

Abstract
MGCD0103 is an isotype-selective inhibitor of histone deacetylases (HDACs) targeted to isoforms 1, 2, 3, and 11. In a phase 1 study in patients with leukemia or myelodysplastic syndromes (MDS), MGCD0103 was administered orally 3 times weekly without interruption. Twenty-nine patients with a median age of 62 years (range, 32-84 years) were enrolled at planned dose levels (20, 40, and 80 mg/m2). The majority of patients (76%) had acute myelogenous leukemia (AML). In all, 24 (83%) of 29 patients had received 1 or more prior chemotherapies (range, 0-5), and 18 (62%) of 29 patients had abnormal cytogenetics. The maximum tolerated dose was determined to be 60 mg/m2, with dose-limiting toxicities (DLTs) of fatigue, nausea, vomiting, and diarrhea observed at higher doses. Three patients achieved a complete bone marrow response (blasts ≤ 5%). Pharmacokinetic analyses indicated absorption of MGCD0103 within 1 hour and an elimination half-life in plasma of 9 (± 2) hours. Exposure to MGCD0103 was proportional to dose up to 60 mg/m2. Analysis of peripheral white cells demonstrated induction of histone acetylation and dose-dependent inhibition of HDAC enzyme activity. In summary, MGCD0103 was safe and had antileukemia activity that was mechanism based in patients with advanced leukemia.
Introduction
Despite many advances in the management of acute leukemia, patients with acute myelogenous leukemia (AML) who are refractory to conventional therapy, or who have relapsed after conventional therapy, have a poor prognosis for survival. In addition, the inability to tolerate or benefit from induction chemotherapy due to advanced age or comorbidities is associated with poor clinical outcomes. Novel therapeutic strategies focusing on tumor-related alterations in chromatin structure and epigenetic silencing are currently being explored.
In eukaryotic cells, histone acetylation/deacetylation has an important role in the control of gene transcription regulation.1 Transcriptionally active genes are characterized by hyperacetylated chromatin, whereas repressed genes are typically in a hypoacetylated state.2,3 This process is mediated by a complex interplay of proteins with histone acetyltransferases and deacetylases.2,3 Histone deacetylases (HDACs) are divided into 4 general classes. Class I includes HDACs 1, 2, 3, and 8; class II HDACs, 4, 5, 6, 7, 9, and 10; and class III, Sirt 1 to 7; the latter group of enzymes is not targeted by HDAC inhibitors.4,5 Class IV includes HDAC 11, which is distinct from the other classes.
Small molecule inhibitors of HDACs are a novel therapeutic class of drugs with anticancer potential.6 Although not fully understood, the clinical activity of these inhibitors is thought to be mediated in part by induction of histone acetylation, resulting in a permissive or more open chromatin configuration and potential reactivation of aberrantly suppressed genes (eg, tumor suppressor genes). The changes in gene expression lead to inhibition of cell proliferation, induction of apoptosis, and/or cell differentiation.7
HDAC inhibitors can be grouped into different subclasses, such as hydroxamic acids8–11and aminophenylbenzamides,12,13 based on their chemical structure. MGCD0103 is an isotype-specific aminophenylbenzamide that was synthesized through medicinal chemistry optimization of small molecule HDAC inhibitors. MGCD0103 inhibits HDAC isotypes 1, 2, 3, and 11.14,15 This greater selectivity allows for targeting of the HDAC isotypes that are thought to be linked to cancer. Indeed, the number of genes with expression induced by MGCD0103 is dramatically smaller than that induced by nonspecific hydroxamate HDAC inhibitors, yet efficacy in preclinical cancer models is maintained or increased).16 Therefore, we hypothesize that the selective targeting of specific HDAC isotypes by MGCD0103 may improve the therapeutic window in cancer patients.
Preclinical studies have demonstrated that MGCD0103 is orally bioavailable with significant in vitro antineoplastic activity at submicromolar concentrations against a broad spectrum of human cancers, including various leukemia cell lines and xenografts).16,17 Of importance, the half-life of histone acetylation after MGCD0103 exposure in animals and in patient peripheral blood mononuclear cells (PBMCs) appears to outlasts pharmacokinetic (PK) exposure.17 This prolonged pharmacodynamic (PD) effect appears to allow for less frequent dosing of MGCD0103. Based on this information, we conducted this open-label, nonrandomized, dose-escalation, multicenter phase 1 trial of oral MGCD0103 administered 3 times a week in patients with leukemia and myelodysplastic syndromes (MDSs).
Methods
The objectives of the study were to assess the safety and tolerability of increasing doses of MGCD0103 when administered to patients with acute or chronic leukemia or MDS, and to determine the maximum tolerated dose (MTD) and dose-limiting toxicities (DLTs). Secondarily, the clinical activity, effects on several potential clinical biomarkers, and PK characteristics of the compound were also evaluated in this patient population.
Patient selection
Patients enrolled in this study were 18 years or older with a diagnosis of relapsed or refractory AML, chronic myelogenous leukemia (CML), acute lymphocytic leukemia (ALL), or MDS (World Health Organization [WHO] classification criteria). Patients 60 years or older with previously untreated AML or MDS, who refused or were not candidates for induction chemotherapy, were also eligible. Patients were required to have an Eastern Cooperative Oncology Group (ECOG) performance status score of 2 or less; have adequate hepatic function (total bilirubin ≤ 34.2 μM [2 mg/dL]; aspartate aminotransferase [AST] or alanine aminotransferase [ALT] ≤ 3× the upper limit of normal); and have adequate renal function (serum creatinine ≤ 176.8 μM [2.0 mg/dL] or a calculated creatinine clearance > 0.8335 mL/s [50 mL/min]; or proteinuria < 2+ on urine dipstick). Patients were excluded from the trial if any of the following criteria were present: other active malignancies or suspicion of central nervous system involvement; pregnancy or breast feeding; serious intercurrent illnesses, medical conditions, or other medical history (including known HIV or hepatitis B or C), which, in the investigator's opinion, would be likely to interfere with a patient's participation in the study or interpretation of the results; treatment with any investigational drug within 30 days before study initiation; concurrent treatment with other experimental drugs or anticancer therapy; known hypersensitivity to any of the components of MGCD0103; and prior treatment with known HDAC inhibitors. The study was conducted at 3 centers in North America: Princess Margaret Hospital (Toronto, ON), Jewish General Hospital, (Montréal, QC), and M. D. Anderson Cancer Center (Houston TX). The study was approved by the local institutional ethics committee of each institution. Informed consent was obtained from all patients per institutional guidelines and in accordance with the Declaration of Helsinki.
Study treatment
The active pharmaceutical ingredient of MGCD0103 was prepared by Torcan (Toronto, ON), and the drug product was prepared by Patheon (Toronto, ON). Finished drug product was provided in 2-mg, 10-mg, and 25-mg gel caps. Patients were instructed to take the intended dose orally 3 times per week on an every other day schedule with 200 mL of a low pH beverage (eg, carbonated drinks). A cycle was defined as 21 days. The initial dose level was 20 mg/m2.
The MTD was defined as the maximum dose at which less than 2 of 6 patients experienced a DLT. If 1 of 3 patients experienced a DLT at a specified dose level, 3 more patients were treated at that dose to confirm that the DLT was not observed in more than 1 of 6 patients. If 0 of 3 or 1 of 6 patients experienced a DLT, accrual began at the next higher dose level. All patients who received 1 dose of drug were evaluable for the safety and PK assessments, and all toxicity assessment for DLT occurred with respect to the first cycle of therapy. For the purpose of DLT and MTD determination, each patient was counted once, at their initial assigned dose. All patients who received at least 1 cycle of therapy were eligible for response assessment unless disease progression had occurred. Patients were allowed to receive supportive and palliative care as clinically indicated throughout the study. The following treatments were not permitted during the study: other anticancer treatment including chemotherapy and radiotherapy, other investigational therapy or antineoplastic agents, or growth factor support for prophylactic use or as a substitute for a scheduled dose reduction.
Toxicities were graded according to the National Cancer Institute (NCI, Bethesda, MD) Common Toxicity Criteria for Adverse Events (CTCAE) version 3.0. Leukemia-specific blood/bone marrow toxicity was used to classify hematologic toxicity. Nonhematologic DLT was defined as any drug-related grade 3 or higher nonhematologic toxicity, except for grade 3 nausea, vomiting, or diarrhea associated with suboptimal premedication and/or management, grade 3 or lower ALT for more than 7 consecutive days, or any drug-associated toxic effect leading to 2 or more missed doses per cycle, or any drug-associated toxic effect resulting in the delay of the subsequent cycle by more than 7 consecutive days. Hematologic DLT was defined as prolonged myelosuppression after therapy administration defined by an absolute neutrophil count of 500 × 109/L (500 000/μL) or less and a platelet count of 30 × 109/L (30 000/μL) or less with a bone marrow cellularity 5% or less without evidence of leukemia involvement lasting for more than 42 days.
Response criteria
Response was assessed using bone marrow aspirates collected before treatment and before the end of cycle 2, or as clinically indicated. A complete response (CR) required an absolute neutrophil count of 1 × 109/L or more, platelet count of 100 × 109/L or more, no blasts in the peripheral blood, bone marrow cellularity of 20% or more with normal trilineage maturation, bone marrow blasts of 5% or less, and absence of extramedullary involvement. PR was considered if there was normalization of peripheral blood counts as for CR, and the complete disappearance of peripheral blasts was observed with more than 5%, but less than 25% blasts in the marrow. A complete marrow response was considered if marrow blasts were 5% or less independent of peripheral counts.
Pharmacokinetic evaluation
The plasma concentration measurements for MGCD0103 were obtained from patients who received oral doses of 20, 40, 60, 70, or 80 mg/m2 3 times weekly for 3 weeks (1 cycle). Total daily doses ranged from 36 mg to 170 mg. Blood samples for evaluation of MGCD0103 PK were collected during cycle 1 on day 1 and day 12 before dosing and at 0.5, 1, 1.5, 2, 3, 4, 6, 8, and 24 hours after dose, and on day 3 of cycle 1 before dosing. Whole blood was collected into a 5-mL sodium heparin Vacutainer tube (Franklin Lakes, NJ) and centrifuged at 2g for 10 minutes at 4°C. Plasma was aliquoted and stored at − 40°C or lower. Samples were analyzed using a validated high-performance liquid chromatography/mass spectrometry (HPLC/MS) method with a detection limit of 0.5 ng/mL. Plasma concentrations of MGCD0103 versus time profiles were generated, and PK parameters were derived using noncompartmental methods with WinNonlin Professional (Pharsight, Mountain View, CA). The following PK parameters were determined: maximum times (Tmax, h) and maximum concentrations (Cmax, ng/mL) of drug in plasma, area under the plasma concentration-time curve (AUC0-24h, ng*h/mL), and elimination half-life of drug in plasma (t½, h).
Isolation of human mononuclear cells
Blood samples were obtained before treatment and on days 1, 3, 8, and 11 of cycle 1; days 1 and 8 of cycles 2 and 4; and at the end of treatment. Whole blood was collected in sodium heparin tubes and shipped at ambient temperature to a central laboratory at MethylGene within 24 hours. Human PBMCs were separated using standard procedures.
Histone acetylation
Histone acetylation analysis, by enzyme-linked immunosorbent assay (ELISA), was performed using nuclear lysate or acid precipitated extracts from patient peripheral white cells. Isolated peripheral white cells for nuclear lysate extraction were washed in phosphate-buffered saline (PBS) and then lysed on ice for 10 minutes in buffer A (10 mM Tris-HCl, pH 8.0, 1.5 mM MgCl2, 5 mM KCl, 0.5% NP-4, protease inhibitors, and sodium butyrate). After centrifugation at 350g for 15 minutes, the nuclear pellet was washed in buffer A and then lysed in nuclear lysis buffer (50 mM HEPES, pH 7.5, 500 mM NaCl, 1% NP-40, 1 mM EDTA, 10% glycerol, protease inhibitors, and sodium butyrate). Samples were sonicated and centrifuged, and the nuclear lysate was transferred to a fresh tube. Isolated peripheral white cells for acid extraction were lysed in buffer A (10 mM Tris-HCl, pH 8.0, 1.5 mM MgCl2, 5 mM KCl, 0.5% NP-4, protease inhibitors, and sodium butyrate). After centrifugation at 350g for 15 minutes, the nuclear pellet was washed in buffer A and resuspended in cold water. Nonhistone proteins were precipitated with 3.3% H2SO4 for 1 hour, then cleared by centrifugation. Acid-soluble proteins were recovered by overnight acetone precipitation and resuspended in H2O.
Sandwich ELISAs from nuclear lysate extracts were performed for determination of histone protein acetylation. ELISA plates (black Maxisorp; Nunc, Rochester, NY) were coated with anti–pan-histone antibody (MAB052; Chemicon Millipore, Billerica, MA) for 2 hours at room temperature. The plates were washed with PBS and blocked with 1% bovine serum albumin plus 0.1% TritonX-100 in PBS. Nuclear lysate extracts (5 μg) were incubated in the plate with rabbit anti–acetyl-H3 antibodies (Chemicon Millipore, Billerica, MA). Detection was with HRP-conjugated goat antirabbit antibody (Sigma-Aldrich, Oakville, ON). The HRP substrate Amplex-Red (Invitrogen Canada, Burlington, ON) was used according to the manufacturer's instructions. ELISAs from purified histones (black Maxisorp, Nunc) were coated with antihistone antibodies (Chemicon Millipore) and blocked with 1% bovine serum albumin plus 0.1% TritonX-100 in PBS. For the H3Ac ELISA, purified histones (2 μg) were incubated in the plate with rabbit anti–acetyl-H3 (Millipore, Billerica, MA); for the total H3 ELISA, purified histones (0.5 μg) were mixed with rabbit anti-H3 (Abcam, Cambridge, MA). For both H3Ac and H3, the detection antibody was HRP-coupled goat antirabbit (Sigma-Aldrich). The HRP substrate Amplex-Red (Invitrogen Canada) was used according to the manufacturer's instructions.
Whole-cell HDAC enzyme activity
Whole-cell HDAC enzyme assays were performed in 96-well plates (Corning, Lowell, CA) by seeding 8 × 105 isolated peripheral white cells per well, in a 50-μL reaction volume. These cells were incubated with 0.3 mM Boc-Lys(ϵ-Ac)-AMC (Bachem, Torrance, CA), a membrane-permeable HDAC substrate. After 1 hour at 37°C with 5% CO2, the reaction was quenched with 1 μM trichostatin A (TSA; BioMol, Plymouth Meeting, PA), cells were lysed with 1% NP-40, and the deacetylated product was cleaved with 1:60 diluted Fluor-de-Lys-Developer (KI-105; BioMol). The reaction was allowed to develop for at least 15 minutes at 37°C with 5% CO2; then the fluorescent signal was detected at Ex 360, Em 470, with a cutoff of 435 on a fluorometer (GeminiXS; Molecular Devices, Sunnyvale, CA). A standard curve of Boc-Lys-AMC (Bachem) allowed the conversion of fluorescent signal into micromoles of deacetylated product.
Statistical methods
PK parameters were calculated based on actual blood sample collection times. MGCD0103 plasma concentrations and PK parameters were summarized using descriptive statistics.
Results
Patient characteristics
In all, 29 patients were enrolled in this study between February 2005 and July 2006. Patient characteristics are shown in Table 1. All 29 patients were eligible for the safety and PK analyses; only 23 patients were eligible for response assessment due to intercurrent illness (3 patients), toxicity (1 patient), or investigator/patient request (2 patients). Patients not eligible for the efficacy evaluation were removed from the study early during the first cycle of treatment. PK parameters were calculated for 27 patients. The most common diagnosis was AML in 22 patients (76%), followed by MDS in 5 patients (17%). Two other patients presented with either ALL or CML. Twenty-four (83%) patients had more than 1 previous chemotherapy regimen including 4 patients (14%) who had also undergone allogeneic stem cell transplantation. Median age was 62 years (range, 32 to 84 years). Cytogenetics were diploid in 11 (38%) of 29 patients, and abnormal in the 18 (62%) of 29 patients, including 2 patients with t(8;21) and 1 with t(9;22).
Table 1.
Patient characteristics, n = 29
| Characteristic | Value |
|---|---|
| Median age, y (range) | 65 (32-84) |
| Female:male | 13:16 |
| Diagnosis, no. (%) | |
| AML | 22 (75.9) |
| MDS | 5 (17.2) |
| ALL | 1 (3.4) |
| CML | 1 (3.4) |
| Median baseline WBC, ×109/L (range) | 5.7 (0.3-57.8) |
| Cytogenetics, n (%) | |
| Diploid | 11 (39) |
| Other | 15 (47) |
| t(8;21) | 2 (8) |
| t(9;22) | 1 (4) |
| Prior treatment | |
| Median (range) | 2 (0-5) |
| Untreated (%) | 5 (17) |
| 1 (%) | 8 (28) |
| 2 or more (%) | 16 (55) |
AML indicates acute myelogenous leukemia; MDSs, myelodysplastic syndromes; ALL, acute lymphocytic leukemia; and CML, chronic myelogenous leukemia.
MGCD0103 dose escalation and toxicities
The most frequently reported adverse events were fatigue (22/29 patients, 75.9%), nausea (20/29 patients, 69%), diarrhea (18/29 patients, 62%), vomiting (14/29 patients, 48.3%), and dyspnea (13/29 patients, 44.8%). The number of drug-related toxicities during cycle 1 of therapy by dose of MGCD0103 is shown in Tables 2,3. No DLTs were observed with 20- or 40-mg/m2 doses. Grade 3 diarrhea, vomiting, and/or fatigue/weakness were observed in 3 of 4 patients receiving a dose of 80 mg/m2 3 times per week. Therefore, 80 mg/m2 was considered to have exceeded the MTD. A dose of 60 mg/m2 3 times per week was then evaluated. Three new patients were enrolled at 60 mg/m2 per day with no significant toxicities observed. The 60 mg/m2 cohort was expanded to include 8 additional patients. No significant toxicities were observed in any patient at this dose level. Therefore, a new cohort was opened at 70 mg/m2 per protocol. Of 6 patients treated at this dose level, 3 experienced DLTs (mucositis, acid reflux/gastritis, hip/leg pain in 1 patient each). Two of the patients treated at 80 mg/m2 continued therapy at a reduced dose of 60 mg/m2 without further significant toxicity.
Table 2.
Nonhematologic drug-related toxicities in patients treated with MGCD0103
| MGCD0103, mg/m2 | No. of patients | No. patients with 1st cycle DLT | NCI-CTCAE grade, no. of patients by worst grade—all cycles |
||||
|---|---|---|---|---|---|---|---|
| 0 | 1 | 2 | 3 | 4 | |||
| 20 | 3 | 0 | 1 | 2 | 0 | 0 | 0 |
| 40 | 5 | 0 | 3 | 2 | 0 | 0 | 0 |
| 60 | 11 | 0 | 3 | 0 | 6 | 2 | 0 |
| 70 | 6 | 3 | 1 | 0 | 2 | 3 | 0 |
| 80 | 4 | 3 | 0 | 0 | 1 | 3 | 0 |
| Total | 29 | 6 | 8 | 4 | 9 | 8 | 0 |
Total number of treatment cycles in the study was 56; the median number of treatment cycles was 1 (range, 1-6). Other drug-related grade 3 nonhematologic toxicities occurring in 2 or fewer patients included gastrointestinal reflux disease in 2 patients and mucosal inflammation, abdominal distension, arthralgia, bacteremia, constipation, gastritis, hematuria, lower GI hemorrhage, and extremity pain occurring in 1 patient each. No grade 4 or 5 toxicities were reported.
Table 3.
Most common (≥ 10%) drug-related nonhematologic toxicities, n = 29
| Toxicity | Total | NCI-CTCAE grade, no. of patients by worst grade |
|||
|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | ||
| Nausea | 15 | 7 | 7 | 1 | 0 |
| Diarrhea | 14 | 6 | 7 | 1 | 0 |
| Fatigue | 14 | 2 | 5 | 7 | 0 |
| Vomiting | 6 | 1 | 4 | 1 | 0 |
| Abdominal pain | 6 | 1 | 3 | 2 | 0 |
| Anorexia | 3 | 2 | 1 | 0 | 0 |
Nonhematologic drug-related adverse events are also listed in Tables 2 and 3. Fatigue (7 patients) and abdominal pain (2 patients) were the most common grade 3 (severe) nonhematologic drug-related toxicities observed in patients receiving MGCD0103. Grade 3 nausea, vomiting, or diarrhea occurred in 1 patient each. There were no grade 4 (life-threatening) nonhematologic drug-related adverse events.
Clinical activity
Twenty-three patients were evaluable for efficacy. One patient receiving 60 mg/m2 and 2 patients initially receiving 80 mg/m2 followed by a dose reduction to 60 mg/m2 achieved a complete bone marrow response (Tables 4,5). The time to response was 1 to 2 cycles, and responses lasted for 1 to 3 cycles. No responses were observed in patients in dose groups receiving 20 mg/m2 and 40 mg/m2 MGCD0103 3 times a week. A summary of the characteristics of the patients who responded to MGCD0103 are also listed in Tables 4 and 5. Of the 3 responders, 1 patient had refractory anemia with excess blasts (RAEBs) and had received no prior therapy, and the other 2 patients had refractory AML and had received prior chemotherapy. For 2 of the responding patients the cytogenetics were diploid, and they were complex for the other responding patient. The 2 patients initiated at the 80-mg/m2 dose of MGCD0103 were subsequently reduced to 60 mg/m2 for the majority of their treatment period due to gastrointestinal toxicity. The total duration of therapy ranged from 4 to 5 cycles for responders. Therapy was eventually discontinued in the responding patients for disease progression or lack of correction of peripheral blood counts. All 3 responding patients exhibited histone acetylation and HDAC inhibition in peripheral blood cells.
Table 4.
Clinical response to MGCD0103
| MGCD0103, mg/m2 | No. | Evaluable,* no. | Marrow response,† no. |
|---|---|---|---|
| 20 | 3 | 3 | 0 |
| 40 | 5 | 5 | 0 |
| 60 | 11 | 9 | 1 |
| 70 | 6 | 3 | 0 |
| 80 | 4 | 3 | 2 |
| Total | 29 | 23 | 3 |
More than 1 week of treatment.
Marrow response indicates complete bone marrow response, blasts 5% or less.
Table 5.
Characteristics of patients responding to MGCD0103
| Patient number |
|||
|---|---|---|---|
| 1 | 2 | 3 | |
| Age, y | 76 | 52 | 58 |
| Sex | Male | Female | Male |
| Disease | MDS; INT-1 | AML | AML |
| Cytogenetics | Diploid | Complex, 43-46, X, −X, add (3) (q35), +10, t(11;12) (p13;p12) | Diploid |
| Prior treatment | None | Ida + Ara-C; high-dose Ara-C | Decitabine; Ida + Ara-C; Fludara + Ara-C |
| Treatment course | 80 to 60 mg/m2 | 80 to 60 mg/m2 | 60 mg/m2 |
| Time to response | 2 cycles | 2 cycles | 1 cycle |
| Response duration | No repeat aspirate | 3 cycles | 1 cycle |
| On study duration | 4 cycles | 5 cycles | 4 cycles |
| Max HDAC inhibition, %* | 23 | 63 | 38 |
| Max histone acetylation, %* | 149 | 337 | 338 |
Inhibition of HDAC activity and histone acetylation were measured in peripheral white cells collected from the patients (“Histone acetylation”).
Pharmacokinetics
Plasma MGCD0103 concentrations were measured serially up to 24 hours after dosing on day 1 to obtain single dose profiles, and on day 12 to obtain “steady-state” profiles. PK data were available from all 27 patients on day 1 and from 13 patients on day 12. MGCD0103 plasma concentration-time profiles for day 1 are presented in Figure 1A,B, and for day 12 in Figure 1C,D. MGCD0103 was rapidly absorbed. On day 1, the maximum plasma concentrations occurred between 0.5 hour and 1 hour after dose. The drug concentration decreased biphasically and remained quantifiable at 24 hours. Similar profiles were observed after dosing on day 12.
Figure 1.

Blood samples for evaluation of MGCD0103 pharmacokinetics. Samples were collected during cycle 1 on day 1 and day 12 before dosing and at 0.5, 1, 1.5, 2, 3, 4, 6, 8, and 24 hours after dose. Samples were analyzed using a validated HPLC/MS method. Dosing of MGCD0103 was 2 times per week. (A,B) Plasma concentration-time profiles for day 1; n = 27 patients. (C,D) Plasma concentration-time profiles for day 12; n = 13 patients.
MGCD0103 plasma PK parameters are presented in Figure 2 and Table S1 (available on the Blood website; see the Supplemental Materials link at the top of the online article). Cmax values and exposure to drug (AUC) increased with dose up to the 60-mg/m2 dose level. Although data were sparse at the higher dose levels, Cmax and AUC values appeared to plateau or, in some cases, even decrease following doses of 70 and 80 mg/m2. The drug had an elimination half-life in plasma of 9 plus or minus 2 hours. Median Tmax ranged from 0.5 hour to 1.2 hours after single or multiple doses of drug, whereas mean t½ values ranged from 7 to 11 hours. Mean clearance (CL/F) was 123 (± 47) L/hour (%CV = 38) on day 1. Individual patient Cmax (Figure 2A,B), AUC(0-24h) (Figure 2C,D), t½ (Figure 2E,F), and Tmax (Figure 2G,H) values are plotted versus dose (expressed in mg/m2 and in mg). These plots (Figure 2A-D) show a general trend of an increase in exposure as a function of dose, and illustrate the apparent plateau or downward trend at the higher dose levels. T½ and Tmax (Figure 2E-H) appear independent of dose, as would be expected. Some interpatient variability was evident.
Figure 2.
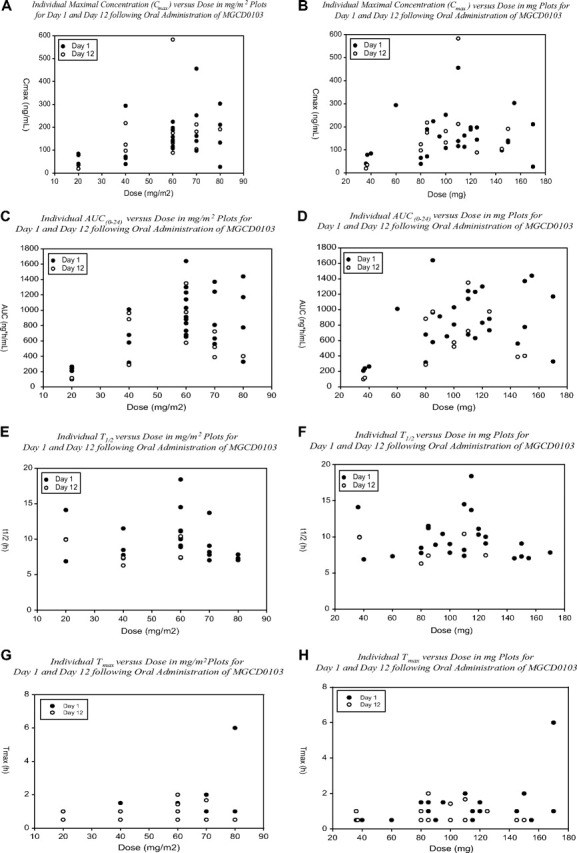
Relationships of MGCD0103 Cmax and AUC with dose. Pharmacokinetic parameters were derived using noncompartmental methods with WinNonlin Professional (Pharsight). (A,B) Cmax dose profiles for day 1 and day 12. (C,D) Individual AUC(0-24) dose relationships for day 1 and day 12 after MGCD0103 treatment 3 times per week. (E,F) Individual T½ profiles on day 1 and day 12. (G,H) Individual Tmax profiles on day 1 and day 12.
An evaluation of PK exposure across all MGCD0103 phase 1 studies was performed (data not shown) comparing predicted exposure (based on actual dose given) with body surface area (BSA). There was a minor trend toward inverse correlation between exposure and BSA. However, this was insignificant relative to the interpatient variability and is felt not to be clinically significant to predicting a given patient's likely exposure. In this evaluation, patients with a very low BSA (≤ 1.6) were not more likely to have excess drug exposure, whereas larger patients (BSA ≥ 2.2) were not more likely to be underexposed. When the pharmacokinetic parameters AUC or Cmax were compared in patients not experiencing first-cycle DLT with patients experiencing first-cycle DLT (Figure 3A,B), a clear correlation was not observed. However, the first-cycle DLT in patients did correlate with the total daily dose they received (Figure 3C).
Figure 3.

Correlation of AUC, Cmax, and total daily dose with DLT. Individual patients are plotted along the x-axis sorted by AUC (A), Cmax (B), or total daily dose (C). Patients with no DLT during the first cycle of treatment are represented by X; patients who experienced a DLT during the first cycle are represented by a ●.
Histone acetylation and inhibition of whole-cell HDAC enzyme activity
Histone H3 acetylation levels and whole-cell HDAC enzyme activity were measured before and after treatment in peripheral white cells as potential PD markers for MGCD0103 activity. In cycle 1, there was induction of average histone H3 acetylation observed 24 hours after an initial dose of MGCD0103, with maximal levels at 60 mg/m2, and these levels reached a plateau after administration of a dose of 60 mg/m2, the MTD (1.3- to 1.5-fold induction, Figure 4A). For all groups more than 40 mg/m2, 1 of 3 patients of each group was positive for induction. The average percentage inhibition of HDAC enzyme activity during cycle 1 was increased in a dose-dependent manner up to the 40-mg/m2 dose (group at 20 mg/m2 vs group at 40 mg/m2; P = .085), where it reached a plateau at approximately 20% to 25% inhibition compared with the baseline (Figure 4B).
Figure 4.
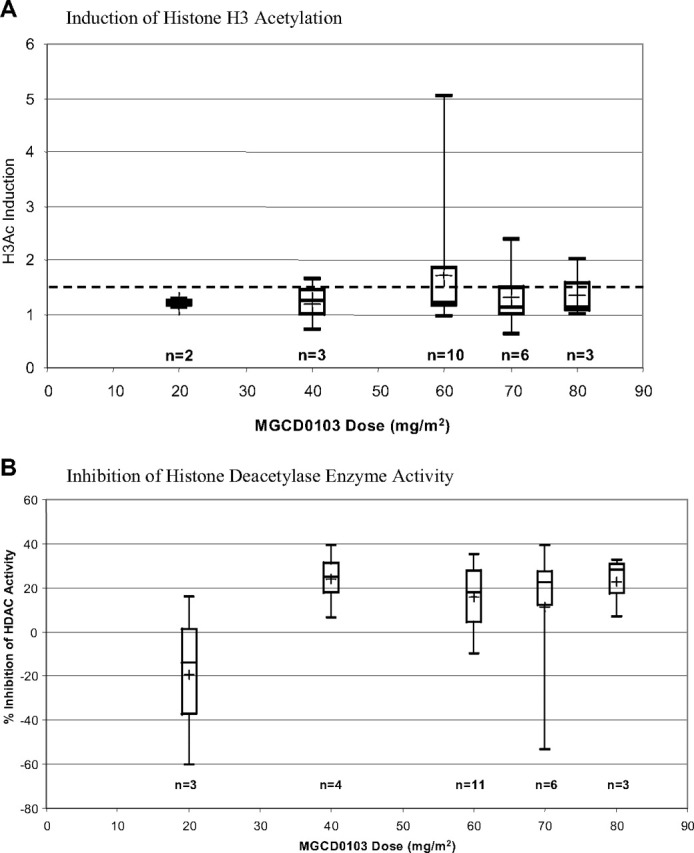
Blood samples from patients were obtained before treatment and on days 1, 3, and 8 of cycle 1. Whole blood was collected in sodium heparin Vacutainer tubes and shipped at ambient temperature to a central laboratory at MethylGene within 24 hours. Peripheral white cells were isolated using standard procedures. (A) Average histone H3 acetylation. Induction of histone acetylation was analyzed using an enzyme-linked immunosorbent assay (ELISA). Nonbiased data with standard error are shown. (B) Average percentage inhibition of HDAC enzyme activity. Whole-cell HDAC enzyme assays were performed using 8 × 105 isolated peripheral white cells per well, which were incubated with 0.3 mM Boc-Lys(ϵ-Ac)-AMC, a membrane-permeable HDAC substrate. After 1 hour at 37°C with 5% CO2, the reaction was quenched with 1 μM TSA, cells were lysed with 1% NP-40, and the deacetylated product was cleaved with Fluor-de-Lys-Developer. The reaction was allowed to develop for at least 15 minutes at 37°C with 5% CO2; then the fluorescent signal was read at Ex 360, Em 470, with cutoff of 435 nm on a fluorometer. Nonbiased data with standard error are shown.
Discussion
In preclinical studies, several structurally diverse HDAC inhibitors, including MGCD010317; depsipeptide18; hydroxamic acid HDAC inhibitors (SAHA [vorinostat],11,19,20 LAQ824,21 and PXD10122); and aminobenzamide HDAC inhibitors (CI-99423,24and MS-27512) have been found to have potent antitumor activities, tumor specificity, and promising therapeutic potential in early phase clinical trials. SAHA is a class I and class II nonspecific hydroxamic acid HDAC inhibitor approved by FDA for the treatment of cutaneous manifestations of advanced refractory cutaneous T-cell lymphoma.25 Phase 1 clinical trials with these agents have determined that the more common toxicities with HDAC inhibitors are fatigue, gastrointestinal side effects, and dose-related transient cytopenias, and, to a lesser extent in a subset of these agents, cardiac toxicity (eg, QTc prolongation). At the present time, several HDAC inhibitors are in development both in leukemias and other indications. Structurally and at the biologic level, these agents are quite diverse, and it is not currently known whether one class of agents is better than others.
This study demonstrates that MGCD0103 administered orally 3 times a week is safe and active in patients with relapsed or refractory leukemia. Dose-limiting toxicities included primarily fatigue, nausea, vomiting, and diarrhea, a symptom cluster that has been observed previously with this class of agents.11 These toxicities, however, were non–life threatening and were effectively addressed by dosing delay and dose reduction. There were no grade 4 drug-related adverse events noted in this study. The MTD was 60 mg/m2 with this dosing regimen, which is approximately equivalent to a 110-mg flat dose. The findings that the PK reached a plateau at higher doses, and that the incidence of DLTs tracked most closely with total daily dose rather than PK exposure, collectively suggest that unabsorbed drug may be limiting at very high doses.
We also demonstrated that MGCD0103 had favorable PK properties in this patient population with this dosing schedule. The drug was rapidly absorbed within 1 hour after oral administration, with a median Tmax ranging from 0.5 hour to 1.2 hours after single or multiple doses. Importantly, the drug had a long elimination half-life in plasma of 9 hours, and remained quantifiable at 24 hours after a single dose. Drug exposure increased with doses up to 60 mg/m2, and then did not increase significantly with further increases in dose. PK exposure at the 60 mg/m2 dose level exceeded the efficacious exposure in mouse xenograft models.17 These properties, along with the biologic effect of prolonged histone acetylation, allowed for the thrice weekly dosing schedule.
The PD activity of HDAC inhibition was explored in this study. In addition to the traditional method of measuring histone acetylation in cell lysates, a novel whole-cell enzyme assay was developed to monitor HDAC activity in peripheral white cells obtained from the clinical setting.25 This assay used a cell-permeable substrate with a fluorescent readout, thus allowing evaluation of HDAC activity in live-cell populations. Considering the intricate complexity of transcriptional complexes that are disrupted in the process of measuring histone acetylation by traditional methods, this novel whole-cell HDAC activity assay maintains the integrity of these transcriptional and other protein-DNA complexes, and significantly contributes to the armamentarium of tools to study the PD effects of HDAC inhibitors. This assay has been validated in a variety of systems, and in patients with solid tumors, HDAC enzyme inhibition was dose dependent and correlated well with drug accumulation in plasma.17,26 The true HDAC inhibition in peripheral white cells in leukemia patients may in fact be underestimated, as the apoptosis of peripheral blast cells may reduce the cell population most sensitive to drug treatment, particularly at time points after prolonged exposure to drug. Once further validated, this method could be adapted into a high throughput clinical system to follow HDAC activity in real time and help facilitate therapeutic decision-making. Other assays exist, however, that could be used to asses HDAC inhibitory activity such as Western blotting,27,28 ELISA,20 or new assays.29 From the cumulative data published so far, it appears that histone acetylation is universal with these potent HDAC inhibitors, but that histone acetylation does not show a simple correlation with response.20,27,28 It should be noted that in this study, we did not analyze histone H4 acetylation. The results of the assays used for pharmacodynamic analysis are difficult to correlate with the pharmacokinetic characteristics of the drug, and it appears that there is a dissociation between the PD properties and the PK characteristics of MGCD010330 and other HDAC inhibitors.20 This PD/PK dissociation has implications for clinical trial development, as other schedules (daily or more prolonged exposure) may be beneficial. Studies examining these issues are ongoing. It is possible that cell selection, for example CD34+ cells, may provide a more precise assessment of molecular effects secondary to therapy. Although using these cells is possible, prior phase 1 studies conducted by our group have failed to detect differences between peripheral and bone marrow biomarker expression in patients with AML or high-risk MDS.20,27,28
Clinical activity with MGCD0103 was observed in this population of patients with refractory, relapsed acute leukemia and MDS. Three patients on the study achieved a complete bone marrow response at doses of 60 mg/m2 and higher, suggesting a possible dose response at these levels. Of significance, 2 of the 3 responding patients had been previously treated with chemotherapy for refractory AML. The time to response observed in this study was 1 to 2 cycles, with the duration of response ranging from 1 to 3 cycles. All 3 patients had histone acetylation and HDAC inhibition as measured by the PD assays, but these limited observations could not necessarily be statistically correlated with response. Although preliminary, the single agent activity of this oral, isotype-specific HDAC inhibitor in such a high-risk, refractory population is notable and should be investigated further in efficacy trials.
In summary, we demonstrate that MGCD0103 is safe, and has clinical activity when administered orally as a single agent in patients with heavily pretreated AML, and in a patient with untreated intermediate-1 (INT-1) MDS. It is important to emphasize that MGCD0103 is highly selective for HDACs 1, 2, 3, and 11, with negligible ability (IC50 > 10 μM) to inhibit class II HDACs. Because preclinical work demonstrates distinct biologic sequelae from different spectra of HDAC inhibition and a significant body of evidence implicates HDAC 1 in cancer, we hypothesize that an isotype-selective HDAC inhibitor will improve the therapeutic window, thus allowing greater efficacy for a given amount of toxicity when HDACs are targeted. The PK properties of MGCD0103 allow for administration in a thrice weekly schedule and maintenance of the biologic effect of histone acetylation. Further preclinical and clinical studies with MGCD0103 alone and in combination with other cytotoxic and or targeted therapies will help define the best timing and possible synergistic activity. Subsequent controlled studies will be necessary to characterize the magnitude of this activity.
Acknowledgments
This work was supported by MethylGene (Montreal, QC). G.G.-M. is supported in part by a Physician-Scientist Award from the Commonwealth Cancer Foundation for Research (Houston, TX) and the Leukemia & Lymphoma Society of America (New York, NY).
Footnotes
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: G.G.-M. and M.M. designed the study, wrote the paper, treated patients on study, and analyzed data; S.A., J.C., Z.E., H.K., W.M.N., W.H.M., and C.R. treated patients on study and analyzed data; H.Y., A.K., C.B., M.D., T.-A.P., Z.L., J.M.B., G.R., and E.L. performed correlative studies and/or analyzed data; and R.E.M. designed the study, analyzed data, and wrote the paper.
Conflict-of-interest disclosure: A.K., C.B., M.D., T.-A.P., Z.L., J.M.B., G.R., and R.E.M. are employees of MethylGene, and E.L. is an employee of Pharmion. The remaining authors declare no competing financial interests.
Correspondence: Guillermo Garcia-Manero, Department of Leukemia, University of Texas M. D. Anderson Cancer Center, 1515 Holcombe Blvd, Houston, TX 77030; e-mail: ggarciam@mdanderson.org.
References
- 1.Csordas A. On the biological role of histone acetylation. Biochem J. 1990;265:23–38. doi: 10.1042/bj2650023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Grunstein M. Histone acetylation in chromatin structure and transcription. Nature. 1997;389:349–352. doi: 10.1038/38664. [DOI] [PubMed] [Google Scholar]
- 3.Hassig CA, Schreiber SL. Nuclear histone acetylases and deacetylases and transcriptional regulation: HATs off to HDACs. Curr Opin Chem Biol. 1997;1:300–308. doi: 10.1016/s1367-5931(97)80066-x. [DOI] [PubMed] [Google Scholar]
- 4.Kouzarides T. Histone acetylases and deacetylases in cell proliferation. Curr Opin Genet Dev. 1999;9:40–48. doi: 10.1016/s0959-437x(99)80006-9. [DOI] [PubMed] [Google Scholar]
- 5.Karagiannis TC, El-Osta A. Will broad-spectrum histone deacetylase inhibitors be superseded by more specific compounds? Leukemia. 2007;21:61–65. doi: 10.1038/sj.leu.2404464. [DOI] [PubMed] [Google Scholar]
- 6.Bolden JE, Peart MJ, Johnstone RW. Anticancer activities of histone deacetylase inhibitors. Nat Rev Drug Discov. 2006;5:769–784. doi: 10.1038/nrd2133. [DOI] [PubMed] [Google Scholar]
- 7.Johnstone RW. Histone-deacetylase inhibitors: novel drugs for the treatment of cancer. Nat Rev Drug Discov. 2002;1:287–299. doi: 10.1038/nrd772. [DOI] [PubMed] [Google Scholar]
- 8.Meinke PT, Liberator P. Histone deacetylase: a target for antiproliferative and antiprotozoal agents. Curr Med Chem. 2001;8:211–235. doi: 10.2174/0929867013373787. [DOI] [PubMed] [Google Scholar]
- 9.Plumb JA, Finn PW, Williams RJ, et al. Pharmacodynamic response and inhibition of growth of human tumor xenografts by the novel histone deacetylase inhibitor PXD101. Mol Cancer Ther. 2003;2:721–728. [PubMed] [Google Scholar]
- 10.Remiszewski SW. The discovery of NVP-LAQ824: from concept to clinic. Curr Med Chem. 2003;10:2393–2402. doi: 10.2174/0929867033456675. [DOI] [PubMed] [Google Scholar]
- 11.Kelly WK, Richon VM, O'Connor O, et al. Phase I clinical trial of histone deacetylase inhibitor: suberoylanilide hydroxamic acid administered intravenously. Clin Cancer Res. 2003;9:3578–3588. [PubMed] [Google Scholar]
- 12.Gojo I, Jiemjit A, Trepel JB, et al. Phase 1 and pharmacologic study of MS-275, a histone deacetylase inhibitor, in adults with refractory and relapsed acute leukemias. Blood. 2007;109:2781–2790. doi: 10.1182/blood-2006-05-021873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ryan QC, Headlee D, Acharya M, et al. Phase I and pharmacokinetic study of MS-275, a histone deacetylase inhibitor, in patients with advanced and refractory solid tumors or lymphoma. J Clin Oncol. 2005;23:3912–3922. doi: 10.1200/JCO.2005.02.188. [DOI] [PubMed] [Google Scholar]
- 14.Li Z, Zhou N, Fournel M, et al. Antitumor activities of MGCD0103, a novel and isotype-selective histone deacetylase inhibitor. Eur J Cancer Supp Mol Targets Cancer Ther. 2004;2:83. Abstract. [Google Scholar]
- 15.Moradei O, Leit S, Zhou N, et al. Substituted N-(2-aminophenyl)-benzamides, (E)-N-(2-aminophenyl)-acrylamides and their analogues: novel classes of histone deacetylase inhibitors. Bioorg Med Chem Lett. 2006;16:4048–4052. doi: 10.1016/j.bmcl.2006.05.005. [DOI] [PubMed] [Google Scholar]
- 16.Fournel M, Bonfils C, Hou Y, et al. MGCD0103, a novel isotype-selective histone deacetylase inhibitor, has broad spectrum antitumor activity in vitro and in vivo. Mol Cancer Ther. 2008;7:759–768. doi: 10.1158/1535-7163.MCT-07-2026. [DOI] [PubMed] [Google Scholar]
- 17.Bonfils C, Kalita A, Liu J, et al. Evaluation of the pharmacodynamic effect of MGCD0103 using a whole cell histone deacetylase enzyme assay. Clin Cancer Res. 2008;14:3441–3449. doi: 10.1158/1078-0432.CCR-07-4427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Byrd JC, Marcucci G, Parthun MR, et al. A phase 1 and pharmacodynamic study of depsipeptide (FK228) in chronic lymphocytic leukemia and acute myeloid leukemia. Blood. 2005;105:959–967. doi: 10.1182/blood-2004-05-1693. [DOI] [PubMed] [Google Scholar]
- 19.Kelly WK, O'Connor OA, Krug LM, et al. Phase I study of an oral histone deacetylase inhibitor, suberoylanilide hydroxamic acid, in patients with advanced cancer. J Clin Oncol. 2005;23:3923–3931. doi: 10.1200/JCO.2005.14.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Garcia-Manero G, Yang H, Bueso-Ramos C, et al. Phase 1 study of the histone deacetylase inhibitor vorinostat (suberoylanilide hydroxamic acid [SAHA]) in patients with advanced leukemias and myelodysplastic syndromes. Blood. 2008;111:1060–1066. doi: 10.1182/blood-2007-06-098061. [DOI] [PubMed] [Google Scholar]
- 21.Kato Y, Salumbides BC, Wang XF, et al. Antitumor effect of the histone deacetylase inhibitor LAQ824 in combination with 13-cis-retinoic acid in human malignant melanoma. Mol Cancer Ther. 2007;6:70–81. doi: 10.1158/1535-7163.MCT-06-0125. [DOI] [PubMed] [Google Scholar]
- 22.Qian X, LaRochelle WJ, Ara G, et al. Activity of PXD101, a histone deacetylase inhibitor, in preclinical ovarian cancer studies. Mol Cancer Ther. 2006;5:2086–2095. doi: 10.1158/1535-7163.MCT-06-0111. [DOI] [PubMed] [Google Scholar]
- 23.Prakash S, Foster BJ, Meyer M, et al. Chronic oral administration of CI-994: a phase 1 study. Invest New Drugs. 2001;19:1–11. doi: 10.1023/a:1006489328324. [DOI] [PubMed] [Google Scholar]
- 24.Richards DA, Boehm KA, Waterhouse DM, et al. Gemcitabine plus CI-994 offers no advantage over gemcitabine alone in the treatment of patients with advanced pancreatic cancer: results of a phase II randomized, double-blind, placebo-controlled, multicenter study. Ann Oncol. 2006;17:1096–1102. doi: 10.1093/annonc/mdl081. [DOI] [PubMed] [Google Scholar]
- 25.Duvic M, Talpur R, Ni X, et al. Phase 2 trial of oral vorinostat (suberoylanilide hydroxamic acid, SAHA) for refractory cutaneous T-cell lymphoma (CTCL). Blood. 2007;109:31–39. doi: 10.1182/blood-2006-06-025999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Siu L, Pili R, Duran I, et al. A phase I study of MGCD0103 given as a three-times per week oral dose in patients with advanced solid tumors. J Clin Oncol. 2008;26:1940–1947. doi: 10.1200/JCO.2007.14.5730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Soriano AO, Yang H, Faderl S, et al. Safety and clinical activity of the combination of 5-azacytidine, valproic acid, and all-trans retinoic acid in acute myeloid leukemia and myelodysplastic syndrome. Blood. 2007;110:2302–2308. doi: 10.1182/blood-2007-03-078576. [DOI] [PubMed] [Google Scholar]
- 28.Garcia-Manero G, Kantarjian HM, Sanchez-Gonzalez B, et al. Phase ½ study of the combination of 5-aza-2′-deoxycytidine with valproic acid in patients with leukemia. Blood. 2006;108:3271–3279. doi: 10.1182/blood-2006-03-009142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chung EJ, Lee MJ, Lee S, Trepel JB. Assays for pharmacodynamic analysis of histone deacetylase inhibitors. Expert Opin Drug Metab Toxicol. 2006;2:213–230. doi: 10.1517/17425255.2.2.213. [DOI] [PubMed] [Google Scholar]
- 30.Zhou N, Moradei O, Raeppel S, et al. Discovery of N-(2-aminophenyl)-4-[(4-pyridin-3-ylpyrimidin-2-ylamino)methyl]benzamide (MGCD0103), an orally active histone deacethylase inhibitor. J Med Chem. 2008;51:4072–4075. doi: 10.1021/jm800251w. [DOI] [PubMed] [Google Scholar]