

Abstract
AIM: To investigate the effect of a high-fat diet in the formation of the precursors of colorectal cancer using an animal model.
METHODS: Wistar rats were divided into two groups that were fed either a high-fat diet (HFD) or a normal-fat diet (ND), and 1,2-dimethylhydrazine was administered at a dose of 40 mg/kg for 10 wk. The body weight/liver weight/epididymal fat weight were recorded after rats were sacrificed, and the formation of colonic adenoma was also observed. The levels of insulin, leptin, tumor necrosis factor (TNF)-α, insulin-like growth factor (IGF)-1 and triglycerides were determined by enzyme-linked immunosorbent assay in order to compare the altered levels of biochemical indices and inflammatory cytokines in the serum between rats fed an ND and HFD. Cell proliferation activity (Ki-67) was determined by immunohistochemical analysis. Western blot and immunofluorescence staining were used to examine the expression of proliferating cell nuclear antigen (PCNA), cyclooxygenase (COX)-2, cyclin D1, β-catenin and nuclear factor (NF)-κB proteins in the adenoma and comparative control tissues.
RESULTS: The number of colonic adenomas and the colonic epithelial Ki-67 were significantly higher in the HFD group than in the ND group. The HFD group also had increased body weight, liver weight and epididymal fat weight, which were associated with increased levels of serum insulin, leptin, TNF-α, IGF-1 and triglycerides. HFD induced upregulation of PCNA, COX-2, cyclin D1, β-catenin and NF-κB proteins, as revealed by Western blot and immunofluorescence staining.
CONCLUSION: HFD promotes the formation of colonic adenoma through inflammation, metabolic abnormalities, and increases cell cycle progression.
Keywords: High-fat diet, Colonic adenomas, Inflammation, Adipokines
Core tip: This study was undertaken to explore the effect of a high-fat diet on the incidence of colonic adenoma induced by intraperitoneal injection of 1,2-dimethylhydrazine in Wistar rats. We demonstrated that a high-fat diet (HFD) increased the proliferative activity of colonic epithelial cells, and we conclude that HFD-mediated tumor growth could be associated with inflammation and increased cell cycle progression. This study might be conducive to understanding the nature of the relationship between high fat intake and the increased risk of colorectal carcinoma.
INTRODUCTION
Colorectal cancer (CRC), accounting for about 10% of the incident cases of cancer, is a major cause of morbidity and mortality worldwide, with > 1 million new cases and > 600000 deaths annually, making it the fourth leading cause of cancer deaths globally[1,2]. Genetic predisposition seems to explain only a small proportion of cases, while many of the risk factors for CRC are associated with a Western lifestyle. Recently, many epidemiological studies have provided evidence of a relationship between dietary fat intake and an increased risk of CRC[3]. Most animal studies have shown that a high-fat diet (HFD) leads to an increased number of chemically induced aberrant crypt foci (ACFs)[4], which are identifiable lesions in experimental colon carcinogenesis and tumors[5]. In addition, high intake of dietary fat is known to cause obesity in humans and rodents[6,7] and has also been implicated in colon cancer[8].
A recent study investigating the molecular mechanisms of diet-induced obesity in a mouse model of colitis-associated CRC demonstrated that an HFD results in a two-fold higher number of colonic tumors compared to animals fed a normal-fat diet (ND)[9]. In the pathophysiological state of obesity, adipose tissue is an active endocrine and metabolic organ. Adipose tissue secretes various hormones and growth factors that have a role in CRC development[10]. Evidence for a positive relationship between circulating insulin concentration and colon cancer risk exists, and a recent meta-analysis showed an increased risk of CRC with high C-peptide/insulin levels (relative risk = 1.31) in humans[11]. Chronically elevated concentrations of insulin decrease insulin-like growth factor (IGF)-1 binding protein levels, which, in turn, increase the circulating concentration of IGF-1[12]. IGF-1 is a key mitogen required for cell cycle progression to increase the risk of cellular transformation by enhancing cell turnover[13]. The adipocyte-derived hormone leptin is also elevated in individuals with obesity, and synthesis of leptin in adipose tissue is influenced by insulin[14]. Despite a lack of direct evidence that increased leptin concentration is a risk factor for colon cancer, studies have proposed that higher leptin concentrations may form a possible link between obesity and colon cancer or colorectal adenoma[15]. Adipose tissue in obese individuals is infiltrated with an increased number of macrophages, which appear to be responsible for the production of many inflammatory cytokines[16]. Since the initial discovery of escalated levels of tumor necrosis factor (TNF)-α in blood and adipose tissue by Hotamisligil and colleagues[17], many other adiposity-related inflammatory molecules, such as interferon-γ and interleukin-1, have been identified in adipose tissue, and in some instances, systemically[18]. Biomarkers of systemic inflammation other than adipokines include pro-inflammatory cytokines (e.g., IL-6) and chemokines (e.g., CXCL-10) which have been suggested to be closely related to excess adipose tissue accumulation[19]. It is now widely accepted that obesity is associated with a state of chronic, low-grade inflammation[20], which may represent an additional mechanism linking increased adiposity to colorectal carcinogenesis[21].
Although the link between an HFD or diet-induced obesity and an increased risk of CRC has been investigated in population-based observational studies, as well as in vitro (experimental) studies (mostly focused on the incidence of ACFs or the change in growth of xenografted human colon cancer cells), there is little understanding concerning the effect of an HFD in the formation of the precursors (colonic adenomas) of CRC. Therefore, the present study was undertaken to explore the effect of HFD on the incidence of 1,2-dimethylhydrazine (DMH)-induced in situ colon carcinogenesis.
MATERIALS AND METHODS
Animals and chemicals
In this study, 4-wk-old male Wistar rats (180-200 g) that were obtained from Shanghai Shriek Laboratory Animal Corporation were utilized. All animals were housed in plastic cages (four or five rats/cage) under controlled conditions of humidity (44 ± 5%), light (12 h light/dark cycle) and temperature (22 ± 2 °C). DMH was purchased from Sigma-Aldrich (St. Louis, MO, United States) and freshly prepared before use in 1 mmol/L EDTA-saline, with pH adjusted to 7.0 using dilute NaOH solution.
Experimental procedures
Twenty 4-wk-old male Wistar rats were divided into two groups: an ND group (n = 10) and an HFD group (n = 10). These rats were acclimatized to the rodent normal diet and water was given ad libitum for 1 wk. After acclimation, the animals were fed either an ND (13.5% of calories coming from fat, 58% from carbohydrates, and 28.5% from protein, RD5001) or an HFD (45% of calories being supplied from fat, 35% from carbohydrates, and 20% from protein, D12451) purchased from LabDiet (St. Louis, MO, United States) and Research Diets (New Brunswick, NJ, United States). The diets were stored at -20 °C and fresh diets were provided daily to allow the rats free access to food. Daily food intake was monitored throughout the study. Rats at 6 wk of age from the two groups were intraperitoneally injected with DMH (40 mg/kg) once weekly for 10 consecutive weeks and were euthanized at 16 wk (colonic adenomas were found from weeks 14 to 18, according to prior studies by Peng et al[22]). The majority of these tumors harbored mutations in the β-catenin gene, which is similar to hereditary non-polyposis colorectal cancer. These mutations affected the N-terminal amino acids of the β-catenin gene product, making the protein resistant to regulatory degradation, stabilizing β-catenin, and increasing WNT signaling to drive tumorigenesis. The colonic adenoma experimental protocol is shown in Figure 1A. Animal body weights in all groups were recorded once weekly until the termination of the experiment. Figure 1B is a representative picture showing the incidence of colon tumors in rats fed either NDs or HFDs. The experimental protocol was approved by the Animal Care and Use Committee and the Ethics Committee of Shanghai Jiao Tong University.
Figure 1.
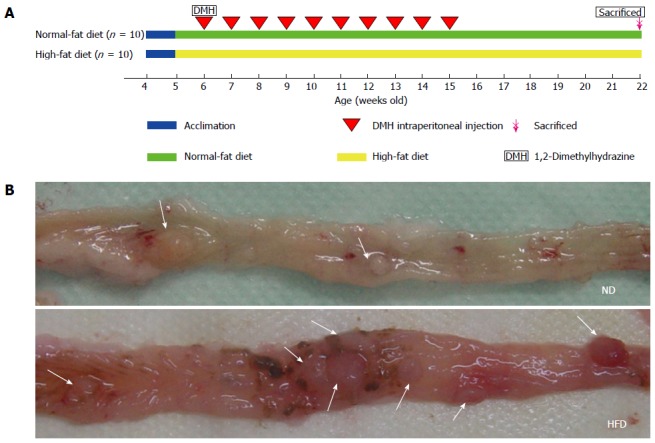
Experiment protocol and representative images of colonic adenomas. A: Colonic adenoma experiment protocol. Rats (4-wk-old) were divided into a normal-fat diet (ND) group (n = 10) and a high-fat diet (HFD) group (n = 10). Sixteen weeks after the start of 1,2-dimethylhydrazine (DMH) intraperitoneal injection, the rats were euthanized; B: Representative images of adenomas in colon specimens from the ND group (above) and HFD group (below) when the colons were opened longitudinally. The location of the adenomas is indicated by arrows.
Necropsy and tumor enumeration
The day before animals were euthanized they were kept on overnight fasting with drinking water ad libitum and euthanized on the next day. After they were anesthetized (ketamine 100 mg/kg + xylazine 15mg/kg, i.p.), blood samples were taken by heart puncture[23]. Samples were centrifuged for 15 min at 4000 rpm, the serum was separated and aliquoted, and frozen at -80 °C until further analysis. The entire colon was removed, opened longitudinally, and rinsed in ice-cold saline. The colons were laid flat on an aseptic towel, and the total number of tumors in the entire colon was enumerated. After that, the tumors were cut into halves; one was prepared for histological examination, and the other was placed in a cryotube and frozen immediately in liquid nitrogen for Western blot. The liver and epididymal fat were also collected and weighed.
Oil red O staining
Liver tissue sections from the ND and HFD groups were fixed for 24 h in 4% paraformaldehyde, 0.2% picric acid, and 0.5% glutaraldehyde in 0.2 mol/L phosphate buffer (pH = 7.4) at 4 °C. After being washed for 1 d with 15% sucrose at 4 °C, the sections were incubated for 1 h in an Oil Red O dye bath to stain for fat accumulation. After the stain was removed and the sections were washed with 60% isopropanol, the image of each group was photographed.
Enzyme-linked immunosorbent assay
The levels of serum triglycerides, cholesterol, insulin, IGF-1, leptin and TNF-α, IL-6, CXCL-10 were measured using corresponding enzyme-linked immunosorbent assay (ELISA) kits (RD, Minneapolis, MN, United States) (n = 10 from each group) according to the manufacturer’s instructions.
Histological examinations and immunohistochemistry
The colon tissues were fixed in 10% neutral phosphate-buffered formalin, routinely processed, sectioned at 5 μm, and stained with hematoxylin and eosin for light microscopic examination. For immunohistochemistry, formalin-fixed paraffin-embedded colonic sections were deparaffinized, rehydrated, and incubated for 5 min in 3% H2O2 to deactivate endogenous peroxidase. The sections were blocked with 5% bovine serum albumin, incubated successively with anti-Ki-67 antibody (1:200; Abcam, Cambridge, MA, United States) and biotinylated horse anti-mouse secondary antibody (Vector Laboratories, Burlingame, CA, United States) in accordance with the manufacturer’s instructions, and finally counter-stained with hematoxylin. The staining of Ki-67 was evaluated by two independent investigators using bright-field light microscopy.
Immunofluorescence staining
To visualize the expression of cyclooxygenase (COX)-2 and cell cycle-related proteins such as proliferating cell nuclear antigen (PCNA), immunofluorescence was performed. Paraffin sections were blocked for endogenous peroxide and treated for optimal antigen retrieval as described before. Sections were incubated with PCNA and COX-2 antibodies (Cell Signaling Technology, Danvers, MA, United States). The sections were further probed with a cyanine-3 conjugated anti-IgG secondary antibody (Jackson ImmunoResearch Laboratories, Philadelphia, PA, United States). Cell nuclei were stained with 4,6-diamidino-2-phenylindole (DAPI). Incubation without the first antibody served as a negative control. Images of tumor sections were acquired under a DSY5000X microscope using the Nikon D200 camera and imaging system.
Western blot
The colonic tissues were homogenized, sonicated, and transferred into ice-cold lysis buffer containing a protease inhibitor. The protein concentration was determined using the BCA Protein Assay Kit (Pierce Biotechnology, Rockford, IL, United States). The extracted proteins were separated on 10% SDS-PAGE, and transferred onto polyvinylidene difluoride membranes (Millipore, Bedford, MA, United States). The membranes were then incubated at 4 °C overnight with the appropriate primary antibodies specific for PCNA, cyclin D1, β-catenin, and NF-κB (1:1000; Cell Signaling Technology). After that, the membranes were washed three times (20 min each) with Tris-buffered saline (TBS) containing 0.1% Tween 20 (TBS-T), and then incubated for 1 h with the appropriate horseradish-peroxidase-conjugated secondary antibody (1:2000) in TBS-T for 4 h at 4 °C. Finally, the proteins were visualized by the enhanced chemiluminescence method (ECL kit, Pierce) according to the manufacturer’s instructions. β-actin was used as an internal loading control.
Statistical analysis
Quantitative variables were analyzed using Student’s t test or Mann-Whitney test. The data are presented as mean ± SD. The confidence intervals throughout the study were set at 95%. A two-sided P value < 0.05 was regarded as statistically significant. The statistical analyses were performed using GraphPad Prism version 5.01 (GraphPad, San Diego, CA, United States) and SPSS for Windows version 19.0.
RESULTS
HFD consumption significantly increases total body weight, liver weight and epididymal fat weight
All rats from both groups survived to the end of the study (16 wk). Then, we measured the total body weight, liver weight and epididymal fat weight. As shown in Figure 2, the body weight was significantly higher in the HFD group than in the ND group after 1 wk of treatment (i.e., at 6-wk-old), and this difference was maintained until the end of the study (Figure 2A). At the beginning of the study, the average body weight was 181.5 ± 9.3 g and 187.7 ± 6.5 g in the ND and HFD groups, respectively (P = 0.054). By the study end at 16 wk, the average body weight significantly increased to 317.4 ± 8.2 g in the ND group and 368.6 ± 13.1 g in the HFD group (P < 0.01). In addition, there was also a significant increase in liver weight and epididymal fat weight in the HFD and ND groups. At the end of the study, the average liver weight significantly increased to 8.43 ± 1.13 g in the ND group and 11.61 ± 1.28 g in the HFD group (1.38-fold increase, P < 0.01), and the Oil Red O examination showed that there were more lipid droplets in the liver cells (Figure 2B, C). Weight of epididymal fat significantly increased to 3.19 ± 0.26 g in the ND group and 5.13 ± 0.45 g in the HFD group (1.61-fold increase, P < 0.01) (Figure 2D).
Figure 2.
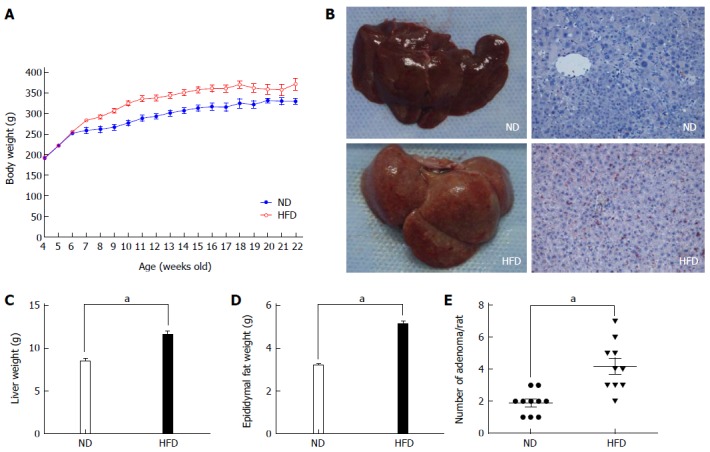
Changes in body weight and representative images of the liver. A: Changes in body weight of rats fed a normal-fat diet (ND) (blue line, n = 10) and high-fat diet (HFD) (red line, n = 10). Significant differences were observed between the ND and HFD groups at all time points except for the initial 2 wk; B: Representative images of the liver from the ND group (left) and HFD group (right). The liver from the HFD group appeared to have a fatty liver-like appearance, and the Oil Red O staining also present that there are more lipid droplets in the cytoplasm of liver cells. Imaging was documented at × 200 magnification; C: Average weight of the liver from the two groups. Each column represents the mean ± SD of 10 rats/group; D: Average weight of epididymal fat from the two groups. Each column represents the mean ± SD of 10 rats/group. aP < 0.01, ND vs HFD; E: Average number of adenomas for rats fed an ND and HFD.
Enhanced formation of colonic adenoma and increased cell proliferation in rats fed an HFD
After the rats were euthanized, we found that tumors were successfully induced in both groups, although there were differences in tumor number. We determined the accurate tumor number by visual counting, as well as their histological appearance (only those with pathologically confirmed adenomas were included). The number of colonic adenomas was significantly higher in the HFD group than in the ND group (Figure 2E). The mean number of adenomas in the HFD group was 4.2 per rat, while the ND group had only 1.9 tumors per rat (P < 0.01). The microscopic characteristics of the colonic adenomas in the ND and HFD groups are shown in Figure 3A and B, respectively, and it should be noted that no morphological differences in the adenomas were observed between the ND and HFD groups. To examine the facilitative effect of an HFD on the formation of colonic adenoma, the precursor of colorectal carcinogenesis, the proliferative activity of the colonic epithelial cells was determined using Ki-67 staining. As shown in Figure 3C-F, which includes adjacent normal epithelial and adenoma tissues, there was a significant increase in Ki-67 staining in the adenomas in the HFD group compared with the ND group. In particular, we found that in Ki-67+ crypts from adjacent normal epithelium, Ki-67+ cells were mainly restricted to the bottom of the colonic crypt in the ND-fed rats, while Ki-67+ cells in the positive crypts of HFD-fed rats were mostly present throughout the height of the crypt. Therefore, these results indicated that the proliferative activity of the colonic epithelial cells was promoted by the HFD, which resulted in an increase in the formation of colonic adenoma.
Figure 3.
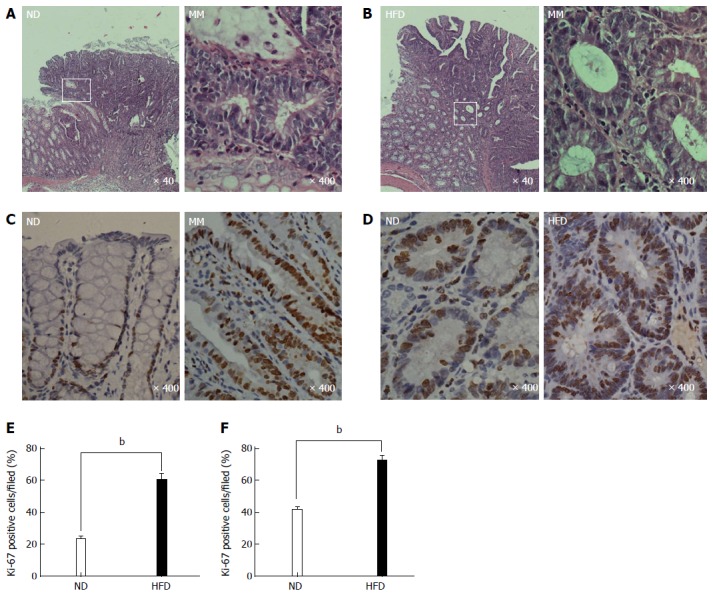
Representative photomicrographs showing the histological characteristics and proliferative state of colonic adenoma. A and B: Histological characteristics of colonic adenoma in the normal-fat diet/high-fat diet (ND/HFD) groups, and the magnified micrograph (right) highlights the area from the corresponding white rectangle (left); C and D: Representative immunohistochemical staining of proliferative activity (Ki-67, brown) of colonic epithelial cells in the ND/HFD groups; C: Adjacent normal epithelial cells; D; Colonic adenoma tissue; E and F: Ki-67-positive cells were quantified for adjacent normal epithelium (E) and adenoma tissue (F) by counting the positive cells per field (n = 5 per group, bP < 0.01, HFD-fed vs ND-fed). The magnification is marked in the lower right of the pictures, respectively. MM: Magnified micrograph.
HFD consumption alters the levels of biochemical indices and inflammatory cytokines in serum
As stated in the Introduction, it is widely accepted that adipose tissue is an active endocrine organ responsible for the production of many inflammatory cytokines. So, we examined the proinflammatory cytokine TNF-α, which has been linked to an increased risk of CRC. Significant increases in the levels of TNF-α were observed in the HFD group compared with the ND group (P < 0.05, Figure 4A). We next investigated the serum levels of various metabolites in the ND and HFD groups. The concentrations of serum insulin, leptin, IGF-1, triglycerides cholesterol IL-6, and CXCL-10 increased in the HFD-fed group compared with the ND-fed group to a different extent (Figure 4B-H).
Figure 4.
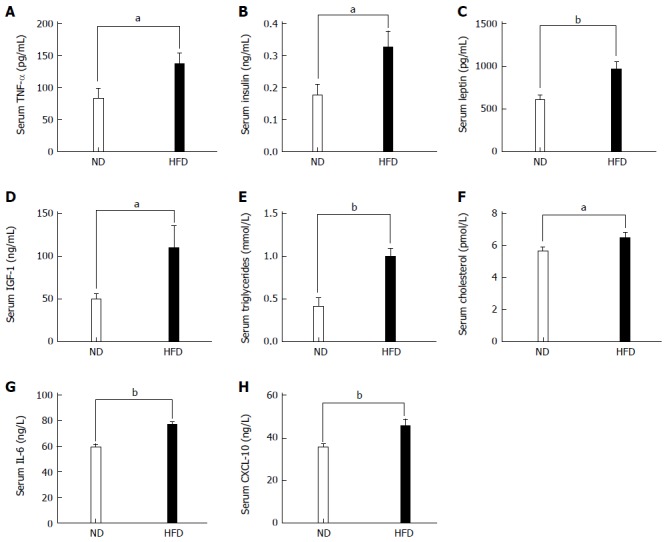
Levels of biochemical indices and inflammatory cytokines in serum between normal-fat diet and high-fat diet groups. Each column represents the mean ± SD; (n = 10/group). A: Levels of TNF-α (86.62 ± 15.77 vs 136.60 ± 17.60, P = 0.034); B: Levels of insulin (0.176 ± 0.03 vs 0.326 ± 0.05, P = 0.024); C: Levels of leptin (608.70 ± 172.0 vs 966.90 ± 239.0, P = 0.002); D: Levels of IGF-1 (49.46 ± 16.74 vs 110.0 ± 26.29, P = 0.045); E: Levels of triglycerides (0.406 ± 0.108 vs 0.992 ± 0.097, P = 0.002); F: Levels of cholesterol (5.645 ± 0.82 vs 6.486 ± 1.06, P = 0.044); G: Levels of IL-6 (59.08 ± 8.405 vs 76.83 ± 7.310, P = 0.004); H: Levels of CXCL-10 (35.47 ± 5.04 vs 45.57 ± 9.45, P = 0.008). TNF-α: Tumor necrosis factor α; IGF-1: Insulin growth factor 1. IL-6: Interlukin-6; CXCL-10: Chemokine-10. (aP < 0.05, bP < 0.01, ND vs HFD).
HFD enhances expression of PCNA and COX-2 in colonic adenomas
We hypothesized that HFD could promote the formation of colonic adenomas. As described above, we observed that the HFD group had more tumors than the ND group, and the proliferative activity in the HFD group was higher than that in the ND group. We further investigated whether HFD could modulate the expression of the inflammatory biomarker COX-2 and another proliferation biomarker PCNA in tumor tissues. Through immunofluorescent staining, we observed that HFD significantly augmented expression of PCNA (Figure 5A) and COX-2 (Figure 5B) in tumor tissues. Meanwhile, the expression of these two biomarkers in normal colon tissues was slightly elevated in the HFD group than in the ND group, although their expression levels remain lower than the tumor tissues. In order to investigate the probable mechanism of action, we determined the expression of NF-κB, β-catenin, cyclin D1 and PCNA in adenoma tissues using Western blot (Figure 6A, B). We found that HFD consumption significantly increased the nuclear levels of cell cycle modulator proteins such as PCNA and cyclin D1 in adenoma tissues (P < 0.01). In addition, HFD consumption also increased the nuclear levels of transcription factor NF-κB (P < 0.01) and β-catenin (P < 0.001). These results demonstrated that consumption of HFD could alter the expression of cyclin D1 and COX-2 proteins, which were correlated with tumor growth in colorectal carcinogenesis. The molecular mechanisms were through the upregulation of β-catenin and NF-κB proteins, respectively[24].
Figure 5.
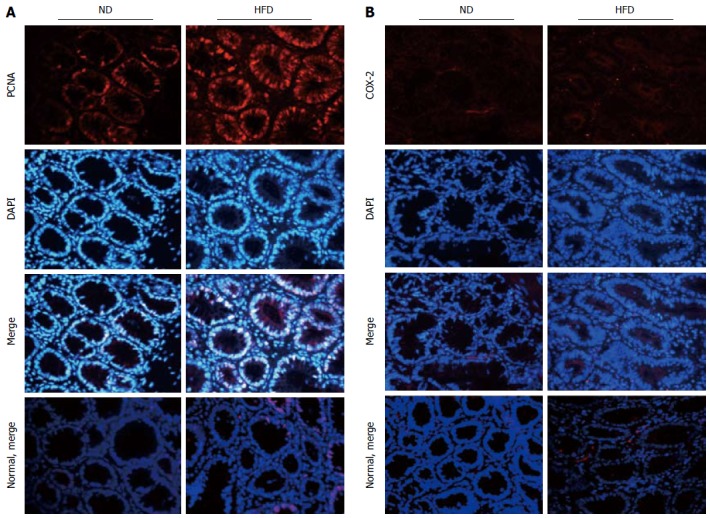
Expression of proliferating cell nuclear antigen and cyclooxygenase -2 in colonic adenoma and normal control tissue for both normal-fat diet and high-fat diet groups by immunofluorescence staining. A: Representative images of immunofluorescence staining for proliferating cell nuclear antigen (PCNA); B: Representative images of immunofluorescence staining for cyclooxygenase -2 (COX-2). The proteins are stained red with corresponding secondary antibody conjugated with Cy3, and cell nuclei are stained blue with 4,6-diamidino-2-phenylindole (DAPI). Imaging was documented at × 400 magnification. ND: Normal-fat diet; HFD: High-fat diet.
Figure 6.
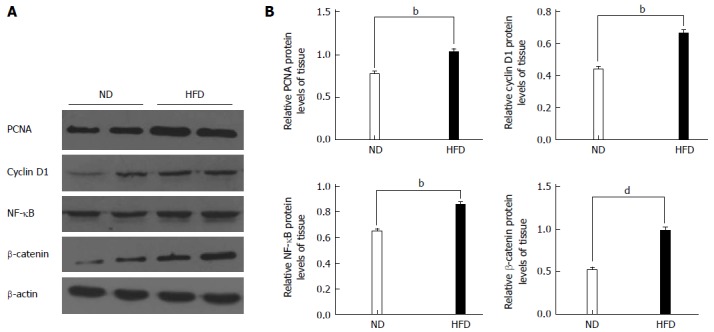
Western blot analyses for proliferating cell nuclear antigen, cyclin D1, nuclear factor-kappa B and β-catenin. A: Representative Western blotting images are shown in the left panel (n = 8/group); B: Right panel: graphs showing the expression ratio of each protein over β-actin in the high-fat diet (HFD) group compared with the normal-fat diet (ND) group. bP < 0.01, dP < 0.01, HFD group vs ND group. PCNA: Proliferating cell nuclear antigen; NF-κB: Nuclear factor-kappa B.
DISCUSSION
It has been reported that dietary factors are thought to account for about 30% of cancers in Western countries, and the contribution of diet to cancer risk in developing countries is lower, perhaps about 20%[25]. A diet-cancer question that has become contentious in recent years is HFD and the role it might play in the development of human CRC[26]. In animal models, the dysplastic aberrant crypts or intraepithelial neoplasia have the potential to progress to advanced adenoma, which has a significant potential to transform into adenocarcinoma through a series of genetic and epigenetic alterations, the adenoma-carcinoma sequence[27]. Therefore, this study was undertaken to explore the effect of an HFD on the incidence of colonic adenoma induced by intraperitoneal DMH injection in Wistar rats.
The DMH model of colon carcinogenesis is a valid, well-appreciated, and widely used model of experimental colon carcinogenesis. It shares many similarities to human sporadic CRC[28], and nowadays is widely used for the evaluation of environmental, dietary, and chemopreventive agents in the colon cancer process. Because the susceptibility to DMH-induced colorectal carcinogenesis is sex and age dependent, 6-wk-old male F344, Wistar and Sprague-Dawley rats are most frequently used[29], we chose Wistar rats in our experiments. We found that tumors were successfully induced in both groups, although there were differences in tumor number. We did not find colon obstruction or hemorrhage in any of the rats when they were sacrificed, and we deduced that this can be explained by the small tumor volume and its early stage.
There are recent reports on the role of HFD in colon carcinogenesis. A previous study by Endo et al[30] indicated that the number of ACFs and colonic epithelial cell proliferative activity were significantly higher in the HFD than in the ND group, and colonic cell proliferation was promoted via the JNK pathway in the presence of an HFD. Morotomi et al[31] demonstrated the enhancing effect of an HFD on ACFs and colonic tumor formation in azoxymethane-injected F344 rats. Park et al[32] evaluated whether HFD could increase colon cancer growth and metastasis in an xenograft tumor model in which CT26 colon cancer cells were subcutaneously injected into BABL/c mice, and confirmed that solid tumor growth as well as the number and volume of tumor nodules in the lungs were increased markedly in the HFD group than in the ND group. In accordance with previous studies, we showed a significant increase in the formation of colonic adenoma (P < 0.01) and an increase in the proliferative activity (Ki-67) of colonic epithelial cells in the HFD group compared with the ND group. It is known that hyperproliferation increases the risk of endogenous mutations in tumor suppressor genes and oncogenes and thus the cancer risk[33]. These results suggest that HFD consumption accelerates tumor growth in a chemical-induced colonic adenoma model.
Our results also showed that the average body weight, liver weight and epididymal fat weight were significantly higher in rats fed an HFD, which indicates that fat tissue mass reflects excess energy consumption in this animal model. Increased caloric intake, usually including an HFD, often results in elevated epididymal fat pads, which leads to obesity in many strains of mice, which also resembles human obesity[34]. Choe et al[35] have also indicated that visceral obesity is a risk factor for colorectal adenoma formation, although it does not have an additional effect on its further progress to colorectal carcinoma. This can be indirectly reflected by the high concentrations of serum triglycerides in the HFD group compared with the ND group. In addition, Nieman et al[36] reported that adipose tissue was characterized by a state of low-grade inflammation. The close link between chronic inflammation and the development of CRC is supported by epidemiological studies. TNF-α, first identified as an antitumor agent, is now also recognized as a tumor-promoting cytokine that links inflammation and cancer[37]. It has been reported that TNF-α plays a prominent role in obesity-induced inflammation and has been shown to be elevated in the circulation as well as in some tissues of obese humans and rodents[38]. In this study, we found that the serum concentration of TNF-α was higher in the HFD group than in the ND group. This cytokine has the ability to promote cell proliferation and survival[37], therefore, our data might conclude that HFD-induced obesity increases the risk of colonic adenoma by inducing chronic inflammation. The importance of TNF-α and its downstream signaling pathways in obesity-mediated colon cancer growth has also been confirmed by Flores et al[39] and Liu et al[40] in a different colon cancer model. Furthermore, adipocytes can secrete adipokines and cytokines, which are known to promote tumor development, such as IGF-1, insulin, leptin, and interleukin-6[41]. These adipocyte-associated molecules are implicated in cell growth, proliferation, differentiation and cell cycle control, as described in the Introduction. The mechanisms for the involvement of these molecules in diet-induced obesity in colon carcinogenesis have been preliminary investigated by Park et al[9]. Their results have led to speculation that HFD-induced obesity facilitates colon tumor formation, possibly by regulating downstream targets of circulating adiposity-related factors via receptor-mediated signaling of the phosphatidylinositol 3-kinase/Akt and extracellular signal-regulated (ERK)1/2 pathways. Our data indicated that HFD-fed rats produced significantly higher amounts of insulin, IGF-1 and leptin. This may explain why there are more adenomas and higher proliferative state in the HFD group compared with the ND group.
It is reported that NF-κB inhibitor protein (Iκ-Bα) is phosphorylated by Iκ-Bα kinase and is subjected to ubiquitin-mediated degradation. Degradation of Iκ-Bα allows the nuclear translocation of activated NF-κB and upregulation of COX-2 gene in cancer cells. Vaish et al[42] have confirmed that COX-2 is a downstream target for NF-κB in inhibiting apoptosis and promoting neoplasia. Overexpression of COX-2 is also frequently observed in the inflammation and progression of colorectal tumors[43,44]. During the proliferation of colon cancer cells, cell cycle-related proteins such as cyclin D1 and PCNA are major regulators of cell cycle progression and DNA replication, respectively[45,46]. In the present study, immunofluorescence staining indicated that HFD consumption could augment the expression of PCNA and COX-2 proteins in tumor tissues. To confirm our findings, we further examined the expression of these malignant biomarkers using Western blot. We found that HFD consumption induced expression of PCNA and cyclin D1 proteins in tumor tissues. The molecular actions of HFD consumption were also associated with upregulation of transcription factors such as β-catenin and NF-κB. These results suggest an important role for β-catenin and NF-κB in regulating the expression of cyclin D1 and COX-2 during HFD-mediated tumor growth, respectively. Furthermore, we can conclude that HFD-mediated tumor growth is associated with inflammation and increased cell cycle progression, although we did not refer to the exact molecular mechanisms involved in this relationship.
In summary, our study demonstrated that HFD consumption could promote the formation of colonic adenoma through inflammation, metabolic abnormalities, and increased cell cycle progression in a DMH-induced tumor model. In the future, continued investigations are needed to elucidate the precise molecular mechanisms in the process of colorectal carcinogenesis under HFD conditions.
COMMENTS
Background
Colorectal cancer (CRC) is a major cause of morbidity and mortality worldwide. Although the link between a high-fat diet (HFD) and an increased CRC risk has been investigated in population-based observational studies, as well as in vitro studies, there is little understanding concerning the effects of HFD in the formation of the precursor (colonic adenoma) of CRC. Therefore, this study was undertaken to explore the effect of HFD on the incidence of 1,2-dimethylhydrazine (DMH)-induced colon carcinogenesis.
Research frontiers
Despite accumulating evidence, the molecular mechanisms underlying the influence of a high intake of dietary fat on the promotion of colorectal carcinogenesis are not fully understood. The possibility that elevated plasma insulin level promotes CRC has also been debated. Some studies support the hypothesis that insulin acts as an important growth factor for colonic epithelial cells. In addition, activation of the inflammation pathway is likely correlated with HFD consumption. These findings suggest that HFD could mediate the development of tumor inflammation through upregulation of cyclo-oxygenase (COX)-2 and activation of multiple signaling cascades including nuclear factor (NF)-κB cascade.
Innovations and breakthroughs
Although most animal experimental studies have shown that an HFD leads to an increased number of chemically induced aberrant crypt foci, which are identifiable lesions in experimental colon carcinogenesis and colon tumors, there is little understanding concerning the effects of HFD in the formation of the precursor (colonic adenoma) of CRC. This is believed to be the first study to report that HFD can promote the formation of colonic adenoma in a DMH-induced tumor model. The authors demonstrated an important role of β-catenin and NF-κB in regulating the expression of cyclin D1 and COX-2 during HFD-mediated tumor growth, respectively.
Applications
By understanding how HFD promotes colon carcinogenesis, the identification and evaluation of the relationship between colonic adenoma and high intake of dietary fat will be crucial for the development of preventive strategies against CRC in the near future.
Terminology
DMH is the most commonly used carcinogen to induce and promote colorectal tumors in rats and mice. DMH is an alkylating agent that is typically injected intraperitoneally or subcutaneously over several weeks to induce development of tumors in the colon. In addition, tumor incidence and multiplicity can be altered by both genetic background and by diet. This makes the models useful for the study of gene-gene and gene-environment interactions that influence the pathogenesis of CRC.
Peer review
This is a good study in which authors investigated the facilitative effect of HFD on the formation of colonic adenoma induced by DMH in Wistar rats. The results are reliable and suggest a significant role of HFD in the development of colon adenoma. This study represents a further step in trying to understand the mechanisms involved in this relationship.
Footnotes
Supported by the National Natural Science Foundation of China, No. 81230057
P- Reviewers: Goetze TO, Kim K, Matusiewicz M S- Editor: Qi Y L- Editor: Wang TQ E- Editor: Liu XM
References
- 1.O'Keefe SJ, Ou J, Aufreiter S, O’Connor D, Sharma S, Sepulveda J, Fukuwatari T, Shibata K, Mawhinney T. Products of the colonic microbiota mediate the effects of diet on colon cancer risk. J Nutr. 2009;139:2044–2048. doi: 10.3945/jn.109.104380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pignone M, Rich M, Teutsch SM, Berg AO, Lohr KN. Screening for colorectal cancer in adults at average risk: a summary of the evidence for the U.S. Preventive Services Task Force. Ann Intern Med. 2002;137:132–141. doi: 10.7326/0003-4819-137-2-200207160-00015. [DOI] [PubMed] [Google Scholar]
- 3.Giovannucci E, Willett WC. Dietary factors and risk of colon cancer. Ann Med. 1994;26:443–452. doi: 10.3109/07853899409148367. [DOI] [PubMed] [Google Scholar]
- 4.Lasko CM, Bird RP. Modulation of aberrant crypt foci by dietary fat and caloric restriction: the effects of delayed intervention. Cancer Epidemiol Biomarkers Prev. 1995;4:49–55. [PubMed] [Google Scholar]
- 5.Nör JE, Christensen J, Liu J, Peters M, Mooney DJ, Strieter RM, Polverini PJ. Up-Regulation of Bcl-2 in microvascular endothelial cells enhances intratumoral angiogenesis and accelerates tumor growth. Cancer Res. 2001;61:2183–2188. [PubMed] [Google Scholar]
- 6.Buettner R, Schölmerich J, Bollheimer LC. High-fat diets: modeling the metabolic disorders of human obesity in rodents. Obesity (Silver Spring) 2007;15:798–808. doi: 10.1038/oby.2007.608. [DOI] [PubMed] [Google Scholar]
- 7.Lin S, Thomas TC, Storlien LH, Huang XF. Development of high fat diet-induced obesity and leptin resistance in C57Bl/6J mice. Int J Obes Relat Metab Disord. 2000;24:639–646. doi: 10.1038/sj.ijo.0801209. [DOI] [PubMed] [Google Scholar]
- 8.Campos FG, Logullo Waitzberg AG, Kiss DR, Waitzberg DL, Habr-Gama A, Gama-Rodrigues J. Diet and colorectal cancer: current evidence for etiology and prevention. Nutr Hosp. 2005;20:18–25. [PubMed] [Google Scholar]
- 9.Park SY, Kim JS, Seo YR, Sung MK. Effects of diet-induced obesity on colitis-associated colon tumor formation in A/J mice. Int J Obes (Lond) 2012;36:273–280. doi: 10.1038/ijo.2011.83. [DOI] [PubMed] [Google Scholar]
- 10.Rajala MW, Scherer PE. Minireview: The adipocyte--at the crossroads of energy homeostasis, inflammation, and atherosclerosis. Endocrinology. 2003;144:3765–3773. doi: 10.1210/en.2003-0580. [DOI] [PubMed] [Google Scholar]
- 11.Pisani P. Hyper-insulinaemia and cancer, meta-analyses of epidemiological studies. Arch Physiol Biochem. 2008;114:63–70. doi: 10.1080/13813450801954451. [DOI] [PubMed] [Google Scholar]
- 12.Renehan AG, Frystyk J, Flyvbjerg A. Obesity and cancer risk: the role of the insulin-IGF axis. Trends Endocrinol Metab. 2006;17:328–336. doi: 10.1016/j.tem.2006.08.006. [DOI] [PubMed] [Google Scholar]
- 13.Pais R, Silaghi H, Silaghi AC, Rusu ML, Dumitrascu DL. Metabolic syndrome and risk of subsequent colorectal cancer. World J Gastroenterol. 2009;15:5141–5148. doi: 10.3748/wjg.15.5141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fantuzzi G. Adipose tissue, adipokines, and inflammation. J Allergy Clin Immunol. 2005;115:911–919; quiz 920. doi: 10.1016/j.jaci.2005.02.023. [DOI] [PubMed] [Google Scholar]
- 15.Chia VM, Newcomb PA, Lampe JW, White E, Mandelson MT, McTiernan A, Potter JD. Leptin concentrations, leptin receptor polymorphisms, and colorectal adenoma risk. Cancer Epidemiol Biomarkers Prev. 2007;16:2697–2703. doi: 10.1158/1055-9965.EPI-07-0467. [DOI] [PubMed] [Google Scholar]
- 16.Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW. Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest. 2003;112:1796–1808. doi: 10.1172/JCI19246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hotamisligil GS, Shargill NS, Spiegelman BM. Adipose expression of tumor necrosis factor-alpha: direct role in obesity-linked insulin resistance. Science. 1993;259:87–91. doi: 10.1126/science.7678183. [DOI] [PubMed] [Google Scholar]
- 18.Fischer-Posovszky P, Wabitsch M, Hochberg Z. Endocrinology of adipose tissue - an update. Horm Metab Res. 2007;39:314–321. doi: 10.1055/s-2007-976539. [DOI] [PubMed] [Google Scholar]
- 19.Bae YJ, Kim SH, Chung JH, Song SW, Kim KS, Kim MK, Kwon O, Choi MS, Sung MK. Evaluation of adiposity-related biomarkers as metabolic syndrome indicators. Clin Nutr Res. 2013;2:91–99. doi: 10.7762/cnr.2013.2.2.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wellen KE, Hotamisligil GS. Obesity-induced inflammatory changes in adipose tissue. J Clin Invest. 2003;112:1785–1788. doi: 10.1172/JCI20514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pischon T, Nöthlings U, Boeing H. Obesity and cancer. Proc Nutr Soc. 2008;67:128–145. doi: 10.1017/S0029665108006976. [DOI] [PubMed] [Google Scholar]
- 22.Peng J, Zhang Q, Ma Y, Wang Y, Huang L, Zhang P, Chen J, Qin H. A rat-to-human search for proteomic alterations reveals transgelin as a biomarker relevant to colorectal carcinogenesis and liver metastasis. Electrophoresis. 2009;30:2976–2987. doi: 10.1002/elps.200900203. [DOI] [PubMed] [Google Scholar]
- 23.Bertkova I, Hijova E, Chmelarova A, Mojzisova G, Petrasova D, Strojny L, Bomba A, Zitnan R. The effect of probiotic microorganisms and bioactive compounds on chemically induced carcinogenesis in rats. Neoplasma. 2010;57:422–428. doi: 10.4149/neo_2010_05_422. [DOI] [PubMed] [Google Scholar]
- 24.Tetsu O, McCormick F. Beta-catenin regulates expression of cyclin D1 in colon carcinoma cells. Nature. 1999;398:422–426. doi: 10.1038/18884. [DOI] [PubMed] [Google Scholar]
- 25.Armstrong B, Doll R. Environmental factors and cancer incidence and mortality in different countries, with special reference to dietary practices. Int J Cancer. 1975;15:617–631. doi: 10.1002/ijc.2910150411. [DOI] [PubMed] [Google Scholar]
- 26.Giovannucci E, Goldin B. The role of fat, fatty acids, and total energy intake in the etiology of human colon cancer. Am J Clin Nutr. 1997;66:1564S–1571S. doi: 10.1093/ajcn/66.6.1564S. [DOI] [PubMed] [Google Scholar]
- 27.Vogelstein B, Fearon ER, Hamilton SR, Kern SE, Preisinger AC, Leppert M, Nakamura Y, White R, Smits AM, Bos JL. Genetic alterations during colorectal-tumor development. N Engl J Med. 1988;319:525–532. doi: 10.1056/NEJM198809013190901. [DOI] [PubMed] [Google Scholar]
- 28.Corpet DE, Pierre F. How good are rodent models of carcinogenesis in predicting efficacy in humans? A systematic review and meta-analysis of colon chemoprevention in rats, mice and men. Eur J Cancer. 2005;41:1911–1922. doi: 10.1016/j.ejca.2005.06.006. [DOI] [PubMed] [Google Scholar]
- 29.Perše M, Cerar A. Morphological and molecular alterations in 1,2 dimethylhydrazine and azoxymethane induced colon carcinogenesis in rats. J Biomed Biotechnol. 2011;2011:473964. doi: 10.1155/2011/473964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Endo H, Hosono K, Fujisawa T, Takahashi H, Sugiyama M, Yoneda K, Nozaki Y, Fujita K, Yoneda M, Inamori M, et al. Involvement of JNK pathway in the promotion of the early stage of colorectal carcinogenesis under high-fat dietary conditions. Gut. 2009;58:1637–1643. doi: 10.1136/gut.2009.183624. [DOI] [PubMed] [Google Scholar]
- 31.Morotomi M, Sakaitani Y, Satou M, Takahashi T, Takagi A, Onoue M. Effects of a high-fat diet on azoxymethane-induced aberrant crypt foci and fecal biochemistry and microbial activity in rats. Nutr Cancer. 1997;27:84–91. doi: 10.1080/01635589709514507. [DOI] [PubMed] [Google Scholar]
- 32.Park H, Kim M, Kwon GT, Lim do Y, Yu R, Sung MK, Lee KW, Daily JW, Park JH. A high-fat diet increases angiogenesis, solid tumor growth, and lung metastasis of CT26 colon cancer cells in obesity-resistant BALB/c mice. Mol Carcinog. 2012;51:869–880. doi: 10.1002/mc.20856. [DOI] [PubMed] [Google Scholar]
- 33.Kinzler KW, Vogelstein B. Lessons from hereditary colorectal cancer. Cell. 1996;87:159–170. doi: 10.1016/s0092-8674(00)81333-1. [DOI] [PubMed] [Google Scholar]
- 34.Lee YS, Li P, Huh JY, Hwang IJ, Lu M, Kim JI, Ham M, Talukdar S, Chen A, Lu WJ, et al. Inflammation is necessary for long-term but not short-term high-fat diet-induced insulin resistance. Diabetes. 2011;60:2474–2483. doi: 10.2337/db11-0194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Choe EK, Kim D, Kim HJ, Park KJ. Association of visceral obesity and early colorectal neoplasia. World J Gastroenterol. 2013;19:8349–8356. doi: 10.3748/wjg.v19.i45.8349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nieman KM, Romero IL, Van Houten B, Lengyel E. Adipose tissue and adipocytes support tumorigenesis and metastasis. Biochim Biophys Acta. 2013;1831:1533–1541. doi: 10.1016/j.bbalip.2013.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Balkwill F. Tumour necrosis factor and cancer. Nat Rev Cancer. 2009;9:361–371. doi: 10.1038/nrc2628. [DOI] [PubMed] [Google Scholar]
- 38.Tilg H, Moschen AR. Adipocytokines: mediators linking adipose tissue, inflammation and immunity. Nat Rev Immunol. 2006;6:772–783. doi: 10.1038/nri1937. [DOI] [PubMed] [Google Scholar]
- 39.Flores MB, Rocha GZ, Damas-Souza DM, Osório-Costa F, Dias MM, Ropelle ER, Camargo JA, de Carvalho RB, Carvalho HF, Saad MJ, et al. Obesity-induced increase in tumor necrosis factor-α leads to development of colon cancer in mice. Gastroenterology. 2012;143:741–753.e1-4. doi: 10.1053/j.gastro.2012.05.045. [DOI] [PubMed] [Google Scholar]
- 40.Liu Z, Brooks RS, Ciappio ED, Kim SJ, Crott JW, Bennett G, Greenberg AS, Mason JB. Diet-induced obesity elevates colonic TNF-α in mice and is accompanied by an activation of Wnt signaling: a mechanism for obesity-associated colorectal cancer. J Nutr Biochem. 2012;23:1207–1213. doi: 10.1016/j.jnutbio.2011.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yehuda-Shnaidman E, Schwartz B. Mechanisms linking obesity, inflammation and altered metabolism to colon carcinogenesis. Obes Rev. 2012;13:1083–1095. doi: 10.1111/j.1467-789X.2012.01024.x. [DOI] [PubMed] [Google Scholar]
- 42.Vaish V, Tanwar L, Sanyal SN. The role of NF-κB and PPARγ in experimentally induced colorectal cancer and chemoprevention by cyclooxygenase-2 inhibitors. Tumour Biol. 2010;31:427–436. doi: 10.1007/s13277-010-0051-7. [DOI] [PubMed] [Google Scholar]
- 43.Raufman JP, Shant J, Guo CY, Roy S, Cheng K. Deoxycholyltaurine rescues human colon cancer cells from apoptosis by activating EGFR-dependent PI3K/Akt signaling. J Cell Physiol. 2008;215:538–549. doi: 10.1002/jcp.21332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Baldassarre G, Nicoloso MS, Schiappacassi M, Chimienti E, Belletti B. Linking inflammation to cell cycle progression. Curr Pharm Des. 2004;10:1653–1666. doi: 10.2174/1381612043384691. [DOI] [PubMed] [Google Scholar]
- 45.Arber N, Doki Y, Han EK, Sgambato A, Zhou P, Kim NH, Delohery T, Klein MG, Holt PR, Weinstein IB. Antisense to cyclin D1 inhibits the growth and tumorigenicity of human colon cancer cells. Cancer Res. 1997;57:1569–1574. [PubMed] [Google Scholar]
- 46.Qiao L, Shiff SJ, Rigas B. Sulindac sulfide inhibits the proliferation of colon cancer cells: diminished expression of the proliferation markers PCNA and Ki-67. Cancer Lett. 1997;115:229–234. doi: 10.1016/s0304-3835(97)04740-x. [DOI] [PubMed] [Google Scholar]