

Background: KCC2 is a potassium-chloride cotransporter essential for hyperpolarizing neurotransmission and is associated with multiple neurological disorders.
Results: Thr934 and Ser937 are major regulatory sites of KCC2 activity, and their status influences other activation processes.
Conclusion: Thr934 and Ser937 phosphorylation increases KCC2 transport kinetics.
Significance: This study identifies a novel C-terminal KCC2 stimulatory phosphorylation site.
Keywords: Cell Culture, Chloride Transport, Evolution, Neurophysiology, Phosphorylation, Post-translational Modification, Protein Conformation, Cation Chloride Cotransporter, Mutation
Abstract
The neuron-specific cation chloride cotransporter KCC2 plays a crucial role in hyperpolarizing synaptic inhibition. Transporter dysfunction is associated with various neurological disorders, raising interest in regulatory mechanisms. Phosphorylation has been identified as a key regulatory process. Here, we retrieved experimentally observed phosphorylation sites of KCC2 from public databases and report on the systematic analysis of six phosphorylated serines, Ser25, Ser26, Ser937, Ser1022, Ser1025, and Ser1026. Alanine or aspartate substitutions of these residues were analyzed in HEK-293 cells. All mutants were expressed in a pattern similar to wild-type KCC2 (KCC2WT). Tl+ flux measurements demonstrated unchanged transport activity for Ser25, Ser26, Ser1022, Ser1025, and Ser1026 mutants. In contrast, KCC2S937D, mimicking phosphorylation, resulted in a significant up-regulation of transport activity. Aspartate substitution of Thr934, a neighboring putative phosphorylation site, resulted in a comparable increase in KCC2 transport activity. Both KCC2T934D and KCC2S937D mutants were inhibited by the kinase inhibitor staurosporine and by N-ethylmaleimide, whereas KCC2WT, KCC2T934A, and KCC2S937A were activated. The inverse staurosporine effect on aspartate versus alanine substitutions reveals a cross-talk between different phosphorylation sites of KCC2. Immunoblot and cell surface labeling experiments detected no alterations in total abundance or surface expression of KCC2T934D and KCC2S937D compared with KCC2WT. These data reveal kinetic regulation of transport activity by these residues. In summary, our data identify a novel key regulatory phosphorylation site of KCC2 and a functional interaction between different conformation-changing post-translational modifications. The action of pharmacological agents aimed to modulate KCC2 activity for therapeutic benefit might therefore be highly context-specific.
Introduction
The K+-Cl− cotransporter (KCC)2 2 is a neuron-specific secondary-active plasma membrane protein (1). In the mature brain, KCC2 is a very effective outward-directed K+ and Cl− cotransporter (2, 3). Its activity generates a Cl− reversal potential more negative than the resting membrane potential (4, 5). As a consequence, the opening of ligand-gated ionotropic GABA and glycine receptors leads to a Cl− influx mediating hyperpolarization of the neuron (3, 6, 7). Therefore, the protein is essential for fast synaptic inhibition. Mice with disruption of the gene Slc12a5 encoding KCC2 die shortly after birth due to motor deficits, including respiratory failure (7). In addition to its transport activity, transport-independent roles in synaptogenesis, neuronal differentiation, and migration have been identified, making KCC2 a multifunctional protein (8, 9).
Sequence similarities and functional properties have assigned KCC2 to the Slc12 family of cation chloride cotransporters. In mammals, the family consists of the inward transporters NCC, NKCC1, and NKCC2, the chloride outward transporters KCC1–4, and the chloride transporter-interacting protein CIP1 (10–12). Furthermore, the orphan transporter CCC93 might be part of this group (10). Paralog KCCs share a sequence identity of >67%, with KCC1 and KCC3 and with KCC2 and KCC4 forming sister groups (12, 13). Paralog NKCC and NCCs share a sequence identity of >50% (12, 13). Phylogenetic analyses revealed the presence of KCC2 in all vertebrates (10, 11), and KCC-like proteins also occur across Eukaryota (10, 11).
Dysregulation of KCC2 is associated with various human neurological disorders (11, 14), such as epileptic activity (15–17), neuropathic pain (18, 19), spasticity (20), ischemic insults (21), and brain trauma (22).
These severe consequences of altered KCC2 function raised interest in mechanisms regulating its activity (5). On the cellular level, location in membrane rafts was shown to modify KCC2 transport activity (23, 24). On the molecular level, interaction partners such as the ATPase subunit α2 (25), CIP1 (26), Neto2 (27), and several protein kinases, including brain-specific creatine kinase (28), SPAK (29), OSR1 (30), and WNKs (31) were identified. The regulatory role of phosphorylation is in line with previous pharmacological studies. The kinase inhibitors lavendustin A, genistein (32), or staurosporine (33) altered KCC2 transport activity in cultured hippocampal neurons, and calyculin (34) and okadaic acid (35), two potent phosphatase inhibitors, blocked activation of KCCs by cell swelling. The important role of phosphorylation was corroborated by the regulatory role of several identified phosphorylation sites in KCC2 (36). Phosphorylation of human threonines Thr906 and Thr1007 reduced the intrinsic transport activity of KCC2 (37, 38). In contrast, phosphorylated Ser940 increased surface expression and KCC2 transport function (39) as well as membrane clustering (40). Finally, the phosphorylation status of Tyr903 and Tyr1087 also regulates KCC2 activity, although the functional consequences of phosphorylation appear to be context-dependent and require further investigation (5, 23, 36, 41, 42).
Here, we wished to further examine the role of phosphorylation for KCC2 activity. Mining of large scale phospho-proteomics data revealed several phosphorylated serines not yet analyzed. To characterize their importance, these amino acids were systematically substituted by either alanine or aspartate in the rat KCC2b isoform to mimic the dephosphorylated state (alanine) or the phosphorylated state (aspartate), respectively. Expression and transport activity of the mutants were then determined in HEK-293 cells. This approach identified Ser937 and the neighboring Thr934 as major novel regulatory phosphorylation sites for KCC2 transport activity.
MATERIALS AND METHODS
Bioinformatic Analyses
To identify native KCC2 phosphorylation from proteomics approaches, the two databases PhosphoSitePlus (43) and PHOSIDA (44) were screened using the term KCC2 as entry. Both databases contain curated data of experimentally observed post-translational modifications, primarily of human and mouse proteins, which were obtained by high resolution mass spectrometric analyses.
KCC protein sequences for a diverse selection of organisms were obtained from a combination of BLAST searches against GenBankTM and data mining of the Ensembl database and the Joint Genome Institute (www.jgi.doe.gov). We used the protein sequences of human KCC1 (NP_005063.1), KCC2 (NP_065759.1), KCC3 (NP_598408.1), and KCC4 (NP_006589.2) as query. For each protein in each target species, we saved all sequences with an E-value of at least 10−2. These sequences were then reverse-blasted (BLASTp or translated BLAST) against the Homo sapiens protein database, and only those protein sequences were retained that showed the same CCC protein sequence of H. sapiens that was used as a query sequence as the best hit (E-value of at least 10−2). Each obtained sequence was then aligned at the amino acid level using the default settings in MUSCLE (45), as implemented in SeaView version 4.4.2 (46) and manually improved by eye thereafter.
Construction of Expression Clones
Wild-type rat KCC2b (GenBankTM accession number NM_134363) and HA-tagged mouse KCC2b (GenBankTM accession number NM_020333) expression clones with an HA tag at the N terminus or in the second extracellular loop were reported previously (47, 48). Site-directed mutagenesis of KCC2b cDNA was performed according to the QuikChange mutagenesis system (Stratagene, Heidelberg, Germany). Forward oligonucleotides for the generation of the mutations are given in Table 1. All generated clones were confirmed by sequencing.
TABLE 1.
Forward primer for site-directed mutagenesis
The abbreviations used are as follows: for, forward; mm, Mus musculus; other primers are designed for KCC2 from Rattus norvegicus.
| Phosphosite | Sequence 5′ to 3′ |
|---|---|
| S25Afor | CGGCAATCCCAAGGAGGCCAGCCCCTTCATCAAC |
| S25Dfor | CGGCAATCCCAAGGAGGACAGCCCCTTCATCAAC |
| S26Afor | CAATCCCAAGGAGAGCGCCCCCTTCATCAACAGC |
| S26Dfor | GCAATCCCAAGGAGAGCGACCCCTTCATCAACAGCA |
| S25A/S26Afor | GACGGCAATCCCAAGGAGGCCGCCCCCTTCATCAACAGCAC |
| S25D/S26Dfor | TGACGGCAATCCCAAGGAGGACGACCCCTTCATCAACAGCACG |
| T906Afor | GCATACACCTACGAGAAGGCATTGGTAATGGAACAAC |
| T1007Afor | CAGAGAAGGTGCATCTCGCCTGGACCAAGGATAAG |
| T934Afor | GGAGATCCAGAGCATCGCAGATGAATCTCGGGG |
| T934Dfor | CGGGAGATCCAGAGCATCGATGATGAATCTCGGGGCTCC |
| S937Afor | TCCAGAGCATCACAGATGAAGCTCGGGGCTCC |
| S937Dfor | GGAGATCCAGAGCATCACAGATGAAGATCGGGGCTCCATT |
| T934A/S937Afor | GGAGATCCAGAGCATCGCAGATGAAGCTCGGGGCTCC |
| S1022Afor | AGAACAAAGGCCCCGCTCCCGTCTCCTCGG |
| S1022Dfor | GAAGAACAAAGGCCCCGATCCCGTCTCCTCGGAG |
| S1025Afor | CCAGTCCCGTCGCCTCGGAGGGG |
| S1025Dfor | CCCCAGTCCCGTCGACTCGGAGGGGATC |
| S1026Afor | CAGTCCCGTCTCCGCGGAGGGGATCAA |
| S1026Dfor | CCCCAGTCCCGTCTCCGATGAGGGGATCAAGGACT |
| mmT934Dfor | CGGGAGATCCAGAGCATCGATGACGAGTCTCGGGGCTCC |
| mmS937Dfor | GGAGATCCAGAGCATCACAGATGAAGATCGGGGCTCCATT |
Determination of K+-Cl− Cotransport
Transport activity was determined by measuring Cl−-dependent uptake of Tl+ in HEK-293 cells. Uptake measurements were done as described previously (49, 50, 47). Cells were transiently transfected with the respective construct, using TurboFect (Fermentas, Schwerte, Germany), according to the protocol provided. Briefly, 150 μl of Opti-MEM (Invitrogen), 6 μl of TurboFect (Fermentas, Karlsruhe, Germany), and ∼3 μg of DNA were mixed and incubated for 20 min at room temperature prior to transfection. 24 h after transfection, HEK-293 cells were plated in a black-walled 96-well culture dish (Greiner Bio-One, Frickenhausen, Germany) at a concentration of 100,000 cells/well. The remainder of the cells were plated on a glass coverslip. After ∼18 h, coverslips were processed for immunocytochemical analysis to determine transfection rates, which were routinely between 20 and 30% (Fig. 2). Cell cultures with lower transfection rates were omitted from subsequent flux measurements. The HEK-293 cells in 96-well culture dishes were processed for flux measurements by replacing the medium with 80 μl of preincubation buffer (100 mm N-methyl-d-glucamine, 5 mm KCl, 2 mm CaCl2, 0.8 mm MgSO4, 5 mm glucose, 5 mm HEPES, pH 7.4) with or without 2 μm FlouZin-2 AM dye (Invitrogen) plus 0.2% (w/v) Pluronic F-127 (Invitrogen). After incubation for 48 min at room temperature, cells were washed three times with 80 μl of preincubation buffer and incubated for 15 min with 80 μl of preincubation buffer plus 0.1 mm ouabain to block Na+/K+ ATPases. Thereafter, the culture dish was inserted into a fluorometer (Fluoroskan Accent, Thermo Scientific, Bremen, Germany), and the wells were injected with 40 μl of 5× thallium stimulation buffer (12 mm Tl2SO4, 100 mm N-methyl-d-glucamine, 5 mm HEPES, 2 mm CaSO4, 0.8 mm MgSO4, 5 mm glucose, pH 7.4). The fluorescence across the entire cell population in a single well was measured in a kinetic dependent manner (excitation 485 nm, emission 538 nm, 1 frame in 5 s in a 200-s time span). The activity was calculated with the initial values of the slope of Tl+-stimulated fluorescence increase by using linear regression. At least two independent DNA preparations were used per construct, giving similar results.
FIGURE 2.

Transfection rates of KCC2 expression constructs. KCC2WT and the indicated KCC2 mutants were expressed in HEK-293 cells. To monitor transfection rates, immunocytochemistry was performed against the transporter (green). In parallel, cell cultures were stained with DAPI (blue). Typically, transfection rates between 20 and 30% were obtained. Scale bar, 100 μm.
The effect of the thiol-alkylating reagent N-ethylmaleimide (NEM) or the kinase inhibitor staurosporine was determined by adding either 1 mm NEM or 8 μm staurosporine to the preincubation buffer 15 min prior to flux measurements on the cells. For statistical analysis, data groups were compared using a Student's t test, and p < 0.05 was considered as statistically significant.
Immunocytochemistry
For immunocytochemistry, HEK-293 cells were seeded on 0.1 mg/ml poly-l-lysine-coated coverslips and incubated for 36 h. After fixation for 10 min with 4% paraformaldehyde in 0.2 m phosphate buffer and three washes in PBS, cells were incubated with blocking solution (0.3% Triton X-100, 3% bovine serum albumin, 10% goat serum in PBS) for 30 min. All steps were performed at room temperature. Primary antibody solution (anti-cKCC2, directed against the C-terminal part of KCC2, 1:1,000) (51) was added in blocking solution for 30 min. After three wash steps with PBS for 5 min, the secondary antibody was added, which was conjugated to a fluorescent probe (1:1,000; Alexa Fluor 488 goat anti-rabbit (Invitrogen)). After washing, cells were mounted onto glass slides with Vectashield Hard Set (Vector Laboratories, Burlingame, CA). Photomicrographs were taken using a Laser scanning microscope (Leica TCS SP2).
Cell Surface Labeling of KCC2-HA N-term Constructs
To determine the cell surface expression of mouse KCC2WT-HA 2nd loop, KCC2T934D-HA, or KCC2S937D-HA, these constructs were expressed in HEK-293 cells. After 36 h, cells were kept at 4 °C for 15 min and washed with a chilled washing solution containing 150 mm NaCl, 2.5 mm KCl, 2 mm CaCl2, 2 mm MgCl2, 2.5 mm HEPES, and 2 g glucose/liter. Primary antibody (mouse anti-HA, 1:250 (Covance, Heidelberg, Germany)) was added for 25 min at 4 °C, and cells were washed three times. The secondary antibody (Alexa Fluor 488 goat anti-mouse, 1:250 (Invitrogen)) was diluted in preheated (37 °C) washing solution, and cells were incubated for 10 min at 37 °C. After washing with preheated solution, cells were fixed with 4% paraformaldehyde. Fixed cells were stained as described above with anti-cKCC2 and an Alexa Fluor 594 goat anti-rabbit to detect all KCC2 protein.
For image analysis, 2048 square pixels containing 8-bit xyz-confocal pictures were taken using a Leica TCS SP5 device with an adjustable 20-fold immersion objective (oil n = 1.514) and a pulsed white light laser with wavelengths adjusted to 498 nm (for Alexa Fluor 488, 10% power) and 590 nm (for Alexa Fluor 594, 4% power). The following settings were applied to all images: 100% detection of fluorescence emission with linear working HyD detectors from 507 to 548 nm (for Alexa Fluor 488) and from 610 to 646 nm (for Alexa Fluor 594) with gating restriction at 498 nm (0.30 and 6.50 ns) and 590 nm (0.30 and 6.50 ns) (Leica-microsystems.com); a pinhole of 300 μm (recorded specimen thickness = 13.3 μm); and 2-fold line and frame averages. Ten to 21 images of each experiment were evaluated for mean intensity per pixel with pre-arranged FIJI software. Three to five biological replica were performed for each construct.
Immunoblot Analyses
To quantify the expression of KCC2 mutants, cells were lysed in a buffer containing 150 mm NaCl, 15 mm Tris, 1% dodecyl β-d-maltoside, and 0.4% iodoacetamide. After incubation for 5 min at 25 °C, samples were centrifuged for 5 min at 125,000 × g. Protein amount of the supernatant was determined using the Bradford assay. 10 μg of each sample were loaded onto a 10% SDS-polyacrylamide gel system. After separation and electrotransfer onto PVDF membranes, membranes were incubated with anti-cKCC2 (dilution 1:2,000). After incubation for 2 h at room temperature, membranes were washed four times with TBS-T (20 mm Tris, 150 mm NaCl, 1% Tween, pH 7.5), and the secondary antibody donkey anti-rabbit IgG-HRP (Santa Cruz Biotechnology, Heidelberg, Germany) was applied for 1 h. After washing, bound antibodies were detected using an enhanced chemiluminescence assay (GE Healthcare) and a LAS-3000 documentation system (Fujifilm, Düsseldorf, Germany). Quantification of bands within the linear range of exposure was performed using the MultiGauge software version 3.1 (Fujifilm). Three biological replicas were performed for each construct. Data are given as means ± S.E. Significant differences between the groups were analyzed by a Student's t test.
RESULTS
Database Mining and Phylogenetic Pattern of KCC2 Phosphorylation Sites
To identify bona fide phosphorylation sites in KCC2, the databases PhosphoSitePlus and PHOSIDA were searched for KCC2 entries. These databases contain experimentally observed phosphorylation sites identified largely by mass spectrometry-based proteomic studies (43, 44). In addition to the known phosphorylation sites Thr906, Ser940, and Thr1007, Ser25, Ser26, Ser937, Ser1022, Ser1025, Ser1026, Thr34, and Thr1009 were reported to be phosphorylated in KCC2 in both databases (numbering according to the rat KCC2b protein) (Table 2). Three of the amino acids, Ser25, Ser26, and Thr34, reside in the cytoplasmic N terminus, and the remainder are located in the C-terminal part (1). Here, we focused on the six phosphoserines Ser25, Ser26, Ser937, Ser1022, Ser1025, and Ser1026. Phylogenetic analyses of the KCC subfamily revealed that they cover different patterns of phylogenetic conservation. Ser1022, Ser1025, and Ser1026 are highly specific to tetrapod KCC2 (Fig. 1). Ser937 is present only in vertebrate KCC2 and in non-therian KCC4. Finally, Ser25 and Ser26 showed the broadest occurrence with a presence in the most vertebrate KCC1, KCC2, and KCC4 family members (Fig. 1). Notably, the previously reported amino acids Thr906 and Thr1007, which had been implicated in developmental activation of KCC2 during brain maturation (37), are highly conserved throughout metazoans (Fig. 1). This suggests that their phosphorylation does not only play a role in vertebrate KCCs but also in those transporters from more ancient species like the choanoflagellate Monosiga brevicolis, which forms a sister group to the metazoan (52). Analysis of the neighboring amino acid sequences demonstrated conservation patterns very similar to those observed for the phosphorylated amino acid residues (Fig. 1). This indicates that the respective phosphorylation can also occur in other species.
TABLE 2.
Phosphorylation sites in PhosphoSitePlus and PHOSIDA
| PhosphoSitePlus hsKCC2 | PhosphoSitePlus mmKCC2 | PhosphoSitePlus rnKCC2 | PHOSIDA mmKCC2 |
|---|---|---|---|
| Ser25 | Ser25 | Ser25 | Ser25 |
| Ser26 | Ser26 | Ser26 | Ser26 |
| Thr34 | Thr34 | Thr34 | Thr34 |
| Thr906 | Thr906 | Thr906 | Thr906 |
| (Thr934) | |||
| Ser937 | Ser937 | Ser937 | Ser937 |
| Ser940 | Ser940 | Ser940 | Ser940 |
| Thr1007 | Thr1006 | Thr1007 | Thr1006 |
| Thr1009 | Thr1008 | Thr1009 | Thr1008 |
| Ser1022 | Ser1021 | Ser1022 | Ser1021 |
| Ser1025 | Ser1024 | Ser1025 | (Ser1024) |
| Ser1026 | Ser1025 | Ser1026 | Ser1025 |
The abbreviations used are as follows: hs, H. sapiens; mm, M. musculus; rn, R. norvegicus.
FIGURE 1.

Evolutionary conservation of phosphorylation sites and their neighboring amino acids in KCC subgroups. Ser25 and Ser26 are present in most vertebrates and KCC1, KCC2, and KCC4 family members. The amino acids Thr906 and Thr1007 are highly conserved throughout the animal kingdom, whereas Ser1022, Ser1025, and Ser1026 are highly specific to tetrapod KCC2. Ser937, Thr934, and Ser940 display similar phylogenetic conservation. They are only present in vertebrate KCC2 and in non-therian KCC4. In addition to the phosphorylation sites, the five amino acids flanking either side of the corresponding phosphorylated amino acid residue are shown. Their pattern of conservation closely matched that of the respective phosphorylated amino acid residue. The abbreviations used are as follows: mm, M. musculus; hs, Homo sapiens; rn, Rattus norvegicus; md, Monodelphis domestica; gg, Gallus gallus; tg, Taeniopyga gutta; ac, Anolis carolinensis; xt, Xenopus tropicalis; tr, Takifugu rubripes; Dr, Danio rerio.
Expression Analyses of KCC2 Phospho-mutants
To study the role of the phosphorylated serines Ser25, Ser26, Ser937, Ser1022, Ser1025, and Ser1026, we generated two mutants of rat KCC2b for each phosphorylation site. This isoform was chosen because it is the major KCC2 protein in the mature brain (53). In the following, we will refer to it as KCC2. Amino acid residues were mutated to either aspartate or alanine to mimic phosphorylated or dephosphorylated states of the respective serine. In addition, T906A and T1007A (corresponding to mouse and human Thr906 and Thr1006) mutants were generated. They were previously shown to result in increased KCC2 activity and served as positive controls (37, 38). This resulted in 14 mutants. To check whether these mutations effect KCC2 expression, all constructs were transfected into HEK-293 cells. This cell line is most widely used to study KCC2 transport activity in a mammalian heterologous expression system (2, 37, 39, 47, 54). Immunocytochemical analyses revealed expression of all 14 mutants (Figs. 2 and 3). Immunoreactivity was detected for all mutants both at the plasma membrane and inside the cells (Fig. 3). Only the nucleus was spared. Compared with wild-type KCC2 (KCC2WT), no difference in subcellular localization was obvious upon visual inspection. Thus, all mutants were well expressed in HEK-293 cells.
FIGURE 3.
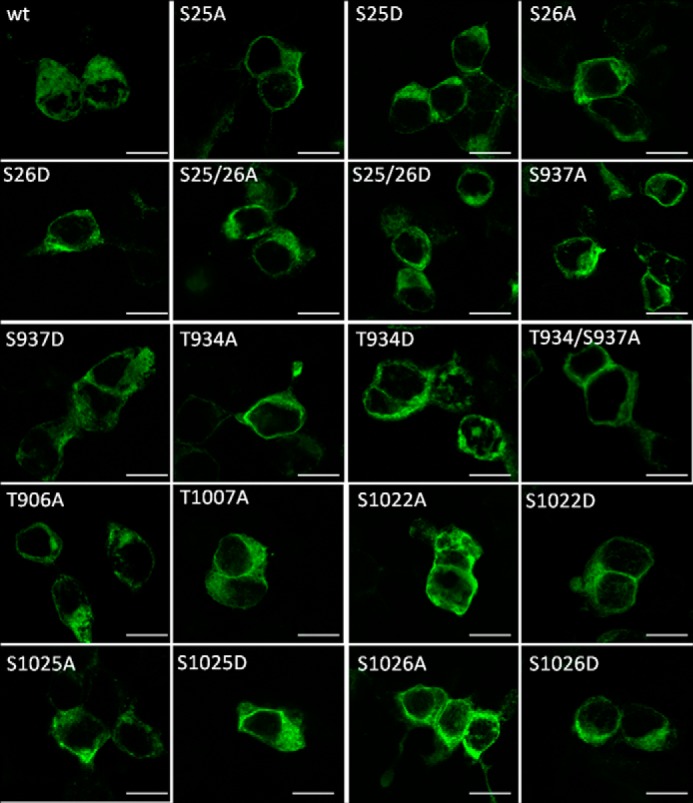
Expression of KCC2 phospho-mutants in HEK-293 cells. KCC2WT and the indicated KCC2 mutants were expressed in HEK-293 cells and efficiently delivered to the plasma membrane. The nucleus was devoid of staining. Upon visual inspection, the subcellular distribution of KCC2 mutants was indistinguishable from KCC2WT. Photomicrographs were taken with a confocal laser scanning microscope. Scale bar, 10 μm.
Transport Activity of KCC2 Phospho-mutants
In a first series of experiments, we focused on the two N-terminal Ser25 and Ser26 phosphorylation sites and determined their activity in HEK-293 cells using Tl+ flux measurements. All four mutants, KCC2S25A, KCC2S25D, KCC2S26A, and KCC2S26D, as well as KCC2WT mediated increased Tl+ flux compared with mock-transfected cells (Fig. 4 and Table 3). Furthermore, 2 mm furosemide blocked most of the flux, demonstrating that the transport activity was largely mediated by KCC2 (Fig. 4 and Table 3) (2).
FIGURE 4.
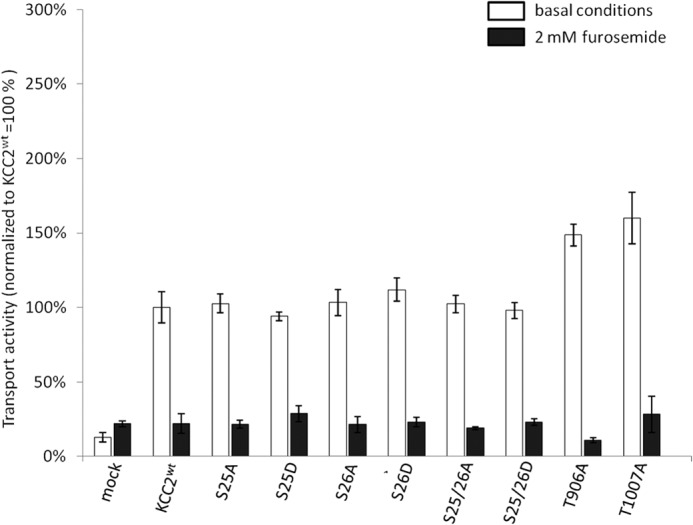
Transport activity of N-terminal KCC2 mutants. HEK-293 cells were transfected with KCC2WT and KCC2 mutant constructs, and transport activity was determined by performing Tl+ flux measurements. Compared with KCC2WT, the N-terminal single mutants KCC2S25A, KCC2S25D, KCC2S26A, and KCC2S26D and the double mutants KCC2S25A/S26A and KCC2S25D/S26D showed no significant differences in transport activity. Confirming previous studies, KCC2T906A and KCC2T1007A displayed a significantly higher transport activity compared with KCC2WT. In the presence of furosemide, the Tl+ flux was blocked. Graphs represent mean ± S.E. of at least three independent measurements, normalized to KCC2WT. Statistical analysis is presented in Table 3.
TABLE 3.
Transport activity under basal conditions and in the presence of furosemide
| Basal conditions (significance in comparison with KCC2WT) | 2 mm furosemide (significance in comparison with untreated samples) | |
|---|---|---|
| HEK-293 | 13 ± 3.2%a | 21.9 ± 1.8% (NS)b |
| KCC2WT | 100 ± 10. 6% | 22.3 ± 6.5%a |
| S25A | 102.8 ± 6.6% (NS) | 21.6 ± 2.6%a |
| S25D | 94.1 ± 3% (NS) | 28.8 ± 5.4%a |
| S26A | 103.4 ± 8.9% (NS) | 21.4 ± 5.5%a |
| S26D | 112 ± 7.8% (NS) | 23.1 ± 3%a |
| S25A/S26A | 102.28 ± 5.6% (NS) | 19 ± 1.1%c |
| S25D/S26D | 97.9 ± 5.4% (NS) | 23.1 ± 2%c |
| T906A | 148.7 ± 7.3%a | 10.9 ± 1.6%d |
| T934A | 97.3 ± 2.1% (NS) | 20.5 ± 3.8%d |
| T934D | 226.6 ± 13%d | 29.6 ± 8.6%d |
| S937A | 106.2 ± 10.2% (NS) | 21.6 ± 4.1%d |
| S937D | 186.9 ± 12%d | 16.1 ± 1.4%d |
| T934A/S937A | 119.4 ± 9.8% (NS) | 15.2 ± 2.9%d |
| T1007A | 159.9 ± 17.4%c | 28.3 ± 12.1%a |
| S1022A | 91.3 ± 11.3% (NS) | 22.5 ± 2.7%c |
| S1022D | 110.8 ± 18.1% (NS) | 20.7 ± 5.8%c |
| S1025A | 102.2 ± 1.6% (NS) | 21 ± 1.9%d |
| S1025D | 99.6 ± 6.9% (NS) | 18 ± 0.37%a |
| S1026A | 124.2 ± 7.8% (NS) | 14 ± 0.9%d |
| S1026D | 115 ± 8.7% (NS) | 15.4 ± 3.1%d |
a p < 0.01.
b NS means not significant.
c p < 0.05.
d p < 0.001.
No difference in activity was detected between KCC2WT and KCC2 mutants or between alanine and aspartate substitutions. Previously, analysis of three N-terminal phosphorylated threonines in NKCC2 had revealed that only compound mutants resulted in altered behavior of the transporter (55). We therefore also generated the double mutants KCC2S25A/S26A and KCC2S25D/S26D. Both mutants are well expressed in HEK-293 cells (Figs. 2 and 3) and showed flux activities similar to KCC2WT (Fig. 4 and Table 3). To ensure that our system detect differences in flux, we also determined the flux of KCC2T906A and KCC2T1007A. These mutants were shown to facilitate KCC2 activity (38). In keeping with the previous report, both mutants displayed a significantly increased transport activity, compared with KCC2WT (Fig. 4 and Table 3). These results revealed that our system was well suited to detect altered KCC2 transport activity. The flux data therefore demonstrate that Ser25 and Ser26 single or combined substitutions do not influence KCC2 transport activity in HEK-293 cells.
We next focused on the C-terminal phosphorylation sites. Substitution of Ser1022, Ser1025, and Ser1026 by alanine or aspartate resulted in no change in transport activity compared with KCCWT (Fig. 5 and Table 3). In contrast, KCC2S937D mediated a furosemide-sensitive Tl+ flux, which was significantly increased compared with KCC2WT (Fig. 5 and Table 3). The phosphorylation-blocking mutant KCC2S937A showed transport activity similar to KCC2WT (Fig. 5 and Table 3). This result indicates that Ser937 is not phosphorylated in KCC2WT when expressed in HEK-293 cells.
FIGURE 5.
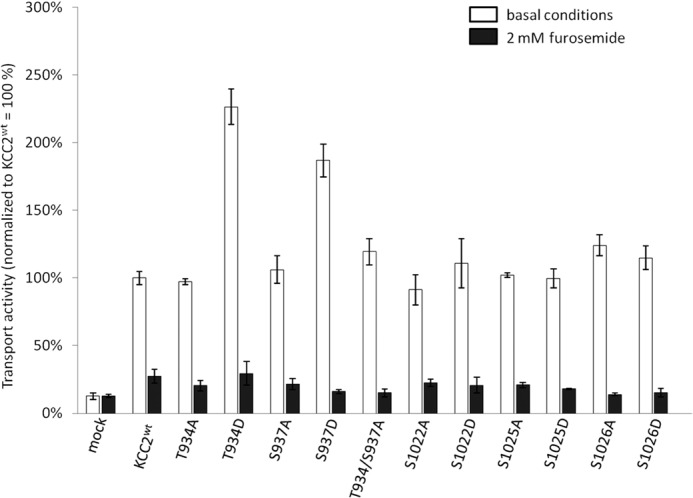
Transport activity of C-terminal KCC2 mutants. HEK-293 cells were transfected with KCC2WT and KCC2 mutant constructs, and transport activity was determined by performing Tl+ flux measurements. The Tl+ flux mediated by KCC2S1022A, KCC2S1022D, KCC2S1025A, KCC2S1025D, KCC2S1026A, and KCC2S1026D was similar to the flux of KCC2WT. In contrast, the phospho-mimetic mutants KCC2T934D and KCC2S937D showed a significantly increased transport activity. The mutants KCC2T934A, KCC2S937A, and KCC2T934A/S937A, which mimicked the dephosphorylated state, exhibited transport levels comparable with KCC2WT. In the presence of furosemide, Tl+ flux was blocked. Graphs represent the mean ± S.E. of at least three independent measurements, normalized to KCC2WT. Statistical analysis is presented in Table 3.
We noticed that the phosphopeptide harboring Ser937 also contains Thr934 in close proximity. In some mass spectrometric analyses of the corresponding phosphopeptide, the precise location of the phosphate group could not be determined, leading to an ambiguous status of Thr934 as a phosphorylation site (Table 2) (56, 57). BecauseThr934 and Ser937 demonstrated exactly the same degree of evolutionary conservation (Fig. 1), we mutated Thr934 as well and determined the transport activity of KCC2T934A and KCC2T934D. Intriguingly, KCC2T934D-mediated flux was significantly facilitated compared with KCC2WT, whereas KCC2T934A transport activity was similar to the control (Fig. 5 and Table 3). Thus, both Thr934 mutants closely matched the results observed for the corresponding Ser937 mutants (Fig. 5). This suggests that Thr934 also represents a regulatory phosphorylation site. To exclude that transport activity of the single alanine mutants KCC2T934A or KCC2S937A was due to residual phosphorylation of the nonmutated Ser937 or Thr934, respectively, we also generated the double mutant KCC2T934A,S937A. The transport activity of this mutant was indistinguishable from that of the single mutants and of KCC2WT (Fig. 5 and Table 3). These data support the notion that both amino acids are not phosphorylated in HEK-293 cells and that their phosphorylation does not contribute to the basal activity of KCC2 in this cell system. We conclude that phosphorylation of Thr934 or Ser937 represents the effective mechanisms to regulate KCC2 activity.
NEM and Staurosporine Effects Reverse upon Phosphorylation of Thr934 or Ser937
NEM is one of the earliest described activators of KCC transport (2, 58). Its mode of action is still unknown, but presumably it occurs via changes in C-terminal phosphorylation (58, 59). To investigate whether Thr934 or Ser937 are involved in the NEM effect, we determined the transport activity of KCC2T934A, KCC2T934D, KCC2S937A, KCC2S937D, and KCC2T934A,S937A in the presence of 1 mm NEM. In agreement with previous studies, this concentration of NEM significantly increased KCC2WT transport activity (Fig. 6 and Table 4). Similarly, all three phosphorylation-blocking alanine mutants KCC2T934A, KCC2S937A, and KCC2T934A,S937A were significantly stimulated by NEM (Fig. 6 and Table 4). This demonstrates that NEM is not acting via phosphorylation of Thr934 or Ser937. Unexpectedly, the two phospho-mimetic mutants KCC2T934D and KCC2S937D were significantly inhibited by NEM and showed transport activities similar to KCC2WT (Fig. 6 and Table 4). This suggests that the NEM effect is dependent on the conformational state of KCC2.
FIGURE 6.
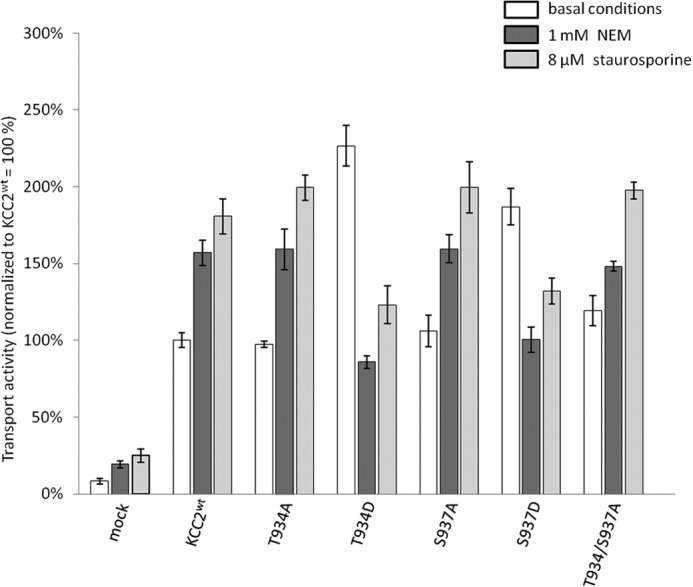
Effect of NEM and staurosporine on the transport activity of Thr934 and Ser937 mutants. HEK-293 cells were transfected with KCC2WT and KCC2 mutant constructs and transport activity was determined by performing Tl+ flux measurements. NEM and staurosporine were added 15 min prior to measurement. KCC2WT, the single mutants KCC2T934A and KCC2S937A, and the double mutant KCC2T934A/S937A were significantly activated by NEM and staurosporine. In contrast, the activity of the mutants KCC2T934D and KCC2S937D was significantly reduced in the presence of NEM and staurosporine. Graphs represent mean ± S.E. of at least three independent measurements. Statistical analysis is presented in Table 4.
TABLE 4.
Transport activity of mutant KCC2Thr-934 and KCC2Ser-937 in the presence of NEM or staurosporine
| Basal conditions (significance in comparison with KCC2WT) | 1 mm NEM (significance in comparison with untreated samples or KCC2WT under basal conditions) | 8 μm staurosporine (significance in comparison to the untreated samples) (or KCC2WT under basal conditions) | |
|---|---|---|---|
| HEK-293 | 8.3 ± 1.9%a | 19.2 ± 2.2%b | 24.8 ± 4.4%b |
| KCC2WT | 100 ± 4.9% | 157 ± 8.2%b | 180.7 ± 11.5%b |
| T934A | 97.3 ± 2.1% (NS)c | 159.3 ± 13%b | 199.5 ± 8.3%d |
| T934D | 226.6 ± 13%a | 86 ± 4.1%a (NS) | 123 ± 12.6%a (NS) |
| S937A | 106.2 ± 10.2% (NS) | 159.6 ± 9.3%d | 199.5% ± 16.7%b |
| S937D | 186.9 ± 12%a | 100 ± 8.2%a (NS) | 132.1 ± 8.5%d (NS) |
| T934A/S937A | 119.4 ± 9.8% (NS) | 148.2 ± 3.2%b | 198% ± 5.4%a |
a p < 0.001.
b p < 0.05.
c NS, not significant.
d p < 0.01.
To test whether this observation is true for other stimuli as well, we applied staurosporine. This alkaloid acts as a kinase inhibitor that activates KCCs (33, 60). Staurosporine closely mimicked the effect of NEM. KCC2WT as well as all three alanine mutants, KCC2T934A, KCC2S937A, and KCC2T934A,T937A, were significantly stimulated by this kinase inhibitor, whereas the transport activity of KCC2T934D and KCC2S937D was significantly reduced to the level of KCC2WT (Fig. 6 and Table 4). From these results, we conclude that the effect of both agents is strongly dependent on the conformational state of KCC2. Furthermore, staurosporine does not act via Thr934 or Ser937, because the transport activities of both the alanine and the aspartate mutants could still be modified by this kinase inhibitor. Finally, the concordant effects of NEM and staurosporine on the mutants suggest that both agents act via a similar mechanism. This is in agreement with the previous observation that both agents were unable to stimulate KCCs in ATP-depleted cells (61) and that deficiency of Src family kinases Fgr and Hck obliterates NEM stimulation of KCCs in red blood cells (62). Thus, NEM likely modifies KCC phosphorylation pattern in a way similar to staurosporine.
No Change in Abundance and Cell Surface Expression of KCC2T934D and KCC2S937D Mutants
Finally, we addressed the underlying mechanism of increased transport activity of KCC2T934D and KCC2S937D. Altered phosphorylation might affect abundance, cell surface expression, or the transport kinetics of KCC2. To address the abundance of the KCC2 mutants, immunoblot analyses were performed for KCC2T934A, KCC2T934D, KCC2S937A, and KCC2S937D. Quantitative analysis of the immunoblots revealed no significant change in protein level (Fig. 7).
FIGURE 7.
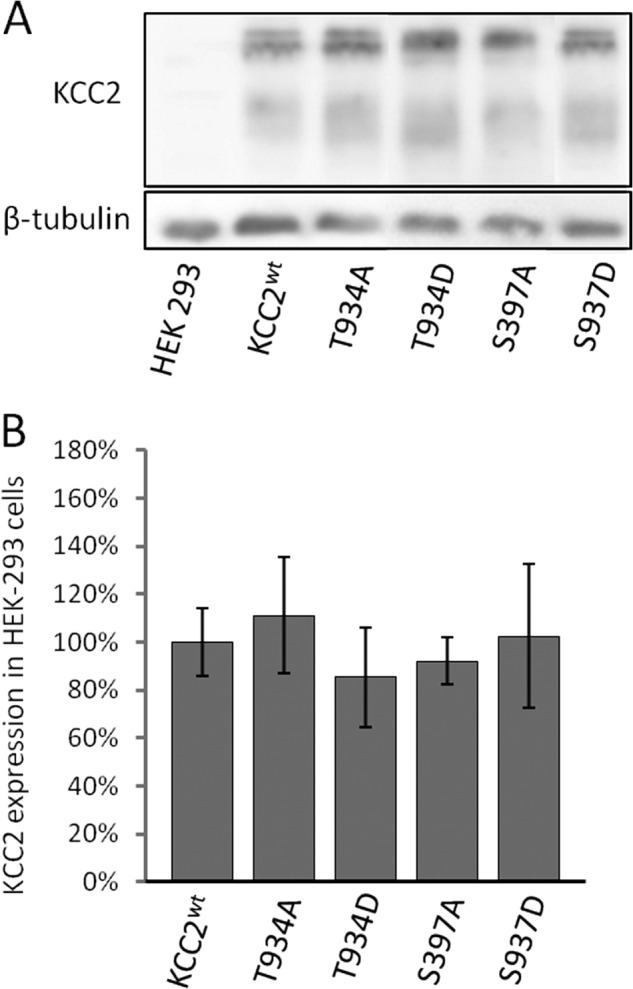
Immunoblot analysis of KCC2WT, KCC2T934A/T934D, and KCC2S937A/S937D. A, KCC2 constructs were transfected into HEK-293 cells, and proteins were isolated 36 h later. 10 μg of protein were loaded onto a 10% SDS-polyacrylamide gel system, and the amount of KCC2 was determined by immunoblotting using an anti-cKCC2 antibody. KCC2-IR was detected for KCC2WT and KCC2 mutants, but not for mock-transfected cells, whereas β-tubulin, serving as a loading control, was present in all lanes. The figure shows one experiment out of three biological replicates with similar results. B, KCC2 bands were quantified and normalized to the expression of β-tubulin. This revealed no differences in the expression level between KCC2WT and KCC2 mutants in HEK-293 cells. Graphs represent mean ± S.E. of three independent experiments.
To investigate cell surface expression of the mutants, we used a previously reported KCC2 expression construct with an extracellular HA tag in the second extracellular loop between transmembrane domains 3 and 4 (48). We substituted Thr934 or Ser937 by aspartate resulting in the two new constructs KCC2T934D-HA 2nd loop and KCC2S937D-HA 2nd loop. Transfection of either of the two constructs into HEK-293 cells resulted in an increased KCC2 activity, compared with KCC2WT-HA 2nd loop, demonstrating that the HA tag did not interfere with mutations in Thr934 or Ser937 (data not shown). Surface expression of the KCC2 variants was determined in HEK-293 cells by a sandwich protocol. As a control, we used the construct KCC2WT-HA N-term with the HA tag at the intracellularly located N terminus. Surface-expressed KCC2 was labeled by incubating unfixed cells prior to permeabilization with antibodies against the extracellular HA tag for 25 min. Subsequently, cells were fixed and permeabilized, and total KCC2 was monitored using a KCC2 antibody. HEK-293 cells transfected with KCC2WT-HA N-term showed only poor labeling with anti-HA, whereas anti-cKCC2 antibodies demonstrated expression of the protein (Fig. 8A). In cells transfected with the HA tag in the second extracellular loop, incubation with the anti-HA antibody resulted in cell surface staining for all three constructs, KCC2WT, KCC2T934D-HA, and KCC2S937D-HA (Fig. 8A). Quantitative analyses of the signal ratio of surface staining to total expression revealed no difference between wild-type and mutant constructs (Fig. 8B), whereas significant differences in surface staining were obtained between KCC2 WT-HA N-term and KCC2WT-HA 2nd loop (p = 0.018), KCC2T934D-HA 2nd loop (p = 0.011), and KCC2S937D-HA 2nd loop (p = 0.027). From this observation, we conclude that the mutations do not affect surface expression but affect the intrinsic transport activity of KCC2.
FIGURE 8.

Surface expression of KCC2 mutants in HEK-293 cells. HEK-293 cells were transfected with the indicated constructs. Surface KCC2 (green) was detected using an HA antibody under nonpermeabilizing conditions and prior to fixation. After fixation and permeabilization, total KCC2 (red) was detected with anti-cKCC2 antibody. A, images show representative examples for each construct. Scale bar, 10 μm. B, ratio of surface signals to total expression signals of KCC2 was measured for each construct. Ratio of KCC2WT-HA 2nd loop was compared with KCC2WT-HA N-term and mutant KCC2HA 2nd loop. Mutant KCC2HA 2nd loop showed no significant difference in the ratio compared with KCC2WT-HA 2nd loop. In contrast, the ratio for the negative control KCC2WT-HA N-term was significantly reduced compared with all three other constructs. Graphs represent mean ± S.D. of at least three independent measurements.
DISCUSSION
Here, we report the identification of novel potent KCC2 phosphorylation sites encompassing Ser937 and Thr934, which are involved in kinetic regulation of the transport activity. Phospho-mimetic substitution of these two amino acid residues inverted the action of staurosporine and NEM. These results hint to a cross-talk between different phosphorylation sites and reveal the impact of the state of the transporter on the action of post-translational modifications.
Modern mass spectrometry-based in vivo phospho-proteomics studies have identified thousands of bona fide phosphorylation sites of brain proteins (56, 57, 63). So far, these publicly available data have been poorly exploited. Here, we started to investigate the possible role of such experimentally verified phosphorylation sites for regulation of KCC2 transport activity. Mutational analyses in combination with flux measurements identified a hitherto unknown potent regulatory role of Thr934 and Ser937 (Fig. 5). Substitution of these amino acids by the phospho-mimetic amino acid aspartate significantly facilitated KCC2 transport in HEK-293 cells. Thus, phosphorylation of these amino acids will enhance KCC2 transport activity. This is in line with the observation of phosphorylated Ser937 and likely Thr934 in adult brain tissue (57).
So far, phosphorylation mainly affected trafficking or the half-lives of KCC2 (5, 36). In contrast to these findings, the increased activity of KCC2T934D or KCC2S937D was not paralleled by an increase in total amount or cell surface expression of the transporter (Figs. 6 and 7). These results reveal a kinetic up-regulation of the transport activity by phosphorylation of these two amino acid residues. Kinetic regulation of the intrinsic transport activity of KCC2 is also suggested for Thr906 and Thr1007, based on studies of the homologous amino acid residues Thr991 and Thr1048 in KCC3 (37). Surface biotinylation experiments demonstrated that dephosphorylation of these two threonines activated KCC3 without an increase in plasma membrane localization (37). Taken together, these data suggest a model, in which the intrinsic transport activity of KCC2 is increased by phosphorylation of Thr934 or Ser937 (this study) and decreased by phosphorylation of Thr906 and Thr1007 (37).
The prevailing notion holds that phosphorylation activates NKCCs and inactivates KCCs (31, 58, 65, 66). This elegant concept of reciprocal regulation of Cl−-inward and Cl−-outward transporters was originally based on the observations that cell swelling, NEM, staurosporine, and serine-threonine phosphatase 1 inhibited NKCC1 (67–69) and activated KCCs (33, 58, 67). This view was subsequently substantiated by the reciprocal effect of WNKs on KCCs and NKCC/NCC transport activities (31, 70) and by the fact that dephosphorylation of Thr906 and Thr1007 facilitates KCC transport (37) and phosphorylation of Tyr903 and Tyr1087 increases KCC2 degradation in lysosomes (41). However, the recent characterization of the Ser940 phosphorylation site of KCC2 already challenged this concept. Dephosphorylation of Ser940 decreased cell surface expression of KCC2 (39) and concomitantly caused depolarizing action of GABA in neurons (71). Our observation of up-regulated activity in KCC2T934D and KCC2S937D supports the emerging concept that KCC2 can both be activated and deactivated by phosphorylation, depending on the amino acid residues involved. However, the staurosporine and NEM experiments indicate that the targeted amino acid residue is not the sole determinant for the effect of phosphorylation/dephosphorylation. Both agents can have opposite effects, depending on the phosphorylation state of Thr934 and Ser937 (Fig. 6). In the dephosphorylated state, mimicked by alanine substitutions, KCC2 transport was facilitated by these two agents, and in the phosphorylated state, represented by the aspartate mutants, the effect was the opposite. These results indicate that the conformational state of KCC2 has an impact on the functional consequences of phosphorylation/dephosphorylation of a given amino acid residue. This is reminiscent of red blood cells, where low concentrations of NEM at 0 °C activated and high concentration of NEM at 37 °C inhibited KCC-mediated transport (72).
Because of the rapid effects within minutes, NEM or staurosporine are assumed to regulate the intrinsic activity of KCCs, similar to phosphorylation of Thr934 or Ser937 (33, 58, 59). A likely explanation for the opposite effect of both agents on Thr934 and Ser937 mutants is because they induce conformational changes that differ depending on the conformational state of KCC2. In KCC2WT, KCC2T934A, and KCC2S937A, they cause a state with high transport activity, whereas in KCC2T934D and KCC2S937D, which have already adopted the state of high transport activity, further conformational changes imposed by indirect action of NEM or staurosporine convert KCC2 into a state of basal activity. An alternative explanation is that the conformation of KCC2T934D and KCC2S937D deoccludes a hidden site, upon which NEM or staurosporine indirectly act in a manner different from their action in KCC2WT (58). In either case, the data reveal a functional cross-talk between phosphorylation of Thr934 and Ser937 and another staurosporine/NEM-modulated phosphorylation site. Thus, the effect of phosphorylation can itself be modulated by phosphorylation of other sites. This might be important for fine-tuning the operational mode of KCC2 and to integrate different signaling pathways. These findings also imply that drugs aimed to modify KCC2 phosphorylation for therapeutic benefit (36, 73) have to be investigated under different conditions as their effect might depend on the conformational state of KCC2 and therefore be highly context-specific. Functional cross-talk between different regions has also been suggested for NKCC1, where phosphorylation of the N terminus causes movement of the C terminus, which in turn entails altered interactions between different transmembrane domains (74).
Among the phosphorylated amino acid residues in KCC2, Thr934 and Ser937 showed an intermediate level of evolutionary conservation. The two residues are only present in the KCC2 subgroup of vertebrate KCCs and in non-therian vertebrate KCC4 family members (Fig. 1). This is in stark contrast to Thr906 and Thr1007, which are strongly conserved throughout evolution. Phosphorylation of Ser26, which is present in most vertebrate KCC1, KCC2 and KCC4 family members, had no influence on KCC2 activity. Taken together, these data establish evolutionary conservation as a poor predictor for regulatory phosphorylation sites in KCCs. This might be expected, because subgroup-specific phosphorylation sites enable differential regulation of the diverse KCCs within the same cell. This is likely an important step favoring subfunctionalization of the different family members during vertebrate evolution (10), as they can be co-expressed in brain areas (75).
Our analyses failed to detect a regulatory role of Ser25, Ser26, Ser1022, Ser1025, and Ser1026 in HEK-293 cells. Ser25 and Ser26 are located in the cytoplasmic N terminus of KCC2. Previous investigations have revealed an important role of this sequence for CCC transport activity. Phosphorylation of Ser96 in KCC3 is involved in cell swelling-related activation of the transporter (76). Furthermore, N-terminal truncation disrupted transport activity of KCC1 (77) and KCC2 (9). In the recently identified KCC2a isoform, a SPAK-binding site with a yet unknown functional role is located in the N terminus (53). In NKCC1 and NKCC2, three N-terminally located phosphothreonines modulate transport activity (55). Interestingly, in NKCC2, mutation of either of the three threonines to alanine had no effect; mutations of two threonines resulted in reduced activity, and mutations of all three threonines blocked hypertonic activation (55). It might therefore be that mutation of more than one phosphorylated amino acid residue in the N terminus of KCC2 is required to observe a phenotype. However, the double mutants KCC2S25D/S26D and KCC2S25A/S26A demonstrated KCC2WT-like activity as well (Fig. 4), indicating that phosphorylation of these two serines do not influence KCC2 activity in HEK-293 cells. This does not exclude a role in other processes such as trafficking of KCC2 in neurons. The C-terminal Ser1022, Ser1025, and Ser1026 are located within the so-called ISO domain of KCC2, which confers KCC2 activity under isotonic conditions (78–80). The observation that this isotonic activity of KCC2 is not influenced by the serine-threonine phosphatase inhibitor calyculin A in Xenopus laevis oocytes (78) supports our conclusion that phosphorylation of Ser1022, Ser1025, and Ser1026 is not required for basal KCC2 transport activity.
CONCLUSION
At least three different regulatory states of KCC2 can exist in the plasma membrane as follows: an inactive conformation, a conformation conferring basal activity, and a conformation with high transport rate. Our data reveal that Thr934 and Ser937 participate in interconversion between these conformational states and, together with Thr906 and Thr1007, may represent key residues for kinetic regulation of KCC2. Furthermore, the varying effect of staurosporine and NEM provide evidence for a cross-talk between different phosphorylation sites. This might have implications for drug research aimed to modify KCC2 function via changes in phosphorylation.
Acknowledgments
We acknowledge the excellent technical support by M. Reents and J. Schröder. We thank F. K. Bedford for the HA-tagged KCC2 construct, J. Brocher from Biovoxxel for help with the image analyses, and Jens Schindler for help in initial experiments.
This work was supported by Deutsche Forschungsgemeinschaft Grants No428/3-1 and No428/4-2 (to H. G. N.) and Ha6338/2-1 (to A.-M. H).
This article was selected as a Paper of the Week.
CCCP9 is a cation chloride cotransporter-interacting protein 1.
- KCC
- K-Cl cotransporter
- NCC
- Na-Cl cotransporter
- NKCC
- Na-K-Cl cotransporter
- NEM
- N-ethylmaleimide
- HA
- human influenza hemagglutinin.
REFERENCES
- 1. Payne J. A., Stevenson T. J., Donaldson L. F. (1996) Molecular characterization of a putative K-Cl cotransporter in rat brain. A neuronal-specific isoform. J. Biol. Chem. 271, 16245–16252 [DOI] [PubMed] [Google Scholar]
- 2. Payne J. A. (1997) Functional characterization of the neuronal-specific K-Cl cotransporter: implications for [K+]o regulation. Am. J. Physiol. 273, C1516–C1525 [DOI] [PubMed] [Google Scholar]
- 3. Rivera C., Voipio J., Payne J. A., Ruusuvuori E., Lahtinen H., Lamsa K., Pirvola U., Saarma M., Kaila K. (1999) The K+/Cl− co-transporter KCC2 renders GABA hyperpolarizing during neuronal maturation. Nature 397, 251–255 [DOI] [PubMed] [Google Scholar]
- 4. Blaesse P., Airaksinen M. S., Rivera C., Kaila K. (2009) Cation-chloride cotransporters and neuronal function. Neuron 61, 820–838 [DOI] [PubMed] [Google Scholar]
- 5. Medina I., Friedel P., Rivera C., Kahle K. T., Kourdougli N., Uvarov P., Pellegrino C. (2014) Current view on the functional regulation of the neuronal K-Cl cotransporter KCC2. Front. Cell. Neurosci. 8, 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Balakrishnan V., Becker M., Löhrke S., Nothwang H. G., Güresir E., Friauf E. (2003) Expression and function of chloride transporters during development of inhibitory neurotransmission in the auditory brainstem. J. Neurosci. 23, 4134–4145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hübner C. A., Stein V., Hermans-Borgmeyer I., Meyer T., Ballanyi K., Jentsch T. J. (2001) Disruption of KCC2 reveals an essential role of K-Cl cotransport already in early synaptic inhibition. Neuron 30, 515–524 [DOI] [PubMed] [Google Scholar]
- 8. Horn Z., Ringstedt T., Blaesse P., Kaila K., Herlenius E. (2010) Premature expression of KCC2 in embryonic mice perturbs neural development by an ion transport-independent mechanism. Eur. J. Neurosci. 31, 2142–2155 [DOI] [PubMed] [Google Scholar]
- 9. Li H., Khirug S., Cai C., Ludwig A., Blaesse P., Kolikova J., Afzalov R., Coleman S. K., Lauri S., Airaksinen M. S., Keinänen K., Khiroug L., Saarma M., Kaila K., Rivera C. (2007) KCC2 interacts with the dendritic cytoskeleton to promote spine development. Neuron 56, 1019–1033 [DOI] [PubMed] [Google Scholar]
- 10. Hartmann A. M., Tesch D., Nothwang H. G., Bininda-Emonds O. R. (2014) Evolution of the cation chloride cotransporter family: ancient origins, gene-losses, and subfunctionalization through duplication. Mol. Biol. Evol. 31, 434–447 [DOI] [PubMed] [Google Scholar]
- 11. Gagnon K. B., Delpire E. (2013) Physiology of SLC12 transporters: lessons from inherited human genetic mutations and genetically engineered mouse knockouts. Am. J. Physiol. Cell Physiol. 304, C693–C714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gamba G. (2005) Molecular physiology and pathophysiology of electroneutral cation-chloride cotransporters. Physiol. Rev. 85, 423–493 [DOI] [PubMed] [Google Scholar]
- 13. Di Fulvio M., Alvarez-Leefmans F. J. (2009) in Physiology and Pathology of Chloride Transporters and Channels in the Nervous System: From Molecules to Diseases (Alvarez-Leefmans F. J., Delpire E., eds) pp. 169–208, Elsevier/Academic Press, Inc., London, UK [Google Scholar]
- 14. Coull J. A., Gagnon M. (2009) The manipulation of cation-chloride co-transporters as a novel means to treat persistent pain, epilepsy and other neurological disorders. Curr. Opin. Investig. Drugs 10, 56–61 [PubMed] [Google Scholar]
- 15. Huberfeld G., Wittner L., Clemenceau S., Baulac M., Kaila K., Miles R., Rivera C. (2007) Perturbed chloride homeostasis and GABAergic signaling in human temporal lobe epilepsy. J. Neurosci. 27, 9866–9873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Rivera C., Li H., Thomas-Crusells J., Lahtinen H., Viitanen T., Nanobashvili A., Kokaia Z., Airaksinen M. S., Voipio J., Kaila K., Saarma M. (2002) BDNF-induced TrkB activation down-regulates the K+-Cl− cotransporter KCC2 and impairs neuronal Cl− extrusion. J. Cell Biol. 159, 747–752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Palma E., Amici M., Sobrero F., Spinelli G., Di Angelantonio S., Ragozzino D., Mascia A., Scoppetta C., Esposito V., Miledi R., Eusebi F. (2006) Anomalous levels of Cl− transporters in the hippocampal subiculum from temporal lobe epilepsy patients make GABA excitatory. Proc. Natl. Acad. Sci. U.S.A. 103, 8465–8468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Coull J. A., Boudreau D., Bachand K., Prescott S. A., Nault F., Sík A., De Koninck P., De Koninck Y. (2003) Trans-synaptic shift in anion gradient in spinal lamina I neurons as a mechanism of neuropathic pain. Nature 424, 938–942 [DOI] [PubMed] [Google Scholar]
- 19. Janssen S. P., Truin M., Van Kleef M., Joosten E. A. (2011) Differential GABAergic disinhibition during the development of painful peripheral neuropathy. Neuroscience 184, 183–194 [DOI] [PubMed] [Google Scholar]
- 20. Boulenguez P., Liabeuf S., Bos R., Bras H., Jean-Xavier C., Brocard C., Stil A., Darbon P., Cattaert D., Delpire E., Marsala M., Vinay L. (2010) Down-regulation of the potassium-chloride cotransporter KCC2 contributes to spasticity after spinal cord injury. Nat. Med. 16, 302–307 [DOI] [PubMed] [Google Scholar]
- 21. Papp E., Rivera C., Kaila K., Freund T. F. (2008) Relationship between neuronal vulnerability and potassium-chloride cotransporter 2 immunoreactivity in hippocampus following transient forebrain ischemia. Neuroscience 154, 677–689 [DOI] [PubMed] [Google Scholar]
- 22. Shulga A., Thomas-Crusells J., Sigl T., Blaesse A., Mestres P., Meyer M., Yan Q., Kaila K., Saarma M., Rivera C., Giehl K. M. (2008) Posttraumatic GABA(A)-mediated [Ca2+]i increase is essential for the induction of brain-derived neurotrophic factor-dependent survival of mature central neurons. J. Neurosci. 28, 6996–7005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Watanabe M., Wake H., Moorhouse A. J., Nabekura J. (2009) Clustering of neuronal K+-Cl− cotransporters in lipid rafts by tyrosine phosphorylation. J. Biol. Chem. 284, 27980–27988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hartmann A. M., Blaesse P., Kranz T., Wenz M., Schindler J., Kaila K., Friauf E., Nothwang H. G. (2009) Opposite effect of membrane raft perturbation on transport activity of KCC2 and NKCC1. J. Neurochem. 111, 321–331 [DOI] [PubMed] [Google Scholar]
- 25. Ikeda K., Onimaru H., Yamada J., Inoue K., Ueno S., Onaka T., Toyoda H., Arata A., Ishikawa T. O., Taketo M. M., Fukuda A., Kawakami K. (2004) Malfunction of respiratory-related neuronal activity in Na+, K+-ATPase α2 subunit-deficient mice is attributable to abnormal Cl− homeostasis in brainstem neurons. J. Neurosci. 24, 10693–10701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wenz M., Hartmann A. M., Friauf E., Nothwang H. G. (2009) CIP1 is an activator of the K+-Cl− cotransporter KCC2. Biochem. Biophys. Res. Commun. 381, 388–392 [DOI] [PubMed] [Google Scholar]
- 27. Ivakine E. A., Acton B. A., Mahadevan V., Ormond J., Tang M., Pressey J. C., Huang M. Y., Ng D., Delpire E., Salter M. W., Woodin M. A., McInnes R. R. (2013) Neto2 is a KCC2 interacting protein required for neuronal Cl− regulation in hippocampal neurons. Proc. Natl. Acad. Sci. U.S.A. 110, 3561–3566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Inoue K., Yamada J., Ueno S., Fukuda A. (2006) Brain-type creatine kinase activates neuron-specific K+-Cl− co-transporter KCC2. J. Neurochem. 96, 598–608 [DOI] [PubMed] [Google Scholar]
- 29. Piechotta K., Garbarini N., England R., Delpire E. (2003) Characterization of the interaction of the stress kinase SPAK with the Na+-K+-2Cl− cotransporter in the nervous system–evidence for a scaffolding role of the kinase. J. Biol. Chem. 278, 52848–52856 [DOI] [PubMed] [Google Scholar]
- 30. Piechotta K., Lu J., Delpire E. (2002) Cation chloride cotransporters interact with the stress-related kinases Ste20-kelated proline-alanine-rich kinase (SPAK) and oxidative stress response 1 (OSR1). J. Biol. Chem. 277, 50812–50819 [DOI] [PubMed] [Google Scholar]
- 31. Kahle K. T., Rinehart J., de Los Heros P., Louvi A., Meade P., Vazquez N., Hebert S. C., Gamba G., Gimenez I., Lifton R. P. (2005) WNK3 modulates transport of Cl− in and out of cells: implications for control of cell volume and neuronal excitability. Proc. Natl. Acad. Sci. U.S.A. 102, 16783–16788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kelsch W., Hormuzdi S., Straube E., Lewen A., Monyer H., Misgeld U. (2001) Insulin-like growth factor 1 and a cytosolic tyrosine kinase activate chloride outward transport during maturation of hippocampal neurons. J. Neurosci. 21, 8339–8347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Khirug S., Huttu K., Ludwig A., Smirnov S., Voipio J., Rivera C., Kaila K., Khiroug L. (2005) Distinct properties of functional KCC2 expression in immature mouse hippocampal neurons in culture and in acute slices. Eur. J. Neurosci. 21, 899–904 [DOI] [PubMed] [Google Scholar]
- 34. Song L., Mercado A., Vázquez N., Xie Q., Desai R., George A. L., Jr., Gamba G., Mount D. B. (2002) Molecular, functional, and genomic characterization of human KCC2, the neuronal K-Cl cotransporter. Mol. Brain Res. 103, 91–105 [DOI] [PubMed] [Google Scholar]
- 35. Mercado A., Song L., Vazquez N., Mount D. B., Gamba G. (2000) Functional comparison of the K+-Cl− cotransporters KCC1 and KCC4. J. Biol. Chem. 275, 30326–30334 [DOI] [PubMed] [Google Scholar]
- 36. Kahle K. T., Deeb T. Z., Puskarjov M., Silayeva L., Liang B., Kaila K., Moss S. J. (2013) Modulation of neuronal activity by phosphorylation of the K-Cl cotransporter KCC2. Trends Neurosci. 36, 726–737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Rinehart J., Maksimova Y. D., Tanis J. E., Stone K. L., Hodson C. A., Zhang J., Risinger M., Pan W., Wu D., Colangelo C. M., Forbush B., Joiner C. H., Gulcicek E. E., Gallagher P. G., Lifton R. P. (2009) Sites of regulated phosphorylation that control K-Cl cotransporter activity. Cell 138, 525–536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Inoue K., Furukawa T., Kumada T., Yamada J., Wang T., Inoue R., Fukuda A. (2012) Taurine inhibits K+-Cl− cotransporter KCC2 to regulate embryonic Cl− homeostasis via with-no-lysine (WNK) protein kinase signaling pathway. J. Biol. Chem. 287, 20839–20850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lee H. H., Walker J. A., Williams J. R., Goodier R. J., Payne J. A., Moss S. J. (2007) Direct protein kinase C-dependent phosphorylation regulates the cell surface stability and activity of the potassium chloride cotransporter KCC2. J. Biol. Chem. 282, 29777–29784 [DOI] [PubMed] [Google Scholar]
- 40. Chamma I., Heubl M., Chevy Q., Renner M., Moutkine I., Eugène E., Poncer J. C., Lévi S. (2013) Activity-dependent regulation of the K/Cl transporter KCC2 membrane diffusion, clustering, and function in hippocampal neurons. J. Neurosci. 33, 15488–15503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lee H. H., Jurd R., Moss S. J. (2010) Tyrosine phosphorylation regulates the membrane trafficking of the potassium chloride co-transporter KCC2. Mol. Cell. Neurosci. 45, 173–179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wake H., Watanabe M., Moorhouse A. J., Kanematsu T., Horibe S., Matsukawa N., Asai K., Ojika K., Hirata M., Nabekura J. (2007) Early changes in KCC2 phosphorylation in response to neuronal stress result in functional downregulation. J. Neurosci. 27, 1642–1650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Hornbeck P. V., Kornhauser J. M., Tkachev S., Zhang B., Skrzypek E., Murray B., Latham V., Sullivan M. (2012) PhosphoSitePlus: a comprehensive resource for investigating the structure and function of experimentally determined post-translational modifications in man and mouse. Nucleic Acids Res. 40, D261–D270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Gnad F., Gunawardena J., Mann M. (2011) PHOSIDA 2011: the post-translational modification database. Nucleic Acids Res. 39, D253–D260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Edgar R. C. (2004) MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Gouy M., Guindon S., Gascuel O. (2010) SeaView version 4: A multiplatform graphical user interface for sequence alignment and phylogenetic tree building. Mol. Biol. Evol. 27, 221–224 [DOI] [PubMed] [Google Scholar]
- 47. Hartmann A. M., Wenz M., Mercado A., Störger C., Mount D. B., Friauf E., Nothwang H. G. (2010) Differences in the large extracellular loop between the K+-Cl− cotransporters KCC2 and KCC4. J. Biol. Chem. 285, 23994–24002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Zhao B., Wong A. Y., Murshid A., Bowie D., Presley J. F., Bedford F. K. (2008) Identification of a novel di-leucine motif mediating K+/Cl− cotransporter KCC2 constitutive endocytosis. Cell. Signal. 20, 1769–1779 [DOI] [PubMed] [Google Scholar]
- 49. Hartmann A.-M., Nothwang H. G. (2011) Opposite temperature effect on transport activity of KCC2/KCC4 and N(K)CCs in HEK-293 cells. BMC Res. Notes 4, 526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Delpire E., Days E., Lewis L. M., Mi D., Kim K., Lindsley C. W., Weaver C. D. (2009) Small-molecule screen identifies inhibitors of the neuronal K-Cl cotransporter KCC2. Proc. Natl. Acad. Sci. U.S.A. 106, 5383–5388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Blaesse P., Guillemin I., Schindler J., Schweizer M., Delpire E., Khiroug L., Friauf E., Nothwang H. G. (2006) Oligomerization of KCC2 correlates with development of inhibitory neurotransmission. J. Neurosci. 26, 10407–10419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. King N., Westbrook M. J., Young S. L., Kuo A., Abedin M., Chapman J., Fairclough S., Hellsten U., Isogai Y., Letunic I., Marr M., Pincus D., Putnam N., Rokas A., Wright K. J., Zuzow R., Dirks W., Good M., Goodstein D., Lemons D., Li W., Lyons J. B., Morris A., Nichols S., Richter D. J., Salamov A., Sequencing J. G., Bork P., Lim W. A., Manning G., Miller W. T., McGinnis W., Shapiro H., Tjian R., Grigoriev I. V., Rokhsar D. (2008) The genome of the choanoflagellate Monosiga brevicollis and the origin of metazoans. Nature 451, 783–788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Uvarov P., Ludwig A., Markkanen M., Pruunsild P., Kaila K., Delpire E., Timmusk T., Rivera C., Airaksinen M. S. (2007) A novel N-terminal isoform of the neuron-specific K-Cl cotransporter KCC2. J. Biol. Chem. 282, 30570–30576 [DOI] [PubMed] [Google Scholar]
- 54. Döding A., Hartmann A. M., Beyer T., Nothwang H. G. (2012) KCC2 transport activity requires the highly conserved L(675) in the C-terminal β1 strand. Biochem. Biophys. Res. Commun. 420, 492–497 [DOI] [PubMed] [Google Scholar]
- 55. Giménez I., Forbush B. (2005) Regulatory phosphorylation sites in the NH2 terminus of the renal Na-K-Cl cotransporter (NKCC2). Am. J. Physiol. Renal Physiol. 289, F1341–F1345 [DOI] [PubMed] [Google Scholar]
- 56. Schindler J., Ye J., Jensen O. N., Nothwang H. G. (2013) Monitoring the native phosphorylation state of plasma membrane proteins from a single mouse cerebellum. J. Neurosci. Methods 213, 153–164 [DOI] [PubMed] [Google Scholar]
- 57. Wiśniewski J. R., Nagaraj N., Zougman A., Gnad F., Mann M. (2010) Brain phosphoproteome obtained by a FASP-based method reveals plasma membrane protein topology. J. Proteome Res. 9, 3280–3289 [DOI] [PubMed] [Google Scholar]
- 58. Lauf P. K., Adragna N. C. (2000) K-Cl cotransport: properties and molecular mechanism. Cell. Physiol. Biochem. 10, 341–354 [DOI] [PubMed] [Google Scholar]
- 59. Jennings M. L., al-Rohil N. (1990) Kinetics of activation and inactivation of swelling-stimulated K+/Cl− transport. The volume-sensitive parameter is the rate constant for inactivation. J. Gen. Physiol 95, 1021–1040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Holtzman E. J., Kumar S., Faaland C. A., Warner F., Logue P. J., Erickson S. J., Ricken G., Waldman J., Kumar S., Dunham P. B. (1998) Cloning, characterization, and gene organization of K-Cl cotransporter from pig and human kidney and C. elegans. Am. J. Physiol. 275, F550–F564 [DOI] [PubMed] [Google Scholar]
- 61. Flatman P. W., Adragna N. C., Lauf P. K. (1996) Role of protein kinases in regulating sheep erythrocyte K-Cl cotransport. Am. J. Physiol. 271, C255–C263 [DOI] [PubMed] [Google Scholar]
- 62. De Franceschi L., Fumagalli L., Olivieri O., Corrocher R., Lowell C. A., Berton G. (1997) Deficiency of Src family kinases Fgr and Hck results in activation of erythrocyte K/Cl cotransport. J. Clin. Invest. 99, 220–227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Huttlin E. L., Jedrychowski M. P., Elias J. E., Goswami T., Rad R., Beausoleil S. A., Villén J., Haas W., Sowa M. E., Gygi S. P. (2010) A tissue-specific atlas of mouse protein phosphorylation and expression. Cell 143, 1174–1189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Deleted in proof.
- 65. Kahle K. T., Rinehart J., Lifton R. P. (2010) Phosphoregulation of the Na-K-2Cl and K-Cl cotransporters by the WNK kinases. Biochim. Biophys. Acta 1802, 1150–1158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Gagnon K. B., England R., Delpire E. (2006) Volume sensitivity of cation-Cl− cotransporters is modulated by the interaction of two kinases: Ste20-related proline-alanine-rich kinase and WNK4. Am. J. Physiol. Cell Physiol 290, C134–C142 [DOI] [PubMed] [Google Scholar]
- 67. Lytle C., McManus T. (2002) Coordinate modulation of Na-K-2Cl cotransport and K-Cl cotransport by cell volume and chloride. Am. J. Physiol. Cell Physiol. 283, C1422–C1431 [DOI] [PubMed] [Google Scholar]
- 68. Darman R. B., Flemmer A., Forbush B. (2001) Modulation of ion transport by direct targeting of protein phosphatase type 1 to the Na-K-Cl cotransporter. J. Biol. Chem. 276, 34359–34362 [DOI] [PubMed] [Google Scholar]
- 69. George J. N., Turner R. J. (1989) Inactivation of the rabbit parotid Na/K/Cl cotransporter by N-ethylmaleimide. J. Membr. Biol. 112, 51–58 [DOI] [PubMed] [Google Scholar]
- 70. Kahle K. T., Rinehart J., Ring A., Gimenez I., Gamba G., Hebert S. C., Lifton R. P. (2006) WNK protein kinases modulate cellular Cl− flux by altering the phosphorylation state of the Na-K-Cl and K-Cl cotransporters. Physiology 21, 326–335 [DOI] [PubMed] [Google Scholar]
- 71. Lee H. H., Deeb T. Z., Walker J. A., Davies P. A., Moss S. J. (2011) NMDA receptor activity downregulates KCC2 resulting in depolarizing GABAA receptor-mediated currents. Nat. Neurosci. 14, 736–743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Lauf P. K., Adragna N. C. (1995) Temperature-induced functional deocclusion of thiols inhibitory for sheep erythrocyte K-Cl cotransport. Am. J. Physiol. 269, C1167–C1175 [DOI] [PubMed] [Google Scholar]
- 73. Gagnon M., Bergeron M. J., Lavertu G., Castonguay A., Tripathy S., Bonin R. P., Perez-Sanchez J., Boudreau D., Wang B., Dumas L., Valade I., Bachand K., Jacob-Wagner M., Tardif C., Kianicka I., Isenring P., Attardo G., Coull J. A., De Koninck Y. (2013) Chloride extrusion enhancers as novel therapeutics for neurological diseases. Nat. Med. 19, 1524–1528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Monette M. Y., Somasekharan S., Forbush B. (2014) Molecular motions involved in Na-K-Cl cotransporter-mediated ion transport and transporter activation revealed by internal cross-linking between transmembrane domains 10 and 11/12. J. Biol. Chem. 289, 7569–7579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Becker M., Nothwang H. G., Friauf E. (2003) Differential expression pattern of chloride cotransporters NCC, NKCC2, KCC1, KCC3, KCC4 and AE3 in the developing rat auditory brainstem. Cell Tissue Res. 312, 155–165 [DOI] [PubMed] [Google Scholar]
- 76. Melo Z., de los Heros P., Cruz-Rangel S., Vázquez N., Bobadilla N. A., Pasantes-Morales H., Alessi D. R., Mercado A., Gamba G. (2013) N-terminal serine dephosphorylation is required for KCC3 cotransporter full activation by cell swelling. J. Biol. Chem. 288, 31468–31476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Casula S., Shmukler B. E., Wilhelm S., Stuart-Tilley A. K., Su W., Chernova M. N., Brugnara C., Alper S. L. (2001) A dominant negative mutant of the KCC1K-Cl cotransporter–Both N- and C-terminal cytoplasmic domains are required for K-Cl cotransport activity. J. Biol. Chem. 276, 41870–41878 [DOI] [PubMed] [Google Scholar]
- 78. Mercado A., Broumand V., Zandi-Nejad K., Enck A. H., Mount D. B. (2006) A C-terminal domain in KCC2 confers constitutive K+-Cl− cotransport. J. Biol. Chem. 281, 1016–1026 [DOI] [PubMed] [Google Scholar]
- 79. Acton B. A., Mahadevan V., Mercado A., Uvarov P., Ding Y., Pressey J., Airaksinen M. S., Mount D. B., Woodin M. A. (2012) Hyperpolarizing GABAergic transmission requires the KCC2 C-terminal ISO domain. J. Neurosci. 32, 8746–8751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Bergeron M. J., Gagnon E., Caron L., Isenring P. (2006) Identification of key functional domains in the C terminus of the K+-Cl− cotransporters. J. Biol. Chem. 281, 15959–15969 [DOI] [PubMed] [Google Scholar]