

Abstract
Thrombin and angiotensin II (angII) have trophic properties as mediators of vascular remodeling. Focal adhesions and actin cytoskeleton are involved in cell growth, shape, and movement and may be important in vascular remodeling. To characterize mechanisms by which thrombin and angII modulate vessel structure, we studied the effects of these G protein–coupled receptor ligands on focal adhesions in vascular smooth muscle cells (VSMCs). Both thrombin and angII stimulated bundling of actin filaments to form stress fibers, assembly of focal adhesions, and protein tyrosine phosphorylation at focal adhesions, such as p130Cas, paxillin, and tensin. To test whether c-Src plays a critical role in focal adhesion rearrangement, we analyzed cells with altered c-Src activity by retroviral transduction of wild-type (WT) and kinase-inactive (KI) c-Src into rat VSMCs, and by use of VSMCs from WT (src+/+) and Src-deficient (src–/–) mice. Tyrosine phosphorylation of Cas, paxillin, and tensin were markedly decreased in VSMCs expressing KI-Src and in src–/– VSMCs. Expression of KI-Src did not inhibit stress fiber formation by thrombin. Surprisingly, actin bundling was markedly decreased in VSMCs from src–/– mice both basally and after thrombin stimulation, compared with src+/+ mice. We also studied the effect of KI-Src and WT-Src on VSMC spreading. Expression of KI-Src reduced the rate of VSMC spreading on collagen, whereas WT-Src enhanced cell spreading. In conclusion, c-Src plays a critical role in agonist-stimulated cytoskeletal reorganization and signal transduction at focal adhesions in VSMCs. c-Src kinase activity is required for the cytoskeletal turnover that occurs in cell spreading, whereas c-Src appears to regulate actin bundling via a kinase-independent mechanism.
Introduction
Angiotensin II (angII) is one of the key regulators of blood pressure and renal blood flow. Thrombin is a key regulator of the coagulation cascade and plays pivotal roles in thrombosis formation at atherosclerotic lesions or after balloon angioplasty. Both angII and thrombin activate signaling pathways that induce cell proliferation, hypertrophy, and migration of vascular smooth muscle cells (VSMCs) (1–3).
Based on their mitogenic and chemotactic activities, thrombin and angII have been proposed to play pathogenic roles in hypertension, atherosclerosis, and restenosis after balloon angioplasty (4–7). Important components of these diseases are alterations in vessel structure termed vascular remodeling (8, 9). Although the precise mechanisms involved in tissue remodeling are not fully understood, remodeling requires rearrangement of cell alignment and changes in cell shape, as well as cell proliferation and even apoptosis. These cellular phenomena depend on cell–matrix interactions and cell–cell interactions (10–12).
We have previously shown (13) that angII activates c-Src kinase in VSMCs, but the function of c-Src activation in the effects of angII in VSMCs remains to be defined. Recently, we found that c-Src is required for ERK activation by angII in VSMCs by use of retroviral vectors to introduce wild-type (WT) and kinase-inactive (KI) c-Src into VSMCs (14). Because, c-Src is also activated upon integrin-mediated cell adhesion and translocates to focal adhesions (15), it may be important in signaling at focal adhesions. Numerous proteins are tyrosine-phosphorylated upon integrin-mediated cell adhesion (16). Among them, p130Cas (Crk-associated substrate, Cas) and paxillin are thought to serve as adapter molecules at focal adhesions (17, 18). Tensin has actin-binding domains and an SH2 domain and is thought to be involved in actin assembly/disassembly (19). The tyrosine kinases responsible for phosphorylating these proteins are undefined because many tyrosine kinases including focal adhesion kinase (FAK), other Src family kinases, and Csk are recruited to focal adhesions upon integrin activation (20, 21). Thrombin and angII, which are G protein–coupled receptor ligands, also increase protein tyrosine phosphorylation in VSMCs (14, 22). However, the identities of these tyrosine-phosphorylated proteins and their responsive kinases are largely unknown. Turner et al. (23) showed that paxillin is tyrosine-phosphorylated in response to angII in VSMCs. Given that both thrombin and angII activate c-Src, it is likely that thrombin and angII stimulate tyrosine phosphorylation of proteins at focal adhesions and that c-Src mediates the phosphorylation.
In this study, we tested the hypotheses that angII and thrombin stimulate actin cytoskeletal and focal adhesion reorganization through activation of c-Src in VSMCs. We show here that thrombin and angII cause stress fiber formation and focal adhesion assembly and increase tyrosine phosphorylation of Cas, paxillin, and tensin as a result of c-Src activation. In addition, stress fiber formation stimulated by thrombin is impaired in VSMCs isolated from src–/– mice, indicating an essential role for c-Src in VSMC actin rearrangement.
Methods
Reagents.
Cell culture media, protein A–agarose, and protein G–agarose were purchased from GIBCO BRL (Gaithersburg, Maryland, USA). Monoclonal anti-phosphotyrosine antibody (PY20) and polyclonal antibody against Cas (for immunoprecipitation and Western blot analysis) were purchased from Santa Cruz Biotechnology Inc. (Santa Cruz, California, USA). Monoclonal antibodies against tensin and Cas (for Western blot analysis) were purchased from Transduction Laboratories (Lexington, Kentucky, USA). Monoclonal antibody against Src (avian-specific) and polyclonal antibody against Cas (for immunofluorescence and immunoprecipitation) were purchased from Upstate Biotechnology Inc. (Lake Placid, New York, USA). Monoclonal antibody against paxillin was purchased from Zymed Laboratories Inc. (South San Francisco, California, USA). Monoclonal antibodies against vinculin (V-4505) and FITC-phalloidin were purchased from Sigma Chemical Co. (St. Louis, Missouri, USA).
Cell culture.
Rat aortic VSMCs were isolated from the aorta of 200- to 250-g male Sprague-Dawley rats and maintained in DMEM supplemented with 10% calf serum as described (14). Passage-6-to-10 VSMCs at 70%–80% confluence were growth arrested by incubation in DMEM without serum for 24 h before use. Mouse aortic VSMCs were isolated from the aorta of 4- to 6-week-old C57BL (WT, src+/+) and src–/– mice, which are homozygous for a disruption in the c-src gene (24), as described previously (14).
Retrovirus and adenovirus preparation.
The methods for construction of recombinant c-Src retrovirus and VSMC transduction were described previously (14). In brief, cDNA for WT- and KI-Src were cloned into the retroviral vector LXSN to construct the recombinant WT and kinase inactive c-Src retroviral vector (LWTSSN or LKISSN, respectively). Virus packaging was performed according to Miller and Rosman (25) using the PE501 cell line. Virus harvests from the PE/LWTSSN, PE/LKISSN, and PE/LXSN cells were used to infect VSMCs. VSMCs were plated at subconfluence and incubated with the virus harvest for 24 h to infect the cells. VSMCs infected with retrovirus were selected with DMEM/10% calf serum containing 0.6 mg/ml G418.
The methods for construction and preparation of recombinant c-Src adenovirus have been described in detail (M. Okuda et al., manuscript in preparation). In brief, cDNA for KI-Src was cloned into the expression vector pAd1.RSV according to the general protocol of Graham and Prevec (26). This vector, derived from a parent vector pXCJL.1, contains 16% of the adenoviral genome cloned into pBR322. The critical E1A region has been deleted and replaced by the Rous sarcoma virus (RSV) long-terminal repeat, multicloning site, and the bovine growth hormone polyadenylation sequence. KI-Src–containing pAd1.RSV was cotransfected into 293 cells with pJM17, a second plasmid containing the majority of the adenoviral genome, using LipofectAMINE (GIBCO BRL). After homologous recombination in vivo, plaques resulting from viral cytopathic effects were selected and expanded in 293 cells; these cells have been transformed with adenoviral E1A and therefore provide this viral transcription factor in trans. The KI-Src adenovirus (AdKI-Src) and a control adenovirus (AdLacZ) were propagated in 293 cells as described (27). The virus preparation was purified and concentrated from cell lysates by ultracentrifugation over a CsCl gradient, followed by dialysis. Virus titer was determined by OD at 260 nm and by plaque-formation assay using 293 cells, and expressed as plaque formation unit (pfu). VSMCs were infected with 2,000 moi of AdLacZ or AdKI-Src in DMEM. Virus was removed, and cells were serum starved for 24 h before experiments.
Experiments were performed in at least two different cell lines for each virally expressed gene. VSMCs were isolated from at least two mice for each genotype. Data presented in figures are representative of three to five experiments.
Immunofluorescence.
Cells were plated on Lab-Tek Chamber Slide (Nalge Nunc International, Naperville, Illinois, USA), serum starved for 24 h, and stimulated with angII (100 nM) or thrombin (3 U/ml). Cells were washed with ice-cold PBS, fixed with 4% paraformaldehyde for 20 min at room temperature, permeabilized with 0.5% Triton X-100/PBS for 10 min, and washed with PBS, 50 mM NH4Cl, and PBS serially. Then the cells were blocked with 1% BSA/PBS and incubated with primary antibodies in 0.1% BSA/PBS followed by incubation with biotin- and FITC-labeled secondary antibodies. Actin filaments were viewed with 0.2 μM FITC-labeled phalloidin.
Immunoprecipitation and immunoblot analysis.
The methods for immunoprecipitation and immunoblot were described previously (14). In brief, growth-arrested VSMCs were stimulated with angII as indicated in each experiment. Cells were lysed in NP-40 buffer (1% NP40, 25 mmol/l Tris [pH 7.5], 50 mmol/l NaF, 10 mmol/l sodium pyrophosphate, 137 mmol/l NaCl, 10% glycerol, 1 mmol/l sodium orthovanadate, 1 mmol/l phenylmethylsulfonyl fluoride, and 10 μg/ml leupeptin), scraped off the dish, and centrifuged. Lysates containing equal amounts of soluble proteins were precleared and incubated with antibodies overnight at 4°C. Antibody complexes were collected by incubation with protein A–agarose for polyclonal antibody or protein G–agarose for monoclonal antibody. Precipitates were washed in lysis buffer and then resuspended in SDS-PAGE sample buffer. Samples were separated by SDS-PAGE and transferred to nitrocellulose membranes. After incubation in blocking solution, membranes were incubated with primary antibodies. After washing in PBS, the blots were incubated with appropriate secondary antibodies. The membranes were washed and proteins were detected by the ECL system (Amersham Life Sciences Inc., Arlington Heights, Illinois, USA).
Cell spreading.
Cell spreading was measured as described by Richardson et al. (28). Briefly, cells were trypsinized, collected, and washed in DMEM containing 0.1 mg/ml soybean trypsin inhibitor. Cells were then resuspended in DMEM containing 0.1% FCS, and 2 × 105 cells were plated on a 35-mm dish coated with type I collagen (50 μg/ml). For each experiment, five random fields were photographed, and a total of at least 500 cells was counted for each cell line at each time point. Unspread cells were defined as round phase-bright cells; spread cells were defined as those that had extended processes, lacked a rounded morphology, and were not phase-bright. The extent of spreading was assessed blindly by three observers.
Results
Thrombin and angiotensin II induce stress fiber formation and rearrangement of focal adhesions in VSMCs.
To study the effects of thrombin and angII on VSMC cytoskeleton, cells were serum starved for 24 hours and stimulated with 3 U/ml thrombin or 100 nM angII. Cells were fixed and stained with phalloidin to reveal F-actin and with anti-vinculin to identify focal adhesions. Serum-starved cells exhibited thin F-actin fibers (Fig. 1a). There were few focal adhesion patches that were small in area (Fig. 1b), and there was little protein tyrosine phosphorylation (Fig. 1c). Treatment with thrombin for 20 minutes stimulated F-actin bundling (stress fibers) (Fig. 1d). There was also an increase in vinculin staining, indicating that focal adhesions had assembled (Fig. 1e). Thrombin also increased phosphotyrosine staining, especially at focal adhesions, in 5 minutes (Fig. 1f). AngII had similar effects on actin bundling, focal adhesions, and protein tyrosine phosphorylation in VSMCs (Fig. 1, g, h, and i).
Figure 1.

Thrombin and angII increase stress fibers and focal adhesion assembly in VSMCs. Serum-starved VSMCs (a–c) were stimulated with 3 U/ml thrombin (d–f), or 100 nM angII (g–i). Immunofluorescence was performed with FITC-phalloidin (0.1 μg/ml; a, d, and g) to stain actin, anti-vinculin (1:100; b, e, and h), or anti-phosphotyrosine (10 μg/ml; c, f, and i) as described in Methods. Cells were stimulated for 20 min before fixation for actin and vinculin, and for 5 min before fixation for phosphotyrosine.VSMCs, vascular smooth muscle cells.
Thrombin and angiotensin II increase tyrosine phosphorylation of Cas, tensin, and paxillin.
Because both thrombin and angII increased protein tyrosine phosphorylation mainly at focal adhesions in VSMCs, we examined their effects on tyrosine phosphorylation of Cas, tensin, and paxillin, which are thought to be localized to focal adhesions. Both thrombin and angII stimulated tyrosine phosphorylation of all three proteins (Cas 4.2 ± 1.9–fold, paxillin 4.0 ± 1.1–fold, and tensin 4.5 ± 1.6–fold by thrombin; Cas 5.0 ± 1.6–fold, paxillin 4.1 ± 0.8–fold, and tensin 4.3 ± 1.7–fold by angII). The time courses of tyrosine phosphorylation by thrombin and angII were similar, with peaks at the following times: Cas at 1–2 minutes, paxillin at 5 minutes, and tensin at 5–10 minutes (Fig. 2).
Figure 2.

Time courses for thrombin- and angII-induced tyrosine phosphorylation of Cas, paxillin, and tensin. Cells were stimulated with 3 U/ml thrombin or 100 nM angII for the indicated times. Cell lysates were immunoprecipitated (IP) with anti-Cas, anti-paxillin, or anti-tensin, and immunoblotted (IB) with anti-phosphotyrosine (1 μg/ml PY20).
p130Cas is translocated to focal adhesions at the cell periphery in response to thrombin.
To study the mechanisms by which thrombin and angII stimulate focal adhesion assembly, we examined agonist-mediated changes in localization of Cas. Inserum-starved VSMCs, Cas was homogeneously distributed in cytoplasm (Fig. 3a). Vinculin staining showed few focal adhesions (Fig. 3c), and colocalization of Cas and vinculin was minimal (Fig. 3b). After stimulation with thrombin and angII, the subcellular localization of Cas changed with increased presence at the cell periphery (Fig. 3, d and g, respectively). In fact, 56 ± 17% of thrombin stimulated cells showed Cas localization at the cell periphery, whereas only 7 ± 4% of unstimulated cells showed Cas at the periphery. Cas present at the cell periphery colocalized with vinculin especially at the cell protrusion sites (Fig. 3, e and h). These findings indicate that Cas translocates from the cytosol to cell periphery/focal adhesion sites in response to thrombin and angII.
Figure 3.

Translocation of Cas by stimulation of thrombin and angII in VSMCs. Serum-starved VSMCs (a–c) were stimulated with 3 U/ml thrombin (d–f) or 100 nM angII (g–i) for 20 min. Immunofluorescence was performed with anti-Cas (5 μg/ml; a, d, and g) or anti-vinculin (c, f, and i) as described in Methods. Cas staining and vinculin staining were merged digitally (b, e, and h).
Effects of overexpression of WT- and KI-Src on tyrosine phosphorylation of focal adhesion proteins.
c-Src is localized at focal adhesions and has been proposed to play an important role in cell adhesion and tyrosine phosphorylation of focal adhesion proteins. Because c-Src is activated by angII and thrombin (13, 29), it is possible that c-Src mediates tyrosine phosphorylation of Cas, paxillin, and tensin by angII and thrombin. Therefore, we overexpressed WT-Src and KI-Src in VSMCs using a retroviral vector and examined the effect on protein tyrosine phosphorylation induced by angII and thrombin. The expression levels of WT- and KI-Src were equal and abundant compared with those of endogenous c-Src (14). Cells infected with retroviral vector alone (LXSN) showed little tyrosine phosphorylation 16 hours after being seeded on collagen-coated plates (Fig. 4a). Cells expressing KI-Src showed even less phosphotyrosine (Fig. 4b). Expression of WT-Src in VSMCs markedly increased the phosphotyrosine staining at focal adhesions as well as in the cytoplasm (Fig. 4c). Western blot analysis revealed that tyrosine phosphorylation of 66-kDa, 130-kDa, and 220-kDa proteins was increased in the cells overexpressing WT-Src, whereas tyrosine phosphorylation of these proteins was decreased in VSMCs expressing KI-Src (Fig. 4d).
Figure 4.

Effects of wild-type (WT) and kinase-inactive (KI) c-Src on protein tyrosine phosphorylation in VSMCs. VSMCs were infected with retroviral vector alone (a), retrovirus encoding KI c-Src (b), or WT c-Src (c). Cells were fixed and stained with anti-phosphotyrosine 16 h after seeding on type I collagen. (d) Serum-starved cells (C) were stimulated with 3 U/ml thrombin (Th) for 5 min, and then lysates from cells infected with vector alone and cells expressing KI-Src and WT-Src were immunoblotted with anti-phosphotyrosine.
We have identified these proteins as paxillin (66 kDa), Cas (130 kDa), and tensin (220 kDa). Expression of KI-Src almost completely abrogated thrombin- and angII-induced tyrosine phosphorylation of Cas, whereas WT-Src increased Cas tyrosine phosphorylation both basally and after stimulation by thrombin and angII (Fig. 5). Tyrosine phosphorylation of tensin by angII was potently inhibited in cells expressing KI-Src, whereas phosphorylation stimulated by thrombin was inhibited only partially. Tyrosine phosphorylation of paxillin in response to thrombin and angII was inhibited by 50% in cells expressing KI-Src. These findings suggest that there is another agonist-stimulated tyrosine kinase for paxillin besides c-Src.
Figure 5.

Tyrosine phosphorylation of Cas, paxillin, and tensin in VSMCs expressing kinase-inactive and wild-type c-Src. VSMCs were infected with retroviral vector alone, retrovirus encoding kinase-inactive c-Src (KI-Src), or wild-type c-Src (WT-Src) as described in Methods. Serum-starved cells (C) were stimulated with 3 U/ml thrombin (Th), or 100 nM angII (AII) for 2 min for Cas and 5 min for paxillin and tensin. Cell lysates were immunoprecipitated (IP) with anti-Cas, anti-paxillin, or anti-tensin, and immunoblotted (IB) with anti-phosphotyrosine (anti-PY). Membranes were stripped and reprobed with the antibodies used for immunoprecipitation. Immunoblot for tensin is not shown because reprobing for tensin after anti-phosphotyrosine immunoblotting was not successful.
To verify results obtained from retrovirally transduced cells, we also isolated aortic VSMCs from src+/+ and src–/– mice. In response to thrombin, tyrosine phosphorylation of p130Cas and paxillin was markedly decreased in VSMCs from src–/– mice compared with src+/+ mice (Fig. 6). These data show that c-Src is the predominant tyrosine kinase responsible for angII- and thrombin-stimulated tyrosine phosphorylation of the focal adhesion proteins Cas, paxillin, and tensin in VSMCs.
Figure 6.
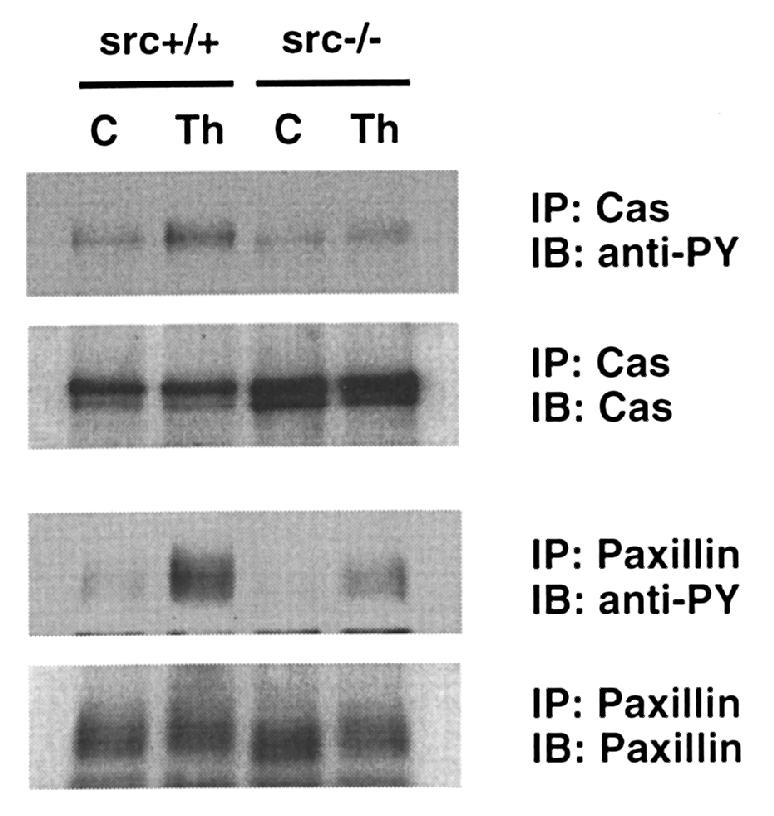
Tyrosine phosphorylation of Cas and paxillin in VSMCs from src–/– and src+/+ mice. Aortic VSMC from src–/– mice and src+/+ mice were isolated and cultured as described in Methods. Serum-starved cells (C) were stimulated with 3 U/ml thrombin (Th) for 2 min for Cas and 5 min for paxillin. Cell lysates were immunoprecipitated (IP) by antibodies with anti-Cas and anti-paxillin and immunoblotted (IB) by anti-phosphotyrosine (anti-PY) antibody. Membranes were stripped and reprobed with the antibodies used for immunoprecipitation.
Effects of overexpression of WT- and KI-Src on Cas association with Crk.
p130Cas acts as a scaffold for assembly of signal-transducing proteins, as shown by the findings that integrin-mediated cell adhesion stimulates binding of proteins to p130Cas such as Crk (30). To obtain insight into the role of Src kinase activity in the function of p130Cas, the association of Cas with Crk was measured by immunoprecipitation with anti-Crk antibody followed by immunoblot with anti-Cas antibody. Thrombin and angII stimulated association of Crk with Cas in VSMCs infected with vector alone (Fig. 7). In contrast, in the cells expressing KI-Src, thrombin- and angII-stimulated Crk association with Cas was markedly inhibited (Fig. 7), indicating that Crk interaction with Cas depends on Src kinase activity.
Figure 7.

The effect of kinase-inactive c-Src on thrombin- and angII-induced Cas–Crk interaction. VSMCs were infected with retroviral vector alone (Vector) or retrovirus encoding kinase-inactive c-Src (KI-Src). Serum-starved cells (C) were stimulated with 3 U/ml thrombin (Th) or 100 nM angII (AII) for 5 min. Cell lysates were immunoprecipitated (IP) with anti-Crk antibody or control mouse IgG and immunoblotted (IB) with anti-Cas antibody. Membranes were stripped and reprobed with anti-Crk.
Comparison of thrombin-stimulated actin rearrangement in src–/– VSMCs and KI-Src–expressing VSMCs.
To determine the role of c-Src in agonist-mediated rearrangement of cytoskeleton, we studied stress fiber formation stimulated by thrombin in VSMCs. In unstimulated cells infected with vector alone, there were few stress fibers (Fig. 8a). In response to thrombin, these cells showed a rapid increase in F-actin bundling to form stress fibers (Fig. 8b). In cells expressing KI-Src, thrombin stimulated formation of stress fibers that was comparable to that observed in cells infected with vector alone (Fig. 8d).
Figure 8.

The effect of retroviral and adenoviral expression of kinase-inactive (KI) c-Src on thrombin-induced stress fibers in VSMCs. Serum-starved VSMC were infected with retroviral vector alone (a and b) or with retrovirus encoding KI-Src (c and d). Cells were then stimulated with 3 U/ml thrombin for 20 min. Cells were fixed and stained with FITC-phalloidin for actin. There was no effect of KI-src on thrombin-mediated actin bundling (b vs. d). VSMCs were infected with AdLazZ (e and g) or AdKI-Src (f and h). After 24-h serum starvation, cells were stimulated with 3 U/ml thrombin for 20 min, fixed, and stained with FITC-phalloidin for actin (e and f). The same cells were also stained with anti–avian Src (g and h). Note that expression of KI-Src varies in individual cells (h), without change in actin staining (f). Again, KI-Src did not alter thrombin-mediated actin bundling.
KI c-Src was also introduced transiently into VSMCs using adenovirus. Thrombin-induced actin stress fibers in cells expressing adenovirally introduced KI-Src (Fig. 8f) were similar to those in cells infected with lacZ-containing adenovirus (Fig. 8e). Additional evidence to support the conclusion that Src kinase activity is not required for stress fiber formation is that among cells expressing different amounts of KI-Src (Fig. 8h), there were no significant differences in stress fibers (Fig. 8f) regardless of KI-Src expression level.
To confirm the results obtained by expression of KI-Src, we examined actin rearrangement in VSMCs from src–/– mice. In unstimulated, serum-starved src+/+ cells (Fig. 9a), there were a few stress fibers distributed throughout the cell. In response to thrombin, src+/+ cells exhibited a large increase in stress fibers similar to that observed in rat VSMCs (Fig. 9b). src–/– VSMCs spread poorly, and basal actin bundling was barely detectable under serum-starved conditions (Fig. 9c). In response to thrombin, src–/– VSMCs showed little increase in stress fibers (Fig. 9d).
Figure 9.

Stress fibers in src+/+ and src–/– VSMCs. VSMCs from src+/+ mice (a and b) and src–/– mice (c and d) were isolated and cultured as described in Methods. Serum-starved cells (a and c) were stimulated with 3 U/ml thrombin for 20 min and fixed and stained with FITC-phalloidin for actin (b and d).
Src–/– VSMCs and KI-Src–expressing VSMCs exhibit impaired spreading on collagen.
To gain further insight into the role of c-Src in VSMC function, we studied the ability of VSMCs to spread on collagen. During cell spreading, there is dynamic actin reorganization that occurs to an even greater extent than in response to hormonal agonists. VSMCs from src+/+ mice spread rapidly, resulting in ∼50% spreading 20 minutes after replating (Fig. 10, a and f). In contrast, src–/– VSMC spreading was significantly slower (Fig. 10, b and f). To confirm the results from src–/– VSMCs, we also studied retrovirally transduced cells. Rat VSMCs infected with vector alone showed rates of spreading that were comparable to those of VSMCs from src+/+ mice (compare Fig. 10, a and c with f and g). However, VSMCs expressing KI-Src (Fig. 10, d and g) exhibited a slower rate of spreading than control (Fig. 10, c and g). In contrast, overexpression of WT-Src accelerated the rate of cell spreading (Fig. 10, e and g). Thus, c-Src kinase activity is an important determinant of the rate of VSMC spreading on collagen. It is possible that c-Src may be involved in cell attachment. Because the mechanisms for cell attachment and spreading largely overlap, we think that separating the two processes is not essential. Therefore, we measured the percentage of cells that spread that reflects the effect of c-Src on both attachment and spreading.
Figure 10.

The role of Src in spreading of VSMCs on collagen-coated dishes. Photographs of VSMCs from src+/+ mice (a), src–/– mice (b), infected with retroviral vector alone (c), retrovirus encoding KI-Src (d), and WT-Src (e) 20 min after being seeded on type I collagen. (f) Time course of spreading of VSMC from src+/+ and src–/– mice. (g) Time course of spreading of VSMCs infected with retroviral vector alone, or retrovirus encoding KI-Src or WT-Src. Data were shown in mean ± SD. Statistics were performed using two-way repeated measures ANOVA. *P < 0.05 vs. vector alone (g); **P < 0.01 vs. src+/+ (f).
Discussion
The major finding of this study is that thrombin and angII stimulate bundling of actin filaments (stress fibers) and focal adhesion assembly in VSMCs that is mediated, in part, by c-Src. As discussed later here, changes in VSMC stress fibers are likely to be important in VSMC functions such as contraction, migration, and proliferation. Thus, understanding the mechanisms by which physiological stimuli promote cytoskeletal rearrangement in VSMCs may provide insight into pathological processes such as hypertension, atherosclerosis, and restenosis. Although focal adhesions have been recognized as dense plaques in VSMCs by electron microscopy, the effects of growth factors on cytoskeletal reorganization in VSMCs have not been well studied. The current paradigm for such cytoskeletal rearrangement is that focal adhesions are formed by activated myosin that bundles F-actin (31, 32). Myosin is activated by Rho kinase/ROCK/ROK, which are effectors of the small G protein Rho. These kinases regulate myosin indirectly by phosphorylating the p130 binding subunit of myosin phosphatase, thereby suppressing the phosphatase activity, and directly by phosphorylating myosin light chain (33, 34). In VSMCs, therefore, stress fiber formation and muscle contraction share common molecular mechanisms. Of great interest, a recent study by Uehata et al. (35) showed that a specific inhibitor of ROCK had a potent vasorelaxing effect in isolated vessels and lowered blood pressure when administered in vivo to hypertensive rats. Because this inhibitor blocks actin bundling in cultured cells, it appears possible that vasoconstrictors such as angII and thrombin work, in part, by activating ROCK and thereby stimulating actin bundling and focal adhesion assembly.
In this study, we focused on tyrosine phosphorylation of focal adhesion proteins and the role of c-Src in VSMC cytoskeletal reorganization. Both thrombin and angII stimulated tyrosine phosphorylation of Cas, paxillin, and tensin. Our data indicate that c-Src is the major tyrosine kinase activated by angII and thrombin that phosphorylates these proteins in VSMCs. Cas and paxillin are thought to be important molecules in cytoskeletal reorganization by serving as linker proteins. Tensin cross-links actin filaments, possesses a barbed-end capping activity, and therefore is thought to be involved in actin assembly/disassembly (19). Cas appears particularly important, as it binds to Src via its SH3 domain and to other signaling molecules such as Crk, Nck, FAK, and PTP-PEST via SH2-binding motifs. Cas is tyrosine-phosphorylated and localizes to focal adhesions upon integrin-mediated cell adhesion. This study shows that both thrombin and angII stimulate association of Cas with Crk. In v-Crk–transformed cells, total cellular tyrosine phosphorylation is markedly increased, and it is thought that Crk potentiates the kinase activity of Src (36). Thus, the association between Cas and Crk induced by thrombin and angII may regulate c-Src kinase activity. Recently, Klemke et al. (37) showed that Cas–Crk association serves as a molecular switch for induction of cell migration. Tyrosine phosphorylation of Cas and tensin by thrombin and angII almost completely depended on c-Src, indicating the importance of c-Src in regulating focal adhesions in VSMCs. In contrast, tyrosine phosphorylation of paxillin was only 50% dependent on c-Src, suggesting that paxillin may also be phosphorylated by other kinases, such as Fyn or Pyk2. FAK is unlikely to be the paxillin kinase in VSMCs because we could not detect tyrosine phosphorylation of FAK in response to thrombin or angII (data not shown).
One of the major findings of this study is that F-actin was poorly bundled in src–/– VSMCs both in the unstimulated state and after thrombin stimulation. In contrast, expression of KI-Src failed to inhibit actin filament bundling. These findings suggest that c-Src may be required for actin filament bundling by serving as a scaffold molecule via its SH2 and SH3 domains, rather than as a tyrosine kinase. In contrast, the kinase activity of c-Src is critical for cell spreading, in which actin turnover is very dynamic. Thus, we propose that c-Src kinase activity and the tyrosine phosphorylation of focal adhesion proteins by c-Src are involved in actin turnover (38). This hypothesis is supported by our findings that Cas association with Crk induced by thrombin and angII also depends on Src kinase activity. Recently, it has been shown that Cas plays an important role in cell migration, which also requires dynamic actin turnover, especially in cooperation with Crk (37, 39). Additional support is provided by Richardson et al. (28), who showed that cell spreading was inhibited by overexpression of FRNK (FAK-related nonkinase) and rescued by overexpression of WT c-Src, but not KI c-Src. However, Kaplan et al. (15) showed that the defect in cell spreading of src–/– fibroblasts could be restored by Src mutants that lack kinase activity. Thus, the role of c-Src in integrin and cytoskeletal reorganization remains controversial. It is possible that the compensation by other Src family kinases may explain the varying results. In fact, the phenomena exhibited in src–/– VSMCs and VSMCs expressing KI-Src became less obvious with serial passage, which correlated with compensatory expression of other Src family kinases (Ishida, T., et al., unpublished data). Introduction of several Src mutants, such as c-Src that lack kinase domain, SH2 domain, and SH3 domain, into src–/– VSMCs would be very informative to determine which domain is critical for the regulation of actin bundling, cell spreading, and cell migration.
Data from v-Src–transformed cells and Csk-deficient (csk–/–) cells have suggested that Src disrupts actin filaments, in contrast to the results of this study (20, 40). In fact, we did not observe defects in actin filaments in VSMCs overexpressing WT c-Src (Ishida, T., et al., unpublished data). There are, however, critical differences between c-Src–overexpressing cells and v-Src–transformed or csk–/– cells. First, in v-Src–transformed cells and cells derived from csk–/– mice, Src kinase activity is constitutively (and dramatically) increased without negative regulation. Second, the SH2 domain of v-Src can interact promiscuously with tyrosine residues of many proteins in these cells, whereas in WT c-Src, the SH2 domain of c-Src is intramolecularly bound to phosphorylated Tyr527 when it is inactive. In addition, v-Src is predominantly distributed in focal adhesions, whereas c-Src localizes in the cytoplasm when it is inactive (15, 41).
Our findings that Cas is translocated from the cytoplasm to focal adhesions after stimulation by thrombin and angII suggest a role for Cas in c-Src function, such as targeting Src to focal adhesions. Cas is localized in the cytoplasm in serum-starved, unstimulated VSMCs as well as in fibroblasts (42). As already discussed here, in unstimulated cells, c-Src is primarily cytosolic and translocates to focal adhesions upon stim ulation (15). In contrast, paxillin and tensin are already present in focal adhesions because of their localization sequences (18, 19). Thus, we propose that Cas is phosphorylated by c-Src in the cytoplasm immediately upon stimulation of VSMCs by angII and thrombin and is then recruited to focal adhesions with c-Src, which then phosphorylates paxillin and tensin.
Rearrangement of actin filaments and focal adhesions plays an essential role in cell growth, survival, locomotion, and shape. For example, cell-cycle progression depends on cell spreading and adhesion, whereas apoptosis is initiated after cells are detached (11, 12). In cardiovascular diseases, such as hypertension, atherosclerosis, and restenosis after angioplasty, vascular remodeling clearly requires rearrangement of VSMC cytoskeleton and focal adhesions (9, 43). Both thrombin and angII are thought to have important roles in remodeling by virtue of their effects on VSMC migration and growth (1, 3, 44). The receptors for both thrombin and angII are highly expressed in atherosclerotic plaque and in restenotic lesions after balloon arterial injury (5–7). In summary, rearrangement of VSMC focal adhesions and actin filaments in response to thrombin and angII may play an important role in vascular remodeling as well as in vascular tone.
Acknowledgments
We thank Byron Gallis for careful reading of this manuscript. This work was supported by a grant from National Institutes of Health (NIH) (HL-44721 to B.C. Berk). T. Ishida was supported by the American Heart Association Washington Affiliate. M. Ishida was supported by Japan Heart Foundation. J. Suero was supported by NIH grant HL-09372. B.C. Berk is an established Investigator of the American Heart Association.
References
- 1.McNamara CA, et al. Thrombin and vascular smooth muscle cell proliferation: implications for atherosclerosis and restenosis. Semin Thromb Hemost. 1996;22:139–144. doi: 10.1055/s-2007-999001. [DOI] [PubMed] [Google Scholar]
- 2.Griendling KK, Ushio F-M, Lassegue B, Alexander RW. Angiotensin II signaling in vascular smooth muscle. New concepts. Hypertension. 1997;29:366–373. doi: 10.1161/01.hyp.29.1.366. [DOI] [PubMed] [Google Scholar]
- 3.Graf K, Xi XP, Hsueh WA, Law RE. Troglitazone inhibits angiotensin II-induced DNA synthesis and migration in vascular smooth muscle cells. FEBS Lett. 1997;400:119–121. doi: 10.1016/s0014-5793(96)01371-3. [DOI] [PubMed] [Google Scholar]
- 4.Ross R. The pathogenesis of atherosclerosis: a perspective for the 1990s. Nature. 1993;362:801–809. doi: 10.1038/362801a0. [DOI] [PubMed] [Google Scholar]
- 5.Nelken NA, et al. Thrombin receptor expression in normal and atherosclerotic human arteries. J Clin Invest. 1992;90:1614–1621. doi: 10.1172/JCI116031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wilcox JN, et al. Characterization of thrombin receptor expression during vascular lesion formation. Circ Res. 1994;75:1029–1038. doi: 10.1161/01.res.75.6.1029. [DOI] [PubMed] [Google Scholar]
- 7.Viswanathan M, Stromberg C, Seltzer A, Saavedra JM. Balloon angioplasty enhances the expression of angiotensin II AT1 receptors in neointima of rat aorta. J Clin Invest. 1992;90:1707–1712. doi: 10.1172/JCI116043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Heagerty AM, Aalkjaer C, Bund SJ, Korsgaard N, Mulvany MJ. Small artery structure in hypertension. Dual processes of remodeling and growth. Hypertension. 1993;21:391–397. doi: 10.1161/01.hyp.21.4.391. [DOI] [PubMed] [Google Scholar]
- 9.Gibbons GH, Dzau VJ. The emerging concept of vascular remodeling. N Engl J Med. 1994;330:1431–1438. doi: 10.1056/NEJM199405193302008. [DOI] [PubMed] [Google Scholar]
- 10.Gumbiner BM. Cell adhesion: the molecular basis of tissue architecture and morphogenesis. Cell. 1996;84:345–357. doi: 10.1016/s0092-8674(00)81279-9. [DOI] [PubMed] [Google Scholar]
- 11.Assoian RK, Marcantonio EE. The extracellular matrix as a cell cycle control element in atherosclerosis and restenosis. J Clin Invest. 1996;98:2436–2439. doi: 10.1172/JCI119059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Meredith JE, Schwartz MA. Integrins, adhesion and apoptosis. Trends Cell Biol. 1997;7:146–150. doi: 10.1016/S0962-8924(97)01002-7. [DOI] [PubMed] [Google Scholar]
- 13.Ishida M, et al. Angiotensin II activates pp60c-src in vascular smooth muscle cells. Circ Res. 1995;77:1053–1059. doi: 10.1161/01.res.77.6.1053. [DOI] [PubMed] [Google Scholar]
- 14.Ishida M, Ishida T, Thomas S, Berk BC. Activation of extracellular signal-regulated kinases (ERK1/2) by angiotensin II is dependent on c-Src in vascular smooth muscle cells. Circ Res. 1998;82:7–12. doi: 10.1161/01.res.82.1.7. [DOI] [PubMed] [Google Scholar]
- 15.Kaplan KB, Swedlow JR, Morgan DO, Varmus HE. c-Src enhances the spreading of src–/– fibroblasts on fibronectin by a kinase-independent mechanism. Genes Dev. 1995;9:1505–1517. doi: 10.1101/gad.9.12.1505. [DOI] [PubMed] [Google Scholar]
- 16.Clark EA, Brugge JS. Integrins and signal transduction pathways: the road taken. Science. 1995;268:233–239. doi: 10.1126/science.7716514. [DOI] [PubMed] [Google Scholar]
- 17.Sakai R, et al. A novel signaling molecule, p130, forms stable complexes in vivo with v-Crk and v-Src in a tyrosine phosphorylation-dependent manner. EMBO J. 1994;13:3748–3756. doi: 10.1002/j.1460-2075.1994.tb06684.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Turner CE, Glenney JR, Jr, Burridge K. Paxillin: a new vinculin-binding protein present in focal adhesions. J Cell Biol. 1990;111:1059–1068. doi: 10.1083/jcb.111.3.1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lo SH, Weisberg E, Chen LB. Tensin: a potential link between the cytoskeleton and signal transduction. Bioessays. 1994;16:817–823. doi: 10.1002/bies.950161108. [DOI] [PubMed] [Google Scholar]
- 20.Thomas SM, Soriano P, Imamoto A. Specific and redundant roles of Src and Fyn in organizing the cytoskeleton. Nature. 1995;376:267–271. doi: 10.1038/376267a0. [DOI] [PubMed] [Google Scholar]
- 21.Parsons JT. Integrin-mediated signalling: regulation by protein tyrosine kinases and small GTP-binding proteins. Curr Opin Cell Biol. 1996;8:146–152. doi: 10.1016/s0955-0674(96)80059-7. [DOI] [PubMed] [Google Scholar]
- 22.Molloy CJ, et al. Thrombin receptor activation elicits rapid protein tyrosine phosphorylation and stimulation of the raf-1/MAP kinase pathway preceding delayed mitogenesis in cultured rat aortic smooth muscle cells: evidence for an obligate autocrine mechanism promoting cell proliferation induced by G-protein–coupled receptor agonist. J Clin Invest. 1996;97:1173–1183. doi: 10.1172/JCI118531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Turner CE, Pietras KM, Taylor DS, Molloy CJ. Angiotensin II stimulation of rapid paxillin tyrosine phosphorylation correlates with the formation of focal adhesions in rat aortic smooth muscle cells. J Cell Sci. 1995;108:333–342. doi: 10.1242/jcs.108.1.333. [DOI] [PubMed] [Google Scholar]
- 24.Soriano P, Montgomery C, Geske R, Bradley A. Targeted disruption of the c-src proto-oncogene leads to osteopetrosis in mice. Cell. 1991;64:693–702. doi: 10.1016/0092-8674(91)90499-o. [DOI] [PubMed] [Google Scholar]
- 25.Miller AD, Rosman GJ. Improved retroviral vectors for gene transfer and expression. Biotechniques. 1989;7:980–982. [PMC free article] [PubMed] [Google Scholar]
- 26.Graham, F.L., and Prevec, L. Manipulation of adenovirus vectors. 1991. In Methods in molecular biology. Gene transfer and expression protocols. Vol. 7. E.J. Murray, editor. Humana Press. Clifton, NJ. 109–128. [DOI] [PubMed]
- 27.Murry CE, Kay MA, Bartosek T, Hauschka SD, Schwartz SM. Muscle differentiation during repair of myocardial necrosis in rats via gene transfer with MyoD. J Clin Invest. 1996;98:2209–2217. doi: 10.1172/JCI119030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Richardson A, Malik RK, Hildebrand JD, Parsons JT. Inhibition of cell spreading by expression of the C-terminal domain of focal adhesion kinase (FAK) is rescued by coexpression of Src or catalytically inactive FAK: a role for paxillin tyrosine phosphorylation. Mol Cell Biol. 1997;17:6906–6914. doi: 10.1128/mcb.17.12.6906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen YH, Pouysségur J, Courtneidge SA, Van Obberghen-Schilling E. Activation of Src family kinase activity by the G protein–coupled thrombin receptor in growth-responsive fibroblasts. J Biol Chem. 1994;269:27372–27377. [PubMed] [Google Scholar]
- 30.Vuori K, Hirai H, Aizawa S, Ruoslahti E. Introduction of p130cas signaling complex formation upon integrin-mediated cell adhesion: a role for Src family kinases. Mol Cell Biol. 1996;16:2606–2613. doi: 10.1128/mcb.16.6.2606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Burridge K, Chrzanowska-Wodnicka M, Zhong CL. Focal adhesion assembly. Trends Cell Biol. 1997;7:342–347. doi: 10.1016/S0962-8924(97)01127-6. [DOI] [PubMed] [Google Scholar]
- 32.Machesky LM, Hall A. Role of actin polymerization and adhesion to extracellular matrix in Rac- and Rho-induced cytoskeletal reorganization. J Cell Biol. 1997;138:913–926. doi: 10.1083/jcb.138.4.913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kimura K, et al. Regulation of myosin phosphatase by Rho and Rho-associated kinase (Rho-kinase) Science. 1996;273:245–248. doi: 10.1126/science.273.5272.245. [DOI] [PubMed] [Google Scholar]
- 34.Amano M, et al. Phosphorylation and activation of myosin by Rho-associated kinase (Rho-kinase) J Biol Chem. 1996;271:20246–20249. doi: 10.1074/jbc.271.34.20246. [DOI] [PubMed] [Google Scholar]
- 35.Uehata M, et al. Calcium sensitization of smooth muscle mediated by a Rho-associated protein kinase in hypertension. Nature. 1997;389:990–994. doi: 10.1038/40187. [DOI] [PubMed] [Google Scholar]
- 36.Sabe H, Okada M, Nakagawa H, Hanafusa H. Activation of c-Src in cells bearing v-Crk and its suppression by Csk. Mol Cell Biol. 1992;12:4706–4713. doi: 10.1128/mcb.12.10.4706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Klemke RL, et al. CAS/Crk coupling serves as a “molecular switch” for induction of cell migration. J Cell Biol. 1998;140:961–972. doi: 10.1083/jcb.140.4.961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chang JH, Gill S, Settleman J, Parsons SJ. c-Src regulates the simultaneous rearrangement of actin cytoskeleton, p190RhoGAP, and p120RasGAP following epidermal growth factor stimulation. J Cell Biol. 1995;130:355–368. doi: 10.1083/jcb.130.2.355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cary LA, Han DC, Polte TR, Hanks SK, Guan JL. Identification of p130Cas as a mediator of focal adhesion kinase-promoted cell migration. J Cell Biol. 1998;140:211–221. doi: 10.1083/jcb.140.1.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Marchisio PC, D’Urso N, Comoglio PM, Giancotti FG, Tarone G. Vanadate-treated baby hamster kidney fibroblasts show cytoskeleton and adhesion patterns similar to their Rous sarcoma virus–transformed counterparts. J Cell Biochem. 1988;37:151–159. doi: 10.1002/jcb.240370203. [DOI] [PubMed] [Google Scholar]
- 41.Kaplan KB, Swedlow JR, Varmus HE, Morgan DO. Association of p60c-src with endosomal membranes in mammalian fibroblasts. J Cell Biol. 1992;118:321–333. doi: 10.1083/jcb.118.2.321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nakamoto T, et al. Requirements for localization of p130cas to focal adhesions. Mol Cell Biol. 1997;17:3884–3897. doi: 10.1128/mcb.17.7.3884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schwartz SM, deBlois D, O’Brien ER. The intima. Soil for atherosclerosis and restenosis. Circ Res. 1995;77:445–465. doi: 10.1161/01.res.77.3.445. [DOI] [PubMed] [Google Scholar]
- 44.Noda H-H, Sobel BE. Vascular smooth muscle cell migration mediated by thrombin and urokinase receptor. Am J Physiol. 1995;268:C1195–C1201. doi: 10.1152/ajpcell.1995.268.5.C1195. [DOI] [PubMed] [Google Scholar]