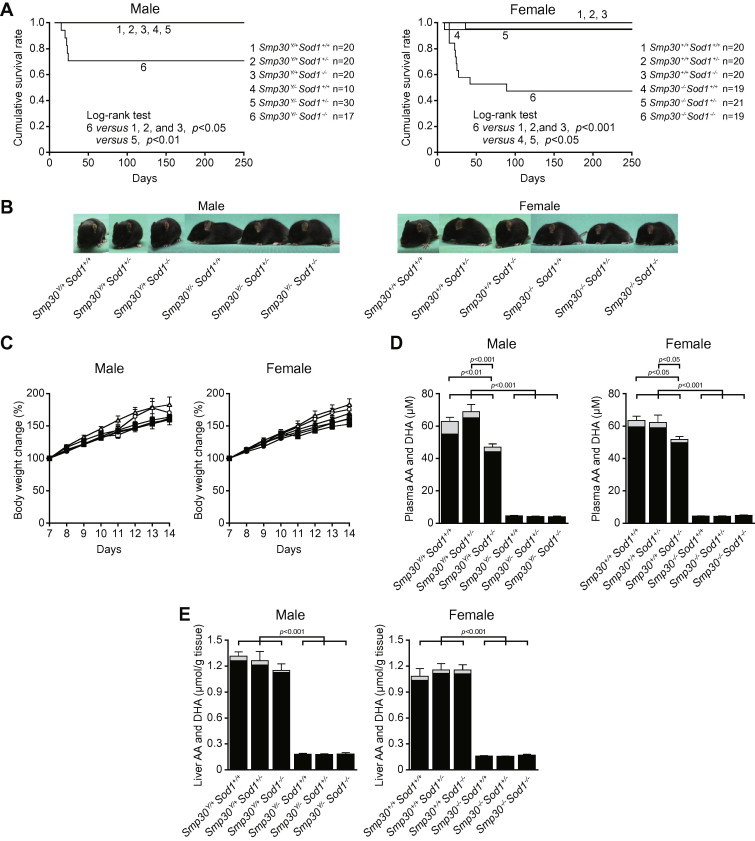
Establishment of Smp30Y/−Sod1−/− male mice and Smp30−/−Sod1−/− female mice. (A) Cumulative survival rate in each experimental group of mice. (B) Appearance of Smp30Y/−Sod1−/− male mice and Smp30−/−Sod1−/− female mice at 14 days of age. (C) Body weight change in each experimental group (n = 10–11) between 7 and 14 days of age. The body weight of mice at 7 days was set at 100%. Open circles, Smp30Y/+Sod1+/+ or Smp30+/+Sod1+/+ mice; open triangles, Smp30Y/+Sod1+/− or Smp30+/+Sod1+/− mice; open squares, Smp30Y/+Sod1−/− or Smp30+/+Sod1−/− mice; filled circles, Smp30Y/−Sod1+/+ or Smp30−/−Sod1+/+ mice; filled triangles, Smp30Y/−Sod1+/− or Smp30−/−Sod1+/− mice; filled squares, Smp30Y/−Sod1−/− or Smp30−/−Sod1−/− mice. Values are given as means ± SEM. (D and E) Ascorbic acid (AA; filled columns) and dehydroascorbic acid (DHA; gray columns) concentration in plasma (D) and livers (E) from each experimental group of mice (n = 7–11) at 14 days. Values are given as means ± SEM (AA plus DHA).
We next determined the AA and DHA levels in the plasma and livers of Smp30Y/−Sod1−/− male and Smp30−/−Sod1−/− female mice at 14 days of age. The AA plus DHA levels in plasma from Smp30Y/−Sod1+/+, Smp30Y/−Sod1+/−, and Smp30Y/−Sod1−/− male mice were 7.3%, 6.7%, and 7.5% that of Smp30Y/+Sod1+/+ mice (62.9 μM), respectively (Fig. 1D). In plasma from Smp30−/−Sod1+/+, Smp30−/−Sod1+/−, and Smp30−/−Sod1−/− female mice, the AA plus DHA levels were 7.0%, 6.8%, and 7.6% that of Smp30+/+Sod1+/+ mice (63.5 μM), respectively. The AA plus DHA levels in Smp30Y/+Sod1−/− male and Smp30+/+Sod1−/− female mice were significantly (25% and 18%, respectively) lower compared with Smp30Y/+Sod1+/+ male and Smp30+/+Sod1+/+ female mice (p < 0.001 and p < 0.05). As shown in Fig. 1E, the AA plus DHA levels of livers from Smp30Y/−Sod1+/+, Smp30Y/−Sod1+/−, and Smp30Y/−Sod1−/− male mice were 13.9%, 13.6%, and 13.9% that of Smp30Y/+Sod1+/+ mice (1.32 μmol/g tissue), respectively. In livers from Smp30−/−Sod1+/+, Smp30−/−Sod1+/−, and Smp30−/−Sod1−/− female mice, the AA plus DHA levels of plasma were 14.8%, 14.3%, and 15.7% of Smp30+/+Sod1+/+ mice (1.08 μmol/g tissue), respectively. The DHA per AA plus DHA in the plasma and livers of the six experimental groups were respectively 5.9–13.7% and 2.5–3.9% in males, and 2.9–9.2% and 1.9–10.0% in females.
2.2. Smp30Y/−Sod1−/− male mice show hypertriglyceridemia and liver injury
The blood biochemical parameters of 14 day old animals in the six experimental groups are presented in Table 1. Plasma TG concentrations were significantly higher in Smp30Y/−Sod1−/− male mice (84.0 ± 10.0 mg/dL) than in Smp30Y/+Sod1+/+ (35.8 ± 5.4 mg/dL, p < 0.001), Smp30Y/+Sod1+/− (33.2±8.9 mg/dL, p < 0.001), Smp30Y/+Sod1−/− (43.0 ± 5.9 mg/dL, p < 0.01), Smp30Y/−Sod1+/+ (41.5 ± 2.7 mg/dL, p < 0.01), and Smp30Y/−Sod1+/− mice (34.0 ± 7.0 mg/dL, p < 0.001). There were no significant differences in plasma T-cho and FFA levels among the six experimental groups. Plasma AST concentrations in Smp30Y/−Sod1−/− male mice (135.9 ± 13.8 IU/L) were significantly higher than those of Smp30Y/+Sod1+/+ (61.7 ± 4.6 IU/L, p < 0.001), Smp30Y/+Sod1+/− (58.6 ± 0.9 IU/L, p < 0.001), Smp30Y/+Sod1−/− (66.3 ± 4.2 IU/L, p < 0.001), Smp30Y/−Sod1+/+ (55.2 ± 3.0 IU/L, p < 0.001), and Smp30Y/−Sod1+/− mice (64.6 ± 11.2 IU/L, p < 0.001). In females, plasma AST concentrations in Smp30−/−Sod1−/− mice (156.7 ± 16.0 IU/L) were significantly higher than Smp30+/+Sod1+/+ (65.0 ± 4.1 IU/L, p < 0.001), Smp30+/+Sod1+/− (60.0 ± 3.5 IU/L, p < 0.001), Smp30+/+Sod1−/− (71.1 ± 3.1 IU/L, p < 0.001), Smp30−/−Sod1+/+ (74.9 ± 5.0 IU/L, p < 0.001), and Smp30−/−Sod1+/− mice (76.8 ± 8.2 IU/L, p < 0.001). The plasma alanine aminotransferase concentration was significantly higher in Smp30Y/−Sod1−/− male mice (22.3 ± 0.7 IU/L) compared with Smp30Y/−Sod1+/+ (16.4 ± 0.4 IU/L, p < 0.05) animals, although there were no significant differences among females in the six experimental groups. Plasma glucose concentrations in Smp30Y/−Sod1−/− male mice (115.0 ± 7.0 mg/dL) were significantly lower than those of Smp30Y/+Sod1+/+ (163.6 ± 10.2 mg/dL, p < 0.05), Smp30Y/+Sod1−/− (181.0 ± 16.7 mg/dL, p < 0.001), and Smp30Y/−Sod1+/+ (164.1 ± 9.0 mg/dL, p < 0.05) mice. There were no significant differences in plasma insulin levels among males in the six experimental groups.
Table 1.
Biochemical parameters of blood plasma in six experimetal groups of male mice.
| Smp30Y/+Sod1+/+ | Smp30Y/+Sod1+/− | Smp30Y/+Sod1−/− | Smp30Y/−Sod1+/+ | Smp30Y/−Sod1+/− | Smp30Y/−Sod1−/− | |
|---|---|---|---|---|---|---|
| Triglyceride (mg/dL) | 35.8 ± 5.4 | 33.2 ± 8.9 | 43.0 ± 5.9 | 41.5 ± 2.7 | 34.0 ± 7.0 | 84.0 ± 10.0†,§,¶,††,§§ |
| Cholesterol (mg/dL) | 104.5 ± 5.7 | 122.2 ± 8.0 | 124.7 ± 11.5 | 119.7 ± 3.4 | 119.9 ± 4.1 | 116.4 ± 10.8 |
| Free fatty acid (mEq/L) | 0.51 ± 0.13 | 0.75 ± 0.31 | 0.92 ± 0.21 | 0.80 ± 0.04 | 0.69 ± 0.08 | 0.84 ± 0.15 |
| Aspartate aminotransferase (IU/L) | 61.7 ± 4.6 | 58.6 ± 0.9 | 66.3 ± 4.2 | 55.2 ± 3.0 | 64.6 ± 11.2 | 135.9 ± 13.8†,§,||,‡‡,§§ |
| Alanine aminotransferase (IU/L) | 19.5 ± 0.9 | 21.8 ± 1.0 | 19.0 ± 0.5 | 16.4 ± 0.4‡ | 19.0 ± 1.9 | 22.3 ± 0.7∗∗ |
| Glucose (mg/dL) | 163.6 ± 10.2 | 153.6 ± 8.1 | 181.0 ± 16.7 | 164.1 ± 9.0 | 147.5 ± 5.2 | 115.0 ± 7.0∗,||,∗∗ |
| Insulin (ng/mL) | 0.15 ± 0.07 | 0.34 ± 0.11 | 0.06 ± 0.04 | 0.34 ± 0.02 | 0.16 ± 0.05 | 0.15 ± 0.05 |
∗p < 0.05 and †p < 0.001 versus Smp30Y/+Sod1+/+, ‡p < 0.01 and §p < 0.001 versus Smp30Y/+Sod1+/−, ¶p < 0.01 and ||p < 0.001 versus Smp30Y/+Sod1−/−, ∗∗p < 0.05, ††p < 0.01, and ‡‡p < 0.001 versus Smp30Y/−Sod1+/+, §§p < 0.001 versus Smp30Y/−Sod1+/−. Values are given as means ± SEM of animals (n = 5–10).
2.3. Increased hepatic lipid levels (TG and T-cho) and lipid peroxidation (TBARS) in Smp30Y/−Sod1−/− male mice
As shown in Fig. 2A and B, histological examination of hepatic tissues revealed increased steatosis in Smp30Y/−Sod1−/− male and Smp30−/−Sod1−/− female mice at 14 days of age. In contrast, apparent histological abnormalities were not observed for the other five experimental groups. Hepatic TG was significantly higher in Smp30Y/−Sod1−/− male mice than those of the other five experimental groups (p < 0.001) (Fig. 3A). Compared with Smp30Y/+Sod1+/+ mice, hepatic TG was significantly higher in Smp30Y/+Sod1−/− mice (p < 0.05). Hepatic TC in Smp30Y/−Sod1−/− mice was significantly higher than that of Smp30Y/+Sod1+/+ animals (p < 0.05) (Fig. 3B). There were no significant differences in hepatic PL and FFA levels among the six experimental groups (Fig. 3C and D).
Fig. 2.
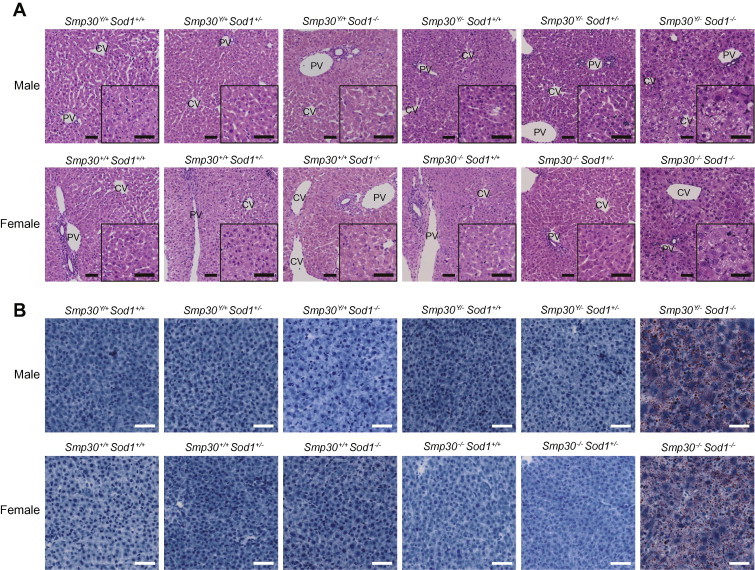
Increased steatosis in liver sections from Smp30Y/−Sod1−/− male mice and Smp30−/−Sod1−/− female mice. Representative images of (A) hematoxylin/eosin staining and (B) oil red O staining in liver sections from each experimental group at 14 days of age. PV, portal vein; CV, central vein. Scale bar is 50 μm.
Fig. 3.
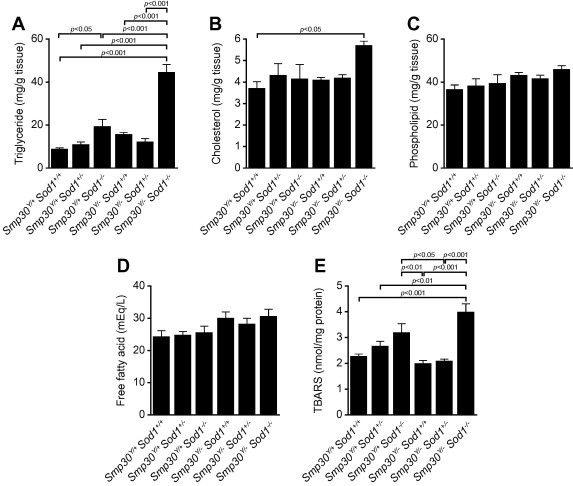
Increased triglyceride, cholesterol, and thiobarbituric acid reactive substances (TBARS) in livers of Smp30Y/−Sod1−/− male mice. (A) Triglyceride, (B) Cholesterol, (C) Phospholipid, (D) Free fatty acid, and (E) TBARS in livers from each experimental group at 14 days of age. Values are given as means ± SEM of five animals.
To examine oxidative stress in livers from Smp30Y/−Sod1−/− mice, a TBARS assay was performed. Hepatic TBARS were significantly higher in Smp30Y/+Sod1−/− mice compared to those of Smp30Y/−Sod1+/+ (p < 0.01) and Smp30Y/−Sod1+/− mice (p < 0.05) (Fig. 3E). Hepatic TBARS in Smp30Y/−Sod1−/− mice were significantly higher than those of Smp30Y/+Sod1+/+ (p < 0.001), Smp30Y/+Sod1+/− (p < 0.01), Smp30Y/−Sod1+/+ (p < 0.001), and Smp30Y/−Sod1+/− mice (p < 0.001). Compared with Smp30Y/+Sod1−/−, hepatic TBARS were higher in Smp30Y/−Sod1−/− mice, although the differences did not reach statistical significance (p = 0.12).
2.4. Increased superoxide anion radical in liver sections from Smp30Y/−Sod1−/− male mice
To evaluate superoxide anion radicals in livers from Smp30Y/−Sod1−/− mice, DHE staining was conducted using frozen liver sections. DHE is oxidized upon reaction with superoxide anion radical to form ethidium bromide, which intercalates in nuclear DNA. DHE fluorescence was increased in liver sections from Smp30Y/+Sod1−/− mice relative to those of Smp30Y/+Sod1+/+, Smp30Y/+Sod1+/−, Smp30Y/−Sod1+/+, and Smp30Y/−Sod1+/− mice (Fig. 4). Furthermore, the DHE fluorescence in Smp30Y/−Sod1−/− mice was markedly increased compared with the other five experimental groups.
Fig. 4.
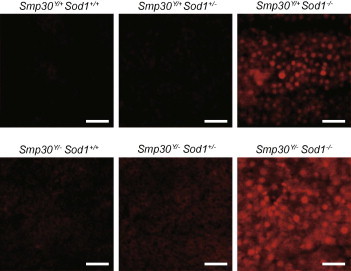
Increased generation of superoxide anion radicals in livers from Smp30Y/−Sod1−/− male mice. Representative images of dihydroethidium staining in liver sections from each experimental group at 14 days of age. Scale bar is 50 μm.
2.5. Decreased ApoB protein levels in livers from Smp30Y/−Sod1−/− male mice
To reveal a mechanism for hepatic steatosis in Smp30Y/−Sod1−/− male mice, we examined liver ApoB protein and mRNA expression levels. Protein levels of ApoB100 and its spliced variant ApoB48 were decreased in liver microsome fractions from Smp30Y/+Sod1−/− mice relative to Smp30Y/+Sod1+/+, Smp30Y/+Sod1+/−, Smp30Y/−Sod1+/+, and Smp30Y/−Sod1+/−animals, whilst these levels were further decreased in Smp30Y/−Sod1−/− mice compared with Smp30Y/+Sod1−/− mice (Fig. 5A). Apob mRNA levels were significantly lower in Smp30Y/−Sod1−/− mice than those of Smp30Y/+Sod1+/+ (p < 0.01), Smp30Y/+Sod1+/− (p < 0.05), Smp30Y/−Sod1+/+ (p < 0.001) and Smp30Y/−Sod1+/− mice (p < 0.001) (Fig. 5B). There were also no differences among the six experimental groups in protein levels of MTP and Grp78, both of which are known to affect ApoB protein levels in VLDL in liver microsome fractions.
Fig. 5.
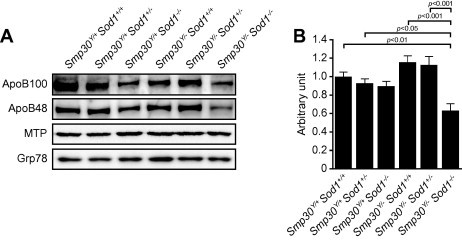
Decreased Apolipoprotein B (ApoB) levels in livers of Smp30Y/−Sod1−/− male mice. (A) Western blot analysis of ApoB100, ApoB48, microsomal triglyceride transfer protein (MTP), and glucose-regulated protein 78 kDa (Grp78) in liver microsome fractions from each experimental group of mice. (B) Apob gene expression levels in the liver. mRNA for the Apob gene was measured using quantitative real-time polymerase chain reaction and normalized to Rplp2, ribosomal protein, large P2, and the value was expressed as arbitrary unit. The values from Smp30Y/+Sod1+/+ male mice were assigned a relative value of 1.0. Values are given as means ± SEM of five animals.
2.6. Altered transcription factor protein levels related to lipid metabolism in livers of Smp30Y/−Sod1−/− male mice
Next, we investigated the hepatic protein levels of transcription factors related to lipid metabolism in Smp30Y/−Sod1−/− male mice. For lipogenesis-related genes, protein levels of the SREBP1c precursor were significantly lower in Smp30Y/−Sod1−/− mice than those of Smp30Y/+Sod1+/+ (p < 0.05), Smp30Y/+Sod1+/− (p < 0.01), Smp30Y/−Sod1+/+ (p < 0.01) and Smp30Y/−Sod1+/− mice (p < 0.05) (Fig. 6A). Protein levels of the SREBP1c precursor in Smp30Y/+Sod1−/− mice were significantly lower relative to Smp30Y/+Sod1+/− mice (p < 0.05). Compared with Smp30Y/+Sod1+/+ and Smp30Y/+Sod1+/− mice, protein levels of mature SREBP1c were significantly lower in Smp30Y/+Sod1−/− (p < 0.001 and p < 0.01), Smp30Y/−Sod1+/+ (p < 0.001 and p < 0.01), and Smp30Y/−Sod1+/− (p < 0.01 versus Smp30Y/+Sod1+/+) mice, as well as Smp30Y/−Sod1−/− mice (p < 0.001 and p < 0.01), although there was no significant difference between Smp30Y/+Sod1+/− and Smp30Y/−Sod1+/− mice (Fig. 6B). Protein levels of mature SREBP2 were significantly lower in Smp30Y/−Sod1+/+ animals than those of Smp30Y/+ mice (Smp30Y/+Sod1+/+, Smp30Y/+Sod1+/−, and Smp30Y/+Sod1−/−) (p < 0.001), and significantly lower in Smp30Y/−Sod1+/− and Smp30Y/−Sod1−/− mice compared with Smp30Y/+Sod1−/− mice (p < 0.05) (Fig. 6C). The SREBP2 precursor was not detected in liver homogenates. Protein levels of PPARα, which is involved in fatty acid catabolism, were significantly lower in Smp30Y/+Sod1−/− (p < 0.05), Smp30Y/−Sod1+/− (p < 0.05), and Smp30Y/−Sod1−/− mice (p < 0.01) compared to that of Smp30Y/+Sod1+/+ mice (Fig. 6D).
Fig. 6.
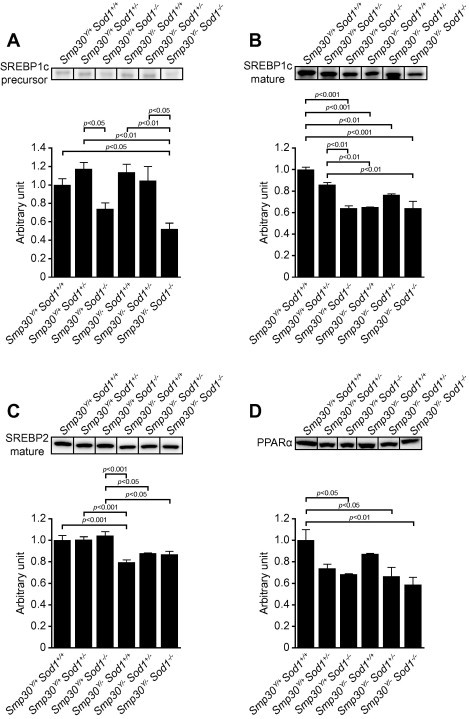
Decreased sterol regulatory element binding protein (SREBP)-1c, SREBP2, and peroxisome proliferator-activated receptor-α (PPARα) levels in livers from Smp30Y/−Sod1−/− male mice. Western blot analysis of (A) precursor (125 kDa) and (B) mature (60 kDa) of SREBP1c, (C) mature SREBP2, and (D) PPARα. Representative images for each experimental group in one blot are shown and the signal intensity for each protein was measured and expressed as an arbitrary unit. The values from Smp30Y/+Sod1+/+ mice were assigned a relative value of 1.0. Values are given as means ± SEM of three animals.
3. Discussion
In the present study we established SMP30/SOD1-DKO mice to assess the possible involvement of SMP30 and SOD1 in NAFLD/NASH. SMP30/SOD1-DKO mice died at 15 days of age without obvious growth retardation, and at 14 days of age exhibited low plasma and hepatic levels of AA, higher plasma levels of TG and AST, higher hepatic TG and T-cho, lower hepatic protein levels of ApoB and PPARα, and higher superoxide anion radicals and TBARS in the liver compared with wild-type, SMP30-KO, and SOD1-KO mice. These results suggest that a concomitant deficiency of SMP30 and SOD1 causes high levels of oxidative stress in the liver that results in abnormal plasma lipid and hepatic lipid accumulation due to impaired VLDL secretion that occurs following decreased expression and protein degradation of ApoB. These defects would result in premature death.
Although nursing of Smp30Y/−Sod1−/− male and Smp30−/−Sod1−/− female pups was frequently observed and these animals showed no growth retardation by 14 days of age, 39% of Smp30Y/−Sod1−/− and 53% of Smp30−/−Sod1−/− mice died by 15–24 and 89 days of age, respectively, although the remaining mice survived more than 250 days (Fig. 1A). During mating and breeding periods, water given to Smp30−/−Sod1+/− dams was supplemented with 1.5 g/L AA. In our previous study, such supplementation was sufficient to maintain plasma and tissue AA to levels in Smp30Y/− mice that were equivalent to Smp30Y/+ mice, even through adulthood [39]. However, plasma and hepatic AA levels in Smp30Y/− male and female pups at 14 days of age were remarkably and unexpectedly lower compared to Smp30Y/+ pups (Fig. 1D and E), presumably because of low AA levels in milk from Smp30−/−Sod1+/− dams. Still, low AA levels were sufficient to prevent scurvy, so that Smp30Y/−Sod1+/+, Smp30−/−Sod1+/+, Smp30Y/−Sod1+/−and Smp30−/−Sod1+/− pups grew normally (Fig. 1A–C). As such, further work will be required to clarify the cause of premature death in Smp30Y/−Sod1−/− and Smp30−/−Sod1−/− mice. Interestingly, plasma AA levels of Smp30Y/+Sod1−/− and Smp30+/+Sod1−/− mice were significantly lower than that of Smp30Y/+Sod1+/+ and Smp30+/+Sod1+/+ mice (Fig. 1D and E). This result might suggest that increased levels of superoxide anion radicals caused by SOD1 deficiency resulted in possible oxidation and degradation of AA in the animals.
A noteworthy finding of this study is that Smp30Y/−Sod1−/− mice showed increased levels of superoxide anion radicals and lipid peroxidation in the liver compared to Smp30Y/+Sod1−/− mice in the postnatal period (Figs. 3E and 4). Elchuri et al. previously reported that compared to Sod1+/+ mice, higher amounts of reactive oxygen species-mediated damage of DNA, protein, and lipids accompanied by lower levels of glutathione peroxidase were observed in adult Sod1−/− mice [12]. We also found increased lipid peroxidation in livers from 50 week old Sod1−/− mice [17]. Furthermore, we observed that AA-depleted Smp30Y/− mice showed higher amounts of oxidized protein that was accompanied by alterations in antioxidant enzymes, higher SOD activity due to increased SOD1 protein levels, and decreased catalase protein levels [40]. In addition, Smp30Y/− mice on a Leprdb/db background exhibited severe fatty liver with increased lipid peroxidation. In this study, cell injury in hepatocytes was apparent in Smp30Y/−Sod1−/− mice as shown by the higher AST levels. Although whether the enzymes and small antioxidant molecules in Smp30Y/−Sod1−/− mice were altered is unclear, this result suggests that SOD1 and SMP30/AA collaboratively exert protective roles against superoxide anion radicals and lipid peroxidation in vivo.
A novel finding of this study is that Smp30Y/−Sod1−/− mice exhibited severe hepatic accumulation of TG and T-cho compared with Smp30Y/+Sod1−/− mice, which also showed TG accumulation (Fig. 3A and B) that likely results from an imbalance between TG synthesis by esterification of FFA and VLDL secretion [15]. Since we observed no alterations in hepatic FFA levels in any experimental group (Fig. 3D), the observed hepatic TG accumulation in Smp30Y/−Sod1−/− mice may have resulted from impaired VLDL secretion in the liver. We previously reported that high levels of oxidative stress enhanced impairment of VLDL secretion due to ApoB degradation in the livers of adult Sod1−/− mice fed normal and high-fat diets [17]. We consistently observed in this study that protein levels of ApoB100 and ApoB48 were lower in the livers of Smp30Y/+Sod1−/− mice relative to those from Smp30Y/+Sod1+/+ animals (Fig. 5A). Andreo et al. showed that the superoxide anion radical is required for lipid peroxidation in docosahexaenoic acid-induced ApoB degradation, although the lipid peroxide levels appear to be correlated with hepatic ApoB100 degradation [41]. Thus, our results suggest that enhanced hepatic ApoB degradation in Smp30Y/−Sod1−/− mice may be attributed to high levels of lipid peroxide and superoxide anion radical due to SMP30 deficiency and AA insufficiency. In addition, we observed that Apob mRNA levels were lower in Smp30Y/−Sod1−/− mice than that of Smp30Y/+Sod1+/+ mice. Interestingly, Huynch et al. reported that mice lacking hepatic leptin signaling decrease hepatic Apob mRNA, and upon re-expression of functional leptin receptors in the liver, hepatic ApoB transcript levels return to wild-type levels [42]. We also reported that Smp30Y/+ and Smp30Y/− mice lacking hepatic leptin signaling show decreased Apob mRNA levels in the livers [38]. Although, it is uncertain whether or not hepatic leptin signaling in fatty livers of Smp30Y/−Sod1−/− mice is obscured, decreased ApoB transcription levels could also account for decreased protein levels of ApoB100 and ApoB48 in the liver.
Decreased VLDL secretion typically leads to low levels of plasma TG. However, compared to the other experimental groups, Smp30Y/−Sod1−/− mice showed higher plasma TG, but not TC and FFA (Table 1), suggesting that uptake of TG-rich VLDL into the liver from blood might decrease in Smp30Y/−Sod1−/− mice. Hepatic FFA levels are determined by uptake, de novo synthesis, and β-oxidation of FFA in livers. Despite the absence of alterations in hepatic and plasma FFA levels among all experimental groups, major changes in master transcription factors involved in regulating expression of hepatic lipid metabolism genes were observed. Protein levels of PPARα, which regulates genes involved in β-oxidation of FFA, were lower in Sod1−/− mice (Smp30Y/−Sod1−/− mice and Smp30Y/+Sod1−/− mice) compared with Smp30Y/+Sod1+/+ mice (Fig. 6D), suggesting that superoxide anion radicals are associated with mitochondrial and peroxisomal FFA β-oxidation. Compared with Smp30Y/+Sod1+/+ mice, the decrease in protein levels of mature SREBP1c that regulates genes that participate in FFA and TG synthesis in Smp30Y/−Sod1+/+, Smp30Y/+Sod1−/−, and Smp30Y/−Sod1−/− mice likely suppresses lipid accumulation (Fig. 6B). Protein levels of mature SREBP2, which regulates genes involved in cholesterol synthesis, were lower in Smp30Y/− mice (Smp30Y/−Sod1−/− mice and Smp30Y/−Sod1+/+ mice) relative to Smp30Y/+Sod1+/+ mice (Fig. 6C). In our previous study Smp30Y/− mice showed hepatic accumulation of TG, T-cho, and PL with age [22]. Taken together, these results suggest that SMP30 deficiency and/or AA insufficiency might be relevant to cholesterol metabolism.
Further studies will be required to clarify the molecular mechanism involved in the alteration of lipid homeostasis and reactive oxygen species metabolism caused by concomitant deficiency of SMP30 and/or AA, and SOD1. However, our data suggest that high levels of oxidative stress caused by defects in the antioxidant system resulted in hepatic lipid accumulation via altered lipid metabolism, predominantly decreased expression and protein degradation of ApoB, and premature death in non-obese mice, and together support that oxidative stress might be one possible cause of NAFLD.
4. Materials and methods
4.1. Animals and experimental protocol
Sod1−/− mice were obtained from the Jackson Laboratory (Bar Harbor, ME, USA) and backcrossed with C57BL/6NCrSlc mice (Japan SLC, Inc., Shizuoka, Japan) for five to six generations. SMP30-KO mice were established and maintained as described previously [21]. The Smp30 gene is located on the X chromosome, so heterozygous SMP30-KO male mice do not exist. Because SMP30 is the penultimate enzyme in the AA biosynthetic pathway, SMP30-KO mice cannot synthesize AA in vivo [23]. To avoid any effects of AA deficiency, SMP30-KO mice were given free access to water supplemented with 1.5 g/L AA (DSM Nutrition Japan, Tokyo, Japan) and 10 μM ethylenediaminetetraacetic acid (EDTA) [39]. Sod1−/− male mice were first crossed with Smp30−/− female mice to produce Smp30Y/−Sod1+/− male mice and Smp30+/−Sod1+/− female mice. Next, Smp30Y/−Sod1+/− male mice and Smp30+/−Sod1+/− female mice were interbred to produce Smp30Y/−Sod1+/− male mice and Smp30−/−Sod1+/− female mice. Studies were conducted on offspring produced by mating Smp30Y/−Sod1+/− male mice with Smp30−/−Sod1+/− female mice supplemented with 1.5 g/L AA and 10 μM EDTA, to give Smp30Y/−Sod1+/+, Smp30Y/−Sod1+/−, and Smp30Y/−Sod1−/− male mice and Smp30−/−Sod1+/+, Smp30−/−Sod1+/−, and Smp30−/−Sod1−/− female mice. To produce control mice, Sod1+/− mice supplemented with 10 μM EDTA water were mated to generate Smp30Y/+Sod1+/+, Smp30Y/+Sod1+/−, and Smp30Y/+Sod1−/− male mice and Smp30+/+Sod1+/+, Smp30+/+Sod1+/−, and Smp30+/+Sod1−/− female mice. All mice were fed an autoclaved commercial chow diet (CRF-1, Oriental Yeast Co., Ltd., Tokyo, Japan). Mice were maintained on a 12-h light/dark cycle in a controlled environment. All experimental procedures using laboratory animals were approved by the Animal Care and Use Committee of the Tokyo Metropolitan Institute of Gerontology (Permit Number: 12016).
4.2. Genotyping
Smp30 genotyping was performed by polymerase chain reaction using genomic DNA isolated from the tail tip as reported previously [21]. Sod1 genotypes were determined as described previously with slight modifications [17]. Polymerase chain reactions to determine both genotypes were performed under the following conditions: 1 cycle of 94 °C for 3 min; 35 cycles of 94 °C for 1 min, 55 °C for 1 min, and 72 °C for 2 min; followed by 1 cycle of 72 °C for 5 min. Mice were maintained on a 12-h light/dark cycle in a controlled environment.
4.3. Collection of blood and liver tissue
All mice were anesthetized at the age of 14 days. Blood was obtained from the inferior vena cava, anticoagulated with EDTA, and subsequently centrifuged at 880×g for 15 min at 4 °C. Mice were systemically perfused with ice-cold phosphate buffered saline through the hepatic portal vein to wash out any remaining blood cells. The livers were then removed and fixed with 10% neutral buffered formalin or snap frozen in Tissue-Tek® OCT compound (Sakura Finetek Japan Co., Ltd., Tokyo, Japan) for histological analysis, or frozen in liquid nitrogen for biochemical analysis. Frozen samples were stored at −80 °C until use.
4.4. Determination of AA and DHA levels in plasma and livers
Amounts of AA and dehydroascorbic acid (DHA), an oxidized form of AA, were determined by high-performance liquid chromatography using an Atlantis dC18 5-μm column (4.6 × 150 mm; Nihon Waters K.K., Tokyo, Japan) as described previously [43]. Plasma was mixed with equal amount of 10% metaphosphoric acid and 1 mM EDTA, and centrifuged at 21,000×g for 15 min at 4 °C. Livers were homogenized in 14 vol 5.4% metaphosphoric acid and 1 mM EDTA, and the homogenate was then centrifuged at 21,000×g for 15 min at 4 °C. To reduce DHA to AA for determination of AA plus DHA, 90 μL of supernatant was incubated with 10 μL 350 mM Tris(2-carboxyethyl)phosphine hydrochloride for 2 h at 4 °C. The mobile phase was 50 mM phosphate buffer (pH 2.8), 0.2 g/L EDTA, and 2% methanol at a flow rate of 1.3 mL/min, and electrical signals were recorded using an electrochemical detector equipped with a glassy carbon electrode at +0.6 V. The value of DHA was determined by subtracting AA from AA plus DHA.
4.5. Biochemical analysis of plasma
Plasma TG, T-cho, FFA, AST, alanine aminotransferase, and glucose levels were measured by enzymatic assay kits (Wako Pure Chemicals Industries, Osaka, Japan). Plasma insulin levels were measured using an enzyme-linked immunoassay system (Ultra sensitive mouse insulin ELISA kit; Morinaga Institute of Biological Science Inc., Kanagawa, Japan).
4.6. Histological examination of liver
To evaluate histological changes, fixed liver sections (3 μm) were subjected to hematoxylin-eosin staining. For lipid staining, frozen sections (10 μm) were prepared on glass slides (New Silane II, Muto Pure Chemicals Col, Ltd., Tokyo, Japan), stained with oil red O, washed with 60% 2-propanol, and counterstained with hematoxylin.
4.7. Measurement of T-cho, TG, PL, and FFA in the liver
Liver tissues were homogenized with 2 vol water using a handy homogenizer (Moji-mojikun; Nippon Genetics Co., Ltd., Tokyo, Japan). Homogenates were added to a chloroform-methanol (2:1; v/v) mixture, incubated for 60 min at 37 °C, and centrifuged at 21,000×g for 10 min at 4 °C. The supernatant organic phase was then collected, dried under nitrogen gas and resolubilized in 2-propanol. TG, T-cho, PL, and FFA concentrations in total lipid extracts were determined using commercial enzymatic kits (Wako Pure Chemical Industries, Ltd., Osaka, Japan)
4.8. Thiobarbituric acid reactive substances (TBARS) assay
Lipid peroxidation was estimated by the amounts of TBARS in the liver that were determined according to the method of Ohkawa et al. [44]. The livers were first homogenized in 10 vol ice-cold 0.1% sodium dodecyl sulfate (SDS) using a handy homogenizer and a sonicator. The homogenates were then centrifuged at 21,000×g for 15 min at 4 °C and the supernatant was used for further assays. Ten microliter of sample was mixed thoroughly with 175 μL stock solution (0.023% (w/v) butylated hydroxytoluene, 0.926% (w/v) SDS, 20% (v/v) acetic acid, pH 3.5), 150 μL 0.8% thiobarbituric acid, and 70 μL distilled water. The mixture was kept on ice for 60 min and then heated for 60 min in a boiling water bath. After cooling, 100 μL distilled water and 500 μL n-butyl alcohol-pyridine (15:1; v/v) were added, and then the mixture was centrifuged at 21,000×g for 5 min at 4 °C. The organic phase supernatant fluorescence was measured using a SPECTRAmax Gemini (Life Technologies Corp., Carlsbad, CA, USA) with an excitation and emission wavelength set of 515 nm and 553 nm, respectively. The TBARS levels are expressed as the equivalent amounts of malondialdehyde produced from 1,1,3,3-tetraethoxypropane.
4.9. Dihydroethidium (DHE) fluorescence of liver tissue
DHE (Wako Pure Chemical Industries, Ltd.), an oxidative fluorescent dye, was used to detect superoxide anion radicals in frozen liver tissue as described previously [45]. Frozen sections (20 μm) were placed on glass slides and incubated with 5 μM DHE at 37 °C for 30 min in a light-protected chamber. Images were obtained using a fluorescence microscope (IX70; Olympus Corp., Tokyo, Japan) with a wideband filter cube (U-MWIG; Olympus Corp.) at excitation and emission wavelengths of 488 nm and 590 nm, respectively.
4.10. Western blot analysis
We used liver microsome fractions for Western blot analysis of Apo B100, ApoB48, microsomal triglyceride transfer protein (MTP), and glucose-regulated protein 78 kDa (Grp78) as described previously [17]. Briefly, livers were homogenized using a Potter-type Teflon homogenizer (model TH-M; Takashima, Tokyo, Japan) in 20 vol of ice cold 20 mM Tris-HCl, pH 7.4, 0.25 M sucrose, and a protease inhibitor cocktail (cOmplete, EDTA-free; Roche Diagnostics GmbH, Mannheim, Germany). Homogenates were centrifuged at 8000×g for 10 min at 4 °C, and the supernatant was further centrifuged at 100,000×g for 60 min at 4 °C. The pellets were suspended in the same buffer. Equal amounts (55 μg for ApoB100 and ApoB48, and 10 μg for MTP and Grp78) of total protein were subjected to 3–8% Tris-acetate gel electrophoresis for ApoB100 and ApoB48 or 7.5% SDS–PAGE for MTP and Grp78, and then electroblotted onto polyvinylidene difluoride membranes, which were then blocked with Block Ace (DS Pharma Biomedical Co., Ltd., Osaka, Japan). The primary antibodies used were: anti-mouse ApoB100 goat polyclonal antibody (1:500; H-15; Santa Cruz Biotechnology, Santa Cruz, CA, USA), anti-human ApoB48 goat polyclonal antibody (1:500; S-18; Santa Cruz Biotechnology), anti-mouse MTP mouse monoclonal antibody (1:2500; 612022; BD Biosciences, San Jose, CA, USA), and anti-human Grp78 mouse monoclonal antibody (1:1000; 610978; BD Biosciences). Immunoreactive ApoB100, ApoB48 and Grp78 were visualized with SuperSignal® West Femto Maximum Sensitivity Substrate (Thermo Fisher Scientific Inc., Waltham, MA, USA) and a LAS-3000 imaging system (GE Healthcare Japan, Tokyo, Japan). Immunoreactive MTP was visualized with ECL (GE Healthcare Japan) and LAS-3000.
Western blot analysis of sterol regulatory element binding protein (SREBP)-1, SREBP2, and peroxisome proliferator-activated receptor-α (PPARα) was performed with liver homogenates. Livers were homogenized in ice cold homogenization buffer (50 mM Tris–HCl (pH 7.6), 150 mM NaCl, 0.1% SDS, and a protease inhibitor cocktail (cOmplete, EDTA-free) for 30 s using a high speed homogenizer (POLYTRON® PT-MR 2100; Kinematica AG, Switzerland). The homogenate was then centrifuged at 21,000×g for 10 min at 4 °C. The supernatants were boiled for 5 min with a lysis buffer containing 0.125 M Tris–HCl (pH 6.8), 4% SDS, 20% glycerol, 10% 2-mercaptoethanol, and 0.2% bromophenol blue at a ratio of 1:1. Total protein (6 μg) equivalents for each sample were separated on a 5–20% polyacrylamide gel and transferred to a polyvinylidene difluoride membrane. The membranes were blocked in Block Ace before incubation with the primary antibody, followed by incubation with a horseradish peroxidase-linked goat anti-mouse IgG (1:5000; Bio-Rad Laboratories, Inc., Hercules, CA, USA) or a horseradish peroxidase-linked goat anti-rabbit IgG (1:5000; Bio-Rad Laboratories, Inc.). The primary antibodies used were: anti-SREBP1 mouse monoclonal antibody (1:500, #MS-1207-P1ABX; Thermo Fisher Scientific Inc.), anti-rat SREBP2 rabbit polyclonal antibody (1:500; NB100-74543; Novus Biologicals, LLC, Littleton, CO, USA), and anti-human PPARα rabbit polyclonal antibody (1:500; sc-9000; Santa Cruz Biotechnology). After washing, the immunoreactivity of SREBP1 and SREBP2 was detected using SuperSignal® West Dura Extended Duration Substrate (Thermo Fisher Scientific Inc.) and LAS-3000. Immunoreactive PPARα was visualized with SuperSignal® West Femto Maximum Sensitivity Substrate and LAS-3000. Chemiluminescence signals were quantified with Multi Gauge ver.3.0 software (GE Healthcare Japan). The mean signals from five Smp30Y/+Sod1+/+ mice were assigned a relative value of 1.0.
4.11. RNA isolation, first-strand cDNA synthesis, and quantitative real-time polymerase chain reaction (qPCR)
Liver tissue was homogenized in ice-cold ISOGEN reagent (Nippon Gene Co., Ltd., Tokyo, Japan) with a handy homogenizer before isolation of total RNA according to the manufacturer’s instructions. RNA concentrations were determined and confirmed as free from protein contamination by measuring absorbance at 260 and 280 nm. Total RNA (2.5 μg) was reverse-transcribed using SuperScript® III Reverse Transcriptase (Life Technologies Corp.) for first-strand cDNA synthesis with Random Primer (hexa-deoxyribonucleotide mixture; Takara Bio Inc., Shiga, Japan) according to the manufacturer’s instructions. Using the Platinum® Quantitative PCR Super-Mix-UDG-with ROX (Life Technologies Corp.) following the manufacturer’s protocol, qPCR reactions were performed. The primers and probes used in the qPCR reactions were PrimeTime® qPCR Assays (Integrated DNA Technologies, Inc.); 5′-CCT GCT TCT CCG ATT ATA TTT GAA C-3′, 5′-CAC TGC AAC CTA TGA ACT CCT AA-3′, and 5′-/56-FAM/ACT GCT GCA/ZEN/CTC CTT CTG CTT GA/3IABkFQ/-3′ for Apob (NM_009693); 5′-GAC TCC TCC TTC TTC TCA TCT TTC-3′, 5′-GTC ATC GCT CAG GGT GTT G-3′, and 5′-/56-FAM/CTG TGG CTG/ZEN/TTT CTG CTG CCC/3IABkFQ/-3′ for Rplp2, ribosomal protein, large P2 (NM_026020). The reactions were performed by using the StepOnePlus™ Real-Time PCR System (Life Technologies Corp.). The product specificity generated for each primer set was examined for gel electrophoresis. The relative expression levels of Apob were normalized to Rplp2 using relative standard method. Signals from Smp30Y/+Sod1+/+mice were assigned a relative value of 1.0. Five animals from each group were examined, and qPCR was run in duplicate for each sample.
4.12. Determination of protein concentration
The protein concentration was determined by the bicinchoninic acid protein assay using bovine serum albumin as a standard [46].
4.13. Statistical analysis
The values are expressed as means ± SEM. Statistical analyses were performed with KaleidaGraph software (Synergy Software Inc., Reading, PA, USA). The significance of differences among the groups was determined by analysis of variance and Turkey’s honestly significant difference test. Survival analysis was conducted by Kaplan–Meier curves and log-rank tests using IBM SPSS Statistics 20 (IBM Japan, Tokyo, Japan). Differences were considered significant at p < 0.05.
Conflict of interest
The authors declare that they have no conflict of interest.
Acknowledgments
This study was supported by the Japan Society for the Promotion of Science (http://www.jsps.go.jp/english/e-grants/index.html) KAKENHI Grant Number 23790122 (Y.K.), 23590441 (N.M.), and 24380073 (A.I.).
References
- 1.Musso G., Gambino R., Cassader M. Non-alcoholic fatty liver disease from pathogenesis to management: an update. Obes. Rev. 2010;11:430–445. doi: 10.1111/j.1467-789X.2009.00657.x. [DOI] [PubMed] [Google Scholar]
- 2.Basaranoglu M., Basaranoglu G., Senturk H. From fatty liver to fibrosis: a tale of “second hit”. World J. Gastroenterol. 2013;19:1158–1165. doi: 10.3748/wjg.v19.i8.1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Novo E., Parola M. Redox mechanisms in hepatic chronic wound healing and fibrogenesis. Fibrogenesis Tissue Repair. 2008;1:5. doi: 10.1186/1755-1536-1-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Imamura Y., Noda S., Hashizume K., Shinoda K., Yamaguchi M., Uchiyama S., Shimizu T., Mizushima Y., Shirasawa T., Tsubota K. Drusen, choroidal neovascularization, and retinal pigment epithelium dysfunction in SOD1-deficient mice. a model of age-related macular degeneration. Proc. Natl. Acad. Sci. U.S.A. 2006;103:11282–11287. doi: 10.1073/pnas.0602131103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hashizume K., Hirasawa M., Imamura Y., Noda S., Shimizu T., Shinoda K., Kurihara T., Noda K., Ozawa Y., Ishida S. Retinal dysfunction and progressive retinal cell death in SOD1-deficient mice. Am. J. Pathol. 2008;172:1325–1331. doi: 10.2353/ajpath.2008.070730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Murakami K., Inagaki J., Saito M., Ikeda Y., Tsuda C., Noda Y., Kawakami S., Shirasawa T., Shimizu T. Skin atrophy in cytoplasmic SOD-deficient mice and its complete recovery using a vitamin C derivative. Biochem. Biophys. Res. Commun. 2009;382:457–461. doi: 10.1016/j.bbrc.2009.03.053. [DOI] [PubMed] [Google Scholar]
- 7.Nojiri H., Saita Y., Morikawa D., Kobayashi K., Tsuda C., Miyazaki T., Saito M., Marumo K., Yonezawa I., Kaneko K. Cytoplasmic superoxide causes bone fragility owing to low-turnover osteoporosis and impaired collagen cross-linking. J. Bone Miner. Res. 2011;26:2682–2694. doi: 10.1002/jbmr.489. [DOI] [PubMed] [Google Scholar]
- 8.Murakami K., Murata N., Noda Y., Tahara S., Kaneko T., Kinoshita N., Hatsuta H., Murayama S., Barnham K.J., Irie K. SOD1 (copper/zinc superoxide dismutase) deficiency drives amyloid beta protein oligomerization and memory loss in mouse model of Alzheimer disease. J. Biol. Chem. 2011;286:44557–44568. doi: 10.1074/jbc.M111.279208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Noda Y., Ota K., Shirasawa T., Shimizu T. Copper/zinc superoxide dismutase insufficiency impairs progesterone secretion and fertility in female mice. Biol. Reprod. 2012;86:1–8. doi: 10.1095/biolreprod.111.092999. [DOI] [PubMed] [Google Scholar]
- 10.Kojima T., Wakamatsu T.H., Dogru M., Ogawa Y., Igarashi A., Ibrahim O.M., Inaba T., Shimizu T., Noda S., Obata H. Age-related dysfunction of the lacrimal gland and oxidative stress: evidence from the Cu, Zn-superoxide dismutase-1 (Sod1) knockout mice. Am. J. Pathol. 2012;180:1879–1896. doi: 10.1016/j.ajpath.2012.01.019. [DOI] [PubMed] [Google Scholar]
- 11.Olofsson E.M., Marklund S.L., Behndig A. Enhanced age-related cataract in copper–zinc superoxide dismutase null mice. Clin. Exp. Ophthalmol. 2012;40:813–820. doi: 10.1111/j.1442-9071.2012.02794.x. [DOI] [PubMed] [Google Scholar]
- 12.Elchuri S., Oberley T.D., Qi W., Eisenstein R.S., Jackson Roberts L., Van Remmen H., Epstein C.J., Huang T.T. CuZnSOD deficiency leads to persistent and widespread oxidative damage and hepatocarcinogenesis later in life. Oncogene. 2005;24:367–380. doi: 10.1038/sj.onc.1208207. [DOI] [PubMed] [Google Scholar]
- 13.Muller F.L., Song W., Liu Y., Chaudhuri A., Pieke-Dahl S., Strong R., Huang T.T., Epstein C.J., Roberts L.J., 2nd, Csete M. Absence of CuZn superoxide dismutase leads to elevated oxidative stress and acceleration of age-dependent skeletal muscle atrophy. Free Radic. Biol. Med. 2006;40:1993–2004. doi: 10.1016/j.freeradbiomed.2006.01.036. [DOI] [PubMed] [Google Scholar]
- 14.Iuchi Y., Okada F., Onuma K., Onoda T., Asao H., Kobayashi M., Fujii J. Elevated oxidative stress in erythrocytes due to a SOD1 deficiency causes anaemia and triggers autoantibody production. Biochem. J. 2007;402:219–227. doi: 10.1042/BJ20061386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Guturu P., Duchini A. Etiopathogenesis of nonalcoholic steatohepatitis: role of obesity, insulin resistance and mechanisms of hepatotoxicity. Int. J. Hepatol. 2012;2012:212865. doi: 10.1155/2012/212865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fisher E.A. The degradation of apolipoprotein B100: multiple opportunities to regulate VLDL triglyceride production by different proteolytic pathways. Biochim. Biophys. Acta. 2012;1821:778–781. doi: 10.1016/j.bbalip.2012.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Uchiyama S., Shimizu T., Shirasawa T. CuZn-SOD deficiency causes ApoB degradation and induces hepatic lipid accumulation by impaired lipoprotein secretion in mice. J. Biol. Chem. 2006;281:31713–31719. doi: 10.1074/jbc.M603422200. [DOI] [PubMed] [Google Scholar]
- 18.Ishigami A., Maruyama N. Significance of SMP30 in gerontology. Geriatr. Gerontol. Int. 2007;7:316–325. [Google Scholar]
- 19.Maruyama N., Ishigami A., Kondo Y. Pathophysiological significance of senescence marker protein-30. Geriatr. Gerontol. Int. 2010;10(Suppl. 1):S88–S98. doi: 10.1111/j.1447-0594.2010.00586.x. [DOI] [PubMed] [Google Scholar]
- 20.Fujita T., Inoue H., Kitamura T., Sato N., Shimosawa T., Maruyama N. Senescence marker protein-30 (SMP30) rescues cell death by enhancing plasma membrane Ca2+-pumping activity in Hep G2 cells. Biochem. Biophys. Res. Commun. 1998;250:374–380. doi: 10.1006/bbrc.1998.9327. [DOI] [PubMed] [Google Scholar]
- 21.Ishigami A., Fujita T., Handa S., Shirasawa T., Koseki H., Kitamura T., Enomoto N., Sato N., Shimosawa T., Maruyama N. Senescence marker protein-30 knockout mouse liver is highly susceptible to tumor necrosis factor-α- and Fas-mediated apoptosis. Am. J. Pathol. 2002;161:1273–1281. doi: 10.1016/s0002-9440(10)64404-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ishigami A., Kondo Y., Nanba R., Ohsawa T., Handa S., Kubo S., Akita M., Maruyama N. SMP30 deficiency in mice causes an accumulation of neutral lipids and phospholipids in the liver and shortens the life span. Biochem. Biophys. Res. Commun. 2004;315:575–580. doi: 10.1016/j.bbrc.2004.01.091. [DOI] [PubMed] [Google Scholar]
- 23.Kondo Y., Inai Y., Sato Y., Handa S., Kubo S., Shimokado K., Goto S., Nishikimi M., Maruyama N., Ishigami A. Senescence marker protein 30 functions as gluconolactonase in l-ascorbic acid biosynthesis, and its knockout mice are prone to scurvy. Proc. Natl. Acad. Sci. U.S.A. 2006;103:5723–5728. doi: 10.1073/pnas.0511225103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Aizawa S., Senda M., Harada A., Maruyama N., Ishida T., Aigaki T., Ishigami A., Senda T. Structural basis of the γ-lactone-ring formation in ascorbic acid biosynthesis by the senescence marker protein-30/gluconolactonase. PLoS One. 2013;8:e53706. doi: 10.1371/journal.pone.0053706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Linster C.L., Van Schaftingen E. Vitamin C. Biosynthesis, recycling and degradation in mammals. FEBS J. 2007;274:1–22. doi: 10.1111/j.1742-4658.2006.05607.x. [DOI] [PubMed] [Google Scholar]
- 26.Pohanka M., Pejchal J., Snopkova S., Havlickova K., Karasova J.Z., Bostik P., Pikula J. Ascorbic acid: an old player with a broad impact on body physiology including oxidative stress suppression and immunomodulation: a review. Mini Rev. Med. Chem. 2012;12:35–43. doi: 10.2174/138955712798868986. [DOI] [PubMed] [Google Scholar]
- 27.Ishikawa Y., Hashizume K., Kishimoto S., Tezuka Y., Nishigori H., Yamamoto N., Kondo Y., Maruyama N., Ishigami A., Kurosaka D. Effect of vitamin C depletion on UVR-B induced cataract in SMP30/GNL knockout mice. Exp. Eye Res. 2012;94:85–89. doi: 10.1016/j.exer.2011.11.010. [DOI] [PubMed] [Google Scholar]
- 28.Arai K.Y., Sato Y., Kondo Y., Kudo C., Tsuchiya H., Nomura Y., Ishigami A., Nishiyama T. Effects of vitamin C deficiency on the skin of the senescence marker protein-30 (SMP30) knockout mouse. Biochem. Biophys. Res. Commun. 2009;385:478–483. doi: 10.1016/j.bbrc.2009.05.104. [DOI] [PubMed] [Google Scholar]
- 29.Amano A., Tsunoda M., Aigaki T., Maruyama N., Ishigami A. Effect of ascorbic acid deficiency on catecholamine synthesis in adrenal glands of SMP30/GNL knockout mice. Eur. J. Nutr. 2014;53:177–185. doi: 10.1007/s00394-013-0515-9. [DOI] [PubMed] [Google Scholar]
- 30.Kashio A., Amano A., Kondo Y., Sakamoto T., Iwamura H., Suzuki M., Ishigami A., Yamasoba T. Effect of vitamin C depletion on age-related hearing loss in SMP30/GNL knockout mice. Biochem. Biophys. Res. Commun. 2009;390:394–398. doi: 10.1016/j.bbrc.2009.09.003. [DOI] [PubMed] [Google Scholar]
- 31.Kondo Y., Sasaki T., Sato Y., Amano A., Aizawa S., Iwama M., Handa S., Shimada N., Fukuda M., Akita M. Vitamin C depletion increases superoxide generation in brains of SMP30/GNL knockout mice. Biochem. Biophys. Res. Commun. 2008;377:291–296. doi: 10.1016/j.bbrc.2008.09.132. [DOI] [PubMed] [Google Scholar]
- 32.Koike K., Kondo Y., Sekiya M., Sato Y., Tobino K., Iwakami S.I., Goto S., Takahashi K., Maruyama N., Seyama K. Complete lack of vitamin C intake generates pulmonary emphysema in senescence marker protein-30 knockout mice. Am. J. Physiol. Lung Cell. Mol. Physiol. 2010;298:L784–792. doi: 10.1152/ajplung.00256.2009. [DOI] [PubMed] [Google Scholar]
- 33.Senmaru T., Yamazaki M., Okada H., Asano M., Fukui M., Nakamura N., Obayashi H., Kondo Y., Maruyama N., Ishigami A. Pancreatic insulin release in vitamin C-deficient senescence marker protein-30/gluconolactonase knockout mice. J. Clin. Biochem. Nutr. 2012;50:114–118. doi: 10.3164/jcbn.11-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hasegawa G., Yamasaki M., Kadono M., Tanaka M., Asano M., Senmaru T., Kondo Y., Fukui M., Obayashi H., Maruyama N. Senescence marker protein-30/gluconolactonase deletion worsens glucose tolerance through impairment of acute insulin secretion. Endocrinology. 2010;151:529–536. doi: 10.1210/en.2009-1163. [DOI] [PubMed] [Google Scholar]
- 35.Yamada S., Saitoh S., Machii H., Mizukami H., Hoshino Y., Misaka T., Ishigami A., Takeishi Y. Coronary artery spasm related to thiol oxidation and senescence marker protein-30 in aging. Antioxid. Redox Signal. 2013;19:1063–1073. doi: 10.1089/ars.2012.4903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mizukami H., Saitoh S., Machii H., Yamada S., Hoshino Y., Misaka T., Ishigami A., Takeishi Y. Senescence marker protein-30 (SMP30) deficiency impairs myocardium-induced dilation of coronary arterioles associated with reactive oxygen species. Int. J. Mol. Sci. 2013;14:9408–9423. doi: 10.3390/ijms14059408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kim H.S., Son T.G., Park H.R., Lee Y., Jung Y., Ishigami A., Lee J. Senescence marker protein 30 deficiency increases Parkinson’s pathology by impairing astrocyte activation. Neurobiol. Aging. 2013;34:1177–1183. doi: 10.1016/j.neurobiolaging.2012.10.008. [DOI] [PubMed] [Google Scholar]
- 38.Kondo Y., Hasegawa G., Okada H., Senmaru T., Fukui M., Nakamura N., Sawada M., Kitawaki J., Okanoue T., Kishimoto Y. Leprdb/db Mice with senescence marker protein-30 knockout (Leprdb/dbSmp30Y/−) exhibit increases in small dense-LDL and severe fatty liver despite being fed a standard diet. PLoS One. 2013;8:e65698. doi: 10.1371/journal.pone.0065698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Iwama M., Shimokado K., Maruyama N., Ishigami A. Time course of vitamin C distribution and absorption after oral administration in SMP30/GNL knockout mice. Nutrition. 2011;27:471–478. doi: 10.1016/j.nut.2010.04.010. [DOI] [PubMed] [Google Scholar]
- 40.Amano A., Sato Y., Kishimoto Y., Takahashi K., Handa S., Aigaki T., Maruyama N., Ishigami A. Effects of ascorbic acid deficiency on protein and lipid oxidation in livers from SMP30/GNL knockout mice. J. Nutr. Sci. Vitaminol. (Tokyo) 2013;59:489–495. doi: 10.3177/jnsv.59.489. [DOI] [PubMed] [Google Scholar]
- 41.Andreo U., Elkind J., Blachford C., Cederbaum A.I., Fisher E.A. Role of superoxide radical anion in the mechanism of apoB100 degradation induced by DHA in hepatic cells. FASEB J. 2011;25:3554–3560. doi: 10.1096/fj.11-182725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Huynh F.K., Neumann U.H., Wang Y., Rodrigues B., Kieffer T.J., Covey S.D. A role for hepatic leptin signaling in lipid metabolism via altered very low density lipoprotein composition and liver lipase activity in mice. Hepatology. 2013;57:543–554. doi: 10.1002/hep.26043. [DOI] [PubMed] [Google Scholar]
- 43.Sato Y., Uchiki T., Iwama M., Kishimoto Y., Takahashi R., Ishigami A. Determination of dehydroascorbic acid in mouse tissues and plasma by using tris(2-carboxyethyl)phosphine hydrochloride as reductant in metaphosphoric acid/ethylenediaminetetraacetic acid solution. Biol. Pharm. Bull. 2010;33:364–369. doi: 10.1248/bpb.33.364. [DOI] [PubMed] [Google Scholar]
- 44.Ohkawa H., Ohishi N., Yagi K. Assay for lipid peroxides in animal tissues by thiobarbituric acid reaction. Anal. Biochem. 1979;95:351–358. doi: 10.1016/0003-2697(79)90738-3. [DOI] [PubMed] [Google Scholar]
- 45.Laing S., Wang G., Briazova T., Zhang C., Wang A., Zheng Z., Gow A., Chen A.F., Rajagopalan S., Chen L.C. Airborne particulate matter selectively activates endoplasmic reticulum stress response in the lung and liver tissues. Am. J. Physiol. Cell Physiol. 2010;299:C736–C749. doi: 10.1152/ajpcell.00529.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Smith P.K., Krohn R.I., Hermanson G.T., Mallia A.K., Gartner F.H., Provenzano M.D., Fujimoto E.K., Goeke N.M., Olson B.J., Klenk D.C. Measurement of protein using bicinchoninic acid. Anal. Biochem. 1985;150:76–85. doi: 10.1016/0003-2697(85)90442-7. [DOI] [PubMed] [Google Scholar]