

Abstract
Marrow stromal cells (MSCs) provide important survival and drug resistance signals to chronic lymphocytic leukemia (CLL) cells, but current models to analyze CLL–MSC interactions are heterogeneous. Therefore, we tested different human and murine MSC lines and primary human MSCs for their ability to protect CLL cells from spontaneous and drug-induced apoptosis. Our results show that both human and murine MSCs are equally effective in protecting CLL cells from fludarabine-induced apoptosis. This protective effect was sustained over a wide range of CLL–MSC ratios (5:1 to 100:1), and the levels of protection were reproducible in 4 different laboratories. Human and murine MSCs also protected CLL cells from dexamethasone- and cyclophosphamide-induced apoptosis. This protection required cell–cell contact and was virtually absent when CLL cells were separated from the MSCs by micropore filters. Furthermore, MSCs maintained Mcl-1 and protected CLL cells from spontaneous and fludarabine-induced Mcl-1 and PARP cleavage. Collectively, these studies define common denominators for CLL cocultures with MSCs. They also provide a reliable, validated tool for future investigations into the mechanism of MSC–CLL cross talk and for drug testing in a more relevant fashion than the commonly used suspension cultures.
Introduction
With the establishment of more effective treatments for patients with chronic lymphocytic leukemia (CLL) over the past decade, complete remissions are no longer the exception.1 Despite these major improvements in CLL treatment, we still consider CLL an incurable disease, because patients generally relapse from minimal residual disease (MRD).2 There is growing evidence suggesting that CLL cells are protected from conventional drugs in tissue microenvironments, such as the bone marrow and secondary lymphoid organs, with facilitation of residual disease that is drug resistant and ultimately paving the way to clonal evolution and relapses. The complex cellular and molecular contexts in the tissues, collectively referred to as the CLL microenvironment, provide signals for the expansion of the CLL clone and for primary drug resistance. This is largely dependent on direct contact between the malignant B cells and stromal cells,3 and therefore has been designated as cell adhesion–mediated drug resistance.4 Disrupting cross talk between leukemia cells and their milieu is an attractive novel but yet incompletely tested strategy for treating CLL. Appropriately, there is growing interest in understanding the biology of CLL-stroma cross talk to find ways to eliminate residual CLL cells that are “hiding” in stromal niches within the marrow and the lymphatic tissues.
Importantly, once CLL cells are removed from the in vivo microenvironment and placed in suspension cultures without supportive stroma, they undergo spontaneous apoptosis, highlighting the importance of external signals from accessory cells.5 Previous studies have shown that CLL cell cocultures with different adherent cell types, collectively referred to as stromal cells, induce leukemia cell survival, migration, and drug resistance. These stromal cells include mesenchymal marrow stromal cells (MSCs),3,6,7 CD68+ nurselike cells derived from monocytes,7–10 and follicular dendritic cells.11 Immunohistochemistry showed that in situ, αSMA+ mesenchymal stromal cells,12 the in vivo counterpart of MSCs, are a dominant stromal cell population in the CLL microenvironment, which is in contrast to other B-cell lymphomas, particularly high-grade lymphomas, which harbor larger numbers of CD68+ hemangiogenic cells.12
MSCs regulate normal hematopoiesis by providing attachment sites and secreted or surface-bound growth factors that constitute the marrow microenvironment.13 During B-cell development in the marrow, programmed cell death regulates B-cell homeostasis by diverting a large fraction of immature B cells into an apoptotic death pathway to eliminate functionless or potentially harmful cells.14,15 Critical factors for the survival of selected B cells are interactions with MSCs in the marrow microenvironment,16–18 expression of surface immunoglobulin molecules, and expression of apoptosis-regulatory proteins, such as Bcl-2.19 In patients with CLL, the marrow invariably is infiltrated with CLL B cells, and the pattern and extent of marrow infiltration correlate with clinical stage and prognosis.20,21 Because MSCs are key regulators in normal B lymphopoiesis and protect CLL cells from spontaneous or drug-induced apoptosis in vitro, it has been proposed that interactions with MSCs play a key role in disease progression or resistance to therapy in CLL,5,22 other mature B-cell malignancies,23,24 and acute lymphoblastic leukemia.25
Previous studies to model the in vivo marrow microenvironment used coculture assays with various MSCs of murine7,26 and human3,6 origins, and it has been of some concern that murine MSCs may introduce confounding factors in these analyses. In addition, it has been discussed whether primary MSCs may be advantageous over MSC lines for studying CLL–MSC interactions. Furthermore, the reproducibility of MSC-based drug-resistance assays, a prerequisite for development of drugs that target CLL–MSC cross talk, has not yet been established. To address these questions, we explored the effects of different human and murine MSC lines as well as primary human MSCs on CLL cell in vitro viability and drug resistance. We established coculture conditions for testing MSC-derived drug resistance that were reproducible in different laboratories. In addition, we explored in part the molecular mechanism related to the broad-based stroma-derived drug resistance. This study therefore provides a basis for further investigation into the biology of MSC–CLL cell interactions and for the development and testing of new drugs or drug combinations that target MSC-derived drug resistance.
Methods
Cell purification, cell lines
After informed consent, peripheral blood samples were obtained from patients fulfilling diagnostic and immunophenotypic criteria for B-cell CLL at the Leukemia Department, University of Texas M. D. Anderson Cancer Center; the Hematology Division, Mayo Clinic; and the Hematology Department, Medical University of Vienna. Patient consent for samples used in this study was obtained in accordance with the Declaration of Helsinki. Approval was obtained from the M. D. Anderson Cancer Center, Mayo Clinic, and the Medical University of Vienna institutional review boards for these studies. Peripheral blood mononuclear cells were isolated by density gradient centrifugation over Ficoll Paque (GE Healthcare). Cells were used fresh or viably frozen in fetal bovine serum (FBS; SAFC Biosciences) plus 10% dimethyl sulfoxide (Sigma-Aldrich) for storage in liquid nitrogen.
The murine stromal cell line M210B4 derived from (C57BL/6J × C3H/HeJ) F1 mouse was purchased from the ATCC. The murine stromal cell lines KUM4 derived from bone marrow of a C3H/He mouse, KUSA-H1 derived from bone marrow of a C3H/He mouse, and the murine stromal cell line ST2 derived from bone marrow of a BALB/c mouse were purchased from the Riken Cell Bank. M210B4, KUM4, KUSA-H1, and ST2 cells were maintained in RPMI 1640 medium supplemented with 2.05 mM l-glutamine (HyClone), 10% FBS (SAFC Biosciences), and penicillin–streptomycin (Cellgro).
The human mesenchymal cell line StromaNKtert derived from bone marrow and immortalized by human telomerase reverse transcriptase (hTERT) and containing also exogene MFG-tsT-IRES-neo was purchased from Riken Cell Bank. Cells were maintained in Minimum Essential Medium Eagle with Earl salts and l-glutamine (α-MEM; HyClone) supplemented with 12.5% FBS (SAFC Biosciences), 12.5% human serum (Cellgro), 1 μM hydrocortisone (Sigma-Aldrich), and 100 μM 2-mercaptoethanol (Sigma-Aldrich). The human mesenchymal cell line UE6E7T-2 derived from bone marrow and immortalized by transformation with HPVs HPV E6 and E7 as well as hTERT was purchased from RIKEN Cell Bank. Cells were maintained in Dulbecco modified Eagle medium with high glucose (4500 mg/L) supplemented with 4 mM l-glutamine (HyClone) and containing 10% FBS (SAFC Biosciences) as well as penicillin–streptomycin (Cellgro). The human mesenchymal cell line UCB408E6E7TERT-33 derived from umbilical cord blood and immortalized by transformation with HPV E6, HPV E7, and hTERT was purchased from Riken Cell Bank. Cells were maintained in mesenchymal stem cell basal medium supplemented with mesenchymal stem cell growth medium single quotes (Poetics; Lonza). Primary human MSCs isolated from bone marrow of patients with CLL were developed by us, as previously described.27 Cells were maintained in α-MEM medium supplemented with l-glutamine, ribonucleosides, and deoxyribonucleosides (HyClone), and 20% FBS (SAFC Biosciences). The morphology of each cell line is displayed in Figure 1, and descriptions of each cell line are provided in the supplemental Table 1 (available on the Blood website; see the Supplemental Materials link at the top of the online article).
Figure 1.
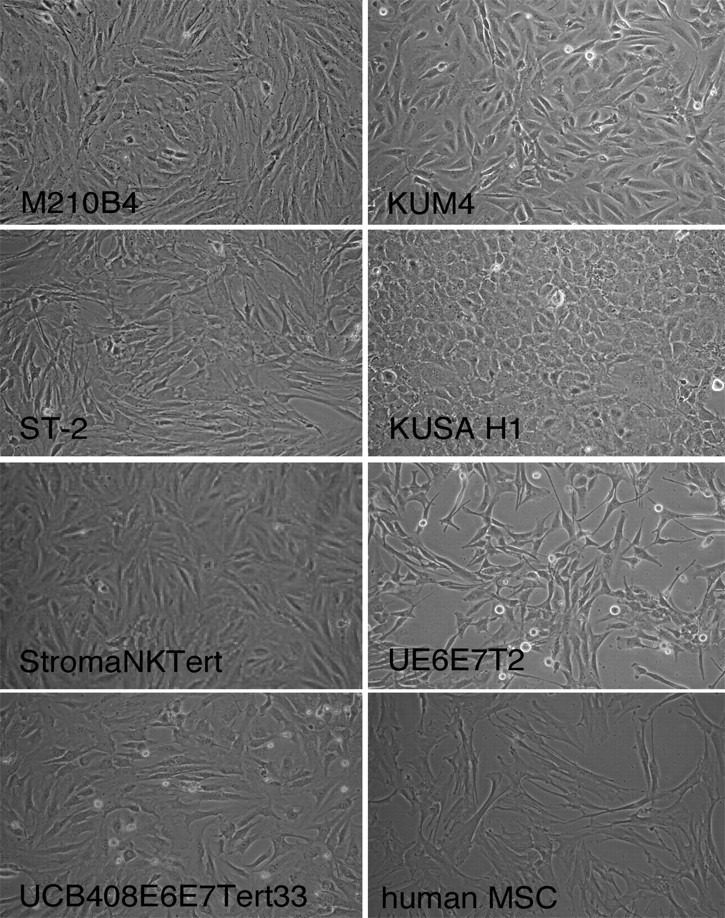
Phenotype of the different MSCs. Figure displays phase-contrast photomicrographs that depict the morphologic appearance of MSC lines and primary MSCs derived from the marrow of a patient with CLL. Cells were imaged in medium using a phase-contrast microscope (Model ELWD 0.3; Nikon) with a 10 × /0.25 NA objective lens. Images were captured with a Nikon D40 digital camera (Nikon Corp) with the use of Camera Control Pro software (Nikon); when necessary, Adobe Photoshop 9.0 (Adobe Systems) was used for image processing.
Coculture experiments
For coculture experiments stromal cells were seeded the day before the experiment onto 48-well plates (Corning Life Sciences) at a concentration of 5 × 104 cells/mL/well and incubated at 37°C in 5% CO2. After confirming the confluence of stromal layer by phase contrast microscopy, CLL cells were added onto the MSC layers at ratios between 5:1 and 100:1. For comparison, CLL cells were also cultured in suspension at a density of 5 × 106 cells/mL. For assessment of MSC-derived drug resistance, CLL cells were treated with 10 μM fludarabine (9-β-D-arabinofuranosyl-2-fluoroadenine; F-ara-A; Sigma-Aldrich), or 10 μM dexamethasone (Sigma-Aldrich), or 60 μM 4-hydroperoxycyclophosphamide (4-HC; kindly provided by Dr M. Colvin and Dr S. Ludeman, Duke University, Durham, NC). At the indicated time points, CLL cells were collected by washing off the CLL cells, leaving the adherent stromal layer intact, and then being assayed for cell viability. For experiments with 4-HC or 4-HC plus F-ara-A, CLL cells were incubated for 2 hours with or without 10 μM F-ara-A, followed by 45 minutes of incubation with 60 μM 4-HC and washing with fresh medium, as previously described.28 After that cells were placed in suspension culture or on top of MSC monolayer (KUSA-H1, StromaNKTert) for 24 hours.
Cell viability testing
Determination of CLL cell viability after treatment with different concentrations of drugs was based on the analysis of mitochondrial transmembrane potential by 3,3′ dihexyloxacarbocyanine iodide (DiOC6; Molecular Probes, Invitrogen) and cell membrane permeability to PI. Determination of CLL viability after 24, 48, and 72 hours of treatment was performed as previously described.7 To differentiate CLL cells and stromal cells, we used a leukocyte gate that excluded large granular stromal cells, based on their forward and side scatter characteristics.
MSC–CLL cell separation experiments
To explore the effect of cell-to-cell contact in mediating drug resistance, we used micropore membranes to separate CLL and stromal cells. Cell culture inserts with pore sizes of 0.4 μm (BD Falcon) were used for noncontact cocultures of CLL cells with MSCs. Control CLL cells were cultured in contact cocultures with MSCs or in suspension cultures without MSCs. CLL cells then were treated with 10 μM F-ara-A and harvested after 24, 48, and 72 hours of incubation, and CLL cell viability was analyzed by flow cytometry.
Immunoblotting
Freshly isolated CLL cells were cultured in suspension or in contact cocultures on confluent layers of 7 different MSC cells lines at a ratio of 20:1 for 48 hours in the presence or absence of 10 μM F-ara-A. Control CLL cells in suspension were analyzed at 0 and 48 hours; for coculture conditions, CLL samples were analyzed after 48 hours. After MSC coculture, CLL cells were washed off the MSC layers and lysed on ice for 30 minutes in lysis buffer containing 25 mM HEPES, 300 mM NaCl, 1.5 mM MgCl2, 0.5% sodium deoxycholate, 20 mM glycerophosphate, 1% Triton X-100, 0.1% SDS, 0.2 mM EDTA, 0.5 mM dithiothreitol, 1 mM sodium orthovanadate, and protease inhibitor. Cells were centrifuged at 14000g for 15 minutes at 4°C, and supernatant was stored at −80°C until use. Protein content was determined using the detergent compatible protein assay kit, according to the manufacturer's instructions (Bio-Rad Laboratories). Aliquots (25 μg) of total cell protein were boiled with Laemmli sample buffer and loaded onto 8% to 12% SDS–polyacrylamide gels and transferred to nitrocellulose membranes (GE Osmonics Labstore). Membranes were blocked for 1 hour in PBS-Tween containing 5% nonfat dried milk and incubated with primary antibodies either overnight or for 2 hours followed by species-specific horseradish peroxidase–conjugated secondary antibody (diluted 1:5000) for 1 hour. The blots were visualized by enhanced chemiluminescence according to the manufacturer's instructions (Pierce Biotechnology) and normalized to the actin levels in each extract. Membranes were probed at 4°C with the following primary antibodies: Mcl-1 antibodies (sc-819; Santa Cruz Biotechnology Inc), PARP (Poly [ADP-ribose] polymerase) antibodies (BD PharMingen International), and β-actin (Cell Signaling). Immunoreactive bands were visualized using peroxidase-conjugated secondary antibodies (GE Healthcare) and enhanced chemiluminescence detection system (Pierce Biotechnology). Cell viability was measured by flow cytometry for each time point and condition.
Data analysis and statistics
Results are shown as mean (± SEM) of at least 3 experiments each. For viability assays, we determined mean relative viabilities to account for variability in spontaneous apoptosis rates in different patients' samples. We define the mean relative viability as the mean CLL cell viability of a particular sample (treated with drug X in the presence or absence of MSCs at a certain time point), divided by the mean cell viability of the same sample at the same time point of control CLL cells, cultured in suspension culture. For statistical comparison between groups, the Student paired t test was used. Analyses were performed with the use of GraphPad Prism 4 software for Macintosh (GraphPad Software). A P value less than .05 was considered as statistically significant. Flow cytometry data were analyzed using the FlowJo software (TreeStar).
Results
Effect of different CLL/MSC ratios
To determine the effect of various CLL-to-MSC ratios on CLL cell survival, we explored a range of different ratios. Thus, we performed titration experiments and tested the following ratios of CLL to MSC cells: 5:1, 10:1, 20:1, 50:1, and 100:1. These conditions were tested in the presence and absence of F-ara-A in cocultures with all 7 MSC lines (stromal characteristics are shown in Figure 1). With the use of 48-well plates, we found that lower ratios (5:1 and 10:1) did not allow for acquisition of sufficient CLL cell numbers by flow cytometry within a time frame that is feasible in larger scale experiments; therefore, we suggest using higher CLL/MSC ratios. Ratios of 20:1 or higher provided optimal conditions for timely and convenient viability testing. There was no significant difference in viability of CLL cells in the presence or absence of 10 μM F-ara-A at 20:1, 50:1, and 100:1 ratios in all MSC cell lines tested (see supplemental Figure 1), indicating optimal stroma-mediated protection within these cell ratios.
Murine and human MSCs protect CLL cells from fludarabine-induced apoptosis
We next examined the effect of coculture with MSCs on the protection of CLL cells from spontaneous and drug-induced apoptosis. For these experiments, CLL cells were treated with 10 μM F-ara-A or left untreated and cultured in the presence or absence of MSC for 24, 48, and 72 hours, and the percentage of viability was measured. As shown in Figure 2, coculture with murine MSCs (M210B4, ST-2, KUM4, and KUSA-H1) protected CLL cells from both spontaneous and fludarabine-induced apoptosis at all time points, the mean relative viabilities (± SEM) and significances are summarized in supplemental Table 3. The protective effect of human MSCs (StromaNKTert, UE6E7T-2, UCB408E6E7 Tert33, hMSCs from 2 patients) on the survival of CLL cells treated with 10μM F-ara-A is depicted in Figure 3. The mean relative viabilities (± SEM) and significances of CLL cell viabilities are summarized in supplemental Table 3. These experiments show that murine MSCs provide significant protection from F-ara-A–induced apoptosis at 48 and 72 hours. In addition, we did not notice any significant differences between different murine MSC lines (Figure 2). In contrast, the levels of protection from F-ara-A–induced apoptosis at 48 and 72 hours with human MSCs generally were lower compared with murine MSCs with the exception of StromaNKtert. Still, all human MSCs provided significant protection from F-ara-A–induced apoptosis compared with CLL cells treated with F-ara-A in suspension with the exception of the primary MSC sample hMSC#2 (Figure 3).
Figure 2.

Murine MSCs protect CLL cells from spontaneous and F-ara-A–induced apoptosis. Displayed are the mean relative viabilities of CLL cells in absence or presence of murine MSCs and/or 10 μM F-ara-A at the time points shown on the horizontal axis. Results represent data for 20:1 CLL/MSC ratios and are the mean (± SEM) relative viabilities, compared with untreated controls (100%) from 5 different CLL patients for each MSC cell line. All murine MSC lines provided significant levels of protection from F-ara-A–induced apoptosis. The names of the murine MSCs are displayed above each diagram.
Figure 3.

Human MSCs protect CLL cells from spontaneous and drug-induced apoptosis. Displayed are the mean relative viabilities of CLL cells in the absence or presence of murine MSCs and/or 10 μM F-ara-A at the time points shown on the horizontal axis. Results represent data for 20:1 CLL/MSC ratios and are the mean (± SEM) relative viabilities, compared with untreated controls (100%) from 3 to 5 different patients with CLL for each MSC line. All human MSC lines (except for the primary hMSC pt#2; see supplemental Table 3) provided significant levels of protection from F-ara-A–induced apoptosis, although the levels of protection generally were lower than those provided by murine MSCs. The names of the human MSCs are displayed above each diagram.
MSCs protect CLL cells from dexamethasone-induced apoptosis
We also tested the cytotoxicity of another chemotherapeutic agent, dexamethasone, in cocultures of CLL cells with murine and human MSCs, using 2 cell lines that displayed high levels of protection in our experiments with F-ara-A. As depicted in Figure 4A, the mean viability of CLL cells treated with 10 μM dexamethasone in the absence of stromal cells was 74.3% (± 6.8%) after 24 hours, 51.9% (± 11.7%) after 48 hours, and 45.3% (± 12.3%) after 72 hours. In presence of murine (KUSA H1) or human MSCs (StromaNKTert), the mean relative viabilities of CLL cells treated with 10 μM dexamethasone were 117.3% (± 7.0%) or 117.4% (± 7.1%) at 24 hours, 110.2% (± 12.0%) or 137.2% (± 10.3%) at 48 hours, and 125.4% (± 13.6%) or 139.7% (± 9.3%) at 72 hours, respectively. Results are presented as the mean (± SEM) relative viability, n = 5. These data indicate that murine and human MSCs provide significant and relatively equivalent protection from dexamethasone-induced apoptosis to CLL cells at all time points.
Figure 4.

Coculture with MSCs protects CLL cells from dexamethasone-induced apoptosis. (A) Bar diagram depicts the mean (± SEM) relative viabilities of CLL cells treated with dexamethasone in the presence or absence of murine (KUSA H) or human (StromaNKTert) MSCs at the time points displayed on the horizontal axis from 5 different patients with CLL. *Significant protection from dexamethasone-induced cytotoxicity compared with control sample (P < .05). (B) Contour plots from a representative CLL sample depict viability of CLL cells, as determined by staining with DiOC6 and PI, after 48 hours of incubation with 10 μM dexamethasone in absence or presence of MSCs, as indicated above each of the plots. The percentage of viable cells is displayed above each of the gates that define viable cells (DiOC6bright PIexclusion).
Combination of 4-HC with F-ara-A partially overcomes MSC-derived drug resistance in CLL
To explore whether cell-adhesion drug resistance applies to other drugs commonly used for CLL treatment, we examined the cytotoxicity of 4-HC, the bioreactive form of cyclophosphamide, alone or in combination with F-ara-A in cocultures of CLL cells with murine and human MSCs. CLL cells were treated with F-ara A, 4-HC, or a combination of these drugs with or without MSCs, and viabilities were determined at 24 hours. As displayed in Figure 5, viability of CLL cells in suspension after treatment with 10 μM F-ara-A was 80.7% (± 3.4%), 7.4% (± 2.5%) after treatment with 60 μM 4-HC, and 4.6% (± 2.0%) after treatment with 10 μM F-ara-A plus 60 μM 4-HC. Coculture of CLL cells with KUSA H1 or StromaNKTert increased the relative viability of CLL cells to 126.5% (± 4.8%) or 137.7% (± 6.2%) in the presence of F-ara-A, 23.1% (± 4.1%) or 44.4% (± 5.9%) after 4-HC, and 12.6% (± 3.9%) or 18.5% (± 5.1%) after the combination of 4-HC with F-ara-A. Results are presented as the mean (± SEM) relative to the viability of untreated controls (n = 8). We conclude that the presence of MSCs improved CLL cell viability after treatment with 4-HC or with 4-HC plus F-ara-A compared with suspension controls. Although F-ara-A had no significant effect on CLL cell viability in MSC cocultures at this time point, the combination of 4-HC plus F-ara-A induced significantly higher levels of apoptosis than did 4-HC alone. This indicates that this mechanism-based combination of a nucleoside analog with an alkylating agent with an alkylating agent28 is also active in CLL–MSC cocultures.
Figure 5.

4-HC and combinations of 4-HC and F-ara-A induce high levels of cytotoxicity, even in the presence of MSCs. CLL cells were cultured with or without MSCs and with 10 μM F-ara-A and/or 60 μM 4-HC. (A) Bar diagram depicts the mean (± SEM) relative viabilities of CLL cells treated with F-ara-A, 4-HC, or a combination of F-ara-A and 4-HC after 24 hours. Results are presented as mean relative viability compared with untreated controls (100%) and are the mean (± SEM) viabilities of CLL samples from 8 different patients. * and ** indicate significant increases in cytotoxicity of the combination of 4-HC plus F-ara-A compared with 4-HC alone (P < .05 or P < .01). (B) Contour plots show the viability of CLL cells after 24 hours of incubation for 1 representative patient. The percentage of viable cells (DiOC6bright PIexclusion) is shown above each of the gates.
MSC-derived drug resistance in CLL cells is dependent on direct cell-to-cell contact
We investigated the role of direct cell-to-cell contact in MSC-derived drug resistance using micropore inserts to separate MSCs and CLL cells. As shown in Figure 6A, the mean viabilities of CLL cells treated with 10 μM F-ara-A and cultured in suspension, in the presence of KUSA H1, or separated from KUSA H1 by inserts was 76.6% (± 3.4%), 139.2% (± 10.5%), or 79.3% (± 3.6%), respectively, at 24 hours; 28.6% (± 6.9%), 110.5% (± 16.4%), or 38.3% (± 7.0%), at 48 hours; and 8.7% (± 4.0%), 74.6% (± 16.3%), or 14.1% (± 4.4%) at 72 hours. As displayed in Figure 6B, the mean viabilities of CLL cells treated with 10 μM F-ara-A after 24 hours was 75.5% (± 4.0%) in suspension, 116.5% (± 4.8%) in contact cocultures with StromaNKTert, and 78.3% (± 4.7%) when separated from StromaNKTert by inserts. After 48 hours the mean viabilities were 26.5% (± 6.5%), 87.3% (± 19.7%), or 28.4% (± 6.1%) for these respective conditions, and 8.5% (± 4.4%), 90.2% (± 17.7%), or 9.9% (± 3.4%) after 72 hours. Results are the mean (± SEM) relative viability of untreated controls in suspension (n = 6). There were no significant difference in CLL cell viabilities between CLL suspension cultures and MSC noncontact cocultures with culture inserts, indicating that CLL cell contact to MSCs is a major contributing factor and that soluble factors have a minor role in MSC-derived protection from F-ara-A–induced apoptosis.
Figure 6.

Direct cell-to-cell contact is essential for cell adhesion–mediated drug resistance. CLL cells were treated with 10 μM F-ara-A and incubated with KUSA H1 (A) and StromaNKTert (B) in the presence or absence of micropore membrane insert for 24, 48, and 72 hours. Bars represent the mean viability of CLL cells compared with untreated control (100%). Data shown are the mean (± SEM) of 6 independent experiments. *Significant protection of CLL cells from F-ara-A–induced apoptosis in direct CLL–MSC contact compared with control sample (P < .05). (C) Presented are contour plots of CLL cells from a representative patient after 48 hours of coculture with StromaNKTert in the conditions indicated above and on the side of the plots. The relative percentages of viable cells are displayed above each of the gates. Direct cell-to-cell contact is essential not only for protection from fludarabine-induced apoptosis but also from spontaneous apoptosis.
Validation of MSC–CLL coculture protocol in different laboratories
Three other laboratories in different regions were used to validate the MSC–CLL coculture protocol in terms of the MSC-derived drug resistance described in “Coculture experiments.” As displayed in supplemental Figure 2, all sites confirmed the efficacy of MSCs in protecting CLL cells from spontaneous and F-ara-A–induced apoptosis. Site no. 1 (Medical University of Vienna) performed the experiments with M210B4 cells and found that the mean viability of CLL cells in suspension treated with 10 μM F-ara-A was 84.3% (± 4.1%), 37.7% (± 4.9%), and 20.5% (± 3.7%) after 24, 48, and 72 hours, respectively. When cocultured with M210B4 without F-ara-A, the mean viability was 125.3% (± 8.3%), 143.7% (± 15.1%), and 140.8% (± 14.7%) at indicated time points. Adding the F-ara-A to coculture conditions resulted in a decrease of the mean viability to 114.9% (± 5.1%) after 24 hours (P < .02 in comparison to CLL cells treated and cultured in suspension), 107.8% (± 8.8%) after 48 hours (P < .001), and 85.3% (± 6.8%) after 72 hours (P < .001). Site no. 2 (Experimental Therapeutics Department, The University of Texas M. D. Anderson Cancer Center) performed the experiments with KUSA H1 cells. The mean viabilities of CLL cells treated with 10 μM F-ara-A and cultured in suspension was 77.5% (± 6.3%) at 24 hours, 38.1% (± 10.5%) at 48 hours, and 13.8% (± 9.6%) at 72 hours. When cultured on KUSA H1 cells without F-ara-A, the mean viabilities of CLL cells were 139.2% (± 4.0%), 151.0% (± 4.3%), and 153.9% (± 8.9%) at these time points and 124.2% (± 5.5%), 98.7% (± 10.8%), and 83.2% (± 19.9%) in the presence of F-ara-A plus MSCs. Results are presented as the mean (± SEM) relative to the viability of untreated controls (n = 5). Site no. 3 (Division of Hematology, Mayo Clinic) tested this protocol in cocultures with M210B4. The mean viabilities of CLL cells in suspension treated with 10 μM F-ara-A were 94.3% (± 1.1%) at 24 hours, 50.9% (± 12.1%) at 48 hours, and 22.7% (± 12.3%) at 72 hours. The addition of F-ara-A to CLL–MSC cocultures resulted in mean viabilities of 93.7% (± 1.7%) after 24 hours, 79.7% (± 6.6%) after 48 hours, and 81.3% (± 10.3%) after 72 hours. When cocultured with MSCs without F-ara-A, the mean viabilities were 94.4% (± 2.4%), 88.2% (± 11.1%), and 96.6% (± 12.6%) at these time points. Results are presented as the mean (± SEM) relative to the viability of untreated controls (n = 4).
MSCs protect CLL cells from spontaneous and F-ara-A–induced apoptosis/effects on Mcl-1 and PARP
To explore and define the molecular pathways of MSC-derived protection of CLL cells from spontaneous and F-ara-A–induced apoptosis, we tested protein levels of Mcl-1 and PARP. These 2 proteins are known to change rapidly when CLL B cells interact with certain stromal cells, but the simultaneous testing of these 2 molecules with our spectrum of stromal cell lines has not been done. Thus, CLL cells were cultured with 7 different MSC cell lines in the presence or absence of 10 μM F-ara-A or in suspension. Culture in suspension for 48 hours (2nd lane from the left) resulted in spontaneous apoptosis (47% viability in Figure 7A and 81% viability in Figure 7B) and was accompanied by the generation of cleaved fragments of Mcl-1 (24 kD) and PARP (85 kDa). Coculture with MSCs induced a significant increase of Mcl-1 expression in CLL cells after 48 hours of incubation in comparison to control in suspension, except for KUSA H1. Treatment with F-ara-A resulted in significant decreases in CLL cell viabilities in suspension after 48 hours, accompanied by a virtually complete cleavage of Mcl-1 and PARP (5th lane from the left). Noticeably, these decreases in full-length Mcl-1 and PARP correlated well with the viability data collected by flow cytometry, as displayed in Figure 7. In contrast, coculture with MSCs resulted in decreased or abrogated spontaneous and F-ara-A–induced cleavage of Mcl-1 and PARP in CLL cells. These results suggest that induction and/or maintenance of Mcl-1 by MSCs plays an important role in the protection of CLL cells from spontaneous and drug-induced apoptosis.
Figure 7.

Mcl-1 and PARP expression in CLL cells cocultured with MSCs. CLL cells were cultured with the MSC lines displayed on the top horizontal axes or in suspension (“Control”) for 48 hours with or without 10 μM F-ara-A (labeled “Ctr” or “+F,” respectively). Then, cleaved and uncleaved Mcl-1 and PARP were analyzed by Western blotting, and the respective immunobands are indicated on the left-hand side. Cell viability for each condition was measured by flow cytometry, and the percentage of viable cells is displayed below each of the blots. In most cases, MSCs coculture up-regulated Mcl-1 and PARP expression compared with CLL cells in suspension (control at 48 hours vs the “Ctr” bands in the presence of MSCs). Suspension culture of CLL cells results in spontaneous apoptosis, with associated Mcl-1 and PARP cleavage (panels A-B 2nd lane from the left), which was paralleled by a decrease in CLL cell viability from 99% to 47% in panel A and from 96% to 81% in panel B. Treatment with fludarabine resulted in cleavage of the majority of Mcl-1 and PARP in the absence of MSCs (panels A-B 5th lane from the left). This was largely inhibited, sometimes almost abrogated (eg, panel A lanes 6-7) by the presence of MSCs. Displayed are Western blots of CLL B-cell lysates from 2 representative patients (A-B).
Discussion
The importance of the marrow microenvironment for protection of CLL and other leukemia cells from drug-induced apoptosis is increasingly recognized, based on a series of in vitro and in vivo studies published over the past decade (reviewed in Burger and Kipps5 and Meads et al22). These studies defined the importance of accessory cells, in particular of MSCs,3,7,23,29 for providing survival, growth, and drug resistance signals, which may explain why the marrow is a preferential site for minimal residual disease2,30 and relapses even after extremely effective conventional treatments.22 Therefore, disrupting the cross talk between CLL cells and their milieu is an attractive and needed novel strategy. For this to be done in a meaningful way, it will be necessary to devise testable in vitro models. We have begun this process by attempting to standardize in vitro culture conditions whereby we are examining stromal/CLL cell interactions. Thus, we systematically examined a series of murine and human MSCs, both cell lines and primary cells, for their capacity to support CLL cells and to protect them from drug-induced apoptosis. We found that both murine and human MSC layers are highly effective in protecting CLL cells over a wide range of CLL/MSC ratios. This primary drug resistance mechanism, also called cell adhesion–mediated drug resistance, is increasingly recognized as a key mechanism accounting for residual disease and relapses after conventional treatments in hematopoietic31 and other malignancies.32 These findings also suggest that the exact match between species is not critical for CLL cell protection. Indeed, we noticed that the levels of protection by murine MSC were more homogenous and higher in some cases, compared with human MSC, with 2 human cell lines and primary human MSC displaying comparably less protective effects from F-ara-A (Figure 3).
These findings parallel previous reports on the importance of MSC for hematopoietic progenitor cell maintenance and expansion in vitro. In the long-term culture systems, stromal feeder layers support the proliferation and differentiation of hematopoietic progenitors in the absence of added cytokines. Originally, these cultures were established from nonseparated marrow cells,33 but subsequent studies established that adherent mesenchymal fibroblast lines from different tissues and species can provide the same supportive function.34 Previous reports comparing the capacity of different human and murine stromal cells to support hematopoietic (long-term culture initiating cells) or B-cell progenitors in vitro have shown that murine MSCs are working as well as human MSCs,34–37 or even better.38 The protective effect of MSCs was largely dependent on direct cell–cell contact between the leukemia cells and the MSCs, and separation of the cells by micropore filters abrogated the protective function of MSCs (Figure 6). This is in accordance with earlier notions that surface molecules and surface-bound growth factors are the key communication factors involved in MSC-derived survival signals.3,4,13 This, however, does not exclude the activity of diffusible factors; for example, the chemokine CXCL12, which is generally regarded as a secreted factor, can be sequestered and retained on the surface of MSCs by glycosaminoglycans and proteoglycans, increasing their local concentration and availability.39
We confirmed the reproducibility of these assays in 4 different laboratories (supplemental Figure 2). This broad-based interactive approach for validation of stromal cell support of CLL B-cell survival will now allow us to move forward with more systematic analyses, including which classes of drugs are subject to significant MSC-derived drug resistance, and importantly toward development of drug combinations that account for MSC-derived drug resistance. The data presented in this report show that MSCs protect CLL cells from F-ara-A–, dexamethasone-, and 4-HC–induced apoptosis. In addition, our data indicate that drug combinations (F-ara-A plus 4-HC) cooperate in inducing CLL cell apoptosis and can, at least partially, overcome MSC-derived drug resistance, a finding that is in accordance with and helps to explain the clinical activity of this drug combination.40 Some examples of novel treatment approaches to overcome MSC-derived drug resistance already have been explored in CLL–MSC cocultures. We reported that AT-101 induced apoptosis in CLL cells as equally as in the presence of MSCs and down-regulated endogenous and MSC-induced Mcl-1.41 CXCR4 antagonists, which disrupt migration and adhesion of CLL cells to MSCs, sensitized CLL cells to F-ara-A–induced apoptosis.42 Moreover, antagonists of phosphoinositide 3-kinases can overcome MSC-derived drug resistance.43 Cyclopamine, a hedgehog signaling inhibitor, blocked MSC-induced survival of CLL cells, suggesting a role for hedgehog signaling in MSC-derived survival in CLL.44 Finally, we have also shown that the timing and combination of epigallocatechin and curcumin can overcome stromal-mediated drug resistance of CLL B cells.45
Our molecular studies have shown that different MSCs (human and murine) induced and effectively maintained Mcl-1 levels in CLL cells and protected CLL cells from spontaneous and F-ara-A–induced apoptosis. Maintenance of intact, noncleaved Mcl-1 was paralleled by high CLL cell viabilities (see Figure 7), suggesting that Mcl-1 is critical for CLL cell viability in MSC cocultures. Among the Bcl-2 family members, Mcl-1 has emerged as one of the most relevant antiapoptotic proteins in normal46 and malignant B cells.47 Mcl-1 is an early response gene that functions as a modulator of cell viability.48 Mcl-1 can undergo rapid up-regulation as well as down-regulation (within 1-3 hours),49 which allows Mcl-1 to provide an acute protective function from apoptosis induced by various factors, including DNA damage,50 growth factor withdrawal,51,52 and treatment with cytotoxic agents.53 Disappearance of Mcl-1 is associated with the onset of apoptosis and is caused by the combination of synthesis blockage and proteasomal degradation.50,54 Cleavage of the nuclear enzyme PARP is another early marker of chemotherapy-induced apoptosis.55 The mechanism through which MSCs maintain Mcl-1 in CLL cells even in the presence of F-ara-A are currently unknown. However, activation of CXCR4 chemokine receptors by its ligand, CXCL12 (stromal cell-derived factor-1/SDF-1) secreted constitutively by MSCs, is a possible mechanism, given the earlier observation of Mcl-1 induction by CXCL12 in hematopoietic cells.56 Another study reported that B-cell–activating factor of the tumor necrosis factor family (BAFF) and a proliferation-inducing ligand (APRIL), but not CXCL12, induced Mcl-1 expression in CLL cells.8 In addition, we have shown that vascular endothelial growth factor, released by stromal cells, also can up-regulate Mcl-1 in CLL cells.27,57,58 Recent studies indicate that Mcl-1 function and regulation is highly complex, integrating several proapoptotic and the prosurvival pathways, such as JNK and AKT54 and regulation by microRNAs.59 Ongoing studies in our laboratory, in which we determine gene expression changes induced in CLL cells by contact coculture with different MSCs, may help to further dissect out the critical pathways in CLL–MSC cross talk.
First trials that are directly targeting the CLL microenvironment are now entering the clinical stage. These trials are largely based on data from in vitro models that allowed us to define key pathways, such as the CXCR4–CXCL12 axis,42,60 VLA-4 adhesion molecules and their respective stromal ligands (VCAM-1, fibronectin),4,61 the phosphoinositide 3-kinases,43,60 and the spleen tyrosine kinase.62 The model system described in this study will help us to move forward with a more standardized approach for testing new and established drugs by in vitro models that resemble the marrow microenvironment. The in vivo validation61,63,64 of targets identified in these models and the association of these findings with the clinical progress in this area emphasize that coculture models are now and will remain an indispensable tool for discovery and dissection of the tumor microenvironment in CLL and beyond.
Acknowledgments
We thank Dr Martin Hilgarth and Dr Deepa Sampath for experimental support and valuable suggestion regarding 4-HC experiments, and Dr Akihiro Umezawa for advice regarding MSC cell lines maintenance.
This work was supported by CLL Global Research Foundation grants (W.G.W., Z.E., V.G., N.E.K., W.P., and J.A.B.), a Sidney Kimmel Foundation for Cancer Research Scholar Award (J.A.B.), an ASCO Career Development Award (J.A.B.), and an Austrian Society of Hematology and Oncology award (M.S.).
Footnotes
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: A.V.K. performed the experiments, analyzed the data, designed the figures, and wrote the paper with J.A.B.; K.B. performed the Western blots with A.V.K. and reviewed the manuscript; R.C., W.D., and S.S. performed experiments to validate the CLL-MSC coculture assays in their respective laboratories; M.P.Q. and M.S. assisted with the experiments and reviewed the manuscript; W.G.W., Z.E., M.J.K., M.S., and U.J. provided patients' samples and reviewed the manuscript; V.G., N.E.K., and W.P. helped with the design of the study and reviewed the manuscript; and J.A.B. designed the research, supervised the study, analyzed the data, and revised the paper.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Jan A. Burger, Department of Leukemia, Unit 428, The University of Texas M. D. Anderson Cancer Center, PO Box 301402, Houston, TX 77230-1402; e-mail: jaburger@mdanderson.org.
References
- 1.Tam CS, O'Brien S, Wierda W, et al. Long-term results of the fludarabine, cyclophosphamide, and rituximab regimen as initial therapy of chronic lymphocytic leukemia. Blood. 2008;112(4):975–980. doi: 10.1182/blood-2008-02-140582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rawstron AC, Kennedy B, Evans PA, et al. Quantitation of minimal disease levels in chronic lymphocytic leukemia using a sensitive flow cytometric assay improves the prediction of outcome and can be used to optimize therapy. Blood. 2001;98(1):29–35. doi: 10.1182/blood.v98.1.29. [DOI] [PubMed] [Google Scholar]
- 3.Lagneaux L, Delforge A, Bron D, De Bruyn C, Stryckmans P. Chronic lymphocytic leukemic B cells but not normal B cells are rescued from apoptosis by contact with normal bone marrow stromal cells. Blood. 1998;91(7):2387–2396. [PubMed] [Google Scholar]
- 4.Damiano JS, Cress AE, Hazlehurst LA, Shtil AA, Dalton WS. Cell adhesion mediated drug resistance (CAM-DR): role of integrins and resistance to apoptosis in human myeloma cell lines. Blood. 1999;93(5):1658–1667. [PMC free article] [PubMed] [Google Scholar]
- 5.Burger JA, Kipps TJ. Chemokine receptors and stromal cells in the homing and homeostasis of chronic lymphocytic leukemia B cells. Leuk Lymphoma. 2002;43(3):461–466. doi: 10.1080/10428190290011921. [DOI] [PubMed] [Google Scholar]
- 6.Panayiotidis P, Jones D, Ganeshaguru K, Foroni L, Hoffbrand AV. Human bone marrow stromal cells prevent apoptosis and support the survival of chronic lymphocytic leukaemia cells in vitro. Br J Haematol. 1996;92(1):97–103. doi: 10.1046/j.1365-2141.1996.00305.x. [DOI] [PubMed] [Google Scholar]
- 7.Burger JA, Tsukada N, Burger M, Zvaifler NJ, Dell'Aquila M, Kipps TJ. Blood-derived nurse-like cells protect chronic lymphocytic leukemia B cells from spontaneous apoptosis through stromal cell-derived factor-1. Blood. 2000;96(8):2655–2663. [PubMed] [Google Scholar]
- 8.Nishio M, Endo T, Tsukada N, et al. Nurselike cells express BAFF and APRIL, which can promote survival of chronic lymphocytic leukemia cells via a paracrine pathway distinct from that of SDF-1alpha. Blood. 2005;106(3):1012–1020. doi: 10.1182/blood-2004-03-0889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Deaglio S, Vaisitti T, Bergui L, et al. CD38 and CD100 lead a network of surface receptors relaying positive signals for B-CLL growth and survival. Blood. 2005;105(8):3042–3050. doi: 10.1182/blood-2004-10-3873. [DOI] [PubMed] [Google Scholar]
- 10.Richardson SJ, Matthews C, Catherwood MA, et al. ZAP-70 expression is associated with enhanced ability to respond to migratory and survival signals in B-cell chronic lymphocytic leukemia (B-CLL). Blood. 2006;107(9):3584–3592. doi: 10.1182/blood-2005-04-1718. [DOI] [PubMed] [Google Scholar]
- 11.Pedersen IM, Kitada S, Leoni LM, et al. Protection of CLL B cells by a follicular dendritic cell line is dependent on induction of Mcl-1. Blood. 2002;100(5):1795–1801. [PubMed] [Google Scholar]
- 12.Ruan J, Hyjek E, Kermani P, et al. Magnitude of stromal hemangiogenesis correlates with histologic subtype of non-Hodgkin's lymphoma. Clin Cancer Res. 2006;12(19):5622–5631. doi: 10.1158/1078-0432.CCR-06-1204. [DOI] [PubMed] [Google Scholar]
- 13.Roberts R, Gallagher J, Spooncer E, Allen TD, Bloomfield F, Dexter TM. Heparan sulphate bound growth factors: a mechanism for stromal cell mediated haemopoiesis. Nature. 1988;332(6162):376–378. doi: 10.1038/332376a0. [DOI] [PubMed] [Google Scholar]
- 14.Osmond DG, Rico-Vargas S, Valenzona H, et al. Apoptosis and macrophage-mediated cell deletion in the regulation of B lymphopoiesis in mouse bone marrow. Immunol Rev. 1994;142:209–230. doi: 10.1111/j.1600-065x.1994.tb00891.x. [DOI] [PubMed] [Google Scholar]
- 15.Melchers F, Rolink A, Grawunder U, et al. Positive and negative selection events during B lymphopoiesis. Curr Opin Immunol. 1995;7(2):214–227. doi: 10.1016/0952-7915(95)80006-9. [DOI] [PubMed] [Google Scholar]
- 16.Dorshkind K. Regulation of hemopoiesis by bone marrow stromal cells and their products. Annu Rev Immunol. 1990;8:111–137. doi: 10.1146/annurev.iy.08.040190.000551. [DOI] [PubMed] [Google Scholar]
- 17.Jacobsen K, Osmond DG. Microenvironmental organization and stromal cell associations of B lymphocyte precursor cells in mouse bone marrow. Eur J Immunol. 1990;20(11):2395–2404. doi: 10.1002/eji.1830201106. [DOI] [PubMed] [Google Scholar]
- 18.LeBien TW. Fates of human B-cell precursors. Blood. 2000;96(1):9–23. [PubMed] [Google Scholar]
- 19.Reed JC. Bcl-2-family proteins and hematologic malignancies: history and future prospects. Blood. 2008;111(7):3322–3330. doi: 10.1182/blood-2007-09-078162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Han T, Barcos M, Emrich L, et al. Bone marrow infiltration patterns and their prognostic significance in chronic lymphocytic leukemia: correlations with clinical, immunologic, phenotypic, and cytogenetic data. J Clin Oncol. 1984;2(6):562–570. doi: 10.1200/JCO.1984.2.6.562. [DOI] [PubMed] [Google Scholar]
- 21.Pangalis GA, Roussou PA, Kittas C, et al. Patterns of bone marrow involvement in chronic lymphocytic leukemia and small lymphocytic (well differentiated) non-Hodgkin's lymphoma. Its clinical significance in relation to their differential diagnosis and prognosis. Cancer. 1984;54(4):702–708. doi: 10.1002/1097-0142(1984)54:4<702::aid-cncr2820540418>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- 22.Meads MB, Hazlehurst LA, Dalton WS. The bone marrow microenvironment as a tumor sanctuary and contributor to drug resistance. Clin Cancer Res. 2008;14(9):2519–2526. doi: 10.1158/1078-0432.CCR-07-2223. [DOI] [PubMed] [Google Scholar]
- 23.Amé-Thomas P, Maby-El Hajjami H, Monvoisin C, et al. Human mesenchymal stem cells isolated from bone marrow and lymphoid organs support tumor B-cell growth: role of stromal cells in follicular lymphoma pathogenesis. Blood. 2007;109(2):693–702. doi: 10.1182/blood-2006-05-020800. [DOI] [PubMed] [Google Scholar]
- 24.Podar K, Chauhan D, Anderson KC. Bone marrow microenvironment and the identification of new targets for myeloma therapy. Leukemia. 2009 Jan;23(1):10–24. doi: 10.1038/leu.2008.259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Iwamoto S, Mihara K, Downing JR, Pui CH, Campana D. Mesenchymal cells regulate the response of acute lymphoblastic leukemia cells to asparaginase. J Clin Invest. 2007;117(4):1049–1057. doi: 10.1172/JCI30235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Edelmann J, Klein-Hitpass L, Carpinteiro A, et al. Bone marrow fibroblasts induce expression of PI3K/NF-kappaB pathway genes and a pro-angiogenic phenotype in CLL cells. Leuk Res. 2008;32(10):1565–1572. doi: 10.1016/j.leukres.2008.03.003. [DOI] [PubMed] [Google Scholar]
- 27.Kay NE, Shanafelt TD, Strege AK, Lee YK, Bone ND, Raza A. Bone biopsy derived marrow stromal elements rescue chronic lymphocytic leukemia B-cells from spontaneous and drug induced cell death and facilitates an “angiogenic switch”. Leuk Res. 2007;31(7):899–906. doi: 10.1016/j.leukres.2006.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yamauchi T, Nowak BJ, Keating MJ, Plunkett W. DNA repair initiated in chronic lymphocytic leukemia lymphocytes by 4-hydroperoxycyclophosphamide is inhibited by fludarabine and clofarabine. Clin Cancer Res. 2001;7(11):3580–3589. [PubMed] [Google Scholar]
- 29.Mudry RE, Fortney JE, York T, Hall BM, Gibson LF. Stromal cells regulate survival of B-lineage leukemic cells during chemotherapy. Blood. 2000;96(5):1926–1932. [PubMed] [Google Scholar]
- 30.Borowitz MJ, Devidas M, Hunger SP, et al. Clinical significance of minimal residual disease in childhood acute lymphoblastic leukemia and its relationship to other prognostic factors: a Children's Oncology Group study. Blood. 2008;111(12):5477–5485. doi: 10.1182/blood-2008-01-132837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Burger JA, Ghia P, Rosenwald A, Caligaris-Cappio F. The microenvironment in mature B-cell malignancies: a target for new treatment strategies. Blood. doi: 10.1182/blood-2009-06-225326. Prepublished on July 27 2009, as DOI 10.1182/blood-2009-06-225326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Meads MB, Gatenby RA, Dalton WS. Environment-mediated drug resistance: a major contributor to minimal residual disease. Nat Rev Cancer. 2009;9(9):665–674. doi: 10.1038/nrc2714. [DOI] [PubMed] [Google Scholar]
- 33.Gartner S, Kaplan HS. Long-term culture of human bone marrow cells. Proc Natl Acad Sci U S A. 1980;77(8):4756–4759. doi: 10.1073/pnas.77.8.4756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sutherland HJ, Eaves CJ, Lansdorp PM, Thacker JD, Hogge DE. Differential regulation of primitive human hematopoietic cells in long-term cultures maintained on genetically engineered murine stromal cells. Blood. 1991;78(3):666–672. [PubMed] [Google Scholar]
- 35.Kurosaka D, LeBien TW, Pribyl JA. Comparative studies of different stromal cell microenvironments in support of human B-cell development. Exp Hematol. 1999;27(8):1271–1281. doi: 10.1016/s0301-472x(99)00067-3. [DOI] [PubMed] [Google Scholar]
- 36.Croisille L, Auffray I, Katz A, Izac B, Vainchenker W, Coulombel L. Hydrocortisone differentially affects the ability of murine stromal cells and human marrow-derived adherent cells to promote the differentiation of CD34++/CD38- long-term culture-initiating cells. Blood. 1994;84(12):4116–4124. [PubMed] [Google Scholar]
- 37.Berardi AC, Meffre E, Pflumio F, et al. Individual CD34+CD38lowCD19-CD10- progenitor cells from human cord blood generate B lymphocytes and granulocytes. Blood. 1997;89(10):3554–3564. [PubMed] [Google Scholar]
- 38.Thiemann FT, Moore KA, Smogorzewska EM, Lemischka IR, Crooks GM. The murine stromal cell line AFT024 acts specifically on human CD34+CD38- progenitors to maintain primitive function and immunophenotype in vitro. Exp Hematol. 1998;26(7):612–619. [PubMed] [Google Scholar]
- 39.Murphy JW, Cho Y, Sachpatzidis A, Fan C, Hodsdon ME, Lolis E. Structural and functional basis of CXCL12 (stromal cell-derived factor-1 alpha) binding to heparin. J Biol Chem. 2007;282(13):10018–10027. doi: 10.1074/jbc.M608796200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.O'Brien SM, Kantarjian HM, Cortes J, et al. Results of the fludarabine and cyclophosphamide combination regimen in chronic lymphocytic leukemia. J Clin Oncol. 2001;19(5):1414–1420. doi: 10.1200/JCO.2001.19.5.1414. [DOI] [PubMed] [Google Scholar]
- 41.Balakrishnan K, Burger JA, Wierda WG, Gandhi V. AT-101 induces apoptosis in CLL B cells and overcomes stromal cell-mediated Mcl-1 induction and drug resistance. Blood. 2009;113(1):149–153. doi: 10.1182/blood-2008-02-138560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Burger M, Hartmann T, Krome M, et al. Small peptide inhibitors of the CXCR4 chemokine receptor (CD184) antagonize the activation, migration, and antiapoptotic responses of CXCL12 in chronic lymphocytic leukemia B cells. Blood. 2005;106(5):1824–1830. doi: 10.1182/blood-2004-12-4918. [DOI] [PubMed] [Google Scholar]
- 43.Niedermeier M, Hennessy BT, Knight ZA, et al. Isoform-selective phosphoinositide 3′-kinase inhibitors inhibit CXCR4 signaling and overcome stromal cell-mediated drug resistance in chronic lymphocytic leukemia: a novel therapeutic approach. Blood. 2009;113(22):5549–5557. doi: 10.1182/blood-2008-06-165068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hegde GV, Peterson KJ, Emanuel K, et al. Hedgehog-induced survival of B-cell chronic lymphocytic leukemia cells in a stromal cell microenvironment: a potential new therapeutic target. Mol Cancer Res. 2008;6(12):1928–1936. doi: 10.1158/1541-7786.MCR-08-0142. [DOI] [PubMed] [Google Scholar]
- 45.Ghosh AK, Shanafelt TD, Cimmino A, et al. Aberrant regulation of pVHL levels by microRNA promotes the HIF/VEGF axis in CLL B cells. Blood. 2009;113(22):5568–5574. doi: 10.1182/blood-2008-10-185686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Opferman JT, Letai A, Beard C, Sorcinelli MD, Ong CC, Korsmeyer SJ. Development and maintenance of B and T lymphocytes requires antiapoptotic MCL-1. Nature. 2003;426(6967):671–676. doi: 10.1038/nature02067. [DOI] [PubMed] [Google Scholar]
- 47.Gandhi V, Balakrishnan K, Chen LS. Mcl-1: the 1 in CLL. Blood. 2008;112(9):3538–3540. doi: 10.1182/blood-2008-07-170241. [DOI] [PubMed] [Google Scholar]
- 48.Kozopas KM, Yang T, Buchan HL, Zhou P, Craig RW. MCL1, a gene expressed in programmed myeloid cell differentiation, has sequence similarity to BCL2. Proc Natl Acad Sci U S A. 1993;90(8):3516–3520. doi: 10.1073/pnas.90.8.3516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yang T, Buchan HL, Townsend KJ, Craig RW. MCL-1, a member of the BLC-2 family, is induced rapidly in response to signals for cell differentiation or death, but not to signals for cell proliferation. J Cell Physiol. 1996;166(3):523–536. doi: 10.1002/(SICI)1097-4652(199603)166:3<523::AID-JCP7>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 50.Cuconati A, Mukherjee C, Perez D, White E. DNA damage response and MCL-1 destruction initiate apoptosis in adenovirus-infected cells. Genes Dev. 2003;17(23):2922–2932. doi: 10.1101/gad.1156903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Huang HM, Huang CJ, Yen JJ. Mcl-1 is a common target of stem cell factor and interleukin-5 for apoptosis prevention activity via MEK/MAPK and PI-3K/Akt pathways. Blood. 2000;96(5):1764–1771. [PubMed] [Google Scholar]
- 52.Le Gouill S, Podar K, Amiot M, et al. VEGF induces Mcl-1 up-regulation and protects multiple myeloma cells against apoptosis. Blood. 2004;104(9):2886–2892. doi: 10.1182/blood-2004-05-1760. [DOI] [PubMed] [Google Scholar]
- 53.Zhou P, Qian L, Kozopas KM, Craig RW. Mcl-1, a Bcl-2 family member, delays the death of hematopoietic cells under a variety of apoptosis-inducing conditions. Blood. 1997;89(2):630–643. [PubMed] [Google Scholar]
- 54.Morel C, Carlson SM, White FM, Davis RJ. Mcl-1 integrates the opposing actions of signaling pathways that mediate survival and apoptosis. Mol Cell Biol. 2009;29(14):3845–3852. doi: 10.1128/MCB.00279-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kaufmann SH, Desnoyers S, Ottaviano Y, Davidson NE, Poirier GG. Specific proteolytic cleavage of poly(ADP-ribose) polymerase: an early marker of chemotherapy-induced apoptosis. Cancer Res. 1993;53(17):3976–3985. [PubMed] [Google Scholar]
- 56.Joo EK, Broxmeyer HE, Kwon HJ, et al. Enhancement of cell survival by stromal cell-derived factor-1/CXCL12 involves activation of CREB and induction of Mcl-1 and c-Fos in factor-dependent human cell line MO7e. Stem Cells Dev. 2004;13(5):563–570. doi: 10.1089/scd.2004.13.563. [DOI] [PubMed] [Google Scholar]
- 57.Kay NE, Bone ND, Tschumper RC, et al. B-CLL cells are capable of synthesis and secretion of both pro- and anti-angiogenic molecules. Leukemia. 2002;16(5):911–919. doi: 10.1038/sj.leu.2402467. [DOI] [PubMed] [Google Scholar]
- 58.Lee YK, Bone ND, Strege AK, Shanafelt TD, Jelinek DF, Kay NE. VEGF receptor phosphorylation status and apoptosis is modulated by a green tea component, epigallocatechin-3-gallate (EGCG), in B-cell chronic lymphocytic leukemia. Blood. 2004;104(3):788–794. doi: 10.1182/blood-2003-08-2763. [DOI] [PubMed] [Google Scholar]
- 59.Mott JL, Kobayashi S, Bronk SF, Gores GJ. mir-29 regulates Mcl-1 protein expression and apoptosis. Oncogene. 2007;26(42):6133–6140. doi: 10.1038/sj.onc.1210436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Burger JA, Burger M, Kipps TJ. Chronic lymphocytic leukemia B cells express functional CXCR4 chemokine receptors that mediate spontaneous migration beneath bone marrow stromal cells. Blood. 1999;94(11):3658–3667. [PubMed] [Google Scholar]
- 61.Matsunaga T, Takemoto N, Sato T, et al. Interaction between leukemic-cell VLA-4 and stromal fibronectin is a decisive factor for minimal residual disease of acute myelogenous leukemia. Nat Med. 2003;9(9):1158–1165. doi: 10.1038/nm909. [DOI] [PubMed] [Google Scholar]
- 62.Quiroga MP, Balakrishnan K, Kurtova AV, et al. B-cell antigen receptor signaling enhances chronic lymphocytic leukemia cell migration and survival: specific targeting with a novel Syk inhibitor, R406. Blood. 2009;114(5):1029–1037. doi: 10.1182/blood-2009-03-212837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sipkins DA, Wei X, Wu JW, et al. In vivo imaging of specialized bone marrow endothelial microdomains for tumour engraftment. Nature. 2005;435(7044):969–973. doi: 10.1038/nature03703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Nervi B, Ramirez P, Rettig MP, et al. Chemosensitization of acute myeloid leukemia (AML) following mobilization by the CXCR4 antagonist AMD3100. Blood. 2009;113(24):6206–6214. doi: 10.1182/blood-2008-06-162123. [DOI] [PMC free article] [PubMed] [Google Scholar]