

Abstract
P-selectin is a leukocyte adhesion receptor present in endothelial cells and platelets. We examined the role of P-selectin in the autologous phase of an accelerated model of anti-glomerular basement membrane (GBM) glomerulonephritis using P-selectin–deficient mice and chimeric mice expressing P-selectin only in platelets or endothelial cells. P-selectin–deficient mice exhibited more severe glomerular damage with increased interstitial mononuclear leukocytic infiltrates, and had significantly increased proteinuria and mortality when compared to wild-type mice. P-selectin on the endothelium was predominantly responsible for protection from the exacerbated disease, because chimeric mice with endothelial P-selectin, and not mice with platelet P-selectin, showed glomerular injury similar to that in wild-type animals. Levels of soluble circulating P-selectin were increased in nephritic wild-type mice and in chimeric mice with endothelial P-selectin, but not platelet P-selectin. Levels of soluble P-selectin, which has been shown to be anti-inflammatory in vitro, were inversely associated with the severity of disease. P-selectin was not expressed in the endothelium of the glomerulus or interstitium. Thus, the protective effect in wild-type mice may be accounted for, in part by soluble P-selectin shed by non-renal endothelial cells, although other endothelial P-selectin–dependent mechanisms cannot be ruled out.
J. Clin. Invest. 103:649–659 (1999)
Introduction
Most forms of glomerulonephritis (GN) result from immunologically mediated glomerular injury, which leads to proteinuria and renal dysfunction. This process is associated with a variety of glomerular histological changes, including leukocyte infiltration, fibrin and platelet deposition, proliferation of intrinsic glomerular cells, mesangial sclerosis, and glomerular basement membrane (GBM) thickening (1–3). Infiltrating leukocytes may contribute to glomerular injury by a variety of mechanisms, including the release of toxic products that can directly damage the GBM and the release of inflammatory mediators that can affect the behavior of glomerular, tubular, and interstitial cells. A number of studies have shown that leukocyte adhesion receptors play a role in leukocyte influx in GN (reviewed in ref. 4), as they do in other inflammatory diseases (5, 6).
P-selectin, a transmembrane protein expressed on the surface of activated endothelial cells and platelets, is an adhesion receptor for neutrophils, monocytes, and T cells. In vivo, antibodies to P-selectin block leukocyte rolling in venules, as well as neutrophil accumulation in tissues and thrombi (7–9). P-selectin–deficient mice exhibit severely diminished early (two to four hours) leukocyte rolling and delayed recruitment of leukocytes in various inflammatory settings (10–12). In contrast, soluble P-selectin, which is present in the plasma of normal subjects (13–15) and elevated in patients with GN (16) and other diseases (13, 15, 17–20), inhibits the adhesion of activated neutrophils to endothelial cells (21), as well as the neutrophil respiratory burst (22).
Although P-selectin has been detected in the glomeruli and interstitium of kidneys from patients with proliferative GN (16), previous studies have been restricted to assessing the role of P-selectin in the acute models of immune-mediated GN, and indeed, inferred differing roles for P-selectin in the glomerular inflammatory response (23–25). In a complement-independent model of acute, passive, heterologous anti-GBM nephritis, we showed that P-selectin–deficient mice had increased glomerular neutrophil accumulation and proteinuria compared with wild-type mice (24), whereas others have reported that functional blocking antibodies to P-selectin diminish one or both of these parameters in acute (less than eight hours) models of heterologous anti-GBM (23) and immune complex nephritis (25).
The objective of the current study was to investigate the role of P-selectin and the relative contribution of platelet and endothelial P-selectin in the autologous phase of an accelerated model of nephrotoxic nephritis, which is associated with prolonged proteinuria and glomerular damage.
Methods
Generation of C57Bl/6 P-selectin–deficient mice, wild-type mice, and mice chimeric for P-selectin expression.
P-selectin–deficient mice, generated by gene targeting (26), were backcrossed seven generations to C57Bl/6 (N7 mice). Thus, theoretically, 99.2% of the unlinked loci will be C57Bl/6. Wild-type C57Bl/6 controls for these animals were purchased from Taconic Farms (Germantown, New York, USA). Eighth-generation C57Bl/6 wild-type and P-selectin–deficient mice were generated in our facility as follows: Seventh-generation C57Bl/6 P-selectin–deficient mice were mated with C57Bl/6 mice purchased from Taconic Farms to produce heterozygous progeny that were then mated to generate wild-type and P-selectin–deficient mice. Wild-type mice pairs and P-selectin–deficient pairs from these matings were bred to produce additional wild-type and P-selectin–deficient mice, respectively (N8 mice). C5-deficient mice and their wild-type controls were purchased from The Jackson Laboratory (Bar Harbor, Maine, USA).
Bone marrow transplantation was undertaken using C57Bl/6 wild-type and P-selectin–deficient mice as donors and/or recipients. Bone marrow was isolated from donor mice sacrificed by CO2 inhalation. Recipient mice were irradiated with 1,200 rad from a cesium source (Center for Blood Research, Harvard Medical School) to destroy their own bone marrow cells, and ∼107 cells of donor bone marrow were injected into the tail vein. In the following 4 weeks, the animals were kept in clean, germ-free microisolator cages to allow full reconstitution.
Mice were bred and maintained in a virus/antibody–free facility at Longwood Medical Research Center (Boston, Massachusetts, USA).
Leukocyte counts and analysis of P-selectin on platelets.
Peripheral blood was collected and total and differential leukocyte counts were determined as described previously (27). Platelets were collected from EDTA-anticoagulated whole blood by density centrifugation and analyzed for P-selectin expression using a polyclonal rabbit anti–mouse P-selectin antibody (provided by M. Berndt, Baker Medical Research Institute, Victoria, Australia) as described previously (26). Samples were subjected to flow cytometry analysis using a FACSCalibur (Becton Dickinson Immunocytometry Systems, San Jose, California, USA).
Induction of GN and collection of urine, serum, and kidneys.
Nephrotoxic serum was prepared by immunizing rabbits with mouse GBM, as described by Schrijver et al. (28). Male mice 8–12 weeks old were immunized subcutaneously with 0.1 mg of rabbit IgG (Pel-Freeze Biologicals, Rogers, Arizona, USA) in incomplete Freund's adjuvant and nonviable desiccated Mycobacterium tuberculosis H37Ra (Difco Laboratories, Detroit, Michigan, USA). Three days later, mice were injected intravenously with heat-inactivated nephrotoxic serum at 5 mg/20 g body weight of anti–mouse globulin. Urine samples were collected in metabolic cages over 24 h before immunization and at day 1, 7, 14, and 21 after injection of nephrotoxic serum. At selected time points, mice were anesthetized with methoxyflurane, and peripheral blood was collected by retro-orbital bleeding into glass tubes to obtain serum. The animals were then euthanized by CO2 inhalation, and both kidneys were harvested.
Histologic studies.
All histological quantitation was performed without prior knowledge of the genotype of the animal. Kidneys were fixed in formalin and paraffin embedded or snap frozen in Optimum Cooling Temperature (OCT) embedding medium (Miles, Elkhart, Indiana, USA). Paraffin-embedded sections (4 μM) were stained with hematoxylin and eosin or with periodic acid-Schiff stain (PAS) for histological analysis. The presence of PAS-positive (PAS+) material within glomeruli was scored semiquantitatively in a minimum of 50 glomeruli per mouse, as described previously (29) and in the legend of Fig. 3. Sections were also stained for dichloroacetate esterase to identify neutrophils (24). The number of neutrophils per 100 glomeruli (50 per kidney) were counted for each animal examined.
Figure 3.

Quantitation of PAS+ deposition as a measure of glomerular injury. Deposition of PAS+ material was quantitated in wild-type and P-selectin–deficient mice at indicated days after induction of disease and given a score of 0–3 as follows: 0 = no deposition of PAS+ material; 1 = up to one-third; 2 = one-third to two-thirds; and 3 = more than two-thirds of the glomerular cross-section stain positive for PAS. Four animals of each genotype and more then 50 glomeruli per animal were evaluated at each time point. *P < 0.05 and **P < 0.005 compared with wild-type controls.
Immunofluorescence microscopy was performed on 4-μm sections cut from OCT-embedded kidneys using previously described antibodies and standard immunofluorescent techniques (30). For a semiquantitative assessment of the glomerular deposition of heterologous (rabbit) or autologous (mouse) antibody, the end point positive titer for detection of staining, assessed in a blinded protocol, was used as described previously (31). A semiquantitative scoring of staining intensity for C3 from 0 to 4+ was used, as detailed previously (32). Glomerular fibrin and platelet deposition were assessed using the scoring system described for the assessment of glomerular PAS+ material. In all cases, a minimum of 50 glomeruli were examined per animal. The degree of interstitial infiltration of CD4+ and CD8+ T cells was quantitated in immunostained frozen sections by counting six high-power fields of cortex and medulla. For a semiquantitative assessment of macrophage infiltration, a score of 0–4+ was given for the amount of positive cells in immunostained sections treated with the specific macrophage antibody F4/80: 0 = no cells stained positive; 1+ = 5–10 cells; 2+ = 10–50 cells; 3+ = 50–200 cells; and 4+ = over 200 cells stained positive per low power field.
Antibodies.
Kidney sections were stained by direct or indirect immunofluorescence with one of the following primary antibodies: FITC-conjugated goat anti–rabbit IgG (Pel-Freeze Biologicals) at four dilutions from 1:200 to 1:1,600; FITC-conjugated goat anti–mouse IgG (Jackson ImmunoResearch Laboratories Inc., West Grove, Pennsylvania, USA) at four dilutions from 1:800 to 1:6,400; FITC-conjugated goat F(ab′)2 fragment to mouse complement C3 ([Cappel] ICN Pharmaceuticals Inc., Aurora, Ohio, USA) at 1:100; FITC-conjugated rabbit anti–human fibrinogen (DAKO Corp., Carpinteria, California, USA) that cross-reacts with mouse fibrin/fibrinogen at 1:80; rat anti–mouse platelet IIb/IIIa monoclonal antibody D9 (10 μg/ml; kindly provided by A.-K. Ng, University of Southern Maine, Portland, Maine, USA) at a dilution of 1:100, followed by FITC-conjugated F(ab′)2 fragment of goat anti–rat antibody (Jackson ImmunoResearch Laboratories Inc.). Staining for macrophages, T cells, and P-selectin was done using a three-layer immunoperoxidase technique as described previously (33). The following antibodies were used: rat anti–mouse mature macrophage (Clone F4/80; Caltag Laboratories Inc., Burlingame, California, USA); rat anti–mouse CD4 (clone L3T4; PharMingen, San Diego, California); rat anti–mouse CD8a (Clone Ly-2; PharMingen) and rat IgG2a isotype control (PharMingen); rat anti–mouse P-selectin (10A10; a gift from B. Wolitsky, Hoffman LaRoche Inc., Nutley, New Jersey, USA). Biotin-conjugated goat anti-rat IgG antibody (Jackson ImmunoResearch Laboratories Inc.) was used as a secondary antibody, followed by incubation with an avidin–biotin complex, development with a 10-min incubation with 0.4% 3-amino-9-ethylcarbazole, and counterstaining with Gill's Hematoxylin No.3 (Polysciences Inc., Warrington, Pennsylvania, USA).
Urinary albumin and urinary and serum creatinine detection.
Urinary albumin was determined by a double-sandwich ELISA as detailed previously (24). Urinary or serum creatinine was quantitated spectrophotometrically using a commercially available kit (Sigma Chemical Co., St. Louis, Missouri, USA). To standardize urine albumin excretion for glomerular filtration rate, proteinuria was expressed as micrograms of urinary albumin per milligram of urinary creatinine (34).
Assessment of circulating mouse anti–rabbit immunoglobulin G antibody.
Serum was prepared from peripheral blood. Microtiter plates (96-well; Dynatech Laboratories Inc., Chantilly, Virginia, USA) were incubated with 100 μg/ml of rabbit IgG (Pel-Freeze Biologicals) in carbonate/bicarbonate buffer (pH 9.5) overnight at 4°C, and the plate was blocked with 1% BSA. Plates were washed with PBS containing 0.1% Tween-20 and then incubated with serial-doubling dilutions of mouse serum, starting at 1:100. After further washing, bound mouse IgG was detected with FITC-conjugated goat anti–mouse IgG (Jackson ImmunoResearch Laboratories Inc.) at a dilution of 1:250. The fluorescence activity was detected by a fluorescence plate reader (IDEXX, Portland, Maine, USA), and results are given in arbitrary units. Serum from nonimmunized mice was used as normal controls.
Detection of soluble P-selectin.
Blood was collected from the retro-orbital plexus of mice into polypropylene tubes containing 0.1 volume of sodium citrate dextrose, and soluble P-selectin was detected by ELISA as described previously (35).
Western blot analysis of P-selectin.
Platelets were lysed in 1% Triton/0.5% deoxycholate in PBS, and 50-μl plasma samples were diluted to 400 μl with platelet lysis buffer. Plasma samples were precleared with Gelatin Sepharose 4B (Amersham Pharmacia Biotech, Piscataway, New Jersey, USA) and a monoclonal antibody to P-selectin (RB40.34; PharMingen) coupled to protein-G beads was then used to concentrate P-selectin in plasma samples. Platelet lysates and immunoprecipitated plasma samples were denatured by boiling and reduced by the addition of β-mercaptoethanol at a final concentration of 5%. Samples were electrophoresed on a 6% SDS-PAGE and transferred onto a Sequi-blot polyvinylidene difluoride membrane using a trans-blot Western transfer apparatus (Bio-Rad Laboratories Inc., Hercules, California, USA). P-selectin was detected with polyclonal rabbit anti–P-selectin IgG (PharMingen). Bound primary antibody was detected by horseradish peroxidase–conjugated goat anti–rabbit IgG (Bio-Rad Laboratories Inc.).
Statistics.
Results are expressed as mean ± SEM. Unpaired Student's t tests were performed for data, and P < 0.05 was considered statistically significant.
Results
Characterization of the accelerated nephrotoxic nephritis model in mice of the C57Bl/6 wild-type strain.
Mice in this study were immunized with rabbit IgG in complete Freund's adjuvant followed by intravenous injection of anti-GBM antibody as described in Methods. The resulting nephrotoxic nephritis in C57Bl/6 mice was characterized by significant proteinuria (day 0: 22.4 ± 5.0 μg albumin/mg creatinine, n = 20; day 14: 7,292.9 ± 2,723.7 μg albumin/mg creatinine, n = 12; day 21: 15,089.7 ± 3,824.6 μg albumin/mg creatinine, n = 8), but minimal serum creatinine elevation. Histological changes consisted of mild focal increases in leukocytes at day 1 and focal mesangial hypercellularity at day 7, which was most prominent at day 14. In addition, at days 7 and 14, there was a mild focal interstitial mononuclear cell infiltrate in some of the animals. Deposits of PAS+ hyaline material in lumina and increases in mesangial matrix were focal and mild, occurring in less than 10% of glomeruli in the most severely affected mice at 14 days (see below for quantitation).
Proteinuria at days 14 and 21 in this model was complement-dependent: C5-deficient mice subjected to the same immunization protocol had minimal proteinuria (day 14: 192.6 ± 125.5 μg albumin/mg creatinine; day 21: 125.0 ± 29.0 μg albumin/mg creatinine) compared with their respective wild-type controls that were similarly treated (day 14: 5,324.2 ± 964.2; day 21: 7,235.6 ± 2,047.6 μg albumin/mg creatinine). As described by others (33), susceptibility to accelerated nephrotoxic nephritis was strain-dependent. In our studies, we directly compared proteinuria in C57Bl/6 and BALB/c mice subjected to nephrotoxic nephritis. Treated BALB/c mice had 80% lesser proteinuria compared with similarly treated C57Bl/6 (data not shown). For this reason, mice of the C57Bl/6 strain were used for subsequent experiments.
P-selectin deficiency exacerbates glomerular injury in the autologous phase of accelerated nephrotoxic nephritis.
To determine the role of P-selectin in this model of nephrotoxic nephritis, disease was induced in wild-type and P-selectin–deficient animals of the C57Bl/6 strain. Animals deficient in P-selectin showed markedly increased albuminuria on days 7 and 14, compared with wild-type animals, and marked elevation of serum creatinine levels at day 14 (Fig. 1). In addition, there was increased mortality in P-selectin–deficient mice: No wild-type animals died during the 21 days of observation, whereas 17% of knockout animals died between 8 and 14 days, and 50% died between 15 and 21 days. Data presented in Figs. 1–5 were collected only through day 14 after induction of nephritis because of the significant mortality of P-selectin–deficient mice between days 15 and 21.
Figure 1.

Mice deficient in P-selectin show increased proteinuria and elevated serum creatinine levels. Proteinuria and serum creatinine levels were quantitated in P-selectin–deficient (white bars) and wild-type (black bars)mice at day 1 (n = 20 per genotype), day 7 (n = 16 per genotype), and day 14 (n = 12 per genotype) after induction of nephritis. Urine albumin excretion (expressed in micrograms) was determined and expressed per milligram of urinary creatinine to standardize for glomerular filtration rate. Serum creatinine levels were measured in four animals of each group at the indicated time points. P-selectin–deficient mice had increased proteinuria at 7 and 14 days and elevated serum creatinine levels at day 14 after administration of nephrotoxic serum, compared with wild-type mice. *P < 0.01 and **P = 0.001 compared with wild-type counterparts.
Figure 5.

Immunohistochemical localization of interstitial macrophage and CD4+ and CD8+ T-cell accumulation. (a) Nephritic wild-type and P-selectin–null mice contained macrophage infiltrates in the interstitium 14 days after induction of nephritis, stained here with antibody F4/80. Macrophages were significantly more abundant in P-selectin–deficient mice compared with wild-type animals (wild-type score: 1.5+; P-selectin–null score: 4+). Note in P-selectin–null mice, two macrophages in the glomerulus (g) beneath the Bowman's capsule (at the upper pole) and throughout the interstitium. (b) Interstitial CD4+ and CD8+ lymphocytes were quantitated at day 7 and day 14 after induction of nephritis. At day 7, the numbers of CD8+ T cells were significantly decreased in P-selectin–deficient mice (white bars) compared with wild-type mice (black bars), whereas no difference was seen in CD4+ T cells. At day 14, there was no longer a difference in the CD8+ T cells. However, significantly more CD4+ T cells were present in the P-selectin–deficient mice compared with the wild-type animals. This was principally because the number of CD4+ T cells decreased in wild-type mice on day 14 compared with day 7, whereas in P-selectin–deficient mice, they remained the same. n = 4 per each genotype at all time points. *P < 0.05.
Histologically, at day 1, P-selectin–deficient mice had over twofold more neutrophils per glomerulus compared with wild-types (wild-type: 0.29 ± 0.06; P-selectin–deficient: 0.69 ± 0.112; P < 0.05), although neutrophil infiltrate was focal and mild in both. At day 14, a dramatic difference in glomerular injury was seen in P-selectin–deficient mice compared with wild-type mice. In contrast to the relatively mild and focal changes described earlier for wild-type mice (Fig. 2a), all kidneys from P-selectin–deficient mice showed extensive glomerular injury (Fig. 2b). We found that 80%–100% of glomeruli in the animals exhibited PAS+, homogenous, and occasionally vacuolated deposits that were often intravascular, but that also occupied mesangial areas and sometimes the Bowman's space (Fig. 2c); 5%–10% of glomeruli in P-selectin–deficient mice showed the formation of small crescents (Fig. 2c). Crescents were not observed in wild-type mice. In addition, a varying proportion of glomeruli in P-selectin–deficient mice showed foci of leukocytic infiltration by neutrophils and mononuclear cells, proliferation of mesangial cells, and increased mesangial matrix occasionally producing a lobular pattern (Fig. 2d). Immunohistochemical studies showed that the mononuclear cells in such glomeruli were macrophages and CD4+ and CD8+ T cells, but the distribution of these leukocytes was focal and small in number, and was therefore not quantitated. In contrast, significant numbers of mononuclear cells were present in the interstitium and were quantitated as described later.
Figure 2.

Analysis of glomeruli from nephritic wild-type and P-selectin–deficient mice. Representative PAS-stained sections from wild-type (a) and P-selectin–deficient (b, c, d) mice at 14 days after induction of disease are shown. (a) Glomerulus from wild-type nephritic mouse at 14 days. This is one of the more severely affected glomeruli, exhibiting increased cellularity, largely in mesangial areas. (b) Glomeruli from P-selectin–deficient nephritic mouse at 14 days. Note PAS-positive deposits distorting glomerular architecture in the lumen, mesangium, and Bowman's space. (c) Higher-power view of a glomerulus to show, in addition to PAS-positive deposits, adhesion to Bowman's capsule, mononuclear infiltrate, and a small crescent (top right). (d) Glomerulus from nephritic P-selectin–deficient mouse showing leukocytic infiltration, predominantly mononuclear, increased mesangial cells, and a capsular adhesion. PAS, periodic acid-Schiff stain.
PAS deposition in P-selectin–deficient and wild-type mice was quantitated as a measure of glomerular injury. The amount of PAS+ material per glomerulus, as well as the number of glomeruli affected, was significantly higher in the P-selectin–deficient animals compared with wild-type mice (Fig. 3). At day 14, glomeruli of P-selectin–deficient mice showed 14-fold more PAS+ material than did wild-type mice; while less than 10% of wild-type glomeruli were affected in the most severely affected animals, 80%–100% of the glomeruli of P-selectin–deficient mice exhibited such damage.
Immunofluorescence stains for fibrin/fibrinogen and platelets revealed a striking increase in glomerular fibrin-containing deposits and large numbers of platelets in the P-selectin–deficient mice compared with wild-type animals (Fig. 4, a and b). Staining for C3 deposition showed higher staining intensity in the P-selectin–deficient mice compared with the wild-type animals at day 7 (Fig. 4a) (wild-type score: 1+; P-selectin–deficient score: 2+), but was the same in both genotypes at day 14 (wild-type score: 2+; P-selectin–deficient score: 2+; data not shown).
Figure 4.

Evaluation of glomerular fibrin/fibrinogen, platelet, and complement C3 deposition. Frozen sections from nephritic P-selectin–deficient (right) and wild-type (left) mice were stained for fibrin/fibrinogen, platelets, and C3 complement. (a) At day 14 after induction of nephritis, fibrin-containing deposits were insignificant in the wild-type animals (less than 10%), whereas over 80% of glomeruli of P-selectin–deficient mice were affected. Platelet deposition coincided with fibrin-containing deposits. Increased complement C3-staining intensity was observed in the glomeruli of P-selectin–deficient mice compared with wild-type mice on day 7 after induction of disease. (b) Quantitation of fibrin-containing deposits in wild-type (black bars) and P-selectin–deficient (white bars) mice using the scoring system described in Fig. 3 at the indicated days after induction of disease. P-selectin–deficient animals had markedly increased glomerular fibrin deposition compared with wild-type animals. Four animals of each genotype and more then 50 glomeruli per animal were evaluated at each time point. *P < 0.05 and **P < 0.005 compared with wild-type mice.
In addition to the glomerular change, there was increased tubulointerstitial leukocytic infiltration in P-selectin–deficient mice, often in a periglomerular distribution. Immunohistochemistry revealed that the infiltrates were predominantly macrophages, which were more abundant in P-selectin–deficient mice compared with wild-type animals on day 14 (Fig. 5a). CD4+ and CD8+ T cells were also present throughout the interstitium. CD4+ T cells were similar between wild-type and P-selectin–deficient mice, but CD8+ cells were reduced by 43% in P-selectin–deficient mice compared with wild-type controls at day 7, but not day 14 (Fig. 5b). On day 14, the number of CD4+ T cells in the knockout mice was significantly higher than that in wild-type (Fig. 5b).
Glomerular injury in mice deficient in platelets or endothelial P-selectin: studies in bone marrow chimeras.
Because P-selectin is expressed in both platelets and endothelium, we examined the contribution of these two cell types to the outcome of GN. Mice expressing P-selectin only in platelets or only in the endothelium were generated by bone marrow transplantation between wild-type and P-selectin–deficient animals. The successful generation of the appropriate animals, i.e., P-selectin–positive in their platelets but negative in endothelium (Pt+/EC–) and vice versa (Pt–/EC+), was determined by flow cytometric analysis of P-selectin expression on platelets isolated from these animals (Fig. 6). To control for any effects of radiation on kidney function, wild-type and P-selectin–deficient mice controls were also generated as follows: wild-type bone marrow was transplanted into wild-type recipient mice (Pt+/EC+) and P-selectin–deficient bone marrow into P-selectin–deficient recipients (Pt–/EC–).
Figure 6.

P-selectin expression in platelets of mice generated by bone marrow transplantation. Mice chimeric for P-selectin expression, wild-type, and P-selectin–deficient mice were generated by transplanting bone marrow between wild-type and P-selectin–deficient mice. Platelets were isolated from peripheral blood of all four sets of animals, activated with thrombin, incubated with antibody to P-selectin, and analyzed by FACS®. Representative histograms from each set of animals is shown. As expected, wild-type or P-selectin–deficient recipients with bone marrow from wild-type donors had P-selectin–positive platelets (a and c), whereas recipients with bone marrow from P-selectin–deficient donors had P-selectin–negative platelets (b and d).
The bone marrow chimeric and control mice were subjected to nephrotoxic nephritis, followed by quantitation of proteinuria and glomerular lesions as described. Bone marrow–transplanted animals exhibited a delay in the glomerular response compared with nontransplanted animals, most likely attributable to the irradiation. Thus the period of observation in these experiments was extended to 21 days after induction of nephritis. At day 14 and day 21, mice with P-selectin expression only in their endothelium (Pt–/EC+) had significantly reduced proteinuria compared with P-selectin–deficient mice (Pt–/EC–), although it was still slightly higher than that seen in wild-type mice (Pt+/EC+) (Fig. 7a). On the other hand, mice that had P-selectin only on their platelets, and not on their endothelium (Pt+/EC–), displayed a phenotype comparable to P-selectin–deficient (Pt–/EC–) mice (Fig. 7a). Glomerular PAS deposition in these animals showed a similar pattern (Fig. 7b): Pt–/EC+ mice had less PAS deposition than Pt+/EC– or Pt–/EC– mice (Fig. 7b). Similarly, mice with endothelial expression of P-selectin (Pt+/EC+ and Pt–/EC+) had much less fibrin-containing deposits and interstitial macrophage accumulation than mice without P-selectin (Pt–/EC– and Pt+/EC–) (data not shown). Consistent with these findings, at day 21 significant differences in serum creatinine levels were found between Pt+/EC+ and Pt–/EC– mice, while levels in Pt+/EC– animals were similar to those in Pt–/EC– mice, and levels in Pt–/EC+ animals were similar to Pt+/EC+ mice (Table 1). Taken together, these results indicate that the markedly increased glomerular damage seen in P-selectin–deficient mice is attributable primarily to the loss of endothelial P-selectin rather than platelet P-selectin.
Figure 7.

Albuminuria and glomerular injury in chimeric, wild-type, and P-selectin–deficient mice. Albuminuria (a) and glomerular PAS+ material (b) in chimeric (Pt+/EC–, hatched bars, n = 11; and Pt–/EC+, cross-hatched bars, n = 10), wild-type (Pt+/EC+, black bars, n = 4), and P-selectin–deficient (Pt–/EC–, white bars, n = 7) mice were determined on days 14 and 21 after nephritis. Mice with endothelial expression of P-selectin (Pt–/EC+ and Pt+/EC+) had lower albuminuria and fewer PAS deposits than those animals with no P-selectin expression on the endothelium (Pt–/EC– and Pt+/EC–). *P < 0.005 compared with Pt–/EC– mice; #P < 0.005 compared with Pt+/EC+ mice.
Table 1.
Serum creatinine (μmol/l) in P-selectin chimeras at indicated days after induction of accelerated glomerulonephritis

All experiments up to this point were conducted on P-selectin–deficient mice backcrossed seven generations to C57Bl/6 mice (therefore 99.2% C57Bl/6) and C57Bl/6 mice that were purchased commercially as controls. To rule out any putative effect of colony environment or background genes in contributing to our results, we generated both wild-type and P-selectin–deficient mice from seventh-generation P-selectin–deficient mice as described in Methods (N8 mice). Bone marrow chimeras were generated from these animals, nephritis was induced, and albuminuria was monitored over a period of 21 days. Results from these studies supported our previous findings: Increased albuminuria was due to a loss of endothelial P-selectin (data not shown).
Analysis of heterologous antibody deposition and autologous antibody production in mice.
Because heterologous antibody deposition in the kidney and the resultant systemic immune response leading to production and deposition of autologous antibodies are determinants of the extent of glomerular damage, we quantitated glomerular deposition of heterologous (rabbit) IgG, autologous antibody production, and autologous antibody deposition. Rabbit IgG deposition, assessed in nephritic kidneys at seven days after induction of disease in both wild-type and P-selectin–deficient mice, was detectable up to a dilution of 1:800 of FITC-conjugated goat anti–rabbit antibody and was similar in both groups of animals at this dilution (data not shown). Circulating levels of mouse anti–rabbit IgG were quantitated in P-selectin–deficient, wild-type, and chimeric mice generated by bone marrow transplantation of the N8 mice. Fourteen days after immunization with rabbit nephrotoxic serum, mouse anti–rabbit antibody titers were similar between all the groups of animals (Table 2), except for Pt–/EC+ mice, which were significantly higher and cannot be explained. However, the higher levels did not lead to increased glomerular IgG deposition. Staining in glomeruli was still detectable at a 1:3,200 dilution of FITC-conjugated goat anti–mouse antibody, but no longer at 1:6400 dilution, and was comparable between all groups of mice (data not shown). Furthermore, the glomerular injury in this group of mice was not higher than that of other groups. In fact, the Pt–/EC+ had indices of glomerular injury that were lower than in mice with lower IgG levels (Pt+/EC–). Furthermore, differences in IgG levels among these groups of mice were not reproduced in N7 mice bone marrow chimeras or wild-type and P-selectin–deficient mice (data not shown).
Table 2.
Mouse anti–rabbit IgG levels on day 14 induction of accelerated glomerulonephritis

Assessment of neutrophilia in mice generated by bone marrow transplantation.
P-selectin–deficient mice were previously shown to have increased circulating neutrophil counts compared with wild-type (Pt+/EC+) animals (26). Peripheral blood neutrophil counts were determined in the bone marrow chimeric (Pt+/EC–, Pt–/EC+), P-selectin–deficient (Pt–/EC–), and wild-type (Pt+/EC+) mice before induction of nephritis. Pt–/EC– mice had a less than twofold increase in neutrophil counts compared with Pt+/EC+, which is consistent with previous results in P-selectin–deficient and wild-type mice (26). Interestingly, both sets of chimeric mice (Pt+/EC– and Pt–/EC+) had neutrophil counts that were lower than those in P-selectin–deficient mice and were not significantly different from those in Pt+/EC+ mice (Fig. 8). Thus P-selectin deficiency in both platelets and endothelial cells is required to produce neutrophilia. Comparison of the extent of neutrophilia (Fig. 8) with albuminuria and deposition of PAS+ material (Fig. 7) in these animals suggests that neutrophilia does not correlate with increased indices of disease.
Figure 8.

Determination of blood neutrophil counts in mice chimeric for P-selectin expression and in P-selectin–null and wild-type mice. Peripheral blood was collected from bone marrow–transplanted mice 4 weeks after engraftment and before induction of nephrotoxic nephritis, and the numbers of neutrophils were determined. P-selectin–deficient mice (Pt–/EC–, white bar, n = 7) had approximately twofold more circulating neutrophils than did wild-type (Pt+/EC+, black bar, n = 4), whereas mice with P-selectin expression only in endothelial cells (Pt–/EC+, cross-hatched bar, n = 10) or only in platelets (Pt+/EC–, hatched bar, n = 11) had levels of circulating neutrophils that were not significantly different than those in wild-type mice. *P < 0.05.
Quantitation of soluble P-selectin in GN.
Soluble P-selectin may attenuate the inflammatory process since it inhibits leukocyte-endothelial interaction and the leukocyte respiratory burst in vitro (21, 22). We reasoned that soluble P-selectin may be required to downregulate the inflammatory response in GN and that its absence leads to the exacerbation of disease. Although plasma P-selectin has been shown to be elevated in human GN (16), it is not known whether P-selectin is elevated in the circulation during experimental GN, or whether it is derived from endothelial cells, platelets, or both. We examined soluble P-selectin levels in the plasma of P-selectin bone marrow chimeras and wild-type and P-selectin–deficient controls before, and 21 days after, induction of disease. Minimal soluble P-selectin was present in unimmunized mice of all four genotypes. Twenty-one days after induction of nephritis, soluble P-selectin was elevated in the plasma of wild-type mice compared with unimmunized mice. Interestingly, Pt–/EC+ bone marrow chimeras had the same levels of circulating soluble P-selectin as did wild-types (Pt+/EC+) (Fig. 9a), while circulating soluble P-selectin levels in Pt+/EC– were only slightly higher than background. The soluble circulating P-selectin in wild-type mice is shed from the surface because it has greater electrophoretic mobility on a gel than the cellular transmembrane form of P-selectin (Fig. 9b). Three conclusions can be made from these studies. First, GN is associated with increased levels of circulating soluble P-selectin. Second, endothelial cells are primarily responsible for contributing to the pool of soluble P-selectin in nephritis. And third, the levels of soluble P-selectin correlate inversely with the degree of observed glomerular damage. Comparison of Figs. 7 and 9a shows that mice with high levels of soluble P-selectin (i.e., Pt+/EC+ and Pt–/EC+ animals) had less severe disease than Pt+/EC– and Pt–/EC– mice, which had low or absent levels of soluble P-selectin, respectively.
Figure 9.

Analysis of circulating soluble P-selectin. Soluble P-selectin levels in plasma generated from untreated (black bars) and 21-day nephritic (white bars) Pt+/EC+, Pt+/EC–, Pt–/EC+, and Pt–/EC– mice were assessed (a), and soluble P-selectin from plasma of mice was characterized by Western blot (b). (a) Levels of soluble P-selectin in plasma were evaluated by an ELISA assay. OD readings from samples obtained from P-selectin–deficient mice (Pt–/EC–) served as the negative control. All four sets of untreated mice had minimal levels of soluble P-selectin. Nephritic wild-type animals (Pt+/EC+) had increased levels compared with untreated mice, as did mice with P-selectin expression on endothelial cells but not on platelets (Pt–/EC+). Mice expressing P-selectin only on platelets (Pt+/EC–) had levels of soluble P-selectin that were not significantly higher than those in the negative control (Pt–/EC–). *P < 0.005 compared with Pt+/EC+ and Pt–/EC+. n = 4 per each group. (b) Western blot analysis of P-selectin in platelets and plasma. Platelet lysates and P-selectin immunoprecipitates from plasma samples of a wild-type and P-selectin–null mouse were reduced and electrophoresed on a 6% polyacrylamide gel, followed by Western blotting using a polyclonal anti–P-selectin antibody. P-selectin in plasma of a normal and a 21-day nephritic wild-type mouse (lanes 1 and 4, respectively) was present as a doublet and had an increased electrophoretic mobility (85–87 kDa) compared with platelet P-selectin (132 kDa) (lane 2). This suggests that plasma P-selectin is a shed form of the transmembrane-bound receptor. No P-selectin was detected in platelets of P-selectin–null mice or in plasma of 21-day nephritic P-selectin–null mice (lanes 3 and 5, respectively).
P-selectin expression in renal tissue.
To determine whether the source of the protective P-selectin was the endothelium in the glomerulus or the systemic circulation, we examined P-selectin in wild-type kidneys before, and 1 hour, 3 hours, 24 hours, 7 days, and 14 days after, induction of nephritis. Lung tissue from a lipopolysaccharide (LPS)-treated wild-type mouse was used to demonstrate the immunoreactivity of the antibody with P-selectin. The lung had strong P-selectin staining on the endothelium and on platelets present in the lumen of the vessel (Fig. 10a). However, in the kidney, there was no endothelial cell staining in the glomeruli or the interstitium either before or after induction of nephritis. After nephritis, a punctate staining was observed in the glomeruli of animals at all the time points tested, a pattern consistent with staining of platelets or platelet clusters. To definitively determine this, we analyzed P-selectin staining in renal tissue harvested from chimeric mice (Pt+/EC– and Pt–/EC+) 14 days after induction of nephritis. Indeed, P-selectin–positive staining was observed in Pt+/EC– mice (Fig. 10b), but not in Pt–/EC+ mice (data not shown), thus confirming our initial interpretation that the P-selectin–positive material in the glomeruli was platelet derived. Because P-selectin is not present in the glomerular or interstitial endothelium, the protective effect of endothelial P-selectin in GN is most likely derived from non-renal endothelium.
Figure 10.
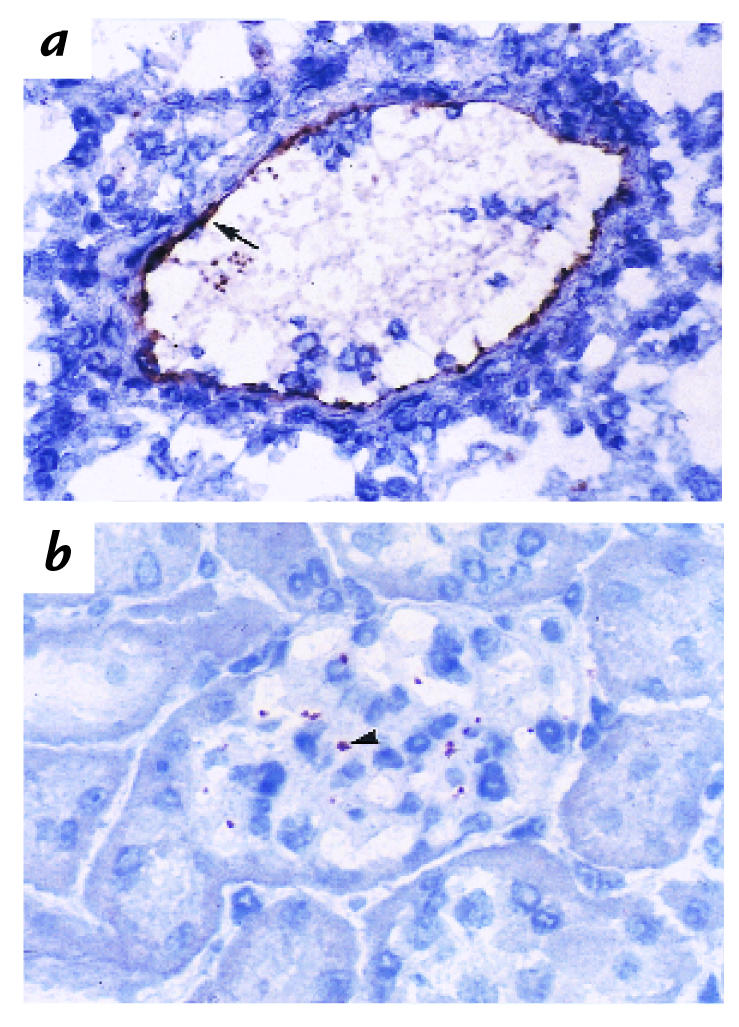
Immunohistochemical analysis of P-selectin expression in the glomerulus. (a) As a positive control, lung tissue from a LPS-treated wild-type mouse was stained with a monoclonal antibody to murine P-selectin. P-selectin was localized to the endothelium (arrow) and to platelets present in the lumen of the vessel. (b) Kidney tissue from a 14-day nephritic Pt+/EC– mouse was stained in parallel. P-selectin was present in a punctate stain, typical of platelets, or platelet clusters (arrowhead). In contrast, no P-selectin staining was observed in renal tissue samples from 14-day nephritic Pt–/EC+ mice (data not shown). Taken together, the data suggest that P-selectin staining in the glomerulus is derived only from platelets.
Discussion
In this study, we have investigated the role of P-selectin in the development of glomerular inflammation in a model of accelerated nephrotoxic nephritis. P-selectin deficiency led to an increase in histological and functional indices of glomerular damage compared with wild-type mice. Notably, glomerular fibrin-containing PAS+ deposits occurred much more significantly in P-selectin–deficient mice, and there was an increase in the focal hypercellularity, mesangial sclerosis, and hyalinosis, as well as the formation of small crescents. Furthermore, interstitial mononuclear infiltrates in the P-selectin–deficient mice, which were largely macrophages, was significantly greater than in wild-type mice. An increase in interstitial mononuclear leukocyte infiltrates has been previously shown by others to be a component of these models and to correlate with glomerular injury (36, 37). P-selectin–deficient mice exhibited markedly increased proteinuria, much higher serum creatinine levels, and increased mortality than did wild-type mice.
Three simple explanations for the exacerbated disease were ruled out in these studies. First, deposition of the heterologous (rabbit) anti-GBM antibody was similar in P-selectin–deficient and wild-type mice. Second, no significant increase in circulating or glomerularly deposited autologous antibody was noted in P-selectin–deficient mice compared with wild-type animals, suggesting that the systemic immune response in wild-type and P-selectin–deficient mice was similar. Thirdly, neutrophilia in unmanipulated P-selectin–deficient mice was not responsible for the increased disease: Pt+/EC– mice had levels of neutrophilia similar to wild-type animals before induction of GN, but developed severe nephritis that was similar to that in P-selectin–deficient mice. With regard to the neutrophilia exhibited in unmanipulated P-selectin–deficient mice, our findings suggest that P-selectin, both on the endothelium and platelets, contributes to this phenotype. Neutrophil clearance is defective in the P-selectin–deficient mice (38), and a recent study suggests that neutrophil clearance may be mediated by P-selectin on hepatic endothelia and platelets (39). Thus, P-selectin on both platelets and endothelium may mediate this clearance. Surprisingly, reconstitution with P-selectin–positive platelets does not repair the significant neutrophilia exhibited by mice deficient in both P- and E-selectin (40). The mechanism(s) for the repair in P-selectin–deficient mice, but not P- and E-selectin–deficient mice, by platelets is unclear and is currently under investigation.
Our studies show that P-selectin on endothelial cells is primarily required to restore the glomerular injury to wild-type levels in terms of both histological and functional indices of disease. This suggests that endothelial P-selectin plays a role in the suppression of the glomerular inflammatory response. P-selectin is stored in the Weibel-Palade bodies of endothelial cells in multiple organs (41) and is upregulated on the surface by agents such as histamine, thrombin, and cytokines (42, 43). However, it is not detected in normal animal kidneys (23, 25, 44). P-selectin is expressed in the glomerulus after experimental GN (23, 25, 44), but we found this expression to be derived from platelets. Our studies suggest that P-selectin is neither stored nor is it inducible in the glomerular endothelium after GN. Thus, it is plausible that nonrenal endothelial sources are responsible for providing the P-selectin that is protective in our model of GN.
What is the mechanism of the protective effect of endothelial P-selectin? One explanation suggested by our studies points to soluble P-selectin, which is markedly elevated in wild-type nephritic mice and is shed from the endothelium. Importantly, its levels are inversely associated with the severity of glomerular damage in the various groups of animals examined. Soluble P-selectin is elevated in the plasma of patients with GN (16) and other diseases (20) and has been shown in vitro to inhibit CD18-dependent adhesion of tumor necrosis factor (TNF)-α–activated neutrophils to endothelium and the superoxide generation of neutrophils (21). Soluble receptors are prevalent among adhesion, TNF-α, hematopoietin, and immunoglobulin receptor superfamilies (45) and may play a role in the normal physiological response to disease. As is the case for other soluble receptors, P-selectin may be generated by proteolytic shedding of the membrane-bound protein from the cell surface. However, in humans, but not in mice, it may also be the result of secretion of polypeptides transcribed from a predicted alternatively spliced mRNA without a transmembrane region (46, 47). In our Western blot analysis of P-selectin, the plasma form had a greater electrophoretic mobility than the platelet transmembrane form, suggesting that, indeed, plasma P-selectin is shed from the cell surface. A similar P-selectin fragment was recently observed in the plasma of mice genetically engineered with P-selectin lacking a cytoplasmic domain, which results in constitutive surface expression of endothelial P-selectin (35). Given our P-selectin expression data, we propose that P-selectin shed from nonrenal endothelium is responsible for the increase in circulating soluble P-selectin after GN and the subsequent attenuation of the inflammatory response in the glomerulus. Soluble P-selectin may inhibit firm leukocyte–endothelial interactions and production of toxic products by leukocytes, thereby limiting endothelial injury; therefore, fibrin and platelet deposition is limited, as is further leukocyte accumulation and glomerular damage resulting in proteinuria. One source of the soluble P-selectin may be the lung, because anti-GBM antibody can lead to inflammation in the lung. Furthermore, inflammatory stimuli such as LPS lead to a greater than 10-fold induction of P-selectin expression in the lung endothelium (26) and a threefold increase in soluble P-selectin levels in the plasma (35). Given that soluble P-selectin levels were inversely associated with disease indices, our studies further suggest that soluble P-selectin may provide a useful marker for predicting the extent of endothelial activation or injury in GN. Levels of other specific soluble receptors in patients have been previously shown to predict outcomes in diseases such as chronic lymphocytic leukemia, Hodgkin's disease, and meningitis (45).
An alternate, or additional, explanation for the increased glomerular injury in the P-selectin–deficient mice may lie in the differential accumulation of CD4+ and CD8+ T cells in P-selectin–deficient vs. wild-type mice. A recent study of GN in CD4+-deficient and CD8+-deficient mice suggests that a decrease in numbers of CD8+ T cells and an increase in CD4+ T cells leads to enhanced levels of glomerular injury. A similar profile of T-cell accumulation was observed in P-selectin–deficient mice. Although CD4+ and CD8+ T cells bind P-selectin (48–50), it is unlikely that P-selectin is directly responsible for the recruitment of these cells to the interstitium because there is no endothelial P-selectin expressed in the kidney. We suggest that the differential T-cell recruitment in P-selectin–null mice compared with wild-type mice may be the result of the increased macrophage accumulation in the interstitium of the P-selectin–deficient mice and the subsequent change in cytokine profiles that affect T-cell recruitment. Another mechanism for the exacerbated disease in P-selectin–null mice may be the observed increase in glomerular C3 deposition in P-selectin–deficient mice in the early phase (day 7) of the disease (but not after 14 days), because proteinuria in this model was complement-dependent. The transmembrane and soluble form of P-selectin contains complement repeats that have homology with regulators of complement activation such as decay-accelerating factor and CR1 (46),suggesting that P-selectin may by important in preventing assembly or promoting disassembly of complement components, although this has not been experimentally demonstrated.
In summary, we have shown an important downregulatory role for a leukocyte adhesion receptor, namely P-selectin, in a model of proliferative GN that mimics aspects of the human disease. We show that endothelial cell P-selectin is largely responsible for the downregulatory effect. Although we have not proven the mechanism for the protective effect of endothelial P-selectin, it may be related to the generation of soluble P-selectin, the decrease in early interstitial accumulation of CD8+ T cells, and/or increased glomerular deposition of complement.
Acknowledgments
We would like to thank George Stavrakis (Department of Pathology, Brigham and Women's Hospital) for excellent assistance in immunohistochemistry. We would like to thank Daqing Hartwell (Center for Blood Research, Harvard Medical School) for performing assays to detect soluble P-selectin, Michael Berndt for providing anti–P-selectin polyclonal antibody, Barry Wolitsky for providing anti–P-selectin monoclonal antibody, Ah-Kau Ng for anti–mouse platelet IIb/IIIa antibody, and Daniel Jones (Department of Pathology, Brigham and Women's Hospital) for helping with the photo-illustration. This research was supported by National Institutes of Health research grants DK-51643 and NS-33296, and by an Erwin Schroedinger scholarship from the Austrian Science Foundation (to A. Rosenkranz).
References
- 1.Couser WG. Pathogenesis of glomerular damage in glomerulonephritis. Nephrol Dial Transplant. 1998;13(Suppl.1):10–15. doi: 10.1093/ndt/13.suppl_1.10. [DOI] [PubMed] [Google Scholar]
- 2.Hricik DE, Chung-Park M, Sedor JR. Glomerulonephritis. N Engl J Med. 1998;339:888–899. doi: 10.1056/NEJM199809243391306. [DOI] [PubMed] [Google Scholar]
- 3.Wilson, C.B. 1996. Renal response to immunologic glomerular injury. In The Kidney. (5th edition).B.M. Brenner, ed. W.B. Saunders, Philadelphia, PA. 1253–1391
- 4.Lefkowith JB. Leukocyte migration in immune complex glomerulonephritis: role of adhesion receptors. Kidney Int. 1997;51:1469–1475. doi: 10.1038/ki.1997.201. [DOI] [PubMed] [Google Scholar]
- 5.Shanley TP, Warner RL, Ward PA. The role of cytokines and adhesion molecules in the development of inflammatory injury. Mol Med Today. 1995;1:40–45. doi: 10.1016/1357-4310(95)80019-0. [DOI] [PubMed] [Google Scholar]
- 6.Bienvenu K, Hernandez L, Granger DN. Leukocyte adhesion and emigration in inflammation. Ann NY Acad Sci. 1992;664:388–399. doi: 10.1111/j.1749-6632.1992.tb39777.x. [DOI] [PubMed] [Google Scholar]
- 7.Granger DN, Kubes P. The microcirculation and inflammation: modulation of leukocyte-endothelial cell adhesion. J Leukoc Biol. 1994;55:662–675. [PubMed] [Google Scholar]
- 8.Furie B, Furie BC. The molecular basis of platelet and endothelial cell interaction with neutrophils and monocytes: role of P-selectin and the P-selectin ligand, PSGL-1. Thromb Haemost. 1995;74:224–227. [PubMed] [Google Scholar]
- 9.McEver RP, Moore KL, Cummings RD. Leukocyte trafficking mediated by selectin-carbohydrate interactions. J Biol Chem. 1995;270:11025–11028. doi: 10.1074/jbc.270.19.11025. [DOI] [PubMed] [Google Scholar]
- 10.Mayadas TN. Gene knockout on P-selectin: its biology and function. Trends Cardiovasc Med. 1995;5:149–157. doi: 10.1016/1050-1738(95)00057-G. [DOI] [PubMed] [Google Scholar]
- 11.Frenette PS, Wagner DD. Insights into selectin function from knockout mice. Thromb Haemost. 1997;78:60–64. [PubMed] [Google Scholar]
- 12.Bullard, D.C., and Beaudet, A.L. 1997. Analysis of selectin deficient mice. In The selectins: initiators of leukocyte endothelial adhesion. D. Vestweber, editor. Hardwood Academic Publishers. Muenster, Germany. 133–142.
- 13.Dunlop LC, et al. Characterization of GMP-140 (P-selectin) as a circulating plasma protein. J Exp Med. 1992;175:1147–1150. doi: 10.1084/jem.175.4.1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ishiwata N, et al. Alternatively spliced isoform of P-selectin is present in vivo as a soluble molecule. J Biol Chem. 1994;269:23708–23715. [PubMed] [Google Scholar]
- 15.Katayama M, et al. Soluble P-selectin is present in normal circulation and its plasma level is elevated in patients with thrombotic thrombocytopenic purpura and haemolytic uraemic syndrome. Br J Haematol. 1993;84:702–710. doi: 10.1111/j.1365-2141.1993.tb03149.x. [DOI] [PubMed] [Google Scholar]
- 16.Segawa C, et al. In situ expression and soluble form of P-selectin in human glomerulonephritis. Kidney Int. 1997;52:1054–1063. doi: 10.1038/ki.1997.428. [DOI] [PubMed] [Google Scholar]
- 17.Chong BH, et al. Plasma P-selectin is increased in thrombotic consumptive platelet disorders. Blood. 1994;83:1535–1541. [PubMed] [Google Scholar]
- 18.Halim A, et al. Plasma P-selectin (GMP-140) and glycocalicin are elevated in preeclampsia and eclampsia: their significances. Am J Obstet Gynecol. 1996;174:272–277. doi: 10.1016/s0002-9378(96)70407-6. [DOI] [PubMed] [Google Scholar]
- 19.Jilma B, et al. Elevated circulating P-selectin in insulin dependent diabetes mellitus. Thromb Haemost. 1996;76:328–332. [PubMed] [Google Scholar]
- 20.Fijnheer R, et al. The origin of P-selectin as a circulating plasma protein. Thromb Haemost. 1997;77:1081–1085. [PubMed] [Google Scholar]
- 21.Gamble JR, Skinner MP, Berndt MC, Vadas MA. Prevention of activated neutrophil adhesion to endothelium by soluble adhesion protein GMP140. Science. 1990;249:414–417. doi: 10.1126/science.1696029. [DOI] [PubMed] [Google Scholar]
- 22.Wong CS, et al. Adhesion protein GMP140 inhibits superoxide anion release by human neutrophils. Proc Natl Acad Sci USA. 1991;88:2397–2401. doi: 10.1073/pnas.88.6.2397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tipping PG, Huang XR, Berndt MC, Holdsworth SR. A role for P-selectin in complement-independent neutrophil-mediated glomerular injury. Kidney Int. 1994;46:79–88. doi: 10.1038/ki.1994.246. [DOI] [PubMed] [Google Scholar]
- 24.Mayadas TN, et al. Acute passive anti-glomerular basement membrane nephritis in P-selectin-deficient mice. Kidney Int. 1996;49:1342–1349. doi: 10.1038/ki.1996.190. [DOI] [PubMed] [Google Scholar]
- 25.Zachem CR, et al. A role for P-selectin in neutrophil and platelet infiltration in immune complex glomerulonephritis. J Am Soc Neprol. 1997;8:1838–1844. doi: 10.1681/ASN.V8121838. [DOI] [PubMed] [Google Scholar]
- 26.Mayadas TN, Johnson RC, Rayburn H, Hynes RO, Wagner DD. Leukocyte rolling and extravasation are severely compromised in P-selectin-deficient mice. Cell. 1993;74:541–554. doi: 10.1016/0092-8674(93)80055-j. [DOI] [PubMed] [Google Scholar]
- 27.Coxon A, et al. A novel role for the b2 integrin CD11b/CD18 in neutrophil apoptosis: a homeostatic mechanism in inflammation. Immunity. 1996;5:653–666. doi: 10.1016/s1074-7613(00)80278-2. [DOI] [PubMed] [Google Scholar]
- 28.Schrijver G, et al. Anti-GBM nephritis in the mouse: Role of granulocytes in the heterologous phase. Kidney Int. 1990;38:86–95. doi: 10.1038/ki.1990.171. [DOI] [PubMed] [Google Scholar]
- 29.Kitching AR, et al. Plasminogen and plasminogen activators protect against renal injury in crescentic glomerulonephritis. J Exp Med. 1997;185:963–968. doi: 10.1084/jem.185.5.963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Quigg RJ, et al. Studies with antibodies to cultured rat glomerular epithelial cells: subepithelial immune deposit formation after in vivo injection. Am J Pathol. 1989;134:1125–1133. [PMC free article] [PubMed] [Google Scholar]
- 31.Tipping PG, Huang XR, Qi M, Van GY, Tang WW. Crescentic glomerulonephritis in CD4- and CD8-deficient mice. Requirement for CD4 but not CD8 cells. Am J Pathol. 1998;152:1541–1548. [PMC free article] [PubMed] [Google Scholar]
- 32.Haas M. IgG subclass deposits in glomeruli of lupus and nonlupus membranous nephropathy. Am J Kidney Dis. 1994;23:358–364. doi: 10.1016/s0272-6386(12)80997-8. [DOI] [PubMed] [Google Scholar]
- 33.Huang X-R, Tipping PG, Shuo L, Holdsworth SR. Th1 responsiveness to nephritogenic antigens determines susceptibility to crescentic glomerulonephritis in mice. Kidney Int. 1997;51:94–103. doi: 10.1038/ki.1997.12. [DOI] [PubMed] [Google Scholar]
- 34.Doi T, et al. Glomerular lesions in nonobese diabetic mouse: before and after the onset of hyperglycemia. Lab Invest. 1990;63:204–212. [PubMed] [Google Scholar]
- 35.Hartwell DW, et al. Role of P-selectin cytoplasmic domain in granular targeting in vivo and in early inflammatory responses. J Cell Biol. 1998;143:1129–1141. doi: 10.1083/jcb.143.4.1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.D’Amico G. Role of interstitial infiltration of leukocytes in glomerular disease. Nephrol Dial Transplant. 1988;3:596–600. doi: 10.1093/oxfordjournals.ndt.a091711. [DOI] [PubMed] [Google Scholar]
- 37.Lan HY, Paterson DJ, Atkins RC. Initiation and evolution of interstitial leukocytic infiltration in experimental glomerulonephritis. Kidney Int. 1991;40:425–433. doi: 10.1038/ki.1991.229. [DOI] [PubMed] [Google Scholar]
- 38.Johnson RC, et al. Blood cell dynamics in P-selectin-deficient mice. Blood. 1995;86:1106–1114. [PubMed] [Google Scholar]
- 39.Shi J, Kokubo Y, Wake K. Expression of P-selectin on hepatic endothelia and platelets promoting neutrophil removal by liver macrophages. Blood. 1998;92:520–528. [PubMed] [Google Scholar]
- 40.Frenette PS, et al. Platelet-endothelial interactions in inflamed mesenteric venules. Blood. 1998;91:1318–1324. [PubMed] [Google Scholar]
- 41.McEver RP, Beckstead JH, Moore KL, Marshall-Carlson L, Bainton DF. GMP-140, a platelet alpha-granule membrane protein, is also synthesized by vascular endothelial cells and is localized in Weibel-Palade bodies. J Clin Invest. 1989;84:92–99. doi: 10.1172/JCI114175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Khew-Goodall Y, et al. Chronic expression of P-selectin on endothelial cells stimulated by the T-cell cytokine, interleukin-3. Blood. 1996;87:1432–1438. [PubMed] [Google Scholar]
- 43.Yao L, Pan J, Setiadi H, Patel KD, McEver RP. Interleukin 4 or oncostatin M induces a prolonged increase in P-selectin mRNA and protein in human endothelial cells. J Exp Med. 1996;184:81–92. doi: 10.1084/jem.184.1.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tipping PG, Huang XR, Berndt MC, Holdsworth SR. P-selectin directs T lymphocyte mediated injury in delayed type hypersensitivity responses: studies in glomerulonephritis and cutaneous delayed type of hypersensitivity. Eur J Immunol. 1996;26:454–460. doi: 10.1002/eji.1830260228. [DOI] [PubMed] [Google Scholar]
- 45.Heaney ML, Golde DW. Soluble receptors in human disease. J Leukoc Biol. 1998;64:135–146. doi: 10.1002/jlb.64.2.135. [DOI] [PubMed] [Google Scholar]
- 46.Johnston GI, Cook RG, McEver RP. Cloning of GMP-140, a granule membrane protein of platelets and endothelium: sequence similarity to proteins involved in cell adhesion and inflammation. Cell. 1989;56:1033–1044. doi: 10.1016/0092-8674(89)90636-3. [DOI] [PubMed] [Google Scholar]
- 47.Sanders WE, Wilson RW, Ballantyne CM, Beaudet AL. Molecular cloning and analysis of in vivo expression of murine P-selectin. Blood. 1992;80:795–800. [PubMed] [Google Scholar]
- 48.Moore KL, Thompson LF. P-selectin (CD62) binds to subpopulations of human memory T lymphocytes and natural killer cells. Biochem Biophys Res Commun. 1992;186:173–177. doi: 10.1016/s0006-291x(05)80790-9. [DOI] [PubMed] [Google Scholar]
- 49.Damle NK, Klussman K, Dietsch MT, Mohagheghpour N, Aruffo A. GMP-140 (P-selectin/CD62) binds to chronically stimulated but not resting CD4+ T lymphocytes and regulates their production of proinflammatory cytokines. Eur J Immunol. 1992;22:1789–1793. doi: 10.1002/eji.1830220718. [DOI] [PubMed] [Google Scholar]
- 50.Luscinskas FW, Ding H, Lichtman AH. P-selectin and vascular cell adhesion molecule 1 mediate rolling and arrest, respectively, of CD4+ T lymphocytes on tumor necrosis factor α-activated vascular endothelium under flow. J Exp Med. 1995;181:1179–1186. doi: 10.1084/jem.181.3.1179. [DOI] [PMC free article] [PubMed] [Google Scholar]