

Abstract
Background
Melanoma is a heterogeneous tumor with subgroups requiring distinct therapeutic strategies. Genetic dissection of melanoma subgroups and identification of therapeutic agents are of great interest in the field. These efforts will ultimately lead to treatment strategies, likely combinatorial, based on genetic information.
Methods
To identify “driver” genes that can be targeted therapeutically, we screened metastatic melanomas for somatic mutations by exome sequencing followed by selecting those with available targeted therapies directed to the gene product or its functional partner. The FBXW7 gene and its substrate NOTCH1 were identified and further examined. Mutation profiling of FBXW7, biological relevance of these mutations and its inactivation, and pharmacological inhibition of NOTCH1 were examined using in vitro and in vivo assays.
Results
We found FBXW7 to be mutated in eight (8.1%) melanoma patients in our cohort (n = 103). Protein expression analysis in human tissue samples (n = 96) and melanoma cell lines (n = 20) showed FBXW7 inactivation as a common event in melanoma (40.0% of cell lines). As a result of FBXW7 loss, we observed an accumulation of its substrates, such as NOTCH1. Ectopic expression of mutant forms of FBXW7 (by 2.4-fold), as well as silencing of FBXW7 in immortalized melanocytes, accelerated tumor formation in vivo (by 3.9-fold). Its inactivation led to NOTCH1 activation, upregulation of NOTCH1 target genes (by 2.6-fold), and promotion of tumor angiogenesis and resulted in tumor shrinkage upon NOTCH1 inhibition (by fivefold).
Conclusions
Our data provides evidence on FBXW7 as a critical tumor suppressor mutated and inactivated in melanoma that results in sustained NOTCH1 activation and renders NOTCH signaling inhibition as a promising therapeutic strategy in this setting.
Metastatic melanoma is a lethal malignancy leading to an estimated 9480 deaths annually in the United States (1). BRAF and NRAS are bona fide oncogenes frequently mutated in melanoma (2). BRAF inhibitors represent the prototype of targeted therapies in melanoma; however they have met with limited success because of rapid emergence of acquired resistance (3). Patients eventually relapse, rendering the disease incurable. Novel therapeutic strategies remain as a great interest in the field. Metastatic melanomas have high mutational load and intricate signaling networks (4,5). Heterogeneity of the disease adds another layer of complexity. It is plausible that undefined genetic events representing novel potential targets are sequestered within the complex landscape of genetic events in melanoma. Thus, beyond recurrent mutated genes with high frequencies, these may be potential targets that are not so obvious but relevant to a subset of patients.
FBXW7 is a member of the F-box protein family (6). The F-box proteins constitute one of the four subunits of ubiquitin protein ligase complex called SCFs (SKP1-cullin-F-box), which function in phosphorylation-dependent ubiquitination and regulate a network of proteins with central roles in cell division, cell growth, and differentiation (7). The FBXW7 protein comprises three functionally critical domains—the dimerization domain (8), the F-box domain that allows the physical interaction of FBXW7 with the SCF complex (7), and the WD40 domain containing eight tandem repeats that form a β-propeller structure that recognizes a specific consensus phosphodegron motif within the target substrate (9,10). Substrates of FBXW7 include known oncoproteins such as NOTCH1 (11–13). Homozygous null mice for FBXW7 are early embryonic lethal, implicating its involvement with critical cellular functions (14).
In this article, we describe identification of FBXW7 as a “driver” genetic event in an exome sequencing screen, characterize its functional impact in melanoma, and highlight its substrate, NOTCH1, as a relevant therapeutic target in this setting.
Methods
Additional methods are available in the Supplementary Methods (available online).
Exome Sequencing
Genomic DNA was extracted from fresh-frozen melanomas and matching peripheral blood lymphocytes (Qiagen, Valencia, CA); this was followed by whole-exome sequencing as previously described (15) using a HiSeq 2500 system (Illumina, San Diego, CA). In eight melanomas with paired blood samples, an average of 42 million reads per sample (n = 32 million –101 million) was found, of which 98.4% mapped to the hg19 genome using Burrows-Wheeler Aligner 0.5.9-r16, followed by the Genome Analysis Tool Kit indel realignment, resulting in an average depth of 11 reads per base covered at depth greater than zero. Using the statistical algorithm for variant frequency identification (16), we called positions with nucleotide mutations. We retained only the variants at positions with depth greater than 10 in both tumor and normal samples and filtered out variants that appeared in normal samples in more than 25% of the reads. We identified a total of 2308 exonic mutations (n = 737 synonymous; n = 1571 nonsynonymous) that consisted of of 1431 missense and 78 nonsense mutations and 62 insertions/deletions. The mean exonic nonsynonymous mutation rate was 10.6 mutations per megabase, with mutation rates varying from 2.8 to 26.7. The majority of nucleotide substitutions were C>T or G>A transitions (73% to 91% of all mutations), indicative of ultraviolet-induced damage. The hot spot mutation, BRAFV600E, was present in six of the eight melanomas.
Selection of FBXW7
After sequencing and variant calling, we used the collection of nonsynonymous and synonymous exome mutations to identify genes with evidence of positive selection for nonsynonymous variants. First, we evaluated whether a gene had more nonsynonymous mutations than would be expected given the number of synonymous mutations. We estimated the expected number of nonsynonymous mutations for each gene using the number of synonymous mutations in that gene, NS,G, and the nonsynonymous to synonymous mutation ratio across all genes, NN/NS, resulting in an expected number of nonsynonymous mutations of NS,G*NN/NS. We evaluated whether the observed number of nonsynonymous mutations, NN,G, was statistically significantly more than this expected value using a Poisson model. Our second test for selection considered whether the number of nonsynonymous mutations per amino acid was elevated to account for coding length. We calculated the probability of the observed findings using a binomial model, where the amino acid length represented the number of trials and the probability of a nonsynonymous mutation was calculated from the average number of nonsynonymous mutations per amino acid across the genes with mutations. Finally, our candidate list contained genes that had P less than .05 in both tests, which provided a list of 23 putative melanoma driver genes. To find interacting partners, we used GeneRIF interactions (17), keeping only those that were documented in human cells and are not based on affinity assays. Then, for each of these interacting partners, we used the Cancer Commons (18) drug target database to assess whether any drugs targeting those genes were documented. Only a subset of the recurrently mutated genes, including FBXW7, had druggable interacting partners.
Quantitative Real-Time Polymerase Chain Reaction
Total RNA was isolated using RNeasy Plus kit (Qiagen). cDNA was synthesized using RNA to cDNA EcoDry Premix (Clontech, Mountain View, CA). SYBR Premix Ex Taq II (Takara, Mountain View, CA) was used for quantitative reverse-transcription polymerase chain reaction (PCR). Amplification and real-time analysis was performed with BioRad CFX96 (BioRad, Hercules, CA). Transcript levels were normalized to the 18S subunit of rRNA levels. The relative mRNA levels were calculated according to the comparative Ct (ΔΔCt) method.
Angiogenesis PCR Array
Changes in expression of angiogenesis-related genes were detected by using RT2 Profiler Mouse Angiogenesis PCR Array (PAMM-024ZD; Qiagen). Total RNA was extracted as explained in the previous section. cDNA was synthesized using RT2 First Strand Kit (Qiagen). RT2 SYBR Green qPCR Master Mix was used for the reaction following manufacturer’s instructions. Amplification and real-time analysis were performed with BioRad CFX96 (BioRad).
Western Blot Analysis
Western blotting was performed using standard protocols. The following commercial antibodies were used in this study: FBXW7 (A301-720A; Bethyl Laboratories, Montgomery, TX), cleaved NOTCH1 (4147; Cell Signaling, Danvers, MA), cyclin E (4129; Cell Signaling), MYC (551101; BD Pharmingen, San Jose, CA), Aurora A (610938; BD Pharmingen), MCL1 (sc-819; Santa Cruz Biotechnology, Santa Cruz, CA), and α-tubulin (CP06-100UG; Oncogene Research Products, Boston, MA).
Immunohistochemistry and Immunofluorescence Analyses
Dual immunofluorescence analyses were conducted on formalin-fixed and paraffin-embedded tissue microarray sections (M1004a; US Biomax, Rockville, MD) using standard protocols.
In Vivo Studies
All animal care was in accordance with institutional guidelines. Five-week-old female NCr nude mice were purchased from Taconic (Hudson, NY). Tumor growth was evaluated after subcutaneous injection of 1×106 melan-a melanocytes expressing NRASG12D together with shScr, shRNA1, or shRNA2 into the flanks of mice. Tumors were measured daily, and mice were killed when tumor surface area reached 1500mm3. The numbers of mice in each group in the in vivo studies is given in the Supplementary Methods (available online).
Statistical Analysis
All quantified data represent an average of triplicate samples or as indicated. Error bars represent standard error of the mean. Statistical significance was determined by the Student t test to compare mean values for continuous variables between the two groups, and the χ2 test was used to compare the frequency distribution of categorical variables. All reported P values were two-sided, and P less than .05 was considered statistically significant.
Results
FBXW7 Mutations in Melanoma
To identify driver genes that can be targeted therapeutically, we developed a strategy that combined defining somatically mutated genes in a discovery cohort, evaluation of potential impact on protein function and therapeutic relevance, validation of mutations in an extension cohort, and functional characterization. Sequencing the exomes of metastatic melanomas identified recurrent somatic mutations in 23 genes (Supplementary Figure 1 and Supplementary Tables 1 and 2, available online). Those with therapies directed to the gene product or its functional partner that are commercially available were selected: BRAF, IL7R, GUCY2C, and FBXW7 (Supplementary Table 3, available online). FBXW7 was further characterized. Substrates of FBXW7 include known oncoproteins such as NOTCH1 (11–13). To assess the prevalence of FBXW7 (NM_018315.4) mutations in melanoma, we sequenced the coding regions of FBXW7 and hot spot regions of BRAF and NRAS in a total of 103 melanomas, including 77 tumor samples and 26 cell lines. We detected nonsynonymous mutations in FBXW7 in eight melanomas (8.1% frequency), including five nonsense, two missense, and one frameshift mutation (Figure 1). This is a statistically significantly elevated mutation rate because the probability of having this number of nonsynonymous mutations given the length of the gene (710 amino acids) and the nonsynonymous mutation rate in our samples (1×10–5) is less than 10−4. Because of the occurrence of the mutations within the WD40 domain of FBXW7 (except the frameshift mutation that is in the F-box domain), it is predicted that these mutations disrupt substrate binding of FBXW7, diminish protein turnover, and thus lead to sustained activation of its substrate oncoproteins. Of note, the presence of mutations in FBXW7 was not associated with BRAF or NRAS mutation status (Supplementary Table 4, available online). Collectively, these findings identify somatic mutations of FBXW7 as a novel recurrent genetic event in melanoma.
Figure 1.
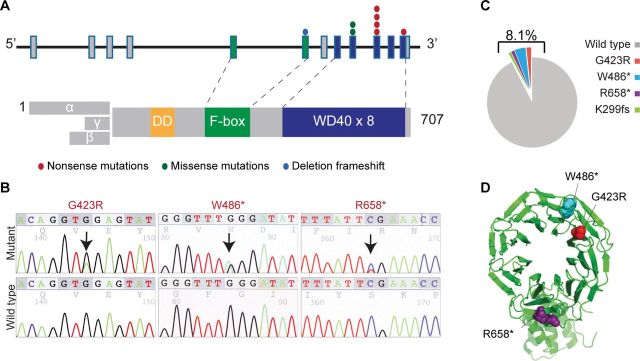
Mutations in the FBXW7 gene in melanoma. A) Schematic diagram of the FBXW7 gene (NM_018315.4) and protein. Exons corresponding to the WD40 and F-box domains are color-coded. Color-coded symbols depict distinct types of mutations. B) Sequencing traces of representative mutated melanoma tumor samples and normal DNA. C) Frequency distribution of FBXW7 mutations among 103 tested samples. D) Crystal structure of the FBXW7 protein showing the G423R (red), W486* (blue), and R658* (purple) mutations.
FBXW7 Expression in Melanoma and NOTCH1 as a Relevant Target
We next examined FBXW7 protein expression by immunofluorescence analysis in tissue microarrays consisting of benign melanocytic nevi (n = 21) and primary (n = 56) and metastatic (n = 19) melanoma samples (total n = 96). There was a statistically significant decrease of nuclear FBXW7 expression in the melanomas (14.7%) as compared with nevi (66.7%) (melanoma vs nevi: odds ratio = 11.63; 95% confidence interval [CI] = 3.83 to 35.31; P < .001 by the χ2 test) (Figure 2, A and B). We also analyzed protein expression in a panel of 20 melanoma cell lines, including eight NRAS-mutant, nine BRAF-mutant, and three wild-type for BRAF and NRAS by western blot analysis. Four lines—WC00125, WM39, WM3702, and WM3862—harbored mutations in FBXW7. Of the 20 melanoma cell lines screened, FBXW7 expression relative to the normal human melanocyte was low (less than onefold) in eight cases (40.0%). In the other twelve cases, expression ranged from 1.06-fold to 2.22-fold (Figure 2, C and D). Protein expression levels were statistically significantly different depending on FBXW7 mutation status; the group of four mutant cell lines had lower levels of expression than the group of sixteen nonmutant cell lines (P = .003 by the t test). Of note, several cell lines were found to express very low or undetectable protein levels in the absence of mutations in the FBXW7 gene. Thus, the fraction of melanoma patients with defective FBXW7 is higher than determined based on mutational profiling. Taken together, these data suggest that FBXW7 inactivation is common in human melanoma.
Figure 2.
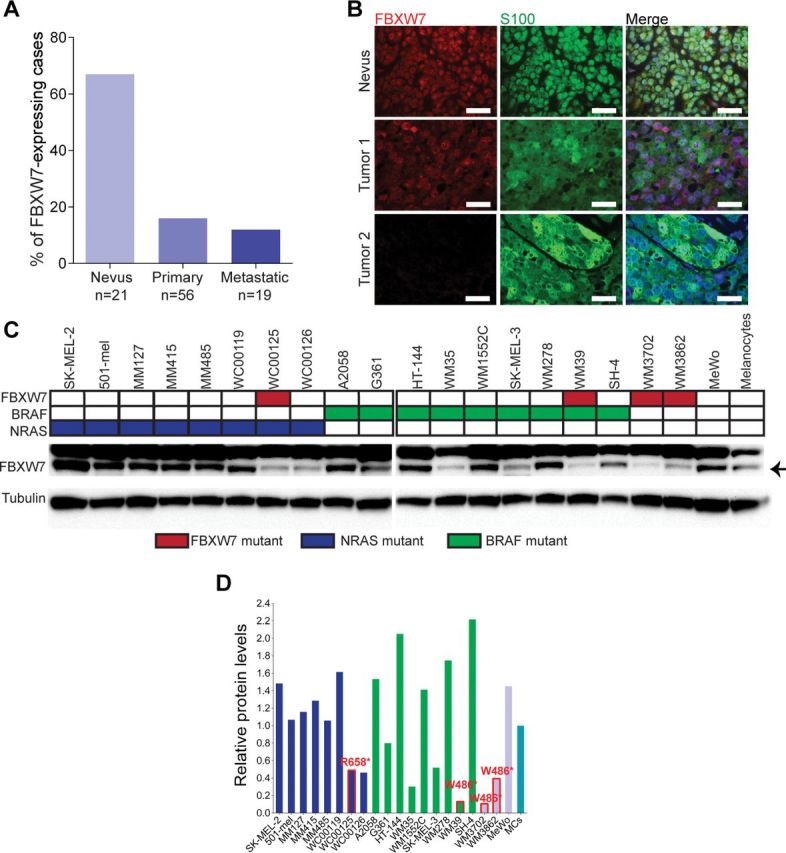
FBXW7 expression in melanoma. A) FBXW7 protein expression in melanoma tissue samples. Immunofluorescence analysis of a tissue microarray consisting of nevi, primary, and metastatic melanomas. The bar graphs indicate percentage of positive nuclear FBXW7 staining. ***P < .001. P values were calculated by the χ2 test. B) Representative cases of tumors (Nevus, Tumor 1, and Tumor 2) stained for FBXW7 (red), S100 (green), and DAPI (blue). Scale bars = 100 μm. FBXW7, S100, and merged images are shown. C) FBXW7 protein expression in human melanocytes and melanoma cells. Western blot analysis carrying wild-type or aberrant FBXW7, BRAF, and NRAS alleles (color-coded as indicated). Tubulin was used as control for protein loading. D) Quantification of western blotting of (C) by densitometry. Arbitrary units are used. Bars are color-coded corresponding to genetic mutations as in (C). FBXW7 mutant cell lines are indicated in red text and red line on the bar graph.
Because FBXW7 has a number of known substrates that have oncogenic activity, we then examined regulation of potential FBXW7 targets in melanoma by silencing FBXW7 using FBXW7-specific siRNA in eight human melanoma cell lines. Loss of FBXW7 resulted in consistent accumulation of the intracellular domain of NOTCH1 across all lines, implicating NOTCH1 as a relevant substrate of FBXW7 in melanoma (Figure 3). Its other known targets—cyclin E, MYC, Aurora A (in some lines), but not MCL1—were elevated upon silencing.
Figure 3.
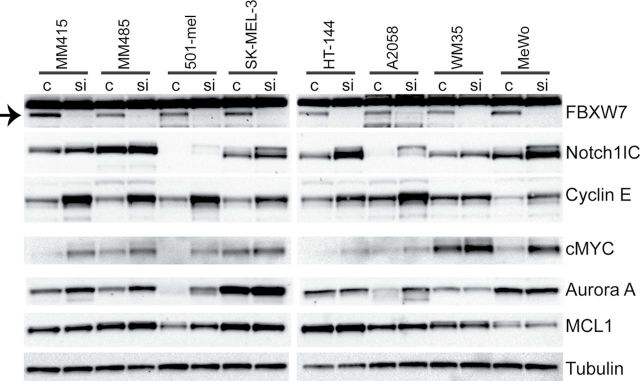
Status of oncoproteins that are substrates of FBXW7 upon downregulation of FBXW7 in melanoma cells. Silencing of FBXW7 in eight melanoma cell lines using nontargeting (c) or FBXW7-specific small interfering RNA (siRNA, si). Western blotting analysis of FBXW7 targets: NOTCH1, cyclin E, MYC, Aurora A, and MCL1. The band corresponding to the FBXW7 protein is indicated with an arrow on the left upper corner.
FBXW7 as a Driver in Melanoma
To determine the functional relevance of FBXW7 inactivation and consequently sustained activation of its target NOTCH1 in melanoma tumorigenesis, we silenced FBXW7 using two distinct shRNAs (shRNA1 and shRNA2) by lentiviral transduction of melan-a melanocytes expressing mutant NRAS (NRASG12D) and examined NOTCH1 target genes in the presence and absence of proteasome inhibition. Silencing of FBXW7 in NRASG12D-expressing melanocytes resulted in statistically significant upregulation of the NOTCH1 target gene, HEY1, with increasing levels after MG-132 treatment (by 2.28-fold to 17.65-fold) (Figure 4A; Supplementary Table 6, available online). These melanocytic cell lines were then transplanted onto immunodeficient mice. FBXW7 loss led to statistically significant acceleration of in vivo tumorigenesis when compared with the control group (tumor volumes (mm3): mean of shScr group = 407.0, 95% CI = 403.5 to 410.4 vs mean of shRNA1 group = 1592.3, 95% CI = 1581.9 to 1602.6, P < .001; mean of shScr group = 407.0, 95% CI = 403.5 to 410.4 vs mean of shRNA2 group = 825.8, 95% CI = 816.4 to 835.3, P = .02) (Figure 4B; Supplementary Table 6, available online). Of note, reduction of FBXW7 enhanced tumor angiogenesis as determined by H&E (Hematoxylin & Eosin) and CD34 staining (Figure 5A) and led to increased levels of genes promoting angiogenesis, including NOTCH1 targets HES1 and HEY1 (Figure 5B). Angiogenesis array analysis of the tumor mRNA showed statistically significant elevation of several angiogenesis-related genes: IL-6, CXCL2, SERPINE1, and PGF1 (by 2.65-fold to 20.16-fold) (Figure 5B; Supplementary Tables 5 and 6, available online). Additionally, Western blot analysis showed increased levels of phosphorylated STAT3 protein as a result of FBXW7 knockdown (3.6-fold). Furthermore, we observed a statistically significant elevation in PD-L1 expression (by 4.89-fold) through transcriptional regulation by STAT3 (19) in FBXW7-silenced tumors (Figure 5C). In an attempt to further validate the impact of FBXW7 mutations in melanoma, we ectopically expressed five representative mutant forms of FBXW7 in NRASG12D-expressing melanocytes and examined the xenograft growth rates. Mutants W486* and R505C led to statistically significant enhanced tumorigenesis when compared with the control group (tumor volumes (mm3): mean of EV group = 675.1, 95% CI = 668.3 to 682.0 vs mean of W486* group = 1609.9, 95% CI = 1599.8 to 1620.0, P < .001; mean of EV group = 675.1, 95% CI = 668.3 to 682.0 vs mean of R505C group = 1657.7, 95% CI = 1637.6 to 1677.8, P = .02) (Figure 6A; Supplementary Table 6, available online). FBXW7 mutant forms have previously been shown to have a dominant-negative activity (they impair the function of the wild-type protein) (20); this corroborates with the phenotype observed in our study. Furthermore, the R505C mutation is of particular interest because this arginine residue is highly conserved among many species and mutations in this residue affect FBXW7’s interaction with many of its substrates, including NOTCH1 (21). These data define FBXW7 as a driver gene in melanoma, and its mutations lead to alterations in many oncogenic signaling pathways.
Figure 4.
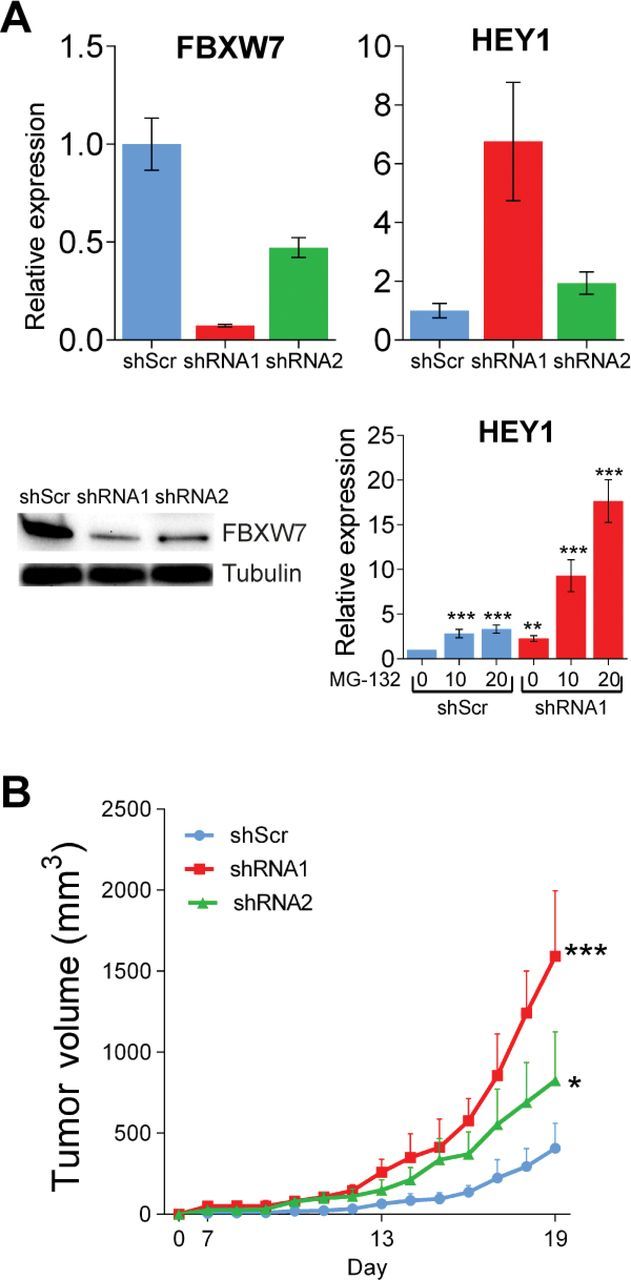
Loss of FBXW7 and its effects in melanomagenesis. A) Silencing FBXW7 in melan-a melanocytes expressing NRASG12D transduced with scrambled (shScr) and two independent FBXW7 short hairpain RNAs (shRNA1 and shRNA2) as determined by quantitative real-time polymerase chain reaction (top, left) and western blotting (bottom, left). The NOTCH1 target gene, HEY1, was assayed by quantitative real-time polymerase chain reaction (top, right). Cells were treated with either 10 μM of dimethyl sulfoxide or 20 μM of MG-132, collected 6 hours later, and examined for HEY1 gene expression (bottom, right). B) Tumor growth curves of these melanocytic lines injected onto Ncr nude mice. *P < .05, **P < .01, ***P < .001. P values were calculated by the two-sided t test; error bars show standard deviation (Supplementary Table 6, available online).
Figure 5.
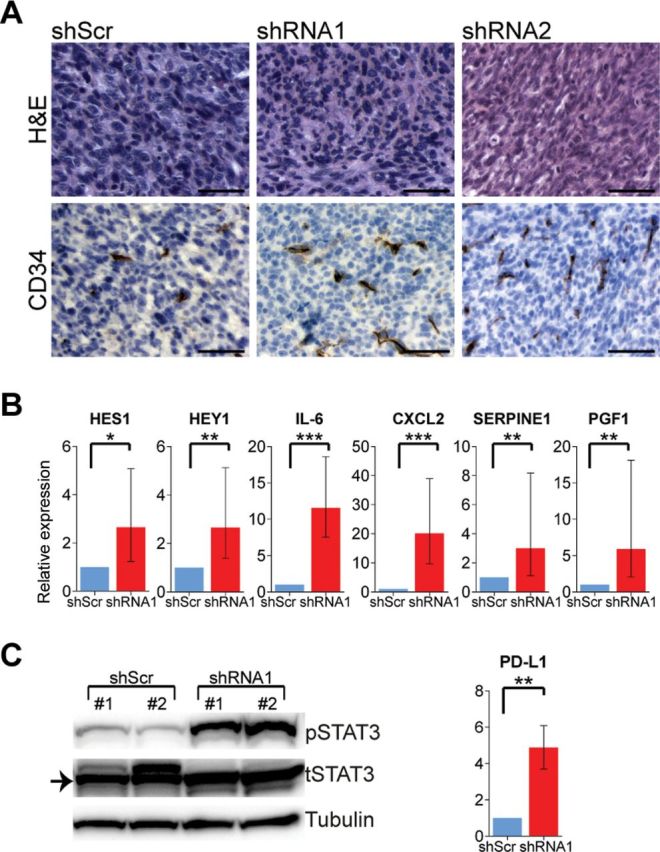
FBXW7 inactive tumors and angiogenic phenotype. A) Tumor morphology and immunohistochemistry analysis for CD34. Scale bars = 50 μm. H&E = Hematoxylin & Eosin. B) Expression of NOTCH1 target genes and angiogenesis-promoting genes of the tumors as assessed by quantitative real-time polymerase chain reaction. C) Western blotting of the tumors for phosphorylated and total STAT3. PD-L1 transcript expression by quantitative real-time polymerase chain reaction. *P < .05, **P < .01, ***P < .001. P values were calculated by the two-sided t test; error bars show standard deviation (Supplementary Table 6, available online).
Figure 6.
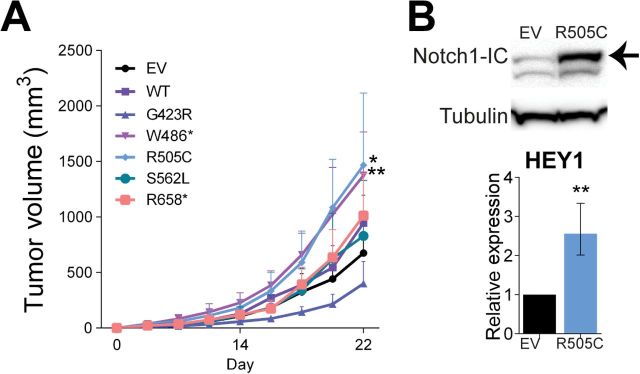
Mutant forms of FBXW7 and their contribution to tumorigenesis. A) Tumor growth curves of melan-a melanocytes expressing NRASG12D transduced with the FBXW7 mutants, wild-type FBXW7, and the empty vector (EV = empty vector control, WT = wild-type FBXW7). B) Western blotting of EV and FBXW7R505C-expressing cells for NOTCH1 levels (top) and HEY1 expression in EV and FBXW7R505C-expressing tumors (bottom). *P < .05, **P < .01. P values were calculated by the two-sided t test; error bars show standard deviation (Supplementary Table 6, available online).
Based on our findings, FBXW7 mutational inactivation represents a mechanism for NOTCH1 activation in melanoma. Of note, we found NOTCH1 and its downstream effector MYC, as well as cyclin E, to be consistently regulated substrates of FBXW7 as opposed to Aurora A (inconsistent) and MCL1 (unregulated) (Figure 3). Therefore, we next examined the effects of inhibiting NOTCH1 and cyclin E alone or in combination in vivo. We used two different γ-secretase inhibitors (dibenzazepine [DBZ] and compound E) for NOTCH1 inhibition and a CDK2 inhibitor (dinaciclib [DNC)] for cyclin E. Mice were grafted with melan-a NRASG12D shRNA1 cells, and once the xenografts reached a volume of 100mm3 they were treated with vehicle, DBZ, compound E, and/or DNC (Figure 7). DBZ treatment resulted in statistically significant reduction of the tumor growth as compared with the vehicle-treated group (tumor volumes (mm3): mean of vehicle group = 2275.3, 95% CI = 2261.7 to 2289.0, vs mean of DBZ group = 453.1, 95% CI = 448.1 to 458.0, P < .001) (Figure 7A; Supplementary Table 6, available online). A similar response in tumor growth was observed with compound E treatment (tumor volumes (mm3): mean of vehicle group = 2275.3, 95% CI = 2261.7 to 2289.0, vs mean of compound E group = 732.8, 95% CI = 728.0 to 737.7, P = .02) (Figure 7B; Supplementary Table 6, available online). There was no major effect of DNC on tumor growth. Combination treatment of DBZ with DNC resulted in mild tumor reduction as compared with the DBZ group alone (not statistically significant) (Figure 7C). Taken together, this study renders anti-NOTCH treatment strategies promising in melanoma as understanding the impact of the spectrum of genomic alterations represented within the melanoma tumors and stratifying patients for treatment based on these aberrations remains the ultimate goal.
Figure 7.
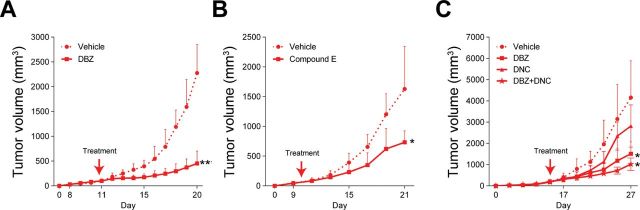
NOTCH1 inhibition of FBXW7 inactive tumors. A–C) Melanocytes expressing NRASG12D transduced with shFBXW7 No. 1 injected onto mice to generate xenografts. Tumor growth curves of mice treated with either vehicle, γ-secretase inhibitors (dibenzazepine [DBZ] and compound E), CDK inhibitor (dinaciclib [DNC]), or the combination of DBZ and DNC. *P < .05, **P < .01. P values were calculated by the two-sided t test; error bars show standard deviation (Supplementary Table 6, available online).
Discussion
Metastatic melanoma is a lethal, incurable malignancy. It represents a heterogeneous tumor with many subgroups that are not well defined. The tumors have high mutational load and complex signaling networks (4,5); however the major focus has so far been on a few driver genes and signaling pathways. Targeted therapies that have been developed, despite effective initial clinical responses, do not lead to sustained clinical remission. Patients eventually relapse, rendering the disease incurable. BRAF and NRAS are proven oncogenes in melanoma critical for tumor initiation (22) and are predictive of response or lack there of, respectively, to RAF inhibition. BRAF inhibitors represent the prototype of targeted therapies in melanoma and prolong survival of patients with BRAF-mutant melanomas; however, resistance emerges after a short period of time (3). By defining a novel melanoma subgroup and characterizing the impact of a new driver gene, FBXW7 tumor suppressor, with potential therapeutic targeting by modulating NOTCH1 signaling, we expect to provide a rationale for mechanism-driven clinical trials and patient stratification based on molecular signatures in future studies.
Recent high-throughput sequencing efforts provide substantial insight into the spectrum of genetic alterations in melanoma (4,5,23–27); however, functional characterization of driver mutations is required for a complete understanding of its pathogenesis. FBXW7 has been found mutated in several exome-sequencing studies (4,5,27); however, experimental validation, functional characterization, and therapeutic implication has not been studied. Thus, the biological relevance of FBXW7 in melanoma has been unknown. Previous exome-sequencing studies in melanoma have identified various MAPK components (24,25), the ERB family member ERBB4 (26), the glutamate receptor GRIN2A (27), G protein-coupled receptor GRM3 (28), and, more recently, a PTEN-interacting protein, PREX2A (23), and RAC1 (5) as mutated and critical for melanoma tumorigenesis. Although these findings require further validation, they represent a viable strategy to dissect the cancer genome.
In this study, we shed light on the genetics of melanoma by identifying FBXW7 as a critical tumor suppressor mutated in this disease. However, our study is not without limitations. It will be important to determine in future studies whether mutations in FBXW7 correlate with poor outcomes in large patient cohorts. Although we found the mutation frequency as 8.1% FBXW7, its protein levels were diminished in 40% of melanoma cell lines. It is thus possible that there are mechanisms other than mutational inactivation (eg, epigenetic) that lead to loss of function and may be relevant for substrate regulation.
Aberrant signaling through NOTCH receptors has been linked to various cancers, including melanoma, making the NOTCH pathway a compelling target for therapeutic intervention (29). New inhibitors are currently being developed and tested (29). NOTCH1 is expressed at high levels in melanoma, although mutations have rarely been documented and validated and have not been studied in depth (4,30). Activated NOTCH1 signaling promotes the progression of early-stage melanoma cell lines in a β-catenin–dependent manner (31). Constitutive NOTCH1 activation confers transforming properties to primary melanocytes in vitro (32), suggesting an oncogenic role for NOTCH1 in melanocytes and melanoma. However, the mechanism of NOTCH1 activation in melanoma is unknown. Moreover, treatment of melanoma with NOTCH1 inhibitors is in a premature stage, and patients are not selected based on molecular signatures.
In summary, we discovered that FBXW7 mutations are recurrent and functionally consequential in melanoma, thus implying that mutations and inactivation of FBXW7 undergo positive selection in human melanoma genesis. We describe, for the first time, a comprehensive mutational spectrum of FBXW7 and its functional impact in melanoma and highlight a new driver gene in melanoma genesis with potential for therapeutic targeting. Additional preclinical studies, as well as clinical trials, of NOTCH1 inhibition in the context of aberrant FBXW7 will be awaited with interest.
Funding
This work was supported by grants from the National Cancer Institute at the National Institutes of Health (R01 CA138678 to JTC; R21 CA158557 to JTC; and R01 CA179044 to RR); the Dow Foundation (to JTC); the Neuberger Dorothy Rodbell Cohen Foundation (to JTC); the Steward Foundation (to RR); and the Partnership for Cure (to RR).
R. D. Melamed and S. J. Adams contributed equally to this work.
The authors have no conflicts of interest to disclose. The study sponsors had no role in the design of the study; the collection, analysis, and interpretation of the data; the writing of the manuscript; and the decision to submit the manuscript for publication.
We respectfully acknowledge Iva Greenwald, PhD, Claire de la Cova, PhD, Bin Zheng, PhD, and Adolfo Ferrando, MD, PhD, for scientific discussions on this study. We appreciate the technical assistance of Rongzhen Chen and Tasneem Fatima.
References
- 1. Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA Cancer J Clin. 2013;63(1):11–30 [DOI] [PubMed] [Google Scholar]
- 2. Gray-Schopfer V, Wellbrock C, Marais R. Melanoma biology and new targeted therapy. Nature. 2007;445(7130):851–857 [DOI] [PubMed] [Google Scholar]
- 3. Sosman JA, Kim KB, Schuchter L, et al. Survival in BRAF V600-mutant advanced melanoma treated with vemurafenib. N Engl J Med. 2012;366(8):707–714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hodis E, Watson IR, Kryukov GV, et al. A landscape of driver mutations in melanoma. Cell. 2012;150(2):251–263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Krauthammer M, Kong Y, Ha BH, et al. Exome sequencing identifies recurrent somatic RAC1 mutations in melanoma. Nat Genet. 2012;44(9):1006–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wang Z, Inuzuka H, Zhong J, et al. Tumor suppressor functions of FBW7 in cancer development and progression. FEBS Lett. 2012;586(10):1409–1418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bai C, Sen P, Hofmann K, et al. SKP1 connects cell cycle regulators to the ubiquitin proteolysis machinery through a novel motif, the F-box. Cell. 1996;86(2):263–274 [DOI] [PubMed] [Google Scholar]
- 8. Zhang W, Koepp DM. Fbw7 isoform interaction contributes to cyclin E proteolysis. Mol Cancer Res. 2006;4(12):935–943 [DOI] [PubMed] [Google Scholar]
- 9. Hao B, Oehlmann S, Sowa ME, Harper JW, Pavletich NP. Structure of a Fbw7-Skp1-cyclin E complex: multisite-phosphorylated substrate recognition by SCF ubiquitin ligases. Mol Cell. 2007;26(1):131–143 [DOI] [PubMed] [Google Scholar]
- 10. Orlicky S, Tang X, Willems A, Tyers M, Sicheri F. Structural basis for phosphodependent substrate selection and orientation by the SCFCdc4 ubiquitin ligase. Cell. 2003;112(2):243–256 [DOI] [PubMed] [Google Scholar]
- 11. Oberg C, Li J, Pauley A, et al. The Notch intracellular domain is ubiquitinated and negatively regulated by the mammalian Sel-10 homolog. J Biol Chem. 2001;276(38):35847–35853 [DOI] [PubMed] [Google Scholar]
- 12. Koepp DM, Schaefer LK, Ye X, et al. Phosphorylation-dependent ubiquitination of cyclin E by the SCFFbw7 ubiquitin ligase. Science. 2001;294(5540):173–177 [DOI] [PubMed] [Google Scholar]
- 13. Welcker M, Orian A, Jin J, et al. The Fbw7 tumor suppressor regulates glycogen synthase kinase 3 phosphorylation-dependent c-Myc protein degradation. Proc Natl Acad Sci U S A. 2004;101(24):9085–9090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Tsunematsu R, Nakayama K, Oike Y, et al. Mouse Fbw7/Sel-10/Cdc4 is required for notch degradation during vascular development. J Biol Chem. 2004;279(10):9417–9423 [DOI] [PubMed] [Google Scholar]
- 15. Frattini V, Trifonov V, Chan JM, et al. The integrated landscape of driver genomic alterations in glioblastoma. Nat Genet. 2013;45(10):1141–1149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Trifonov V, Pasqualucci L, Tiacci E, Falini B, Rabadan R. SAVI: a statistical algorithm for variant frequency identification. BMC Systems Biology. 2013;7(Suppl 2):1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Brown G, Wallin C, Tatusova T, Pruitt K, Maglott D. Gene Help: Integrated Access to Genes of Genomes in the Reference Sequence Collection. Bethesda, MD: National Center for Biotechnology Information; 2005–2006 [Google Scholar]
- 18. Shrager JTJ, Travers M. Cancer commons: biomedicine in the Internet age. In: Elkins S, Hupcey M, Williams A, eds. Collaborative Computational Technologies for Biomedical Research. Hoboken, NJ:, John Wiley & Sons, Inc; 2010 [Google Scholar]
- 19. Marzec M, Zhang Q, Goradia A, et al. Oncogenic kinase NPM/ALK induces through STAT3 expression of immunosuppressive protein CD274 (PD-L1, B7-H1). Proc Natl Acad Sci U S A. 2008;105(52):20852–20857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Akhoondi S, Sun D, von der Lehr N, et al. FBXW7/hCDC4 is a general tumor suppressor in human cancer. Cancer Res. 2007;67(19):9006–9012 [DOI] [PubMed] [Google Scholar]
- 21. Thompson BJ, Buonamici S, Sulis ML, et al. The SCFFBW7 ubiquitin ligase complex as a tumor suppressor in T cell leukemia. J Exp Med. 2007;204(8):1825–1835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Davies H, Bignell GR, Cox C, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417(6892):949–954 [DOI] [PubMed] [Google Scholar]
- 23. Berger MF, Hodis E, Heffernan TP, et al. Melanoma genome sequencing reveals frequent PREX2 mutations. Nature. 2012;485(7399):502–506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Nikolaev SI, Rimoldi D, Iseli C, et al. Exome sequencing identifies recurrent somatic MAP2K1 and MAP2K2 mutations in melanoma. Nat Genet. 2012;44(2):133–139 [DOI] [PubMed] [Google Scholar]
- 25. Stark MS, Woods SL, Gartside MG, et al. Frequent somatic mutations in MAP3K5 and MAP3K9 in metastatic melanoma identified by exome sequencing. Nat Genet. 2012;44(2):165–169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Prickett TD, Agrawal NS, Wei X, et al. Analysis of the tyrosine kinome in melanoma reveals recurrent mutations in ERBB4. Nat Genet. 2009;41(10):1127–1132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wei X, Walia V, Lin JC, et al. Exome sequencing identifies GRIN2A as frequently mutated in melanoma. Nat Genet. 2011;43(5):442–446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Prickett TD, Wei X, Cardenas-Navia I, et al. Exon capture analysis of G protein-coupled receptors identifies activating mutations in GRM3 in melanoma. Nat Genet. 2011;43(11):1119–1126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wu Y, Cain-Hom C, Choy L, et al. Therapeutic antibody targeting of individual Notch receptors. Nature. 2010;464(7291):1052–1057 [DOI] [PubMed] [Google Scholar]
- 30. Bales ES, Dietrich C, Bandyopadhyay D, et al. High levels of expression of p27KIP1 and cyclin E in invasive primary malignant melanomas. J Invest Dermatol. 1999;113(6):1039–1046 [DOI] [PubMed] [Google Scholar]
- 31. Balint K, Xiao M, Pinnix CC, et al. Activation of Notch1 signaling is required for beta-catenin-mediated human primary melanoma progression. J Clin Invest. 2005;115(11):3166–3176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Pinnix CC, Lee JT, Liu ZJ, et al. Active Notch1 confers a transformed phenotype to primary human melanocytes. Cancer Res. 2009;69(13):5312–5320 [DOI] [PMC free article] [PubMed] [Google Scholar]