

Abstract
Pyrrolidine dithiocarbamate (PDTC) can lower the blood glucose level and improve the insulin sensitivity in diabetic rats. However, the mechanisms underlying this effect of PDTC treatment in diabetic rats remained uncertain. In this study, we evaluated the mechanisms by which PDTC conferred protection against oxidative damage to pancreatic islet β-cells in rats with experimental type 2 diabetes mellitus (DM). DM in the rats was elicited by long-term high-fat diet accompanied with a single intraperitoneal (i.p.) injection of a low dose of streptozotocin. After a 7-day administration of PDTC (50 mg/kg/day i.p.), blood glucose levels were measured and pancreatic tissues were collected for the determination of various biochemical and enzymatic activities using immunohistochemistry, immunofluorescence, and western blot techniques. The percentage of apoptotic pancreatic islet β-cells was detected by flow cytometry. The results showed that diabetic rats had elevated blood glucose levels and insulin resistance, accompanied with an increase in malondialdehyde content, nitrotyrosine production, and inducible nitric oxide synthase expression. A decrease in superoxide dismutase and glutathione peroxidase activities was also observed in DM rats, culminating with elevated β-cell apoptosis. PDTC treatment significantly reduced the oxidative damage and the β-cell apoptosis, and also increased the insulin production through down-regulating FoxO1 acetylation and up-regulating nuclear PDX-1 level. These data suggested that PDTC can protect islet β-cells from oxidative damage and improve insulin production through regulation of PDX-1 and FoxO1 in a DM rat model.
Keywords: pyrrolidine dithiocarbamate (PDTC), diabetes, oxidative damage, FoxO1, PDX-1
Introduction
Diabetes mellitus (DM) is a chronic metabolic disease characterized by hyperglycemia due to defects in pancreatic insulin production and/or in insulin action on peripheral tissues [1]. The incidence of DM has dramatically increased worldwide, and the explosion of DM is mainly due to type 2 DM [2]. The pathological basis of type 2 DM is pancreatic β-cell dysfunction and insulin resistance. Accumulating evidence suggests that hyperglycemia-induced production of reactive oxygen species (ROS) and the subsequent oxidative stress contributes to the development and progression of diabetes and related complications [3–7].
ROS are oxygen-free radicals, including superoxide (O2−), hydroxyl radicals (HO•), and hydrogen peroxide (H2O2). They are products of mitochondrial metabolism and function as signaling molecules in cell proliferation, migration, and apoptosis [8]. During normal metabolic processes, ROS are removed rapidly with the help of various endogenous detoxifying enzymes, such as superoxide dismutase (SOD) and glutathione peroxidase (GSH-PX). Normal cellular ROS concentrations are necessary for proper functioning of cells, but excessive, non-physiological concentrations of ROS result in oxidative stress [8]. Elevated glucose levels promote the production of ROS, thus leading to oxidative stress in β-cells [8–11]. Both hyperglycemia and oxidative stress have been reported to activate the transcription factor NF-κB [12,13], which can enhance the transcription of inducible nitric oxide synthase (iNOS), resulting in an increase in the production of NO. NO as reactive nitrogen species induces β-cell apoptosis and can also react with superoxide resulting in the formation of peroxynitrite (ONOO−), which damages the structure and function of β-cells through the formation of nitrotyrosine (NT) in proteins. NT is a stable marker for the formation of peroxynitrite [14,15]. Beside the increase in reactive species in DM, depletion of intracellular antioxidants has been shown to make β-cells more susceptible to oxidative damage [16,17].
Pancreatic duodenal homeobox 1 (PDX-1), a transcription factor that is highly expressed in pancreatic β-cells, plays a central role in β-cell function and survival. PDX-1 activation results in the expression of target genes such as insulin, glucagon-like peptide 1, and glucokinase, which are critical for the functions of β-cells [18]. It has been reported that transient elevation in glucose levels activates PDX-1, and subsequently increases the expression of insulin and enhances the function of β-cells. However, hyperglycemia and hyperlipidemia cause β-cell dysfunction via reduced PDX-1 expression [18]. Binding of the forkhead transcription factor FoxO1 to the PDX-1 promoter negatively regulates transcription of this gene [19]. Oxidative stress regulates FoxO1 activity through various post-translational modifications including phosphorylation, acetylation, and ubiquitination, which, in turn, regulate the subcellular localization and transcriptional activity of FoxO1. FoxO1 is retained in the cytoplasm upon phosphorylation by protein kinase B (PKB/Akt), while a decrease in FoxO1 phosphorylation allows FoxO1 to enter the nucleus. It has been reported that oxidative stress decreases FoxO1 phosphorylation in β-cells, thus allowing its translocation from the cytoplasm to the nucleus [20,21]. FoxO1 acetylation by histone acetyltransferase such as cAMP-response-element-binding (CREB)-binding protein (CBP)/P300 positively regulates FoxO1 transcriptional activity during oxidative stress, thereby protecting it from ubiquitin-mediated degradation [22]. Besides reducing PDX-1 transcription, nuclear localization of FoxO1 induces export of PDX-1 to the cytosol, leading to β-cell dysfunction [19].
Pyrrolidine dithiocarbamate (PDTC) is a low-molecular-weight thiol compound with many biological functions including heavy metal chelation and antioxidant activities [23]. PDTC has been widely used as an inhibitor of NF-κB activation to reduce the generation of proinflammatory cytokines [24–26]. In addition, it has been reported that PDTC lowers blood glucose levels [27], prevents lung injury [24], and reduces abnormal prostanoid signaling [28] in diabetic rats. Our previous study has found that PDTC enhances hepatic glycogen synthesis and reduces FoxO1 transcriptional activity in the liver of diabetic rats [29]. The purpose of this study was to investigate whether PDTC protects β-cells against oxidative damage and improves insulin production through regulation of FoxO1 and PDX-1.
Materials and Methods
Animals and experimental design
Male Wistar rats (180–210 g) were obtained from Hebei Medical University Animal Center (Hebei, China). They were kept at a constant temperature (22 ± 1°C) with 12 h light and dark cycles and were allowed free access to water. Type 2 diabetes was established as described previously [29]. Briefly, rats were divided randomly into two groups: a normal control (NC) group (n = 11) and a high-fat diet (HFD) group (n = 22). Rats in the NC group received a regular diet containing 10.3% fat, and rats in the HFD group received an HFD containing 42% fat for 8 weeks, after which rats in the HFD group were subjected to an oral glucose tolerance test (OGTT) and insulin tolerance test (ITT) to confirm insulin resistance. These determinations were carried out as previously described [29]. Subsequently, HFD rats received an intraperitoneal (i.p.) injection of a single dose of streptozotocin (STZ; Sigma-Aldrich, St Louis, USA) at 27 mg/kg body weight (dissolved in citric acid buffer) to induce type 2 diabetes (T2D). Rats in the NC group were injected with the same volume of citric acid buffer at the same time. Blood glucose levels were measured 72 h after STZ injection in HFD rats. Rats with non-fasting blood glucose levels ≥16.7 mM were considered diabetic, and were included in the study. Twenty-two diabetic rats were randomly divided into two groups: the diabetes group (DM, n = 11) and the PDTC-treated group (PDTC, n = 11). Rats in the PDTC group received PDTC at a dose of 50 mg/kg body weight (dissolved in saline) i.p. once daily for a week, whereas rats in the NC and DM groups were treated with the same volume of saline. After an overnight fast, tail blood samples of NC, DM, and DM + PDTC groups were collected for the measure of fasting blood glucose (FBG). At the end of experiment, all rats were euthanized and the pancreatic tissues were collected for analysis. Animal experimental protocols were approved by the Committee for Animal Experiment at Hebei Medical University.
Detection of SOD, GSH-PX activities, and malondialdehyde content
Pancreatic tissues (200 mg) were homogenized in cold saline and centrifuged at 1300 g for 10–15 min. The supernatants were used for the measurement of SOD, GSH-PX activities, and malondialdehyde (MDA) content by using commercial kits (Jiancheng Biotech, Nanjing, China) according to the manufacturer's instructions.
Apoptosis assay by flow cytometry
Pancreatic tissues were fixed with 70% alcohol for 24 h and single cell suspension was prepared by pressing the tissues through a 100 µm mesh. Cells were then incubated with anti-insulin-FITC (Boster BioTech Co., Wuhan, China) and propidium iodide (PI; Sigma, St Louis, USA) for 30 min at 4°C. The apoptosis rate of islet β-cells was determined using Epics-XL II flow cytometry (Beckman Coulter, Miami, USA). Cells that stained positively for insulin-FITC and with lower DNA (stained with PI) content were identified as apoptotic islet β-cells.
Immunohistochemical staining
Pancreatic tissues were fixed in 10% neutral-buffered formalin and embedded in paraffin. After a xylene wash to remove paraffin and a rehydration step with serial dilutions of alcohol, sections (5 µm thick) were incubated in 0.3% H2O2 for 15 min to block endogenous peroxidases and heated at 98°C for 15 min to retrieve antigen. Sections were then incubated overnight at 4°C with primary antibodies against iNOS (rabbit polyclonal, 1 : 100; Santa Cruz Biotechnologies, Santa Cruz, USA), NT (mouse monoclonal, 1 : 60; Santa Cruz Biotechnologies), insulin (mouse monoclonal, 1 : 50; Boster BioTech Co.), PDX-1 (rabbit polyclonal, 1 : 50; Cell Signaling Technology, Danvers, USA), and FoxO1 (rabbit polyclonal, 1∶100; Cell Signaling Technology). After a series of washes, peroxidase-conjugated secondary antibodies (1 : 100; Santa Cruz Biotechnologies) were applied. The sections were stained with diaminobenzidine solution (Zhongshanjinqiao Biotech Co., Beijing, China), washed, dehydrated, permeabilized, mounted, and viewed by bright-field microscopy.
Immunofluorescence staining
Pancreatic tissues fixed in 4% paraformaldehyde were cryoprotected in 30% sucrose for 48 h, embedded in TissueTek freezing media (Leica, Heidelberg, Germany), frozen on dry ice, and cut into 5 μm thick sections. Sections were incubated with primary antibodies against insulin (1 : 50) and PDX-1 (1 : 50) overnight at 4°C. Then, Cy3- or FITC-conjugated secondary antibodies (1 : 500; Santa Cruz Biotechnologies) were applied. Nuclei were counterstained with 4′,6-diamino-2-phenylindole (DAPI; Sigma-Aldrich). Images were acquired by an inverted confocal laser fluorescence microscope (LSM-510; Carl Zeiss, Jena, Germany) and analyzed using Zeiss LSM Image Examiner.
Nuclear extract preparation and pancreatic tissue homogenization
Nuclear extracts from pancreatic tissues were prepared using the NE-PER™ extraction reagents (Thermo Fisher Scientific Pierce, Rockford, USA) according to the manufacturer's protocol. Protein concentrations were determined using the bicinchoninic acid (BCA) method (Thermo Fisher Scientific Pierce), and extracts were stored at −80°C until further analysis.
Frozen pancreatic tissues (100 mg) were placed on the ice and pulverized with liquid nitrogen in a pre-cooled mortar. Tissues were lysed in 500 μl RIPA buffer (25 mM HEPES, pH 7.4, 134 mM NaCl, 1% NP-40, 0.1% sodium dodecyl sulfate (SDS), 1 mM vanadate, 0.5% sodium deoxycholate, and 100 mM NaF) supplemented with phosphatase and protease inhibitor cocktails (Thermo Fisher Scientific Pierce) and then centrifuged at 10,000 g for 20 min at 4°C. The soluble extracts were collected and stored at −80°C until analysis by western blotting. The determination of protein concentrations was carried out using the BCA method.
Western blot analysis
Soluble lysates (40 μg) and nuclear extracts (20 μg) were separated on 8% SDS–PAGE and transferred to polyvinylidene fluoride membranes (Millipore, Billerica, USA). The membranes were blocked with 5% non-fat milk in TBS-T (10 mM Tris, pH 7.6, 150 mM NaCl, and 0.05% Tween 20) for 2 h at room temperature, and probed with the anti-iNOS antibody (1 : 500), anti-ac-FoxO1 antibody (1 : 1000), and anti-PDX-1 antibody (1 : 1000) overnight at 4°C. Peroxidase-conjugated donkey anti-rabbit IgG or goat anti-mouse IgG (1 : 2000; Santa Cruz Biotechnologies) were applied for 1.5 h at room temperature. Bands were visualized using enhanced chemiluminescence kit (Beyotime Biotech, Haimen, China), and quantified using Image J software (National Institutes of Health, Bethesda, USA).
Statistical analysis
All data were presented as mean ± SEM. Statistical analysis was performed using SPPSS version 13.0 (SPSS, Chicago, USA). One-way analysis of variance was used for multiple comparisons with Bonferroni post hoc test. P-values <0.05 were considered statistically significant.
Results
HFD feeding induced insulin resistance
Our recent work [29] established that significant alterations in the rates of blood glucose clearance occurred during OGTT in HFD rats even though FBG levels were not altered. It was found that after glucose ingestion, the levels of blood glucose increased quickly and peaked at 30 min before gradual decrease to preprandial concentrations in the NC group. In contrast, the blood glucose levels remained elevated 2 h after glucose ingestion in the HFD group. Moreover, HFD feeding was found to induce insulin resistance as assessed by ITT [29].
Anti-oxidative effect of PDTC in diabetic rats
We next examined whether PDTC protects pancreatic β-cells from oxidative damage. In the first series of experiments, SOD and GSH-PX activities were found to be significantly lower in pancreatic tissues of DM rats compared with NC controls, and PDTC treatment significantly increased both enzymatic activities in diabetic rats (Fig. 1A,B). MDA is a well-known marker of oxidative damage whose content in pancreatic tissues was significantly higher in DM rats compared with control rats. A significant decrease in the MDA content was observed in the DM + PDTC cohort (Fig. 1C).
Figure 1.
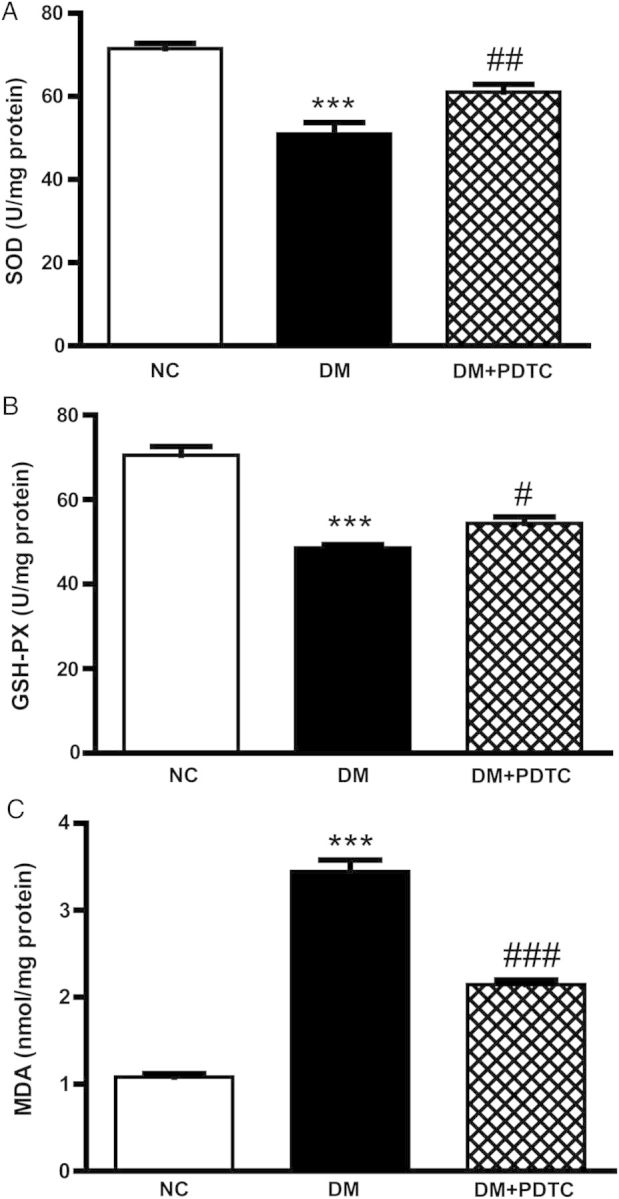
PDTC improves DM-induced impairments in SOD, GSH-PX activities, and MDA content in Wistar rats Pancreatic tissues (200 mg) were homogenized in cold saline and centrifuged, SOD, GSH-PX activities (A,B), and MDA content (C) in the supernatants were measured using commercial kits. ***P < 0.001 vs. NC group. #P < 0.05, ##P < 0.01, ###P < 0.001 vs. DM group.
In a second series of experiments, immunohistochemical staining was carried out and showed that pancreatic islets from DM animals exhibited stronger iNOS expression and higher production of NT as compared with the NC group. As anticipated, there was clear reduction in DM-induced iNOS expression and NT production following PDTC treatment (Fig. 2A). When the expression of iNOS was assessed by western blotting, the results were found to be consistent with the immunohistochemical data (P < 0.001, Fig. 2B,C).
Figure 2.
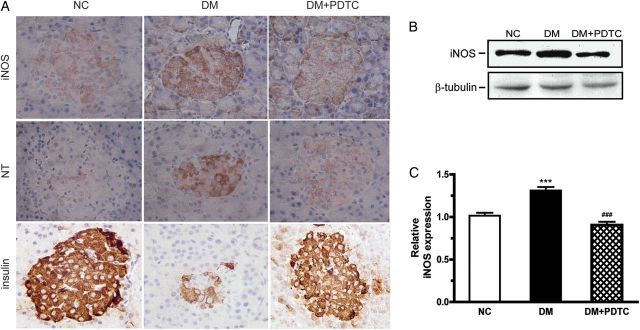
PDTC treatment inhibited the expression of iNOS, NT formation, and improved the production of insulin in diabetic rats (A) Immunohistochemical staining for iNOS, NT, and insulin in pancreatic tissues (magnification: ×400). (B) Western blot analysis for iNOS expression in pancreatic tissues. (C) Quantification of iNOS expression normalized to β-tubulin levels (n = 6 for each group). ***P < 0.001 vs. NC group; ###P < 0.001 vs. DM group.
PDTC lowered FBG levels and inhibited pancreatic β-cell apoptosis in diabetic rats
As previously described [29], after a 1-week PDTC treatment, FBG levels showed a significant reduction as compared with the DM group. Pancreatic β-cell apoptosis was assessed using flow cytometry (Fig. 3). The results in Fig. 3A illustrated that the percentage of apoptotic β-cells was significantly higher in DM rats compared with NC rats (P < 0.001). PDTC treatment significantly lowered the extent of pancreatic β-cell apoptosis in DM rats (P < 0.001).
Figure 3.

PDTC inhibited pancreatic β-cell apoptosis in diabetic rats Single cell suspension was prepared from pancreatic tissues and stained with insulin-FITC antibody and PI. (A) The apoptosis rate of islet β-cells was determined using flow cytometry (n = 6 for each group). ***P < 0.001 vs. NC group; ###P < 0.001 vs. DM group. (B) F, P, and K area represent the apoptotic islet β-cells in NC, DM, and DM + PDTC group, respectively.
PDTC improved insulin production through regulation of PDX-1 and FoxO1
Immunohistochemical staining showed that pancreatic β-cells stained strongly for insulin in NC rats, but weakly in DM animals. Of significance, the number of insulin-positive β-cells increased dramatically in the DM + PDTC group vs. DM group (Fig. 2A).
Active PDX-1 induces efficient production of insulin. Here, the effect of PDTC on the expression of PDX-1 in diabetic rats was examined using immunofluorescence and immunohistochemical techniques. Strong nuclear expression of PDX-1 was shown in islet β-cells of NC pancreatic tissues (Fig. 4A,B). Pancreatic PDX-1 staining was weak in the DM group but was restored to control levels with PDTC treatment (DM + PDTC). We next determined the subcellular localization of PDX-1 among the three experimental groups as it relates to PDX-1 function as a transcriptional regulator. When compared with the NC cohort, there was a significant redistribution of PDX-1 from the nucleus to the cytoplasm in DM pancreatic tissues (Fig. 4C,D). In contrast, PDTC prevented the nuclear export of PDX-1 caused by STZ-induced T2D in HFD rats.
Figure 4.

Effect of PDTC on PDX-1 expression in the pancreatic tissues in diabetic rats (A) Immunofluorescence analysis of PDX-1 expression in pancreatic tissues. β-cells were stained with antibodies against PDX-1 (green) and insulin (red), and then nuclei were counterstained with DAPI (blue). (B) Immunohistochemical staining for PDX-1 in pancreatic tissues. Magnification, ×400. (C) Western blot analysis for the nuclear and cytoplasmic expression of PDX1 in pancreatic tissues. p89TFIIH and β-actin were used as loading controls for nuclear and cytoplasmic fractions, respectively. (D) Quantification of the nuclear and cytoplasmic PDX-1 levels (n = 6 for each group). For the nuclear expression of PDX-1, ***P < 0.001 vs. NC group; ##P < 0.01 vs. DM group. For the cytoplasmic expression of PDX-1, ***P < 0.001 vs. NC group; ###P < 0.001 vs. DM group.
FoxO1 inhibits PDX-1 expression and both transcription factors exhibit mutually exclusive patterns of nuclear localization in pancreatic β-cells. The pancreatic tissues of DM animals exhibited a strong expression of nuclear FoxO1 (Fig. 5A), and a weak expression of nuclear PDX-1 (Fig. 4A,B) in pancreatic β-cells as compared with the NC and DM + PDTC groups. The levels of acetylated FoxO1 in DM pancreatic tissues were markedly increased compared with NC pancreatic tissues. PDTC significantly reduced the marked increase in acetylated FoxO1 levels in DM pancreatic tissues (Fig. 5B,C).
Figure 5.

Effects of PDTC on FoxO1 expression in the pancreatic tissues in diabetic rats (A) Immunohistochemical staining for FoxO1 expression in pancreatic tissues (magnification: ×400). (B) Western blot analysis for the expression of acetylated FoxO1 (ac-FoxO1) in pancreatic tissues. β-Actin was used as a loading control. (C) Quantification of the levels of ac-FoxO1 (n = 6 for each group). ***P < 0.001 vs. NC group; ###P < 0.001 vs. DM group.
Discussion
In this study, we investigated the effects of PDTC on oxidative injury to β-cells and insulin production in T2D rats. The diabetic rat model was established by 8-week HFD followed by an i.p. injection of a single dose of STZ. This rat model exhibits hyperglycemia and insulin resistance, and has been found to resemble the metabolic dysfunction of type 2 DM in human [30]. We found that 1-week PDTC treatment significantly lowered blood glucose levels and improved insulin sensitivity in diabetic rats, in agreement with earlier reports [27,29]. However, the mechanisms underlying the decrease in blood glucose levels in PDTC-treated diabetic rats remained uncertain. Here, PDTC was found to inhibit β-cell apoptosis and prevent oxidative injury to islet β-cells in DM rats. In addition, PDTC promoted the nuclear expression of PDX-1 with concomitant decrease in acetylated FoxO1 levels, both of which leading to increased production of insulin. Our results suggest that PDTC can protect pancreatic β-cells from oxidative injury and improve insulin production through regulation of PDX-1 and FoxO1 in T2D rats.
Oxidative stress plays an important role in the pathogenesis of DM [7–10]. DM-associated elevations in glucose and free fatty acid levels result in the formation of a large amount of ROS, which is detrimental to pancreatic β-cells. ROS bind directly to proteins, lipids, and DNA, and activates several stress-sensitive pathways that can cause cell damage [8,10]. In our experimental model of DM, there was an increase in MDA content and a decrease in antioxidant enzyme activities together with significant elevation in β-cell apoptosis. Treatment with PDTC for 7 days reversed these adverse effects of DM, which led to improved β-cell survival. In addition, ROS can act as second messenger by activating many signaling pathways, including NF-κB [10], whose activation leads to the production of NO through enhanced transcription of iNOS [31]. By acting as a reactive nitrogen species, NO further damages β-cells via formation of NT-protein conjugates. Therefore, NF-κB activation in response to oxidative stress contributes to β-cell dysfunction and insulin resistance [9,10]. Here, the increase in iNOS and NT levels in DM rat β-cells were significantly reduced by PDTC, a potent NF-κB inhibitor [24–26]. Our results illustrate the fact that NF-κB activation contributes to β-cell apoptosis in response to hyperglycemia [32].
The transcription factor PDX-1 induces pancreatic β-cell differentiation and insulin gene expression [18,19]. While transient high glucose concentration stimulates the expression of PDX-1 and increases insulin secretion, chronic hyperglycemia and hyperlipidemia attenuates PDX-1 expression and the secretion of insulin [18,20]. It is likely that the mechanisms underlying the inhibition of PDX-1 expression in response to hyperglycemia and hyperlipidemia stem from increased oxidative stress. During oxidative stress the nuclear export signal within PDX-1 was activated, thereby promoting the translocation of PDX-1 from the nucleus to the cytoplasm, with concomitant decline in target gene transcription [33]. Our results illustrate elevated cytoplasmic PDX-1 levels and low nuclear PDX-1 accumulation in DM rat β-cells, and PDTC treatment blocked the DM-mediated nucleocytoplasmic shuttling of PDX-1. These results indicate that the ability of PDTC to promote insulin production comes from the preservation of a nuclear pool of PDX-1.
Oxidative stress induces translocation of FoxO1 from the cytoplasm to the nucleus, with concomitant nuclear export of PDX-1 [34]. Activation of the JNK signaling pathway in response to oxidative stress leads to inhibition of Akt and subsequent dephosphorylation of FoxO1, which results in nuclear accumulation of FoxO1 [8,21]. Interestingly, the pancreatic tissues from DM rats exhibited significantly higher nuclear FoxO1 levels than that from NC animals, and PDTC treatment conferred resistance to DM-dependent FoxO1 nuclear entry. FoxO1 activity is regulated by multiple post-translational modifications, including phosphorylation, acetylation, and ubiquitination, the latter being dependent on FoxO1 deacetylation [20,35,36]. The fact that PDTC lowered FoxO1 acetylation may explain the reduction in FoxO1 protein levels through an increase in proteasomal degradation in β-cells of DM + PDTC rats.
In summary, we found that PDTC lowered blood glucose levels in HFD-fed, STZ-treated rats. PDTC protected pancreatic β-cells from oxidative injury and improved insulin production through regulation of FoxO1 and PDX-1. PDTC and related compounds may be of therapeutic value in the treatment of metabolic diseases, such as type 2 DM.
Funding
This work was supported by the grants from the Natural Science Foundation of Hebei Province in China (No. C2009001232), the Scientific Research Foundation for the Returned Overseas Chinese Scholars, Ministry of Personnel of China, the Department of Science and Technology of Hebei Province (No. 10276105D-66), the Health Bureau of Hebei Province (No. 20090168), and the Hebei Education Department in China (No. 2008135) and in part, by the Intramural Research Program of the NIH, National Institute on Aging.
References
- 1.American Diabetes Association. Diagnosis and classification of diabetes mellitus. Diabetes Care. 2012;35(Suppl 1):S64–S71. doi: 10.2337/dc12-s064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wild S, Roglic G, Green A, Sicree R, King H. Global prevalence of diabetes: estimates for the year 2000 and projections for 2030. Diabetes Care. 2004;27:1047–1053. doi: 10.2337/diacare.27.5.1047. [DOI] [PubMed] [Google Scholar]
- 3.Giacco F, Brownlee M. Oxidative stress and diabetic complications. Circ Res. 2010;107:1058–1070. doi: 10.1161/CIRCRESAHA.110.223545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rains JL, Jain SK. Oxidative stress, insulin signaling, and diabetes. Free Radic Biol Med. 2011;50:567–575. doi: 10.1016/j.freeradbiomed.2010.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pitocco D, Zaccardi F, Di Stasio E, Romitelli F, Santini SA, Zuppi C, Ghirlanda G. Oxidative stress, nitric oxide, and diabetes. Rev Diabet Stud. 2010;7:15–25. doi: 10.1900/RDS.2010.7.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Singh DK, Winocour P, Farrington K. Oxidative stress in early diabetic nephropathy: fueling the fire. Nat Rev Endocrinol. 2011;7:176–184. doi: 10.1038/nrendo.2010.212. [DOI] [PubMed] [Google Scholar]
- 7.Folli F, Corradi D, Fanti P, Davalli A, Paez A, Giaccari A, Perego C, et al. The role of oxidative stress in the pathogenesis of type 2 diabetes mellitus micro- and macrovascular complications: avenues for a mechanistic-based therapeutic approach. Curr Diabetes Rev. 2011;7:313–324. doi: 10.2174/157339911797415585. [DOI] [PubMed] [Google Scholar]
- 8.Storz P. Forkhead homeobox type O transcription factors in the responses to oxidative stress. Antioxid Redox Signal. 2011;14:593–605. doi: 10.1089/ars.2010.3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Evans JL, Goldfine ID, Maddux BA, Grodsky GM. Are oxidative stress-activated signaling pathways mediators of insulin resistance and beta-cell dysfunction? Diabetes. 2003;52:1–8. doi: 10.2337/diabetes.52.1.1. [DOI] [PubMed] [Google Scholar]
- 10.Newsholme P, Rebelato E, Abdulkader F, Krause M, Carpinelli A, Curi R. Reactive oxygen and nitrogen species generation, antioxidant defenses, and β-cell function: a critical role for amino acids. J Endocrinol. 2012;214:11–20. doi: 10.1530/JOE-12-0072. [DOI] [PubMed] [Google Scholar]
- 11.Brownlee M. Biochemistry and molecular cell biology of diabetic complications. Nature. 2001;414:813–820. doi: 10.1038/414813a. [DOI] [PubMed] [Google Scholar]
- 12.Mohamed AK, Bierhaus A, Schiekofer S, Tritschler H, Ziegler R, Nawroth PP. The role of oxidative stress and NF-kappaB activation in late diabetic complications. Biofactors. 1999;10:157–167. doi: 10.1002/biof.5520100211. [DOI] [PubMed] [Google Scholar]
- 13.Bierhaus A, Schiekofer S, Schwaninger M, Andrassy M, Humpert PM, Chen J, Hong M, et al. Diabetes-associated sustained activation of the transcription factor nuclear factor-kappaB. Diabetes. 2001;50:2792–2808. doi: 10.2337/diabetes.50.12.2792. [DOI] [PubMed] [Google Scholar]
- 14.Andrikopoulos S. Obesity and type 2 diabetes: slow down!—can metabolic deceleration protect the islet beta cell from excess nutrient-induced damage? Mol Cell Endocrinol. 2010;316:140–146. doi: 10.1016/j.mce.2009.09.031. [DOI] [PubMed] [Google Scholar]
- 15.Drel VR, Pacher P, Stevens MJ, Obrosova IG. Aldose reductase inhibition counteracts nitrosative stress and poly(ADP-ribose) polymerase activation in diabetic rat kidney and high-glucose-exposed human mesangial cells. Free Radic Biol Med. 2006;40:1454–1465. doi: 10.1016/j.freeradbiomed.2005.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Maritim AC, Sanders RA, Watkins JB., III Diabetes, oxidative stress, and antioxidants: a review. J Biochem Mol Toxicol. 2003;17:24–38. doi: 10.1002/jbt.10058. [DOI] [PubMed] [Google Scholar]
- 17.Lenzen S. Oxidative stress: the vulnerable beta-cell. Biochem Soc Trans. 2008;36:343–347. doi: 10.1042/BST0360343. [DOI] [PubMed] [Google Scholar]
- 18.Fujimoto K, Polonsky KS. Pdx1 and other factors that regulate pancreatic beta-cell survival. Diabetes Obes Metab. 2009;11(Suppl 4):30–37. doi: 10.1111/j.1463-1326.2009.01121.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kaneto H, Miyatsuka T, Kawamori D, Yamamoto K, Kato K, Shiraiwa T, Katakami N, et al. PDX-1 and MafA play a crucial role in pancreatic β-cell differentiation and maintenance of mature β-cell function. Endocr J. 2008;55:235–252. doi: 10.1507/endocrj.k07e-041. [DOI] [PubMed] [Google Scholar]
- 20.Vogt PK, Jiang H, Aoki M. Triple layer control: phosphorylation, acetylation and ubiquitination of FOXO proteins. Cell Cycle. 2005;4:908–913. doi: 10.4161/cc.4.7.1796. [DOI] [PubMed] [Google Scholar]
- 21.Kitamura YI, Kitamura T, Kruse JP, Raum JC, Stein R, Gu W, Accili D. FoxO1 protects against pancreatic beta cell failure through NeuroD and MafA induction. Cell Metab. 2005;2:153–163. doi: 10.1016/j.cmet.2005.08.004. [DOI] [PubMed] [Google Scholar]
- 22.Daitoku H, Sakamaki J, Fukamizu A. Regulation of FoxO transcription factors by acetylation and protein-protein interactions. Biochim Biophys Acta. 2011;1813:1954–1960. doi: 10.1016/j.bbamcr.2011.03.001. [DOI] [PubMed] [Google Scholar]
- 23.Chabicovsky M, Prieschl-Grassauer E, Seipelt J, Muster T, Szolar OH, Hebar A, Doblhoff-Dier O. Pre-clinical safety evaluation of pyrrolidine dithiocarbamate. Basic Clin Pharmacol Toxicol. 2010;107:758–767. doi: 10.1111/j.1742-7843.2010.00573.x. [DOI] [PubMed] [Google Scholar]
- 24.Eren G, Cukurova Z, Hergunsel O, Demir G, Kucur M, Uslu E, Dalo E, et al. Protective effect of the nuclear factor kappa B inhibitor pyrrolidine dithiocarbamate in lung injury in rats with streptozotocin-induced diabetes. Respiration. 2010;79:402–410. doi: 10.1159/000264920. [DOI] [PubMed] [Google Scholar]
- 25.Nurmi A, Vartiainen N, Pihlaja R, Goldsteins G, Yrjanheikki J, Koistinaho J. Pyrrolidine dithiocarbamate inhibits translocation of nuclear factor kappa-B in neurons and protects against brain ischaemia with a wide therapeutic time window. J Neurochem. 2004;91:755–765. doi: 10.1111/j.1471-4159.2004.02756.x. [DOI] [PubMed] [Google Scholar]
- 26.Muller DN, Dechend R, Mervaala EM, Park JK, Schmidt F, Fiebeler A, Theuer J, et al. NF-kappaB inhibition ameliorates angiotensin II-induced inflammatory damage in rats. Hypertension. 2000;35:193–201. doi: 10.1161/01.hyp.35.1.193. [DOI] [PubMed] [Google Scholar]
- 27.Stosic-Grujicic SD, Miljkovic DM, Cvetkovic ID, Maksimovic-Ivanic DD, Trajkovic V. Immunosuppressive and anti-inflammatory action of antioxidants in rat autoimmune diabetes. J Autoimmun. 2004;22:267–276. doi: 10.1016/j.jaut.2004.01.005. [DOI] [PubMed] [Google Scholar]
- 28.Matsumoto T, Ishida K, Kobayashi T, Kamata K. Pyrrolidine dithiocarbamate reduces vascular prostanoid-induced responses in aged type 2 diabetic rat model. J Pharmacol Sci. 2009;110:326–333. doi: 10.1254/jphs.09116fp. [DOI] [PubMed] [Google Scholar]
- 29.Zhu T, Zhao R, Zhang L, Bernier M, Liu J. Pyrrolidine dithiocarbamate enhances hepatic glycogen synthesis and reduces FoxO1-mediated gene transcription in type 2 diabetic rats. Am J Physiol Endocrinol Metab. 2012;302:E409–E416. doi: 10.1152/ajpendo.00453.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Srinivasan K, Viswanad B, Asrat L, Kaul CL, Ramarao P. Combination of high-fat diet-fed and low-dose streptozotocin-treated rat: a model for type 2 diabetes and pharmacological screening. Pharmacol Res. 2005;52:313–320. doi: 10.1016/j.phrs.2005.05.004. [DOI] [PubMed] [Google Scholar]
- 31.Hu WH, Mo XM, Walters WM, Brambilla R, Bethea JR. TNAP, a novel repressor of NF-kappaB-inducing kinase, suppresses NF-kappaB activation. J Biol Chem. 2004;279:35975–35983. doi: 10.1074/jbc.M405699200. [DOI] [PubMed] [Google Scholar]
- 32.Maedler K, Sergeev P, Ris F, Oberholzer J, Joller-Jemelka HI, Spinas GA, Kaiser N, et al. Glucose-induced beta cell production of IL-1beta contributes to glucotoxicity in human pancreatic islets. J Clin Invest. 2002;110:851–860. doi: 10.1172/JCI15318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kawamori D, Kajimoto Y, Kaneto H, Umayahara Y, Fujitani Y, Miyatsuka T, Watada H, et al. Oxidative stress induces nucleo-cytoplasmic translocation of pancreatic transcription factor PDX-1 through activation of c-Jun NH(2)-terminal kinase. Diabetes. 2003;52:2896–2904. doi: 10.2337/diabetes.52.12.2896. [DOI] [PubMed] [Google Scholar]
- 34.Kawamori D, Kaneto H, Nakatani Y, Matsuoka TA, Matsuhisa M, Hori M, Yamasaki Y. The forkhead transcription factor Foxo1 bridges the JNK pathway and the transcription factor PDX-1 through its intracellular translocation. J Biol Chem. 2006;281:1091–1098. doi: 10.1074/jbc.M508510200. [DOI] [PubMed] [Google Scholar]
- 35.Matsuzaki H, Daitoku H, Hatta M, Aoyama H, Yoshimochi K, Fukamizu A. Acetylation of Foxo1 alters its DNA-binding ability and sensitivity to phosphorylation. Proc Natl Acad Sci USA. 2005;102:11278–11283. doi: 10.1073/pnas.0502738102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yoshimochi K, Daitoku H, Fukamizu A. PCAF represses transactivation function of FOXO1 in an acetyltransferase-independent manner. J Recept Signal Transduct Res. 2010;30:43–49. doi: 10.3109/10799890903517947. [DOI] [PubMed] [Google Scholar]