

Abstract
To maintain genetic integrity, ultraviolet light-induced photoproducts in DNA must be removed by the nucleotide excision repair (NER) pathway, which is initiated by damage recognition and dual incisions of the lesion-containing strand. We intended to detect the dual-incision step of cellular NER, by using a fluorescent probe. A 140-base pair linear duplex containing the (6–4) photoproduct and a fluorophore–quencher pair was prepared first. However, this type of DNA was found to be degraded rapidly by nucleases in cells. Next, a plasmid was used as a scaffold. In this case, the fluorophore and the quencher were attached to the same strand, and we expected that the dual-incision product containing them would be degraded in cells. At 3 h after transfection of HeLa cells with the plasmid-type probes, fluorescence emission was detected at the nuclei by fluorescence microscopy only when the probe contained the (6–4) photoproduct, and the results were confirmed by flow cytometry. Finally, XPA fibroblasts and the same cells expressing the XPA gene were transfected with the photoproduct-containing probe. Although the transfer of the probe into the cells was slow, fluorescence was detected depending on the NER ability of the cells.
Ultraviolet (UV) light causes photochemical reactions of the base moieties in DNA1. The major products are cyclobutane pyrimidine dimers (CPDs) and pyrimidine(6–4)pyrimidone photoproducts ((6–4) photoproducts), formed between adjacent pyrimidine bases. Since these photoproducts induce genetic mutations, which lead to carcinogenesis and cell death, they must be repaired by the nucleotide excision repair (NER) pathway to maintain genetic integrity2. The (6–4) photoproduct is repaired by NER more efficiently than the CPD3, and other lesions that significantly alter the chemical structure of DNA, such as carcinogen adducts, are also removed by NER4.
There are two subpathways in eukaryotic NER: global genome NER (GG-NER) and transcription-coupled NER (TC-NER)2. In GG-NER, the XPE protein, which consists of the DDB1 and DDB2 subunits, recognizes the lesion by forming a specific complex with damaged DNA5, and then the DNA is transferred to the XPC protein, through the ubiquitylation of both proteins by the ubiquitin ligase bound to DDB16. After this damage recognition step, TFIIH, a general transcription factor, and the XPA protein are recruited. TFIIH contains the XPB and XPD proteins, which both have DNA helicase activity, and the XPA protein is considered to stabilize the open complex and to verify the damage2. Subsequently, the ERCC1–XPF complex and the XPG protein, which are both structure-dependent nucleases, cleave the lesion-containing strand on the 5′ and 3′ sides of the damage site, respectively, to produce a fragment with a chain length of about 30 nucleotides. TC-NER differs from GG-NER only at the damage recognition step. This subpathway is initiated by the stalling of RNA polymerase II at the damage site, and other factors such as CSA and CSB are required, instead of the XPE and XPC proteins, to form the open complex containing the TFIIH complex and the XPA protein7. After the dual incisions by ERCC1–XPF and XPG, the repair is completed by DNA polymerase and DNA ligase.
Mutations in genes related to NER result in the diseases known as xeroderma pigmentosum (XP), Cockayne syndrome (CS), and trichothiodystrophy, which are autosomal recessive disorders8,9. Seven genes, XPA–XPG, which encode the proteins functioning in the NER pathway described above, were identified by complementation experiments, and the products of the CS-related genes are required to link the damage recognition to the open complex formation in the TCR subpathway. XP patients are extremely sensitive to sunlight and develop skin cancer at high rates10,11. The relative risks of non-melanoma skin cancer and melanoma in XP patients were recently calculated to be about 10,000-fold and more than 2,000-fold higher, respectively, than those of the general population12. Neurological degeneration is also correlated with mutations in several XP genes13.
Since XP is a hereditary disease in which skin cancer develops at an early age12, its diagnosis is important. Based on the diagnosis, the patients are advised to avoid sunlight to prevent the onset of skin cancer. The most commonly used test is the measurement of unscheduled DNA synthesis (UDS)10. Skin fibroblast cultures are prepared from a biopsy obtained from the subject, and after irradiation with UV light, [3H]thymidine incorporation into DNA is measured by autoradiography or liquid scintillation counting14,15. The XP cells show lower UDS ability, because the cells are deficient in the active proteins that recognize and remove the lesion. However, this diagnostic method is not entirely satisfactory, for several reasons. One is the risk of exposure of the medical staff to radiation in the use of the radioactive compound, although this problem may be solved by using a fluorescent nucleotide16. Another is the considerable suffering of the subject. In this study, we developed a fluorescent method for detecting the cellular ability to incise the damaged strand in the NER pathway, which could be applied to the screening of XP.
Results
Trial of linear DNA as a fluorescent probe
In our previous study17, we developed fluorescent probes to detect base excision repair (BER), which initiates the removal of damaged bases with relatively small changes in their chemical structures. The probes were hairpin-shaped 30-mer oligonucleotides containing an oxidatively damaged base in the center, and bearing a fluorophore and a quencher at the 5′ and 3′ ends, respectively, in the same manner as a molecular beacon18. To detect the BER enzyme reaction in cells, nonspecific degradation was prevented by changing the phosphodiester linkages to nuclease-resistant phosphorothioate analogs in the regions that were not required for enzyme binding. We tried to use this system to detect the NER-specific dual incisions. However, the shortest substrate for NER reported so far was about 100 base-pair (bp) long19, and a 140 bp duplex was used successfully to analyze NER in vitro20, while the above-mentioned BER probe was composed of only 13 base pairs with a loop. Therefore, a strategy of attaching the fluorophore and the quencher to the base moieties in the central part of a long duplex was formulated, with the expectation that the fluorophore-tethered fragment would be released from the duplex after the dual incisions (Fig. 1a). Since the fluorophore and the quencher are generally attached to the end of the stem structure in molecular beacon-type probes, we firstly determined whether such internal labeling could function, using a short duplex shown in Supplementary Fig. S1a. When the two strands were mixed, the fluorescence intensity decreased, depending on the amount of the quencher-containing strand (Supplementary Fig. S1b).
Figure 1. Fluorescent probes to detect the NER dual incisions.
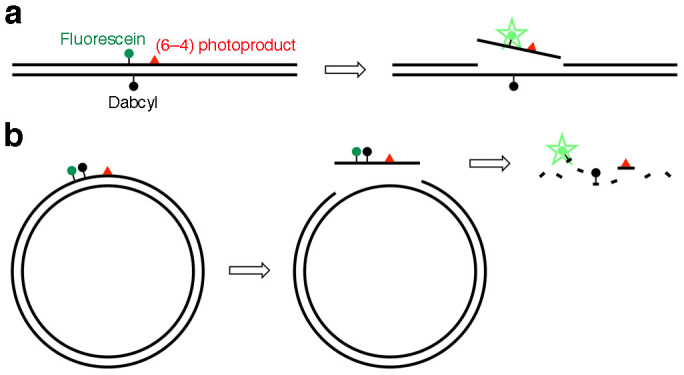
(a) A linear duplex containing the (6–4) photoproduct and the fluorophore–quencher pair. Detection of the dual incisions in cells was not successful due to nonspecific degradation. (b) A plasmid-type probe containing the fluorophore and the quencher in the same strand. Degradation of the dual-incision product by cellular nucleases was expected to obtain the positive signal.
Based on the above results, we designed a 140 bp duplex containing fluorescein, Dabcyl, and the (6–4) photoproduct, as shown in Supplementary Fig. S1c. Since little information is available about the protein–DNA interactions at the atomic level throughout the NER process, only three phosphodiester linkages at both ends in each strand were changed into phosphorothioates to protect the duplex from exonuclease digestion. A duplex containing normal thymines in place of the (6–4) photoproduct was also prepared, as a control. The duplexes with and without the (6–4) photoproduct were treated with a HEK293 cell extract, and the products were analyzed by PAGE. Replication protein A (RPA) was added to one of the reaction mixtures containing each duplex, because this factor reportedly facilitates NER21. A ladder of bands was obtained for the photoproduct-containing duplex, which was presumed to be produced by the dual incisions in the NER pathway. However, other bands were detected at the positions of 15- and 16-mers, in both the presence and absence of the (6–4) photoproduct (Supplementary Fig. S1d). Changes in the fluorescence intensity after a treatment with a HeLa cell extract were also analyzed. As expected from the PAGE results, no difference was observed between the duplexes with and without the photoproduct (Supplementary Fig. S1e). Finally, XPA cells, which are deficient in the NER activity, were transfected with these duplexes. Fluorescence was detected even when the duplex without the photoproduct was used (Supplementary Fig. S1f). These results suggested that the 140 bp duplexes were nonspecifically degraded during the incubation with the cell extracts or within the cells.
Plasmid-type fluorescent probe
Since the linear DNA was found to be too labile to be used as a fluorescent probe for the detection of cellular NER, even with the phosphorothioate modification at both ends, we tried a plasmid, which has often been used as a substrate for NER21,22. In the design of the plasmid-type probe, the fluorophore–quencher system was reconsidered. Since the linear probes were degraded nonspecifically in the cell extracts and in cells, we expected that the dual-incision product, which would be released from the plasmid as an oligonucleotide, would be degraded and fluoresce if the fluorophore and the quencher were attached at adjacent positions in this fragment (Fig. 1b). In preliminary experiments, oligonucleotides bearing fluorescein and Dabcyl at adjacent positions with no or one nucleotide insertion (Supplementary Fig. S2a) were prepared, and their properties were analyzed. When the Fl-Dab and Fl-T-Dab 28-mers in Supplementary Fig. S2a were hybridized to the complementary 28-mer, the quenching efficiencies were 99 and 98%, respectively. Quenching occurred very efficiently even in the single-stranded form (at 0 min in Supplementary Fig. S2b), and the fluorescence intensity increased upon treatment with a HeLa cell extract. Fluorescence was also observed in the cells transfected with these oligonucleotides (Supplementary Fig. S2c). Since the fluorescence intensity was increased more efficiently when an extra nucleotide was inserted between the fluorophore and the quencher (Supplementary Fig. S2b), we adopted this system, and prepared a plasmid designated as pBSII KS (-) FQ64 (Fig. 2a). A 9 bp sequence was inserted into the original plasmid, pBSII KS (-) UV23, to incorporate the modified thymidines bearing the fluorophore and the quencher, and the single-stranded DNA was prepared using VCS-M13 helper phage. A 37-mer oligonucleotide containing the (6–4) photoproduct and the modified thymidines (Fig. 2b) was hybridized to this DNA, and pBSII KS (-) FQ64 was obtained using DNA polymerase and DNA ligase. pBSII KS (-) FQTT, which contained undamaged thymine bases in place of the (6–4) photoproduct, was also prepared as a negative-control probe. The presence of the (6–4) photoproduct in pBSII KS (-) FQ64 was confirmed by UVDE digestion (Fig. 2c) and the repair synthesis in NER (Fig. 2d).
Figure 2. Plasmid-type fluorescent probe to detect the NER dual incisions.

(a) Structure of the plasmid. The green and black circles represent fluorescein and Dabcyl, respectively. The italicized letters represent the BstNI recognition sequence, and the underlined sequence was added to the original pBSII KS (-) UV plasmid23 to incorporate the modified nucleosides. (b) Chemical structures of the (6–4) photoproduct and the modified nucleosides bearing fluorescein and Dabcyl. (c) UVDE treatment of pBSII KS (-) FQTT (TT) and pBSII KS (-) FQ64 ((6–4)). (d) Repair synthesis on pBSII KS (-) FQTT (TT) and pBSII KS (-) FQ64 ((6–4)), followed by BstNI digestion.
Fluorescence detection of NER in cells
HeLa cells were transfected with either pBSII KS (-) FQ64 or pBSII KS (-) FQTT, using Lipofectamine 2000. A transfection reporter bearing Cy5, which was developed in our previous study24, was co-transfected to prevent false negative results, although its structure was substantially different from that of the probe. At 3 h after transfection, the fluorescein emission was detected at the nuclei, depending on the presence of the photoproduct (Fig. 3a). The Cy5 emission in the cells transfected with pBSII KS (-) FQTT demonstrated that the result obtained for these cells was not falsely negative.
Figure 3. Detection of the NER dual incisions in HeLa cells.

(a) Fluorescence images of the cells cultured at 37°C for 3 h, after transfection with pBSII KS (-) FQTT (upper panels) or pBSII KS (-) FQ64 (lower panels). (b) Flow cytometry analysis of the HeLa cells transfected with pBSII KS (-) FQTT (blue) or pBSII KS (-) FQ64 (red), together with the transfection reporter bearing Cy524.
To obtain more quantitative information, the cellular NER reaction was analyzed by flow cytometry. HeLa cells were transfected with pBSII KS (-) FQ64 or pBSII KS (-) FQTT, together with the transfection reporter, in the same manner as above. At 1 h intervals, the cells were harvested by trypsin digestion, and analyzed on a flow cytometer. The Cy5 emission from the transfection reporter was the same between the pBSII KS (-) FQ64- and pBSII KS (-) FQTT-transfected cells, and the transfer of the probes into the cells was apparently completed at 3 h after the transfection (Fig. 3b, lower panels). At this time, a difference in the fluorescein emission was observed between the cells transfected with the probes with and without the (6–4) photoproduct, and this difference disappeared at 4 h, probably due to the nonspecific degradation of the probes (Fig. 3b, upper panels). These results demonstrated that cellular NER could be monitored by flow cytometry, although the background was relatively high at the fluorescein emission wavelength (Supplementary Fig. S3).
Finally, the XPA fibroblasts that were deficient in the NER activity (XP12ROSV) and the same cells in which the XPA gene was expressed (XP12ROSV/XPA)25 were transfected with pBSII KS (-) FQ64. In this case, a longer culture time was required because the transfer of the probe into the cells was apparently slower, as determined by the Cy5 emission from the transfection reporter. However, positive signals were observed only in the experiments using the NER-proficient cells (Fig. 4a). The fluorescence intensities in the microscopic images of the cells were quantified (Fig. 4b), and a significant difference was found between the NER-deficient and -proficient cells, although the background fluorescence, which was observed in the flow cytometry analysis (Supplementary Fig. S3), was included.
Figure 4. Detection of the cellular NER ability.

(a) The XPA cells (upper panels) and the same cells in which the XPA gene was expressed (lower panels) were cultured at 37°C for 6 h after transfection with pBSII KS (-) FQ64. (b) The fluorescein and Cy5 emissions from the pBSII KS (-) FQ64 probe and the transfection reporter, respectively, in the XP12ROSV (NER –) and XP12ROSV/XPA (NER +) cells, shown in panel a, were quantified. The data were statistically analyzed by Student's t test (n = 10; *, p < 0.01), and error bars represent standard deviation. Background correction was not performed.
Discussion
In this study, we intended to develop a fluorescent probe to detect cellular NER, for possible use in the diagnosis of XP. At first, we tried to apply our study on the BER probe, a short hairpin oligonucleotide that emits fluorescence upon strand scission by DNA glycosylase/AP lyase17. However, there was a problem that the substrate for NER must be much longer than that for BER19,20. Since the molecular beacon-type fluorophore–quencher system18 could be used when the two dyes were tethered to the base moieties in the inner region of each strand (Supplementary Figs. S1a and S1b), a 140 bp duplex containing the (6–4) photoproduct (Supplementary Fig. S1c), which is known as a good substrate for NER3, was tested. Contrary to our expectations, degradation of the duplex was observed in in vitro and ex vivo experiments (Supplementary Figs. S1d–f). Therefore, we used a plasmid as a scaffold, and reexamined the fluorophore–quencher system to utilize the nuclease degradation of the probe. The structure of the plasmid in which the fluorophore and the quencher were attached to the same strand (Fig. 2a) facilitated the preparation of the probe, because only the DNA polymerase and DNA ligase reactions were required after a modified oligonucleotide primer was synthesized.
At 3 h and 6 h after transfection of the HeLa cells and the XPA fibroblasts, respectively, fluorescein fluorescence was detected, depending on the presence of the (6–4) photoproduct and the cellular NER ability (Figs. 3a and 4a). Therefore, these signals were attributed to the degradation of the dual-incision fragment produced by NER. At a longer culture time, nonspecific degradation of the plasmid was observed, as detected by flow cytometry (4 h in Fig. 3b). Therefore, control experiments using the plasmid without the (6–4) photoproduct were important. The transfection reporter developed in our previous study24 was also useful to distinguish between the true- and false-negative results. Interestingly, we always observed fluorescence emission from the nuclei. Several types of fluorescent probes were developed to visualize the nuclease degradation of DNA in cells26,27, and our plasmid that lacks the (6–4) photoproduct can be used for this purpose. The half-life of plasmid DNA microinjected into the cytosol is reportedly 50–90 min28, but we never observed fluorescence in the cytosol, suggesting that our probe, which was transferred into the cells using a cationic lipid, was transported into the nuclei, although no nuclear targeting strategy29 was used.
Recently, Sancar and co-workers investigated the fate of the fragments produced by the dual incisions in NER30,31. Their in vitro study30 revealed that the photoproduct is removed in the form of a 30-mer oligonucleotide in complex with TFIIH. The oligonucleotide is dissociated from TFIIH in an ATP-dependent manner, and then RPA binds to the DNA fragment. It was suggested that the RPA protects this oligomer from degradation by nuclease digestion. From our results, however, the dual-incision product appeared to be quite labile in cells. Degradation products shorter than 20-nucleotide long were detected after incubation for 1 h or longer in their study30, and the fluorophore was not located close to the (6–4) photoproduct in our probe. Therefore, the protection by RPA seems to have limited effects at least at the ends of the dual-incision fragment.
Our fluorescent probe may be applied to the screening of XP, which is a hereditary disease caused by cellular NER deficiency. For use as an alternative to the UDS measurement for the diagnosis of XP, the transfection efficiency into fibroblasts or the quantum yield of fluorescence should be improved by changing the transfection reagent or the fluorophore, because the fluorescence intensity was low when the XPA fibroblasts were used (Fig. 4a). Our probe can also be used to compare two types of lesions as substrates for NER in a single cell. When cells are transfected with two plasmids, each containing an individual set of the lesion and the fluorophore, the better substrate can be determined by observing the fluorescence at two wavelengths. Artificial substrates for NER were reported recently32. If this method reveals that they are better substrates than the (6–4) photoproduct, then their incorporation into the plasmid may improve the sensitivity of the probe. Another benefit of our method, in which cells are not irradiated with UV, is that only the cellular GG-NER process is monitored, and thus other effects of UV-related reactions, such as lipid oxidation, are avoided. Therefore, our probe will contribute to both medical and biological studies.
Methods
Synthesis of oligonucleotides
The phosphoramidite building block of the (6–4) photoproduct was synthesized by the previously described method33. To incorporate the fluorescein- and Dabcyl-tethered nucleosides, Fluorescein-dT Phosphoramidite and Dabcyl-dT were purchased from Glen Research. Oligonucleotides were synthesized on an Applied Biosystems 3400 DNA synthesizer. Benzimidazolium triflate was used as an activator for the synthesis of the (6–4) photoproduct-containing oligonucleotide34. After deprotection, the products were purified by HPLC, using a Waters μ-Bondasphere C18 5 μm 300 A column (3.9 × 150 mm) with a linear gradient of acetonitrile in 0.1 M triethylammonium acetate (pH 7.0) at a flow rate of 1.0 ml/min. The transfection reporter was synthesized as described previously24.
Preparation of 140 bp duplexes
For the analysis of the dual incisions by gel electrophoresis, 14-mers (20 pmol) with and without the (6–4) photoproduct, d(CGCGAATTGCGCCC), in which the photoproduct site is underlined, were incubated with [γ-32P]ATP (3.7 MBq) and T4 polynucleotide kinase (Takara Bio) (10 units), in buffer (50 μl) containing 50 mM Tris-HCl (pH 8.0), 10 mM MgCl2, and 5 mM DTT, at 37°C for 30 min. After heating at 95°C for 5 min, the products were purified using an illustra MicroSpin G-25 column (GE Healthcare). Aliquots of each 5′-phosphorylated 14-mer (4.8 pmol) were mixed with four oligonucleotides, d(GsAsCsCTGAACACGTACGGAATTCGATATCCTCGAGCCAGATCTGCGCCAGTTGGCCXACTCGTC) (7.2 pmol), in which s and X represent the phosphorothioate linkage and the fluorescein-tethered nucleoside (Fig. 2b), respectively, p-d(ACAGTCAGTGGCTTGGGCTGCAGCAGGTCGACTCTAGAGGATCCCGGGCGAGCTCGAATTsCsGsC) (7.2 pmol), in which p represents the terminal phosphate, d(TTCGCGGACGAGTAGG) (10 pmol), and d(CACTGACTGTGGGCGC) (10 pmol), in buffer (58 μl) containing 10 mM Tris-HCl (pH 7.9), 50 mM NaCl, 10 mM MgCl2, and 1 mM DTT. The mixtures were heated at 95°C for 2 min, and gradually cooled to 25°C during 45 min. After ATP (12 nmol) and T4 DNA ligase (Takara Bio) (210 units) were added, the mixtures were incubated at 16°C overnight, and then heated at 75°C for 10 min. The products were purified by 10% denaturing PAGE, and desalted using the MicroSpin G-25 column. For fluorescence detection, 10 nmol of the 14-mer was phosphorylated in buffer (100 μl) using ATP (0.2 μmol) and T4 polynucleotide kinase (100 units), and the ligation was performed in buffer (100 μl), using 12 nmol of the 5′- and 3′-side oligonucleotides, 20 nmol of the splint 16-mers, and 3,500 units of T4 DNA ligase, in the presence of 1 mM ATP. The complementary strand was prepared using d(GsCsGsAATTCGAGCTCGCCCGGGATCCTCTAGAGTCGACCTGCTGCAGCCCAAGCCACTGACTGTGGGCGCAAT) (10 nmol), p-d(TCGCGGACGAGYAGGCCAACTGGCGCAGATCTGGCTCGAGGATATCGAATTCCGTACGTGTTCAGsGsTsC) (10 nmol), in which Y represents the Dabcyl-tethered nucleoside (Fig. 2b), and d(GTCCGCGAATTGCGCC) (20 nmol), in the same manner. The amounts of the 140-mer oligonucleotides obtained after the PAGE purification were determined by the radioactivity and the UV absorption, in the cases of the 32P labeling and the large-scale preparation, respectively. The 140 bp duplex (Supplementary Fig. S1c) was formed by mixing the fluorescein-containing strand with a 1.25-fold amount of the Dabcyl-containing strand, in buffer containing 10 mM Tris-HCl (pH 8.0) and 50 mM NaCl, followed by heating at 95°C for 2 min and cooling to 25°C during 45 min.
Experiments using the 140 bp duplexes
Dual-incision assays (Supplementary Fig. S1d) were performed according to the described method35, using the 32P-labeled 140 bp duplexes with and without the (6–4) photoproduct (35 fmol) and the whole cell extracts36 in 50 μl solutions for 2 h, followed by 14% denaturing PAGE and band detection with a Typhoon FLA 7000 image analyzer (GE Healthcare). Fluorescence intensities (Supplementary Fig. S1e) were measured at 37°C, using the same assay solutions after dilution to 200 μl with 1 M Tris-HCl (pH 8.0), on a JASCO FP-6500 spectrofluorometer equipped with an EHC 573 temperature controller. The excitation and emission wavelengths were 494 and 520 nm, respectively, with bandwidths of 3 nm. For the transfection of the XP12ROSV cells25 (Supplementary Fig. S1f), the cells were grown in Dulbecco's modified Eagle's medium (DMEM) supplemented with 15% fetal bovine serum, 100 units/ml penicillin, and 100 μg/ml streptomycin at 37°C in a humidified 5% CO2 incubator. The cells were seeded into a 4-well slide lumox (Greiner Bio-One), grown to about 90% confluence, and transfected with the 140 bp duplexes using Lipofectamine 2000 (Life Technologies), according to the manufacturer's instructions. At 6 h after transfection, the cells were analyzed with an Olympus IX71 fluorescence microscopy system.
Preparation of the plasmid-type probes
A 9-nucleotide sequence for the incorporation of the modified nucleosides was inserted into pBSII KS (-) UV23, using two primers, d(CGAAAAGGCCAAGCCTGGTACCCAGCTTTT) and d(GGCTTGGCCTTTTCGTCAGCATCTTCATCA), in which the inserted sequences are underlined, a PrimeSTAR Mutagenesis Basal Kit (Takara Bio), and HIT-JM109 competent cells (RBC Bioscience), according to the manufacturers' instructions. A single colony, cultured at 37°C for 1 day in Lysogeny Broth medium (4 ml) containing ampicillin, was diluted with the same medium (400 ml), and VCS-M13 helper phage (Stratagene) was added at a concentration of >1.0 × 1011 pfu/ml. After an incubation at 37°C for 1 day, the single-stranded DNA was obtained, as described previously37. The probes, pBSII KS (-) FQ64 (Fig. 2a) and pBSII KS (-) FQTT, were prepared from this DNA using a primer, p-d(AGGCTTGGCCXTYTCGTCAGCATCTTCATCATACAGT), in which X and Y represent the fluorescein- and Dabcyl-tethered nucleosides (Fig. 2b), with and without the (6–4) photoproduct at the underlined site, as described previously35.
UVDE treatment
pBSII KS (-) FQ64 and pBSII KS (-) FQTT (250 ng) were incubated with Schizosaccharomyces pombe UVDE (Trevigen) (1 μl), in buffer (20 μl) containing 20 mM HEPES-KOH (pH 6.5), 10 mM MgCl2, 1 mM MnCl2, and 100 mM NaCl, at 30°C for 1 h. After the addition of loading buffer (2 μl) containing 25% Ficoll, 2% SDS, and 10 mM EDTA (pH 8.0), the products were analyzed by 1% agarose gel electrophoresis, followed by visualization of the bands with ethidium bromide.
Repair synthesis
pBSII KS (-) FQ64 and pBSII KS (-) FQTT (150 ng) were incubated with a HeLa whole cell extract36 (86 μg) and [α-32P]dCTP (15 kBq), in a solution (10 μl) containing 50 mM HEPES-KOH (pH 7.8), 70 mM KCl, 9.8 mM MgCl2, 1.3 mM DTT, 2 mM ATP, 1 μM dNTP (N = A, G, T), 0.1 μM dCTP, 22 mM phosphocreatine, 0.2 mM glycine, 0.4 mM EDTA, 0.36 mg/ml BSA, 50 μg/ml creatine phosphokinase, and 7.8% glycerol, at 30°C for 1 h. To this solution, 0.5 M EDTA (2 μl), 10% SDS (3 μl), and proteinase K (Invitrogen) (20 μg) were added, and the mixtures were incubated at 56°C for 20 min. After phenol-chloroform extraction and ethanol precipitation, the DNA was incubated with BstNI (New England Biolabs) (5 units), in buffer (10 μl) containing 10 mM Tris-HCl (pH 7.9), 10 mM MgCl2, 50 mM NaCl, 1 mM DTT, and 100 μg/ml BSA, at 60°C for 1 h. The products were analyzed by 12.5% denaturing PAGE, and the bands were detected with the FLA 7000 image analyzer.
Transfection of the cells and fluorescence detection
HeLa S3, XP12ROSV, and XP12ROSV/XPA cells were grown in DMEM supplemented with 15% fetal bovine serum, 100 units/ml penicillin, and 100 μg/ml streptomycin, at 37°C in a humidified 5% CO2 incubator. The cells were seeded into 24-well plates (Trueline), grown to about 90% confluence, and transfected with pBSII KS (-) FQ64 or pBSII KS (-) FQTT (20 μg), which was mixed with the transfection reporter24 (10 pmol), using Lipofectamine 2000 (Life Technologies), according to the manufacturer's instructions. At the described intervals after transfection, the cells were analyzed with the Olympus IX71 fluorescence microscopy system. The fluorescence intensities of 10 cells in Fig. 4a were quantified and averaged, using the ImageJ software (National Institutes of Health).
Flow cytometry
HeLa cells were cultured and transfected with the probes, in the same manner as described above. After 1, 2, 3, and 4 h, the culture medium was removed, and the cells were washed with phosphate-buffered saline (PBS). The cells were harvested by trypsin digestion, and suspended in DMEM (2 ml). The suspension was centrifuged at 1,000 rpm for 3 min, and the cell pellet was mixed with 4% paraformaldehyde phosphate buffer solution (1 ml). After 30 min at room temperature, the cells were collected by centrifugation at 1,000 rpm for 3 min, washed with PBS (1 ml), and suspended in PBS (1 ml). The samples were prepared by passing the cells through a cell strainer (BD Falcon). The analysis was performed on a BD Biosciences FACSCalibur flow cytometer controlled by the CellQuest software, version 3.3. The fluorescein and Cy5 emissions were detected using the FL1 and FL3 filters, respectively, and each measurement was performed using 10,000 cells. Each data set was analyzed using the FlowJo software. To reduce the background signal, the gating of the cells was set to analyze more than 96.7% of the total cells.
Author Contributions
S.I. and C.N. designed research; T.T., I.K., S.W., E.N., S.T. and K.S. performed research; I.K. and S.T. analyzed data; S.I. wrote the paper.
Supplementary Material
Supplementary Information
Acknowledgments
This study was supported by a Grant-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science, and Technology of Japan (233948 to T.T.). T.T. was supported by a Research Fellowship for Young Scientists from the Japan Society for the Promotion of Science.
References
- Iwai S. Pyrimidine dimers: UV-induced DNA damage. In: Modified Nucleosides in Biochemistry, Biotechnology and Medicine: Wiley-VCH, 97–131 (2008). [Google Scholar]
- Naegeli H. & Sugasawa K. The xeroderma pigmentosum pathway: decision tree analysis of DNA quality. DNA Repair 10, 673–683 (2011). [DOI] [PubMed] [Google Scholar]
- Szymkowski D. E., Lawrence C. W. & Wood R. D. Repair by human cell extracts of single (6-4) and cyclobutane thymine-thymine photoproducts in DNA. Proc. Natl. Acad. Sci. U.S.A. 90, 9823–9827 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C., Wang L.-E. & Wei Q. DNA repair phenotype and cancer susceptibility – a mini review. Int. J. Cancer 124, 999–1007 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scrima A. et al. Structural basis of UV DNA-damage recognition by the DDB1–DDB2 complex. Cell 135, 1213–1223 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugasawa K. et al. UV-induced ubiquitylation of XPC protein mediated by UV-DDB-ubiquitin ligase complex. Cell 121, 387–400 (2005). [DOI] [PubMed] [Google Scholar]
- Fousteri M. & Mullenders L. H. F. Transcription-coupled nucleotide excision repair in mammalian cells: molecular mechanisms and biological effects. Cell Res. 18, 73–84 (2008). [DOI] [PubMed] [Google Scholar]
- Cleaver J. E., Lam E. T. & Revet I. Disorders of nucleotide excision repair: the genetic and molecular basis of heterogeneity. Nat. Rev. Genet. 10, 756–768 (2009). [DOI] [PubMed] [Google Scholar]
- Thoms K.-M., Kuschal C. & Emmert S. Lessons learned from DNA repair defective syndromes. Exp. Dermatol. 16, 532–544 (2007). [DOI] [PubMed] [Google Scholar]
- Lehmann A. R., McGibbon D. & Stefanini M. Xeroderma pigmentosum. Orphanet J. Rare Dis. 6, 70 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiGiovanna J. J. & Kraemer K. H. Shining a light on xeroderma pigmentosum. J. Invest. Dermatol. 132, 785–796 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradford P. T. et al. Cancer and neurologic degeneration in xeroderma pigmentosum: long term follow-up characterises the role of DNA repair. J. Med. Genet. 48, 168–176 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nouspikel T. Nucleotide excision repair and neurological diseases. DNA Repair 7, 1155–1167 (2008). [DOI] [PubMed] [Google Scholar]
- Stefanini M. et al. Differences in the levels of UV repair and in clinical symptoms in two sibs affected by xeroderma pigmentosum. Human Genet. 54, 177–182 (1980). [DOI] [PubMed] [Google Scholar]
- Lehmann A. R. & Stevens S. A rapid procedure for measurement of DNA repair in human fibroblasts and for complementation analysis of xeroderma pigmentosum cells. Mutat. Res. 69, 177–190 (1980). [DOI] [PubMed] [Google Scholar]
- Limsirichaikul S. et al. A rapid non-radioactive technique for measurement of repair synthesis in primary human fibroblasts by incorporation of ethynyl deoxyuridine (EdU). Nucleic Acids Res. 37, e31 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto N. et al. Fluorescent probes for the analysis of DNA strand scission in base excision repair. Nucleic Acids Res. 38, e101 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyagi S. & Kramer F. R. Molecular beacons: probes that fluoresce upon hybridization. Nat. Biotechnol. 14, 303–308 (1996). [DOI] [PubMed] [Google Scholar]
- Huang J.-C. & Sancar A. Determination of minimum substrate size for human excinuclease. J. Biol. Chem. 269, 19034–19040 (1994). [PubMed] [Google Scholar]
- Matsunaga T., Mu D., Park C.-H., Reardon J. T. & Sancar A. Human DNA repair excision nuclease: analysis of the roles of the subunits involved in dual incisions by using anti-XPG and anti-ERCC1 antibodies. J. Biol. Chem. 270, 20862–20869 (1995). [DOI] [PubMed] [Google Scholar]
- Rapić Otrin V. et al. Relationship of the xeroderma pigmentosum group E DNA repair defect to the chromatin and DNA binding proteins UV-DDB and replication protein A. Mol. Cell. Biol. 18, 3182–3190 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moggs J. G., Yarema K. J., Essigmann J. M. & Wood R. D. Analysis of incision sites produced by human cell extracts and purified proteins during nucleotide excision repair of a 1,3-intrastrand d(GpTpG)-cisplatin adduct. J. Biol. Chem. 271, 7177–7186 (1996). [DOI] [PubMed] [Google Scholar]
- Mei Kwei J. S. et al. Blockage of RNA polymerase II at a cyclobutane pyrimidine dimer and 6–4 photoproduct. Biochem. Biophys. Res. Commun. 320, 1133–1138 (2004). [DOI] [PubMed] [Google Scholar]
- Toga T., Kuraoka I., Yasui A. & Iwai S. A transfection reporter for the prevention of false-negative results in molecular beacon experiments. Anal. Biochem. 440, 9–11 (2013). [DOI] [PubMed] [Google Scholar]
- Hasegawa M., Iwai S. & Kuraoka I. A non-isotopic assay uses bromouridine and RNA synthesis to detect DNA damage responses. Mutat. Res. 699, 62–66 (2010). [DOI] [PubMed] [Google Scholar]
- Srinivasan C., Siddiqui S., Silbart L. K., Papadimitrakopoulos F. & Burgess D. J. Dual fluorescent labeling method to visualize plasmid DNA degradation. Bioconjug. Chem. 20, 163–169 (2009). [DOI] [PubMed] [Google Scholar]
- Gong P. et al. DNase-activatable fluorescence probes visualizing the degradation of exogenous DNA in living cells. Nanoscale 4, 2454–2462 (2012). [DOI] [PubMed] [Google Scholar]
- Lechardeur D. et al. Metabolic instability of plasmid DNA in the cytosol: a potential barrier to gene transfer. Gene Ther. 6, 482–497 (1999). [DOI] [PubMed] [Google Scholar]
- Wagstaff K. M. & Jans D. A. Nucleocytoplasmic transport of DNA: enhancing non-viral gene transfer. Biochem. J. 406, 185–202 (2007). [DOI] [PubMed] [Google Scholar]
- Kemp M. G., Reardon J. T., Lindsey-Boltz L. A. & Sancar A. Mechanism of release and fate of excised oligonucleotides during nucleotide excision repair. J. Biol. Chem. 287, 22889–22899 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu J. et al. Nucleotide excision repair in human cells: fate of the excised oligonucleotide carrying DNA damage in vivo. J. Biol. Chem. 288, 20918–20926 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evdokimov A. et al. New synthetic substrates of mammalian nucleotide excision repair system. Nucleic Acids Res. 41, e123 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwai S., Shimizu M., Kamiya H. & Ohtsuka E. Synthesis of a phosphoramidite coupling unit of the pyrimidine (6-4) pyrimidone photoproduct and its incorporation into oligodeoxynucleotides. J. Am. Chem. Soc. 118, 7642–7643 (1996). [Google Scholar]
- Iwai S. et al. Benzimidazolium triflate-activated synthesis of (6–4) photoproduct-containing oligonucleotides and its application. Nucleic Acids Res. 27, 2299–2303 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shivji M. K. K., Moggs J. G., Kuraoka I. & Wood R. D. Dual-incision assays for nucleotide excision repair using DNA with a lesion at a specific site. Methods Mol. Biol. 113, 373–392 (1999). [DOI] [PubMed] [Google Scholar]
- Manley J. L., Fire A., Cano A., Sharp P. A. & Gefter M. L. DNA-dependent transcription of adenovirus genes in a soluble whole-cell extract. Proc. Natl. Acad. Sci. U.S.A. 77, 3855–3859 (1980). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green M. R. & Sambrook J. Preparation of single-stranded bacteriophage M13 DNA by precipitation with polyethylene glycol. In: Molecular Cloning: A Laboratory Manual, 4th Ed.: Cold Spring Harbor Laboratory Press, 31–33 (2012). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information