

Background: UppP, an integral membrane protein involved in the bacterial cell wall synthesis, catalyzes the dephosphorylation of undecaprenyl pyrophosphate.
Results: The enzyme active site is proposed by modeling, molecular dynamics, and mutagenesis.
Conclusion: The enzyme active-site, composed of (E/Q)XXXE and PGXSRSXXT motifs and a histidine, is proposed to be in the periplasm.
Significance: This study provides a first insight into structure-function relationships of E. coli UppP.
Keywords: Computer Modeling, Enzyme Catalysis, Enzyme Kinetics, Enzyme Structure, Mutagenesis, BacA, UppP, Undecaprenyl Pyrophosphate Phosphatase
Abstract
Undecaprenyl pyrophosphate phosphatase (UppP), an integral membrane protein, catalyzes the dephosphorylation of undecaprenyl pyrophosphate to undecaprenyl phosphate, which is an essential carrier lipid in the bacterial cell wall synthesis. Sequence alignment reveals two consensus regions, containing glutamate-rich (E/Q)XXXE plus PGXSRSXXT motifs and a histidine residue, specific to the bacterial UppP enzymes. The predicted topological model suggests that both of these regions are localized near the aqueous interface of UppP and face the periplasm, implicating that its enzymatic function is on the outer side of the plasma membrane. The mutagenesis analysis demonstrates that most of the mutations (E17A/E21A, H30A, S173A, R174A, and T178A) within the consensus regions are completely inactive, indicating that the catalytic site of UppP is constituted by these two regions. Enzymatic analysis also shows an absolute requirement of magnesium or calcium ions in enzyme activity. The three-dimensional structural model and molecular dynamics simulation studies have shown a plausible structure of the catalytic site of UppP and thus provides insights into the molecular basis of the enzyme-substrate interaction in membrane bilayers.
Introduction
In the cytoplasm, undecaprenyl pyrophosphate (C55-PP)3 is synthesized by undecaprenyl pyrophosphate synthase (UppS) through consecutive condensation reactions of eight molecules of isopentenyl pyrophosphate (Ipp) with farnesyl pyrophosphate (Fpp) (1, 2). The product C55-PP is then dephosphorylated to monophosphate undecaprenyl phosphate (C55-P) in de novo synthesis by an integral membrane protein, undecaprenyl pyrophosphate phosphatase (BacA/UppP) (3, 4). C55-P is an essential carrier lipid in the bacterial cell membrane for the biosynthesis of peptidoglycan and various carbohydrate polymers, such as lipopolysaccharides, teichoic acids, and osmoregulated periplasmic glucans (5). Fig. 1 shows a detailed pathway of the C55-P serving as a carrier lipid for the translocation of the hydrophilic oligosaccharide precursors (lipid II) across the cell membranes via a recently identified flippase (most probably by FtsW proteins) for peptidoglycan assembly in the periplasm (6). After the transfer of the glycan component to the peptidoglycan chain, the C55-P molecules flip back to the cytoplasm via an unknown mechanism in the recycling pathway. Fig. 1 summarizes a generally accepted model of biosynthesis of carrier lipid in de novo synthesis and the recycling pathway.
FIGURE 1.

Biosynthesis of C55-P and cell wall peptidoglycan in E. coli. UppS synthesizes C55-PP, which is dephosphorylated to monophosphate C55-P by the phosphatase BacA/UppP. Subsequently, the sugars-pentapeptide are transferred to C55-P by the enzymes MraY and MurG producing lipid I and lipid II, respectively. Lipid II is then translocated to the periplasm via flippase (FtsW) for peptidoglycan assembly by the penicillin-binding proteins (PBPs). Finally, the peptidoglycan is transferred to the peptidoglycan chain, and the C55-P is flipped back to the cytoplasm to repeat the cycle after being dephosphorylated by enzymes YbjG, PgpB, or YeiU. The blue question mark denotes that it is currently unknown how the dephosphorylated periplasmic C55-P molecules return to the cytoplasm in the recycling pathway. The red question mark denotes that the function of BacA/UppP in the cytosolic compartment or periplasmic space is still unclear. Cell wall active antibiotics are as follows. Bacitracin forms a metal-dependent complex with C55-PP and inhibits its dephosphorylation. Tunicamycin inhibits the transfer of peptidoglycan precursor (phospho-MurNAc-pentapeptide) to the C55-PP. Vancomycin inhibits cell wall synthesis by binding to the two d-Ala residues on the end of the pentapeptide chains. Penicillin and moenomycin A (MmA) inhibit the penicillin-binding proteins.
In Escherichia coli, four genes, uppP (formerly bacA), pgpB, ybjG, and lpxT (formerly yeiU), have been identified encoding integral membrane proteins with C55-PP phosphatase activity. UppP has been suggested to generate 75% of the total cellular C55-PP phosphatase activity, whereas the additional enzymes may account for the remaining 25% phosphatase activity (3, 7). Genomic disruption of each one or in pairs was shown to be nonlethal in E. coli, suggesting that no single enzyme among them is essential for cell growth (3, 4, 7, 8). However, inactivation of all three genes (uppP, ybjG, and pgpB) caused cell lysis due to the depletion of the pool of C55-P as well as the accumulation of peptidoglycan nucleotide used for cell wall synthesis (7).
C55-PP can be produced in two different pathways, by de novo synthesis and by recycling. In both ways, C55-PP must be dephosphorylated before it can be used or reused as a carrier lipid for polymer biosynthesis. It has been suggested that UppP participates in the C55-PP de novo synthesis at the cytoplasmic site, whereas the other three enzymes (PgpB, YbjG, and LpxT) participate in the C55-PP recycling pathway at the periplasmic site. Tatar et al. and Touzé et al. (4, 9) demonstrated that the active sites of LpxT, YbjG, and PgpB are all oriented toward the periplasmic space and might participate in C55-PP recycling based on biochemical evidence and topology analysis. On the contrary, the topological model of UppP revealed a large cytosolic loop that is conserved in bacterial UppP enzymes, suggesting that it may participate in C55-PP de novo synthesis in the cytoplasm (4, 10). To date, however, it is still unclear on which side of the plasma membrane the dephosphorylation of C55-PP molecules occurs. Whether the dephosphorylation of C55-PP occurs on both sides of the plasma membrane or only on one side needs to be determined.
Although UppP, PgpB, YbjG, and LpxT all share the C55-PP phosphatase activity, UppP has no sequence homology to the others. Sequence alignment shows that the bacterial PgpB together with YbjG and LpxT belong to the phosphatidic acid phosphatase type 2 superfamily, which is characterized by three conserved motifs (KX6RPX12–54PSGHX31–54SRX5HX3D) (9–11). These conserved motifs are also found in eukaryotic dolichyl pyrophosphate phosphatases (DOLPP1) and yeast CWH8 enzyme (12–14). Ishikawa et al. (15) previously determined the crystal structure of a soluble phosphatidic acid phosphatase type 2 protein from Escherichia blattae and demonstrated that the catalytic site is composed of the three consensus motifs, suggesting that its catalytic mechanism could be homologous to membrane-embedded phosphatase (PgpB, YbjG, and LpxT) (9, 15).
Because no crystal structures of the bacterial UppP or a similar protein of this family are currently available, structure-function relationships of UppP are therefore unknown. A recent study in topology analysis of UppP showed that two consensus motifs are specific to the UppP enzymes (10). One is located at the putative first transmembrane helix embedded within the plasma membrane, suggesting lipid substrate binding, whereas the other one is located in a large cytosolic loop and is suggested to be a catalytic motif. Sequence alignment also revealed that the latter conserved motif could resemble a tyrosine phosphate phosphatase motif of the tumor suppressor, phosphatase and tensin homolog. However, its exact role in the reaction of the carrier lipid dephosphorylation, including the enzyme catalytic mechanism and structural information, is still not known.
UppP is predicted to be a highly hydrophobic protein with eight transmembrane helices. Therefore, the difficulties in producing sufficient quantities of properly folded and active proteins for structural and functional studies are anticipated. We recently have successfully purified active UppP (N terminus is located in the cytoplasm) from E. coli by using a bacteriorhodopsin as a tag fused at the N terminus of the target proteins (16). This opens new opportunities to investigate the specific amino acids critical to enzymatic catalysis by site-directed mutagenesis. In this study, we predict a two-dimensional structure of UppP, showing that both of the consensus regions, containing (E/Q)XXXE plus PGXSRSXXT motifs and a histidine residue, are localized near the aqueous interface of UppP and oriented toward the periplasmic site, implicating that its biological function is on the outer side of the plasma membrane. We also propose a three-dimensional model for UppP constructed by the Rosetta membrane ab initio modeling program (17). The model was validated by the molecular dynamics (MD) simulation analysis as well as the site-directed mutagenesis of amino acids putatively involved in enzyme catalysis. Our study demonstrates that both mutations (E17A and E21A) of the residues Glu-17 and Glu-21 within the (E/Q)XXXE motif, interacting with the pyrophosphate moiety of Upp through a magnesium ion in the model, result in a decrease of kcat values of ∼5 fold, and the Km value of E17A for Fpp increases ∼4–5-fold compared with the wild type. The double mutation E17A/E21A completely eliminates enzyme activity. In the PGXSRSXXT motif, a putative structural P-loop, Arg-174, also establishes a hydrogen bond with the OH group of the pyrophosphate moiety in the model. The R174A mutant is completely inactive. The conserved His-30 residue is spatially in close proximity to the pyrophosphate moiety in the model. The H30A mutant also results in a severely impaired enzyme activity. Our data demonstrate that the active site of UppP is composed of these two consensus regions. This study provides a first insight into structure-function relationships of UppP in E. coli and probably in other bacterial species.
EXPERIMENTAL PROCEDURES
Site-directed Mutagenesis of UppP
Mutants were constructed using KOD Hot Start Master Mix obtained from Novagen. DNA oligonucleotides were synthesized at Genomics (Taiwan). Sequence verification of the mutagenesis products was performed at Mission Biotech (Taiwan).
Protein Expression and Purification
Expression and purification of the E. coli UppP were performed as described previously (16). Briefly, the expression vector harboring the Hmbop1/D94N-uppP gene was transformed into E. coli C41 (DE3), which was grown at 37 °C in LB medium containing 100 mg/ml ampicillin. When the A600 reached about 0.9, the final concentrations of 0.5 mm isopropyl β-d-thiogalactoside and 5–10 mm all-trans-retinal (Sigma) were added for 5 h of induction at 37 °C. For purification of UppP, the cells were harvested and resuspended in buffer A (50 mm Tris, pH 7.5, 500 mm NaCl). The cells were disrupted by Constant Cell Disruption Systems (Constant Systems Ltd.), and the membrane was collected by ultracentrifugation at 40,000 rpm for 1.5 h. The pellet was solubilized in buffer A with 1% (w/v) n-dodecyl-β-d-maltopyranoside at 4 °C for 2.5 h. The latter solution was centrifuged again (20,000 rpm for 0.5 h at 4 °C), and the supernatant was loaded onto nickel-nitrilotriacetic acid column and washed with buffer A containing 75 mm imidazole and 0.05% DDM. Tobacco etch virus protease was then added for digestion during the buffer dialysis at 4 °C overnight. Finally, the protein solution was loaded onto Ni-NTA column, and the native UppP was eluted by washing with buffer A containing 0.02% DDM. For enzyme long term storage, aliquots of protein were dropped directly in liquid nitrogen and stocked in a −80 °C freezer.
Mass Spectrometry Analysis
The mass spectrometry (MS) analysis was performed at the Core Facility for Protein Structural Analysis in Academia Sinica (Taiwan). The dried gel pieces containing purified UppP proteins were prepared for trypsin digestion, and then the peptide mixtures were purified and desalted for mass analysis. MS analysis was performed on an ABI 4700 TOF-TOF Proteomics Analyzer (Applied Biosystems). Data were searched using GPS Explorer (Version 3.6) with the search engine MASCOT.
Phosphatase Activity Assays
The UppP activity was measured as described previously (16). Basically, the phosphatase activity was determined by using a phosphate colorimetric assay kit (BioVision). The enzymatic assay reaction mixture (200 μl) contains 50 mm Hepes at pH 7.0, 150 mm NaCl, 10 mm MgCl2, 0.02% DDM, 35 mm Fpp (Sigma), and 20 nm purified UppP. The reaction was incubated at 37 °C and then quenched by adding 30 μl of Malachite Green reagent. The released phosphate was measured at 650 nm and quantified based on the phosphate standard curve. The effect of pH on UppP activity was assayed at various pH values, pH 5–6 (sodium acetate), pH 6.5–8 (Hepes), and pH 9 (Tris-HCl). For determination of Fpp kinetic parameters of wild-type and UppP mutants, 0.3–57 μm Fpp were used along with 20–40 nm UppP. All reactions were carried out in 50 mm Hepes at pH 7.0, 150 mm NaCl, 10 mm MgCl2, 0.02% DDM at 37 °C. The initial velocity data were fitted to Michaelis-Menten equation using the KaleidaGraph computer program (Synergy software) to obtain Km and kcat values.
Measurement of Inhibition Constant for Bacitracin
The measurement of the inhibition constant for bacitracin was performed in a reaction mixture containing 20 nm UppP and 35 μm Fpp in the buffer of 50 mm Hepes at pH 7.0, 150 mm NaCl, 10 mm MgCl2, and 0.02% DDM in the presence of various concentrations of the inhibitor. The reaction was incubated at 37 °C and quenched by adding 30 μl of Malachite Green reagent as described previously. The initial rate was obtained from different concentrations (0–100 μm) of the bacitracin, and the Ki value was determined by fitting Equations 1 and 2(18) and calculated by nonlinear regression using the KaleidaGraph computer program (19).
In these equations, A(I) is the enzyme activity with the inhibitor concentration I; A(0) is enzyme activity without inhibitor; I is the inhibitor concentration; Ki is the inhibition constant of the inhibitor; S is Fpp concentration, and Km is Michaelis constant of Fpp.
Assignment of Transmembrane Helical Regions
Multiple sequence alignment of several UppPs was first aligned by CLC sequence viewer software, a bioinformatic tool for multiple sequence alignment and editing. The transmembrane helical regions of UppP were then estimated through the comparison of the secondary structure and membrane protein topology predictions, which were done by Phyre2 (20) and Topcons (21) servers, respectively. Certain known information based on the experiments, such as both the N and C termini of UppP localized in the cytoplasm and the active site comprising regions I and II, were also required in this assignment. These studies are important and provide critical information for model generation (Rosetta membrane ab initio modeling prediction).
Rosetta Membrane ab Initio Modeling Prediction
The atomic three-dimensional model of E. coli UppP was generated by the Rosetta membrane ab initio modeling procedure (17). The lipophilicity of each residue in the defined transmembrane regions of E. coli UppP was first calculated. Two-fragment (3 and 9 amino acids) databases generated by the Rosetta Fragment Libraries server were then utilized to generate the atomic models. One thousand models were generated with the default parameters. The three-dimensional lookup table was used for residue neighbor calculations. No nonhelical secondary structure fragments were in the predicted transmembrane regions in the hydrophobic layer of the membrane. To reduce the calculation time, the total number of Monte Carlo-based membrane normal cycles was set to 40. The magnitude of membrane normal angle search step size was 15°, and the center search step size was 2 Å and sorted according to the Rosetta scoring function. Eighteen candidates were filtered on the basis of the criteria that the Cα positions of three residues Glu-21, His-30, and Arg-174 were in a 10 Å diameter (an average distance of H-bond and the side chain of arginine) of sphere. All the model candidates were checked manually, and only the models composed of an active site pocket were considered. Finally, the best model was chosen and optimized by PyMol software, XtalView (22), and RefMac (23). The optimal model of Upp molecule was also applied to the PPM server to validate the position of E. coli UppP in the lipid bilayers. The three-dimensional structure of Upp substrate was prepared and optimized by the MarvinSketch software for the UppP and Upp complex modeling. The conformation of Upp was carefully adjusted and fitted into the predicted active site along with a Mg2+ ion that was coordinated with the pyrophosphate group and two glutamates in the conserved (E/Q)XXXE motif. The rest part of the 55-carbon chain was docked into the hydrophobic surface composed of TM2, TM3, TM4, and TM5.
Molecular Dynamics Simulation Analysis
To evaluate the stability of E. coli UppP model in the membrane, the MD simulation was performed using NAMD version 2.9 (24). The pre-oriented E. coli UppP model calculated by the PPM server was loaded into the membrane builder module of CHARMM-GUI server (25). The cross-sectional area profile was calculated to assist the alignment of protein and lipid bilayer. The rectangular box was selected, and 1.5 lipid layers between neighboring proteins was set (1.5 lipid layers mean, at least, three lipid molecules will be placed between two proteins in the system). A homogeneous lipid bilayer was built with palmitoyl oleoyl phosphatidylcholine, and the water thickness was 15 Å on the top and bottom of the protein. The E. coli UppP model was then inserted into the pre-equilibrated lipid bilayer with a hole whose size is comparable with the protein size. Finally, the assembled system contained 42,738 atoms (1 protein, 112 palmitoyl oleoyl phosphatidylcholines, 7,826 waters, and 2 Cl− ions). Six steps of system equilibration were performed to ensure gradual equilibration of the initially assembled system. Various restraints were applied to the protein, water, ions, and lipid molecules, and the forces were reduced slowly as the equilibration progresses. The NVT (constant volume and temperature) was used for the first two steps, and the NPAT (constant pressure, area, and temperature) was used for the following four steps at 303.15 K. The first five steps were done on CHARMM-GUI server. Step 6 of system equilibration and the 30 ns of production runs were done on the ALPS1 computing facility of National Center for High Performance Computing, Hsinchu, Taiwan. The output trajectory files were analyzed using VMD (26) to obtain the values of root mean square deviation (r.m.s.d.) and root mean square fluctuation (r.m.s.f.). The sausage representation of r.m.s.f. distribution was performed by PyMOL software.
RESULTS
Comparison of Amino Acid Sequences of UppP Enzymes and Two-dimensional Structure Analysis
The amino acid sequence alignment of UppP enzymes from 12 bacteria is presented in Fig. 2. This alignment reveals two highly conserved regions, region I (residues 17–30) and region II (residues 170–178). Region I contains three highly conserved charged amino acids, including two negatively charged residues Glu-17 and Glu-21, one histidine (His-30), and two conserved polar residues Ser-26 and Ser-27. In this region, a glutamate-rich (E/Q)XXXE motif (residues 17–21) constructed by two highly conserved residues, Glu-17 and Glu-21, is found. This motif could be functionally similar to the aspartate-rich DDXXD motif that has been shown to be essential for the catalytic function and substrate binding of metal ion-complexed pyrophosphate ester by x-ray structural and site-directed mutagenesis studies (27–30). In region II, one proline residue (Pro-170), one positively charged residue Arg-74, three polar residues Ser-173, Ser-175, and Thr-178, ands two glycine residues (Gly-171 and Gly-176) are conserved. This region contains a strongly conserved PGXSRSXXT motif (residues 170–178) that could be a structural P-loop, phosphate-binding loop, commonly found in many phosphate-binding enzymes, such as nucleotide triphosphate hydrolase and the cAMP binding domain (31). In E. coli UppS, for example, the P-loop motif is composed of Gly, Asn, Gly, and Arg located at the N terminus of the α1 helix. Except for the second Gly, all residues are strictly conserved among cis-prenyltransferases. Mutations in this region may affect the substrate affinity and turnover number (32, 33). The P-loop in the bacterial RecA proteins have also been defined by the sequence GPESSGKT (34). Most of the mutations within this motif are from the loss of DNA repair function. Amino acids with a smaller side chain, such as glycine or serine, in the P-loop may play a critical structural role for DNA binding (35, 36). Besides the two conserved regions, we also identified certain charged or polar residues that are highly conserved (Asp-111, Glu-137, Asp-150, and Arg-261) and partially conserved (Glu-41, Asp-43, Glu-49, Gln-53, Arg-189, and Glu-194) among the 12 UppP sequences (Fig. 2). Because the crystal structures of the bacterial UppP or a similar protein of this family are currently unavailable, the structural information of UppP is not currently known. We therefore attempted to employ various computational programs to predict transmembrane helices of UppP, and the results are shown in Table 1. Certain important information about the protein topology based on these experiments, such as the localization of N and C termini of UppP as well as the amino acid composition of its active site, are also considered in this assignment (see “Discussion”). The results, containing a two-dimensional structure of UppP, two consensus regions, and certain not fully identical residues in the UppP family, are presented in Fig. 3. Surprisingly, most of the selected residues, including two consensus regions, are coincidentally localized near the aqueous interface of UppP and oriented toward the periplasmic site. Two exceptions are Glu-137 and Asp-150, located at the aqueous interface of UppP but facing the cytosol (Fig. 3). Because these consensus regions are specific to the UppP family, we speculate, considering evolutionary conservation and arrangement of active sites, that its catalytic site is composed of these consensus regions, and its function is likely in the periplasmic space. To explore their possible roles in enzyme catalysis, site-directed mutagenesis and structural modeling of these regions were undertaken and described below.
FIGURE 2.

Alignment of the amino acid sequences of the UppP enzymes. The E. coli UppP (UPPP_ECOLI; P60932) is aligned with the following: Bacillus subtilis (UPPP_BACSU; P94507); Streptococcus pneumonia (UPPP_STRPN; P60934); Staphylococcus aureus (UPPP_STAAU; Q9KIN5); Yersinia pestis (UPPP_YERPE; Q8ZI65); Pseudomonas putida (UPPP_PSEPK; Q88IY7); Neisseria gonorrhoeae (UPPP_NEIG1; Q5F6K4); Enterococcus faecalis (UPPP1_ENTFA; Q831R1); Marinobacter aquaeolei (UPPP_MARAV; A1U6U1); Vibrio cholerae (UPPP_VIBCH; Q9KUJ4); Geobacter sulfurreducens (UPPP_GEOSL; P60938); Sulfolobus solfataricus (UPPP_SULSO; Q97X94); and Thermotoga maritima (UPPP_THEMA; Q9WZZ5). The conserved (more than 50%) residues are colored in red. The upper cylinders show the helical regions in the model. The transmembrane (TM) helices are shown in light blue and extracellular helix (H1) is in purple. The two consensus regions I and II are underlined in orange. The residues mutated in this study are indicated by asterisks above the corresponding UppP amino acids.
TABLE 1.
The TM prediction of E.coli UppP from various programs
The nomenclature of transmembrane helices is based on the E. coli UppP model proposed in this study. TM1′ denotes the additional helix between TM1 and TM2.
| TMHMM | TOPCONS | TopPred 2 | Phobius | SVMtop | |
|---|---|---|---|---|---|
| Signal peptide | 1–18 | ||||
| TM1 | 3–23 | 2–21 | |||
| TM1′ | 13–35 | 20–40 | 24–44 | ||
| TM2 | 50–69 | 48–68 | 46–66 | 50–69 | 50–72 |
| TM3 | 90–109 | 90–110 | 90–110 | 90–110 | 91–113 |
| TM4 | 119–136 | 116–136 | 119–139 | 116–136 | 117–139 |
| TM5 | 156–176 | 155–175 | 157–175 | 153–175 | |
| TM6 | 187–209 | 194–214 | 192–212 | 195–213 | 191–213 |
| TM7 | 224–246 | 226–246 | 224–244 | 225–246 | 223–247 |
| TM8 | 253–272 | 252–272 | 253–273 | 252–272 | 250–272 |
| TM no. | 7 | 8 | 8 | 7 | 9 |
FIGURE 3.

Two-dimensional structure for UppP. Shown are the eight transmembrane helices in Upp. Circles show positions of conserved or not fully conserved residues mutated in this study. Residues Glu-17 and Glu-21 within (E/Q)XXXE motif as well as Ser-173, Arg-174, Ser-175, and Thr-178 within PGXSRSXXT motif are shown in orange and green, respectively. Residue His-30 is shown in red, and other putative catalytic residues are shown in gray.
Characterization and Enzymatic Activities of UppP
The recombinant wild-type E. coli UppP is purified and shown by SDS-PAGE under reducing conditions (Fig. 4). The major band, marked with an arrow of Fig. 4 on SDS-PAGE, was excised and subjected to mass spectrometry analysis. The result shows that the significant top one hit is UppP with a mascot score of 17,387.92 and a sequence coverage of 54.21% of the whole amino acid sequence of UppP. For testing the phosphatase activity of UppP, experiments are performed in the presence of various substrates, and the amount of released phosphate is measured by using Malachite Green assay. Assignments of determining phosphatase activity have been proposed previously (3, 7). It has been reported that another E. coli undecaprenyl pyrophosphate phosphatase, PgpB, catalyzes the dephosphorylation of Upp with a relatively low efficiency compared with diacylglycerol pyrophosphate and Fpp, probably due to the arrangement difficulties of the long chain isoprenoids in the mixed detergent micelles (9). Therefore, in our test we use Fpp as a model substrate.
FIGURE 4.
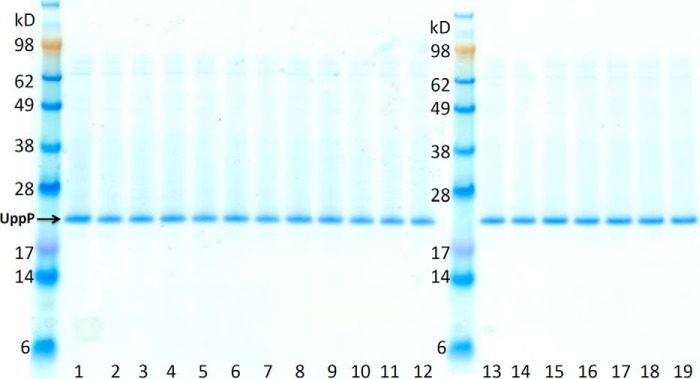
Purified wild-type UppP and mutants. The lanes from left to right are as follows: wild-type UppP (lanes 1 and 19); E17A (lane 2); E21A (lane 3); E17A-E21A (lane 4); H30A (lane 5); E41A (lane 6); D43A (lane 7); E49A (lane 8); Q53A (lane 9); D111A (lane 10); D150A (lane 11); S173A (lane 12); R174A (lane 13); S175A (lane 14); T178A (lane 15); R189A (lane 16); E194A (lane 17), and R261A (lane 18).
The purified UppP proteins show clear activity of dephosphorylation of Fpp. To determine the optimal reaction conditions of UppP, the enzyme at various pH values and detergent concentrations is investigated. As shown in Fig. 5A, the optimal pH value for the phosphatase activity has been found between pH 6.5 and 7.0, and the activities are decreased gradually at pH 5.0 and 8–9, respectively. The phosphatase activity of UppP at pH 7.0 is also determined at various concentrations of DDM (0.02–1%). Although the maximum activity has been found at 0.02% DDM (Fig. 5B), the enzyme presents high activity over 80% between 0.05 and 1% DDM, with an average of 85%. This finding is consistent with the previous conclusion that the PgpB phosphatase activity was not affected by the detergent (9). Subsequent assays of UppP are therefore performed at the optimal pH of 7.0 with 0.02% DDM.
FIGURE 5.

Enzymatic activity analysis. A, effect of pH on UppP activity was assayed at various pH values, pH 5.0–6.0 (sodium acetate), pH 6.5–8.0 (Hepes), and pH 9 (Tris-HCl). B, effect of the detergent concentration on the phosphatase activity. The concentration of DDM was utilized in the range of 0.02 to 1%. C, effect of the EDTA on the UppP phosphatase activity. D, metal ion requirements. The experiment was carried out by incubating 20 nm enzyme and various divalent cations (Mg2+, Mn2+, Zn2+, Co2+, and Ca2+) at a concentration of 10 mm. Control indicates sample without addition of extra metal ions or EDTA.
The polypeptide antibiotic bacitracin has been shown to inhibit the bacterial cell wall synthesis through the sequestration of the pyrophosphate moiety of C55-PP (37, 38). Therefore, the inhibition of E. coli UppP by bacitracin has also been determined (Fig. 6B). The IC50 value of bacitracin in the presence of 0.02 μm enzyme and 35 μm Fpp is ∼33 μm, which corresponds to the 7.8 μm Ki value calculated from Equations 1 and 2 (details are provided under “Experimental Procedures”). The data presented here show that bacitracin can efficiently inhibit the dephosphorylation of lipid pyrophosphate in UppP enzyme.
FIGURE 6.

Kinetic parameters of wild-type and mutations E17A and E21A and the inhibition of bacitracin for wild-type UppP. For the measurements of kinetic parameters of enzymes, 0.3–57 μm Fpp were used along with the 20–40 nm UppP. For the measurement of the inhibition constant for bacitracin, 20 nm UppP and 35 μm Fpp were used in the presence of various concentrations of the bacitracin from 0 to 100 μm. Details are provided under “Experimental Procedures.”
Metal Ion Requirements
The metal ion often plays an essential role in metal-requiring enzymes. In prenyltransferases, for example, the common mechanism may be that a magnesium ion coordinates with the pyrophosphate moiety of Fpp substrate and facilitates the nucleophilic attack by making the pyrophosphate a better leaving group. Therefore, we tested the phosphatase activity in the presence or absence of various divalent cations (Mg2+, Mn2+, Zn2+, Co2+, and Ca2+) after incubation of the enzyme with the metal ion chelator EDTA at 25 °C for 10 min. Fig. 5C shows that the enzyme is completely inhibited in the presence of 5 μm EDTA, whereas the activity is recovered and stimulated by addition of Mg2+, Ca2+, Co2+, and Mn2+ exhibiting100, 125, 39, and 18% relative activities respectively, as compared with that in the presence of Mg2+ ions. On the contrary, Zn2+ failed to activate UppP enzyme, retaining only 2% activity. The enzyme also retains about 50% activity in the absence of divalent metal ions, suggesting that the purified enzyme has retained some bound metal ions (Fig. 5D). This result demonstrates an absolute requirement of Mg2+ or Ca2+ ion for catalytic activity in UppP.
Site-directed Mutagenesis of E. coli UppP
To determine the structure-function relationships of this enzyme, we performed a series of site-directed mutations to examine their possible roles in substrate binding and catalysis. All the conserved residues within the UppP enzyme were mutated to the nonpolar residue alanine to eliminate their hydrogen bonding capability. In region I, mutations were targeted to convert either the first or second glutamate residue (within the (E/Q)XXXE motif) and the conserved residue histidine to alanine (E17A, E21A, and H30A). In region II, four conserved residues, Ser-173, Arg-174, Ser-175, and Thr-178 in the structural P-loop motif and other two highly conserved residues Arg-189 and Arg-261, which could have interactions with the substrate, were also replaced with alanine. The UppP mutants are purified and are shown by SDS-PAGE in Fig. 4, and the mutagenesis results are shown in Table 2. The H30A mutant completely lost its enzyme function, whereas E17A and E21A mutants within the (E/Q)XXXE motif still retain some enzyme activity (26 and 40% of the wild type). We also measured the kinetic parameters of wild-type UppP and E17A and E21A mutants (Fig. 6). The Fpp Km and kcat values of the wild type are 10.8 μm and 2.1 s−1, respectively. In contrast, E17A and E21A within the (E/Q)XXXE motif result in a decrease of kcat values of ∼5-fold compared with the wild type. E17A also shows a Km value ∼4–5-fold higher than that of the wild type (Fig. 6, A, C and D). These data suggest that both glutamates are involved in catalytic function, and Glu-17 is also important for substrate binding. Because this motif might be functionally similar to the DDXXD motif, in which two Asp residues coordinate with the pyrophosphate moiety of substrate via a magnesium ion, a double mutation in this motif was prepared (E17A/E21A). As predicted, mutation E17A/E21A has complete loss of function (<1%), presumably by abolishing substrate binding to the active site. This result is similar to the previous study in farnesyl diphosphate synthase from Bacillus stearothermophilus, which concluded that these aspartate residues within the DDXXD motif are essential in catalysis and substrate binding (29). Mutations within the putative structural P-loop (PGXSRSXXT motif) and R189A and R261A show similar results. Except for S175A and R189A retaining 32 and 11% activity, all mutations have significantly reduced catalytic activity (<1%), suggesting that these residues are important in enzyme function (Table 2).
TABLE 2.
Site-directed mutations generated in E. coli UppP enzyme
ND is not determined. E137A is expressed at levels too low to obtain reliable phosphatase activity data. The P- and C-sites indicate periplasmic and cytoplasmic sites, respectively. TM indicates transmembrane helix.
| UppP mutants | Predicted localization | Relative activities of the wild type | Suggested role |
|---|---|---|---|
| % | |||
| Region I | |||
| E17A | Active site (P-site) | 26 | H-bond with pyrophosphate of UppP via the Mg2+ |
| E21A | Active site (P-site) | 40 | H-bond with pyrophosphate of UppP via the Mg2+ |
| E17A/E21A | Active site (P-site) | <1 | H-bond with pyrophosphate of UppP via the Mg2+ |
| H30A | Active site (P-site) | <1 | Initiating a nucleophilic attack on the phosphorus center |
| Region II (structural P-loop) | |||
| S173A | Active site (P-site) | <1 | The backbone oxygen H-bond with Arg-174 |
| R174A | Active site (P-site) | <1 | H-bond with oxygen atom of α-phosphate of UppP |
| S175A | Active site (P-site) | 32 | Interaction with phosphate group of UppP |
| T178A | Active site (P-site) | <1 | H-bond with the backbone NH of Arg-174 |
| Other mutants | |||
| E41A | Aqueous interface (P-site) | 85 | Loop structural maintenance |
| D43A | Aqueous interface (P-site) | 73 | Loop structural maintenance |
| E49A | Membrane-water interface (P-site) | 36 | TM2 structural maintenance |
| Q53A | Membrane-water interface (P-site) | 14 | TM2 structural maintenance |
| D111A | Membrane-water interface (P-site) | 100 | |
| E137A | At the end of TM4 (C-site) | ND | Salt bridge with Arg-67 and His-94 |
| D150A | Membrane-water interface (C-site) | 64 | |
| R189A | Aqueous interface (P-site) | 11 | Stabilizing His-30 |
| E194A | Aqueous interface (P-site) | 31 | Stabilizing Arg-189 |
| R261A | At the end of TM8 (P-site) | <1 | H-bond with Ser-173 |
In the search for other residues that may play a role in catalysis, we examined eight more charged or polar residues, including highly conserved residues Asp-111, Glu-137. and Asp-150, and partially conserved residues Glu-41, Asp-43, Glu-49, Gln-53, and Glu-194 as we described above. However, most of the mutations (E41A, D43A, E49A, Q53A, D111A, D150A, and E194A) have only a little or no influence on catalytic activity (Table 2). The E137A mutant is expressed at a very low level, implying some instability or folding problems. Therefore, its activity was not determined. These data are in contrast to mutations within regions I and II, which render the enzyme completely inactive. Taken together with these mutagenesis results, we suggest that these polar residues (Glu-137 not included) are not essential for the catalysis and may not be involved in the active site. In other words, residues essential for enzyme activity within the (E/Q)XXXE and PGXSRSXXT motifs as well as His-30 are considered to be the catalytic core of the enzyme.
Structural Model of E. coli UppP
The structural model for E. coli UppP, as proposed here, is constructed by the Rosetta membrane ab initio modeling program. To be able to design a more accurate model of UppP, the transmembrane helical regions of UppP are estimated by comparison of the secondary structure and membrane protein topology predictions as described above, and all eight predicted transmembrane helical regions are required to span the membrane during modeling. Finally, 18 candidates (data not shown) were filtered, and the best model was chosen and optimized (details are provided under “Experimental Procedures”). The final model includes a full-length sequence of 273 amino acids of UppP, composed of eight predicted transmembrane helices (TM1–8), including a short TM5 with 11 amino acids, and a magnesium ion as well as Upp substrate (Fig. 7A).
FIGURE 7.

Overall structure of UppP in complex with Upp and Mg2+. A, transmembrane helices, labeled from 1 to 8, are shown as cylinders and colored in rainbow colors. The extracellular helix is purple and labeled as H1. B, electric surface potential of enzyme-substrate complex model. The Upp substrate is shown using sticks and its pyrophosphate moiety as well as the 55-carbon chain are colored in orange and green, respectively. The Mg2+ ion is presented in red. Boundaries of the lipid bilayer are shown as dotted planes (30.2 Å), calculated by OPM server.
Fig. 7 shows the structural model of UppP in complex with Upp and a magnesium ion. In this model, the substrate-binding pocket is mainly constituted of TM1, TM2, TM4, and TM5, although TM3, TM6, and TM8 are folded on the opposite site to this pocket. The TM7 is located at the outermost layer and lies on the surface of TM3 and TM6 (Fig. 7A). The pyrophosphate moiety of the Upp substrate sits in an active-site pocket surrounded by negatively (Glu-17 and Glu-21) and positively (Arg-174) charged residues, whereas part of its 55-carbon chain (approximately between C16 and C40) lies on a hydrophobic surface mainly composed of Val-51, Ile-52, Gly-55, Ala-59, Val-60, Met-63, and Phe-64 amino acids in the TM2 helix within the membrane region of the enzyme (Figs. 7 and Fig. 8A). The remaining hydrocarbon tail (approximately between C1 and C15) is highly flexible and oriented toward the lipid bilayers. Our structural model of enzyme-substrate complex is similar to the crystal structure of membrane-embedded phospho-MurNAc-pentapeptide translocase (MraY), suggesting an inverted U-shaped hydrophobic groove surrounding TM9b helix (transmembrane 9b) within the membrane region of the protein surface along with a Mg2+ ion-chelated aspartate-rich active site for the substrate binding of the 55-carbon chain undecaprenyl phosphate (39).
FIGURE 8.

Putative active site of E. coli UppP. A, residues surrounding the Upp substrate are represented. The nonpolar residues Val-51, Leu-54, Gly-55, Leu-58, Ala-59, Val-60, Met-63, Phe-64, Trp-65, Leu-68, Leu-168, Trp-169, and Pro-170 are in gray. The polar residues Thr-47, Gln-164, Ser-173, and Ser-175 are in yellow. The positively charged residues His-30 and Arg-174 are in blue. The negatively charged residues Glu-17 and Glu-21 are in purple. B, interactions between Upp pyrophosphate moiety and the nearby amino acids in the catalytic side are shown. The pyrophosphate moiety might be stabilized by Glu-17 and Glu-21 through a Mg2+ ion, and Arg-174 might interact with the α-phosphate of Upp directly. His-30 might catalyze through a water molecule in the reaction of hydrolysis.
The structural details of the conserved (E/Q)XXXE motif, in which two carboxylate groups of Glu-17 and Glu-21 interact with the pyrophosphate moiety of Upp through a magnesium ion and the guanidinium group of Arg-174 in the putative structural P-loop establishes a hydrogen-bonding interaction with the OH group of the α-phosphate of Upp, are shown in Fig. 8B. His-30 is in close proximity to the phosphorus center of Upp in the model and may participate in the reaction of hydrolysis directly (Fig. 8B). Residues Ser-173, Ser-175, and Thr-178 probably participate in stabilizing the positioning of the pyrophosphate moiety on its site (Fig. 9, A and B). Arg-261 is positioned behind the structural P-loop and may stabilize it through the hydrogen bond with Ser-173 (Fig. 9A). Arg-189 may stabilize His-30 by interacting with its main chain, whereas Glu-194 may stabilize the Arg-189-containing loop (Fig. 9B). Our data presented here are consistent with the mutagenesis analysis in which these UppP mutants cause a significant reduction of phosphatase activity (Table 2).
FIGURE 9.

Roles of Glu-41, Asp-43, Glu-49, Gln-53, Glu-137, Ser-173, Ser-175, Thr-178, Arg-189, Glu-194, and Arg-261 in E. coli UppP. A, Ser-173 and Ser-175 may have direct or indirect interactions with Upp substrate. Arg-261 may stabilize the structural P-loop through the Ser-173. B, Thr-178 may have interaction with Arg-174. Arg-189 may stabilize His-30, and Glu-194 may stabilize the Arg-189-containing loop. C, Glu-41 and Asp-43 may stabilize the loop, whereas Glu-49 and Gln-53 may stabilize the TM2. D, Glu-137, at the cytoplasmic end of TM4, may stabilize the TM2 and TM3 through the salt bridges with Arg-67 and His-94, respectively.
The flexible loop composed of amino acids from 41 to 53 comprises part of the active site pocket in the model (Fig. 9C). Mutations E41A and D43A could influence the stability of this loop, whereas E49A and Q53A could change the hydrophobicity of TM2 (Table 2). Glu-137 is located at the cytoplasmic end of TM4 and may stabilize the TM2 and TM3 through the salt bridges with the Arg-67 and His-94, respectively (Fig. 9D). The E137A mutant could eliminate the interaction capabilities among TM2, TM3, and TM4, which render protein unstable during synthesis (Table 2). Asp-111 is located at the periplasmic end of TM4 (at the membrane-water interface) where it is far away from the putative active site, and Asp-150 is located at the aqueous interface oriented toward the cytosolic side. The D111A and D150A mutants still retain high activity (64 and 100%, respectively). The ab initio predicted model provides a structural explanation for inactive mutations covered in two consensus regions as well as some not fully conserved residues.
Molecular Dynamics Simulation of E. coli UppP
The backbone r.m.s.d. of native E. coli UppP shows a steady increase from 2.5 to 3 Å in 30 ns. For the enzyme-substrate complex, the region of the long chain Upp between C16 and C40 is modulated by the hydrophobic surface located in TM2 helix, and its remaining 15-carbon chain is oriented toward the lipid bilayers. The backbone r.m.s.d. slightly increases at the beginning of 7 ns (3.0 to 3.1 Å) and then remains stable over 15 ns (2.8 to 3.1 Å) (data not shown). The transmembrane core of native and complex structures of E. coli UppP in the lipid bilayers shows only small structural change during the simulation analysis (Fig. 10). The distribution of r.m.s.f. indicates that the fluctuation of transmembrane helical regions is 1.9 and 2.1 Å in native UppP and enzyme-substrate complex, respectively, whereas the extracellular loops show a higher flexibility (3.4 and 3.8 Å, respectively) (Fig. 10). Two loops composed of amino acids 31–43 (4.3 Å) and 72–85 (5.8 Å) are flexible in the native UppP but remain stable when substrate is bound. The other three loops 25–30 (6.2 Å), 43–50 (4.6 Å), and 181–186 (5.9 Å) show higher flexibility only in the substrate-bound enzyme complex, whereas the loop 140–150 shows flexibility in both cases (5.2 and 7.1 Å, respectively) (Fig. 10).
FIGURE 10.

Molecular dynamics simulations for E. coli UppP. r.m.s.f. of individual residues of the native E. coli UppP (A) and the enzyme-substrate complex (B) in 30 ns of MD simulation. C, native UppP and the enzyme-substrate complex are shown in red and blue, respectively.
Plausible Catalytic Mechanism
The plausible reaction mechanism of UppP deduced from the present structural model and mutagenesis studies is outlined in Fig. 11. The catalytic event is likely initiated when the conserved residue His-30 acts as a nucleophilic attack on the phosphorus center to form a phosphohistidine intermediate. Then a water ion (or OH− ion) makes a second nucleophilic attack on the phosphate of the phosphohistidine intermediate. The importance of His-30 in catalysis has been shown by a significant decrease in enzyme activity of the H30A mutant (Table 2). Glu-17 and Glu-21 residues within the (E/Q)XXXE motif may also be involved in the catalysis and substrate binding by way of a chelated magnesium ion.
FIGURE 11.

Two possible reaction mechanisms of the dephosphorylation of undecaprenyl pyrophosphate in UppP. A, based on the present model, the Upp dephosphorylation reaction is likely to begin with histidine (His-30) initiating a nucleophilic attacking on the phosphorus center to form a phosphohistidine intermediate. The negative charges of the phosphate groups are chelated by a magnesium ion to facilitate the transferring of the phosphate group to the histidine. Then a water (or OH− ion) makes a second nucleophilic attack on the phosphate of the phosphohistidine intermediate. B, histidine initiates a nucleophilic attack on a water molecule, which acts as a second nucleophilic attack on the phosphate of the Upp substrate.
DISCUSSION
In this study, the wild type and mutated UppP proteins are overexpressed in E. coli, extracted from the membrane, and purified using a bacteriorhodopsin tag fused at the N terminus of UppP. The enzyme dephosphorylating activity is also examined. We identified that a divalent cation, such as magnesium or calcium ions, is essential for enzyme activity. This is also observed by the analysis of eliminating the metal ions from the reaction mixture by adding EDTA, resulting in a completely inactive enzyme (Fig. 5, C and D). In fact, the requirement of metal ions is commonly observed in pyrophosphate moiety substrate-binding protein, such as pyrophosphatases and prenyltransferases. The enzymatic mechanism has been determined by x-ray structural and site-directed mutagenesis analysis. It is believed that the metal ion(s) chelated by acidic residues stabilizes the pyrophosphate moiety of substrate, making the inorganic phosphate or pyrophosphate group a better leaving group. We previously solved the E. coli UppS (undecaprenyl pyrophosphate synthase) crystal structure in complex with farnesyl thiopyrophosphate (an analog of Fpp) and Ipp substrates, and we demonstrated that Mg2+ ion is essential for the enzyme catalysis by facilitating the pyrophosphate elimination (40). Pojer et al. (41) demonstrated that CloQ, a soluble aromatic prenyltransferase involved in clorobiocin biosynthesis, from Streptomyces roseochromogenes requires Mg2+ or Ca2+ ions for its enzymatic activity. Plus, recent reports on the crystal structure of membrane-embedded sodium (or H+)-translocating pyrophosphatase showed that metal ions (Mg2+ or Ca2+) mediate the interactions between pyrophosphate substrate and aspartate residues in the active site for enzymatic hydrolysis (42, 43). The role of the metal ion in these enzymes is apparently required not only for substrate binding but also for catalysis. In E. coli UppP, the metal ion may play a similar role in the catalytic mechanism.
In de novo synthesis, after C55-PP is produced by a soluble UppS, this highly hydrophobic carrier lipid precursor may enable spontaneous diffusion in the membrane before its negatively charged pyrophosphate moiety docks to the active site for hydrolysis. Certain conserved acidic residues, such as Asp and Glu, and basic residues, such as Lys, Arg, and His, surrounding the active site essential for enzyme catalysis and substrate binding can be commonly observed in many phosphate-binding proteins. For example, we previously solved the crystal structure of the trans-type octaprenyl pyrophosphate synthase from Thermotoga maritima and demonstrated that sulfate ions (resembling the pyrophosphate moiety of the Fpp or Ipp substrate) are directly attached to the two aspartate-rich DDXXD motifs via magnesium ions, and the basic residues (arginine, lysine, and histidine) may play an important role for substrate binding (30). In bacterial UppP enzymes, sequence alignment shows two consensus regions, containing a glutamate-rich (E/Q)XXXE motif and a putative structural P-loop PGXSRXXXT motif as well as a conserved histidine residue, that are unique to bacterial UppP family (Fig. 2). Presumably, its active site is composed of these two regions. This hypothesis is supported by the site-directed mutagenesis experiments in which mutations (E17A, E21A, E17A/E21A, H30A, S173A, R174A, S175A, and T178A) within these two regions are essential for enzyme catalysis and might also participate in the substrate binding (Table 2 and Fig. 6). The proposed two-dimensional topology and the structural model of the substrate-bound enzyme complex in this work also provide a plausible suggestion for a structure of the active site of UppP (Figs. 3 and 7–9). However, we do note that a topological model of UppP produced using the TMHMM program predicts only seven transmembrane helices that cause the consensus region I, including (E/Q)XXXE motif and His-30, buried in the plasma membrane, whereas the PGXSRSXXT motif is oriented toward the inner membrane within a large cytosolic loop. Such an arrangement of active sites seems unlikely to work functionally if indeed both regions are identified to be essential in the catalysis and substrate binding. The predicted topology produced using the SVMtop or TopPred 2 program also has a similar problem with the arrangement of the enzyme active site (Table 1). Although the structural information of protein in complex with the 55-carbon long chain Upp molecule is currently not known, we did previously solve the crystal structure of E. coli UppS in complex with sulfate ions and two Triton X-100 molecules, which may mimic the pyrophosphate moiety of the Upp product docking in a hydrophilic active site along with the long chain hydrocarbon moiety of the Upp occupying the entire hydrophobic tunnel in UppS (44). It is therefore reasonable to assume that the (E/Q)XXXE plus PGXSRSXXT motifs and His-30 are involved in the active site of UppP, and these charged or polar residues may directly or indirectly interact with the substrate for catalysis and binding.
The role of the conserved residue histidine in the enzymatic mechanism is intriguing. This amino acid is in close proximity to the pyrophosphate moiety in the model, and its mutation H30A causes severely impaired enzyme activity (<1%), suggesting its importance in catalysis (Table 2). In the proposed catalytic mechanism, it seems plausible that His-30 initiates a direct or indirect nucleophilic attack at an electron-deficient phosphorus center of pyrophosphate moiety of Upp substrate during the hydrolysis (Fig. 11). In our structural model, however, His-30 is located in a flexible loop composed of amino acids 23–50, which would be considered to be less accurate than the rest of the model. Therefore, several charged or polar residues located near the putative active site have also been examined (E41A, D43A, E49A, Q53A, D111A, and E194A) to clarify the role of histidine in enzyme catalysis. However, all mutations have enzyme activity (20–100%), indicating that these nearby residues are postulated to be functionally less important. These data suggest that the histidine residue is involved directly in the catalytic mechanism. In fact, using a histidine as a nucleophile in the catalytic mechanism is commonly found in phosphatase family. For example, the histidine phosphatase superfamily, containing dPGM, PhoE, SixA, fructose-2,6-bisphosphatase, and TIGAR (TP53 (tumor protein 53)-induced glycolysis and apoptosis regulator), used a histidine to initiate a nucleophilic attack and was phosphorylated during the reaction (45–49). Another example is the phosphatidic acid phosphatase type 2 superfamily. As mentioned above, the membrane-embedded undecaprenyl pyrophosphate phosphatases (PgpB, YbjG, and LpxT) belong to this family, characterized by three conserved motifs. This family also contains PhoN protein, vanadium-dependent chloroperoxidases, glucose-6-phosphatase, and lipid phosphate phosphatases (50–53). The reaction mechanism has been well defined in the PhoN protein by x-ray structural and mutagenesis studies (50). It is generally believed that the histidine initiates a nucleophilic attack at the phosphorus center to form a phosphohistidine intermediate, and the second histidine acting as a general acid/base facilitates the release of the phosphate. Interestingly, UppP has no sequence homology to the PgpB, YbjG, and LpxT but does share the activity of C55-PP dephosphorylation. It is highly possible that UppP utilizes a similar catalytic mechanism in E. coli and probably in other bacterial species.
The MD simulation provides a desirable reliability of the proposed E. coli UppP model. The minor variation of the transmembrane core indicates that the predicted structural model might remain stable over time in the lipid bilayers (Fig. 10). Our data suggest that the TM2 helix might play an essential role for the arrangement of the long chain isoprenoid of the substrate in E. coli UppP. This can be observed by the evaluation of an alternative enzyme-substrate complex model, in which the hydrocarbon tail of Upp occupies a hydrophobic groove (composed of TM2 and TM4) instead of lying on the hydrophobic surface of TM2 helix, and its remaining carbon chain (approximately between C1 and C15) is oriented toward the cytosolic compartment rather than in membrane bilayers. The average r.m.s.d., however, increases to 3.8 Å (2.8–3.1 Å in the current substrate-bound enzyme complex model), implicating that the substrate binding is unstable. Our data also suggest that the flexible loops composed of amino acids 25–30 and 43–50 may play an essential role in mediating the substrate binding, whereas loop 181–186 might act as a second lid to protect the hydrophilic environment from the catalytic site. However, more evidence is needed to test this hypothesis.
The structural model of UppP is suggests that its putative active site faces the outer side of the plasma membrane (Figs. 3 and 7). Our model is generated on the basis of four criteria as follows: 1) N terminus of UppP is located in the cytoplasm; 2) C terminus of UppP is located in the cytoplasm; 3) the active site must comprise consensus regions I and II; and 4) the MD simulation analysis. The recombinant UppP is purified using a bacteriorhodopsin tag fused at the N terminus of UppP (16). Bacteriorhodopsin belongs to the seven-transmembrane receptor family, normally its N terminus is at the periplasm and C terminus at the cytoplasm (54). Therefore in our system, the N terminus of UppP should be localized in the cytoplasm to fold with correct topology. UppP with C terminally fused green fluorescent protein (GFP) also exhibits fluorescence activity,4 suggesting that its C terminus is at the cytoplasmic space, too. These results are consistent with the predicted model of UppP with eight transmembrane helices in which both the conserved regions I and II are localized near the aqueous interface oriented toward the periplasm. This conclusion is also validated by the MD simulation analysis in which the backbone r.m.s.d. of native UppP or the enzyme-substrate complex is constantly stable over time in the lipid bilayer membranes (Fig. 10). These data implicate that the enzymatic function of UppP is in the periplasmic space. To date, however, it is still unclear on which side the UppP function localized. Whether the C55-PP dephosphorylation occurs on one side only or on both sides of the plasma membrane remains unknown. Several important studies previously suggested that PgpB together with YbjG and LpxT participate in the recycling of C55-PP with its active site oriented toward the periplasm, but no direct evidence has been shown that UppP is involved in de novo synthesis at the cytoplasm. Our data are suggestive, although not compelling, that UppP mediates the reaction of C55-PP dephosphorylation at the outer side of the membrane instead of in the cytosolic compartment. However, further studies in biochemical and structural analysis are needed to test this hypothesis.
The key remaining questions regarding the functional metabolism of the bacterial cell wall synthesis are as follows. 1) Which side of the plasma membrane C55-PP does dephosphorylation occurs? Is it only at the periplasm or cytoplasm or on both sites? 2) What is the mechanism by which the carrier lipid C55-P (or C55-PP) can cross membranes after the transfer of the glycan component to the peptidoglycan chain? Our UppP model suggests that its biological function is in the periplasmic space, and so are the other three bacterial phosphatases (PgpB, YbjG, and LpxT). However, this raises the question of how the dephosphorylation of de novo synthesized C55-PP occurs in the cytosolic compartment. A recent study on the crystal structure of MraY proposed a Mg2+ ion-chelated aspartate-rich active site, suggesting for the binding of the phosphate group of C55-P, localized in the cytoplasmic site, which clearly shows that the transfer of the phospho-MurNAc-pentapeptide to the carrier lipid C55-P occurs in the cytoplasm (39). Whether an alternative membrane machinery is responsible for the translocation of the carrier lipid C55-PP or C55-P across the cell membrane is not clear. However, an interesting study recently demonstrated that the FtsW (flippase), an essential division protein in bacteria, mediates the translocation of lipid II molecules to the periplasm for peptidoglycan assembly by using a fluorescent-labeled lipid II in the FtsW-reconstituted proteoliposomes (6), but for the transport of the long chain of the carrier lipid it remains to be determined.
In summary, this study is perhaps first report on structure-function relationships of UppP in E. coli. In this study, we successfully purified wild type and a series of mutated UppP proteins that are investigating the impact of mutations within two consensus regions on phosphatase activity, and we identified that the (E/Q)XXXE and PGXSRSXXT motifs as well as His-30 residue are essential in enzyme catalysis and substrate binding, suggesting that its active site may comprise these conserved regions and is located in the periplasmic site. The predicted structural model and molecular dynamics simulation also provide an explanation and a plausible suggestion for a structure of the catalytic site in UppP as well as the enzyme-substrate interaction in the plasma membrane. Further experiments, including structural analysis, combined with biochemical studies will be required to identify the exact role of UppP in the bacterial cell wall synthesis.
Acknowledgments
We thank the National Research Program for Biopharmaceuticals (supported by Grant NSC 10102325-B-492-001) and the National Center for High Performance Computing, National Applied Research Laboratories of Taiwan, for providing computing resources.
This work was supported by Academia Sinica and National Science Council Grant NSC101-2319-B-001-003 (to A. H. J. W.) for Core Facilities for Protein Structural Analysis at Academia Sinica, Taiwan.
H.-Y. Chang, C.-C. Chou, M.-F. Hsu, and A. H. J. Wang, unpublished observations.
- C55-PP
- undecaprenyl pyrophosphate
- UppP
- undecaprenyl pyrophosphate phosphatase
- C55-P
- undecaprenyl phosphate
- UppS
- undecaprenyl pyrophosphate synthase
- Fpp
- farnesyl pyrophosphate
- Ipp
- isopentenyl pyrophosphate
- r.m.s.d.
- root mean square deviation
- r.m.s.f.
- root mean square fluctuation
- MD
- molecular dynamics.
REFERENCES
- 1. Apfel C. M., Takács B., Fountoulakis M., Stieger M., Keck W. (1999) Use of genomics to identify bacterial undecaprenyl pyrophosphate synthetase: cloning, expression, and characterization of the essential uppS gene. J. Bacteriol. 181, 483–492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Shimizu N., Koyama T., Ogura K. (1998) Molecular cloning, expression, and purification of undecaprenyl diphosphate synthase. No sequence similarity between E- and Z-prenyl diphosphate synthases. J. Biol. Chem. 273, 19476–19481 [DOI] [PubMed] [Google Scholar]
- 3. El Ghachi M., Bouhss A., Blanot D., Mengin-Lecreulx D. (2004) The bacA gene of Escherichia coli encodes an undecaprenyl pyrophosphate phosphatase activity. J. Biol. Chem. 279, 30106–30113 [DOI] [PubMed] [Google Scholar]
- 4. Tatar L. D., Marolda C. L., Polischuk A. N., van Leeuwen D., Valvano M. A. (2007) An Escherichia coli undecaprenyl-pyrophosphate phosphatase implicated in undecaprenyl phosphate recycling. Microbiology 153, 2518–2529 [DOI] [PubMed] [Google Scholar]
- 5. Barreteau H., Magnet S., El Ghachi M., Touzé T., Arthur M., Mengin-Lecreulx D., Blanot D. (2009) Quantitative high-performance liquid chromatography analysis of the pool levels of undecaprenyl phosphate and its derivatives in bacterial membranes. J. Chromatogr. B. Analyt. Technol. Biomed. Life Sci. 877, 213–220 [DOI] [PubMed] [Google Scholar]
- 6. Mohammadi T., van Dam V., Sijbrandi R., Vernet T., Zapun A., Bouhss A., Diepeveen-de Bruin M., Nguyen-Distèche M., de Kruijff B., Breukink E. (2011) Identification of FtsW as a transporter of lipid-linked cell wall precursors across the membrane. EMBO J. 30, 1425–1432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. El Ghachi M., Derbise A., Bouhss A., Mengin-Lecreulx D. (2005) Identification of multiple genes encoding membrane proteins with undecaprenyl pyrophosphate phosphatase (UppP) activity in Escherichia coli. J. Biol. Chem. 280, 18689–18695 [DOI] [PubMed] [Google Scholar]
- 8. Cain B. D., Norton P. J., Eubanks W., Nick H. S., Allen C. M. (1993) Amplification of the bacA gene confers bacitracin resistance to Escherichia coli. J. Bacteriol. 175, 3784–3789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Touzé T., Blanot D., Mengin-Lecreulx D. (2008) Substrate specificity and membrane topology of Escherichia coli PgpB, an undecaprenyl pyrophosphate phosphatase. J. Biol. Chem. 283, 16573–16583 [DOI] [PubMed] [Google Scholar]
- 10. Bickford J. S., Nick H. S. (2013) Conservation of the PTEN catalytic motif in the bacterial undecaprenyl pyrophosphate phosphatase, BacA/UppP. Microbiology 159, 2444–2455 [DOI] [PubMed] [Google Scholar]
- 11. Stukey J., Carman G. M. (1997) Identification of a novel phosphatase sequence motif. Protein Sci. 6, 469–472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Rush J. S., Cho S. K., Jiang S., Hofmann S. L., Waechter C. J. (2002) Identification and characterization of a cDNA encoding a dolichyl pyrophosphate phosphatase located in the endoplasmic reticulum of mammalian cells. J. Biol. Chem. 277, 45226–45234 [DOI] [PubMed] [Google Scholar]
- 13. Fernandez F., Rush J. S., Toke D. A., Han G. S., Quinn J. E., Carman G. M., Choi J. Y., Voelker D. R., Aebi M., Waechter C. J. (2001) The CWH8 gene encodes a dolichyl pyrophosphate phosphatase with a luminally oriented active site in the endoplasmic reticulum of Saccharomyces cerevisiae. J. Biol. Chem. 276, 41455–41464 [DOI] [PubMed] [Google Scholar]
- 14. van Berkel M. A., Rieger M., te Heesen S., Ram A. F., van den Ende H., Aebi M., Klis F. M. (1999) The Saccharomyces cerevisiae CWH8 gene is required for full levels of dolichol-linked oligosaccharides in the endoplasmic reticulum and for efficient N-glycosylation. Glycobiology 9, 243–253 [DOI] [PubMed] [Google Scholar]
- 15. Ishikawa K., Mihara Y., Gondoh K., Suzuki E., Asano Y. (2000) X-ray structures of a novel acid phosphatase from Escherichia blattae and its complex with the transition-state analog molybdate. EMBO J. 19, 2412–2423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hsu M. F., Yu T. F., Chou C. C., Fu H. Y., Yang C. S., Wang A. H. (2013) Using Haloarcula marismortui bacteriorhodopsin as a fusion tag for enhancing and visible expression of integral membrane proteins in Escherichia coli. PLoS One 8, e56363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Barth P., Schonbrun J., Baker D. (2007) Toward high-resolution prediction and design of transmembrane helical protein structures. Proc. Natl. Acad. Sci. U.S.A. 104, 15682–15687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Segel I. H. (1993) in Enzyme Kinetics: Behavior and Analysis of Rapid Equilibrium and Steady-state Enzyme Systems (Seqel I. H., ed) pp. 100–118, John Wiley & Sons, Inc., New York [Google Scholar]
- 19. Laskovics F. M., Poulter C. D. (1981) Prenyltransferase; determination of the binding mechanism and individual kinetic constants for farnesylpyrophosphate synthetase by rapid quench and isotope partitioning experiments. Biochemistry 20, 1893–1901 [DOI] [PubMed] [Google Scholar]
- 20. Kelley L. A., Sternberg M. J. (2009) Protein structure prediction on the Web: a case study using the Phyre server. Nat. Protoc. 4, 363–371 [DOI] [PubMed] [Google Scholar]
- 21. Bernsel A., Viklund H., Hennerdal A., Elofsson A. (2009) TOPCONS: consensus prediction of membrane protein topology. Nucleic Acids Res. 37, W465–W468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. McRee D. E. (1999) XtalView/Xfit-A versatile program for manipulating atomic coordinates and electron density. J. Struct. Biol. 125, 156–165 [DOI] [PubMed] [Google Scholar]
- 23. Winn M. D., Ballard C. C., Cowtan K. D., Dodson E. J., Emsley P., Evans P. R., Keegan R. M., Krissinel E. B., Leslie A. G., McCoy A., McNicholas S. J., Murshudov G. N., Pannu N. S., Potterton E. A., Powell H. R., Read R. J., Vagin A., Wilson K. S. (2011) Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 67, 235–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Phillips J. C., Braun R., Wang W., Gumbart J., Tajkhorshid E., Villa E., Chipot C., Skeel R. D., Kalé L., Schulten K. (2005) Scalable molecular dynamics with NAMD. J. Comput. Chem. 26, 1781–1802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Jo S., Lim J. B., Klauda J. B., Im W. (2009) CHARMM-GUI membrane builder for mixed bilayers and its application to yeast membranes. Biophys. J. 97, 50–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Humphrey W., Dalke A., Schulten K. (1996) VMD: Visual molecular dynamics. J. Mol. Graph. 14, 33–38 [DOI] [PubMed] [Google Scholar]
- 27. Joly A., Edwards P. A. (1993) Effect of site-directed mutagenesis of conserved aspartate and arginine residues upon farnesyl diphosphate synthase activity. J. Biol. Chem. 268, 26983–26989 [PubMed] [Google Scholar]
- 28. Song L., Poulter C. D. (1994) Yeast farnesyl-diphosphate synthase: site-directed mutagenesis of residues in highly conserved prenyltransferase domains I and II. Proc. Natl. Acad. Sci. U.S.A. 91, 3044–3048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Koyama T., Tajima M., Sano H., Doi T., Koike-Takeshita A., Obata S., Nishino T., Ogura K. (1996) Identification of significant residues in the substrate binding site of Bacillus stearothermophilus farnesyl diphosphate synthase. Biochemistry 35, 9533–9538 [DOI] [PubMed] [Google Scholar]
- 30. Guo R. T., Kuo C. J., Chou C. C., Ko T. P., Shr H. L., Liang P. H., Wang A. H. (2004) Crystal structure of octaprenyl pyrophosphate synthase from hyperthermophilic Thermotoga maritima and mechanism of product chain length determination. J. Biol. Chem. 279, 4903–4912 [DOI] [PubMed] [Google Scholar]
- 31. Kinoshita K., Sadanami K., Kidera A., Go N. (1999) Structural motif of phosphate-binding site common to various protein superfamilies: all-against-all structural comparison of protein-mononucleotide complexes. Protein Eng. 12, 11–14 [DOI] [PubMed] [Google Scholar]
- 32. Chang S. Y., Ko T. P., Chen A. P., Wang A. H., Liang P. H. (2004) Substrate binding mode and reaction mechanism of undecaprenyl pyrophosphate synthase deduced from crystallographic studies. Protein. Sci. 13, 971–978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Fujihashi M., Zhang Y. W., Higuchi Y., Li X. Y., Koyama T., Miki K. (2001) Crystal structure of cis-prenyl chain elongating enzyme, undecaprenyl diphosphate synthase. Proc. Natl. Acad. Sci. U.S.A. 98, 4337–4342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Logan K. M., Knight K. L. (1993) Mutagenesis of the P-loop motif in the ATP binding site of the RecA protein from Escherichia coli. J. Mol. Biol. 232, 1048–1059 [DOI] [PubMed] [Google Scholar]
- 35. Pai E. F., Krengel U., Petsko G. A., Goody R. S., Kabsch W., Wittinghofer A. (1990) Refined crystal structure of the triphosphate conformation of H-ras p21 at 1.35 A resolution: implications for the mechanism of GTP hydrolysis. EMBO J. 9, 2351–2359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Konola J. T., Logan K. M., Knight K. L. (1994) Functional characterization of residues in the P-loop motif of the RecA protein ATP binding site. J. Mol. Biol. 237, 20–34 [DOI] [PubMed] [Google Scholar]
- 37. Stone K. J., Strominger J. L. (1971) Mechanism of action of bacitracin: complexation with metal ion and C 55-isoprenyl pyrophosphate. Proc. Natl. Acad. Sci. U.S.A. 68, 3223–3227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Economou N. J., Cocklin S., Loll P. J. (2013) High-resolution crystal structure reveals molecular details of target recognition by bacitracin. Proc. Natl. Acad. Sci. U.S.A. 110, 14207–14212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Chung B. C., Zhao J., Gillespie R. A., Kwon D. Y., Guan Z., Hong J., Zhou P., Lee S. Y. (2013) Crystal structure of MraY, an essential membrane enzyme for bacterial cell wall synthesis. Science 341, 1012–1016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Guo R. T., Ko T. P., Chen A. P., Kuo C. J., Wang A. H., Liang P. H. (2005) Crystal structures of undecaprenyl pyrophosphate synthase in complex with magnesium, isopentenyl pyrophosphate, and farnesyl thiopyrophosphate. J. Biol. Chem. 280, 20762–20774 [DOI] [PubMed] [Google Scholar]
- 41. Pojer F., Wemakor E., Kammerer B., Chen H., Walsh C. T., Li S. M., Heide L. (2003) CloQ, a prenyltransferase involved in clorobiocin biosynthesis. Proc. Natl. Acad. Sci. U.S.A. 100, 2316–2321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kellosalo J., Kajander T., Kogan K., Pokharel K., Goldman A. (2012) The structure and catalytic cycle of a sodium-pumping pyrophosphatase. Science 337, 473–476 [DOI] [PubMed] [Google Scholar]
- 43. Lin S. M., Tsai J. Y., Hsiao C. D., Huang Y. T., Chiu C. L., Liu M. H., Tung J. Y., Liu T. H., Pan R. L., Sun Y. J. (2012) Crystal structure of a membrane-embedded H+-translocating pyrophosphatase. Nature 484, 399–403 [DOI] [PubMed] [Google Scholar]
- 44. Chang S. Y., Ko T. P., Liang P. H., Wang A. H. (2003) Catalytic mechanism revealed by the crystal structure of undecaprenyl pyrophosphate synthase in complex with sulfate, magnesium, and Triton. J. Biol. Chem. 278, 29298–29307 [DOI] [PubMed] [Google Scholar]
- 45. Fothergill-Gilmore L. A., Watson H. C. (1989) The phosphoglycerate mutases. Adv. Enzymol. Relat. Areas Mol. Biol. 62, 227–313 [DOI] [PubMed] [Google Scholar]
- 46. Rigden D. J., Bagyan I., Lamani E., Setlow P., Jedrzejas M. J. (2001) A cofactor-dependent phosphoglycerate mutase homolog from Bacillus stearothermophilus is actually a broad specificity phosphatase. Protein Sci. 10, 1835–1846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ogino T., Matsubara M., Kato N., Nakamura Y., Mizuno T. (1998) An Escherichia coli protein that exhibits phosphohistidine phosphatase activity towards the HPt domain of the ArcB sensor involved in the multistep His-Asp phosphorelay. Mol. Microbiol. 27, 573–585 [DOI] [PubMed] [Google Scholar]
- 48. Pilkis S. J., Lively M. O., el-Maghrabi M. R. (1987) Active site sequence of hepatic fructose-2,6-bisphosphatase: homology in primary structure with phosphoglycerate mutase. J. Biol. Chem. 262, 12672–12675 [PubMed] [Google Scholar]
- 49. Bensaad K., Tsuruta A., Selak M. A., Vidal M. N., Nakano K., Bartrons R., Gottlieb E., Vousden K. H. (2006) TIGAR, a p53-inducible regulator of glycolysis and apoptosis. Cell 126, 107–120 [DOI] [PubMed] [Google Scholar]
- 50. Makde R. D., Mahajan S. K., Kumar V. (2007) Structure and mutational analysis of the PhoN protein of Salmonella typhimurium provide insight into mechanistic details. Biochemistry 46, 2079–2090 [DOI] [PubMed] [Google Scholar]
- 51. Renirie R., Hemrika W., Wever R. (2000) Peroxidase and phosphatase activity of active-site mutants of vanadium chloroperoxidase from the fungus CurVularia inaequalis. Implications for the catalytic mechanisms. J. Biol. Chem. 275, 11650–11657 [DOI] [PubMed] [Google Scholar]
- 52. Ghosh A., Shieh J. J., Pan C. J., Sun M. S., Chou J. Y. (2002) The catalytic center of glucose-6-phosphatase. HIS176 is the nucleophile forming the phosphohistidine-enzyme intermediate during catalysis. J. Biol. Chem. 277, 32837–32842 [DOI] [PubMed] [Google Scholar]
- 53. Zhang Q. X., Pilquil C. S., Dewald J., Berthiaume L. G., Brindley D. N. (2000) Identification of structurally important domains of lipid phosphate phosphatase-1: implications for its sites of action. Biochem. J. 345, 181–184 [PMC free article] [PubMed] [Google Scholar]
- 54. Pebay-Peyroula E., Rummel G., Rosenbusch J. P., Landau E. M. (1997) X-ray structure of bacteriorhodopsin at 2.5 angstroms from microcrystals grown in lipidic cubic phases. Science 277, 1676–1681 [DOI] [PubMed] [Google Scholar]