

Background: Lytic polysaccharide monooxygenases (LPMOs) exhibit a copper center that binds dioxygen for catalysis.
Results: We present LPMO structures from Cu(II) to Cu(I) and analyze the transition with quantum mechanical calculations.
Conclusion: Reduction changes the copper coordination state but requires only minor structural and electronic changes.
Significance: These structures provide insight into LPMO catalytic activation for further mechanistic studies.
Keywords: Carbohydrate-binding Protein, Cellulase, Metalloenzyme, Polysaccharide, X-ray Crystallography
Abstract
Lytic polysaccharide monooxygenases (LPMOs) are a recently discovered class of enzymes that employ a copper-mediated, oxidative mechanism to cleave glycosidic bonds. The LPMO catalytic mechanism likely requires that molecular oxygen first binds to Cu(I), but the oxidation state in many reported LPMO structures is ambiguous, and the changes in the LPMO active site required to accommodate both oxidation states of copper have not been fully elucidated. Here, a diffraction data collection strategy minimizing the deposited x-ray dose was used to solve the crystal structure of a chitin-specific LPMO from Enterococcus faecalis (EfaCBM33A) in the Cu(II)-bound form. Subsequently, the crystalline protein was photoreduced in the x-ray beam, which revealed structural changes associated with the conversion from the initial Cu(II)-oxidized form with two coordinated water molecules, which adopts a trigonal bipyramidal geometry, to a reduced Cu(I) form in a T-shaped geometry with no coordinated water molecules. A comprehensive survey of Cu(II) and Cu(I) structures in the Cambridge Structural Database unambiguously shows that the geometries observed in the least and most reduced structures reflect binding of Cu(II) and Cu(I), respectively. Quantum mechanical calculations of the oxidized and reduced active sites reveal little change in the electronic structure of the active site measured by the active site partial charges. Together with a previous theoretical investigation of a fungal LPMO, this suggests significant functional plasticity in LPMO active sites. Overall, this study provides molecular snapshots along the reduction process to activate the LPMO catalytic machinery and provides a general method for solving LPMO structures in both copper oxidation states.
Introduction
Glycoside hydrolases (GHs)6 are responsible for significant turnover of recalcitrant polysaccharides such as cellulose, hemicellulose, and chitin in nature and are thus of major importance in the global carbon and nitrogen cycles. GHs are extremely diverse enzymes and have undergone extensive characterization and classification, often driven by their potential utilization in the growing biofuels industry (1–5). More recently, a new class of enzymes was discovered, classified as lytic polysaccharide monooxygenases (LPMOs), which cleave glycosidic linkages in polysaccharides via a copper-mediated, oxidative mechanism (6–12). LPMOs represent a new enzyme mechanism for the decomposition of recalcitrant polysaccharides and act synergistically with traditional hydrolytic enzymes (6, 13–15). Unlike GHs, LPMO action generally does not involve a decrystallization step to detach single polysaccharide chains from their insoluble and often crystalline substrates, a process that requires a substantial amount of thermodynamic work (16–18). Instead, most LPMOs characterized to date are thought to act directly on surfaces of crystalline polysaccharides (6, 19). In this manner, LPMOs are able to synergize with hydrolytic enzymes because they are hypothesized to make chain breaks in crystalline regions that are typically thought to be inaccessible for endoglucanases. Conversely, an LPMO able to cleave soluble substrates was recently discovered, indicating an increasing diversity in substrate specificities of these enzymes (20).
LPMOs were previously classified as family 33 carbohydrate-binding modules (CBM33s), which range in origin from bacteria to algae, and family 61 glycoside hydrolases (GH61s), which are of fungal origin. CBM33s mined from genomic data often exhibit modular complexity, whereas GH61s are typically either single module enzymes or are bimodular with a catalytic domain and a family 1 CBM (14), similar to many fungal carbohydrate-active enzymes. Recently, Henrissat and co-workers (5) updated the Carbohydrate-Active Enzyme database and classified CBM33s as Auxiliary Activity 10 (AA10) and GH61s as AA9. Another LPMO family was also recently classified as AA11, which exhibits sequence, structural, and electronic characteristics between that of AA9 and AA10 (12).
The chitin-active LPMO from the Gram-negative chitinolytic bacterium Serratia marcescens, CBP21, was the first LPMO to be biochemically characterized (6, 21, 22). CBP21 catalysis was shown to be dependent on molecular oxygen, an external electron donor, and the presence of a metal ion cofactor (6), later identified as copper (19). Copper ions have been identified to activate AA10 (19, 23), AA9 (7, 9, 10), and AA11 LPMOs (12). In addition to CBP21, LPMO activity has only been demonstrated for two other CBM33s so far, a cellulose-active CBM33 from Streptomyces coelicolor (CelS2 (8)) and a chitin-active CBM33 from Enterococcus faecalis (EfaCBM33A (24)), the latter of which is the subject of this study. EfaCBM33A is the only LPMO found in the genome of E. faecalis and constitutes, along with a GH family 18 chitinase (EfaChi18A), the chitinolytic machinery of the bacterium. E. faecalis is an opportunistic pathogen, and both EfaCBM33A and EfaChi18A are virulence factors (25, 26), suggesting a putative second role for these enzymes beyond biomass depolymerization.
The LPMO active site is constituted by two histidine residues (one of which is the N-terminal residue) that coordinate a copper ion in a motif referred to as the “histidine brace” (6, 7, 10, 27). The copper ion is essential for catalytic activity and is likely to be involved in the activation of molecular oxygen (7, 9, 24). Soluble products resulting from lytic oxidation have been identified as aldonic acids (6, 8–10, 28) or as oligomers with an oxidized nonreducing end sugar, i.e. a 4-keto sugar (20, 29), indicating differences in enzyme regioselectivity. The oxidation products were recently definitively confirmed by NMR spectroscopy (20), and some progress has recently been made toward understanding regioselectivity (30). To date, aldonic acids are the only products observed for AA10 and AA11 LPMOs, whereas both aldonic acids and 4-keto sugars have been observed for AA9 enzymes.
The catalytic mechanism of AA9 LPMOs, which to date only are found in fungi, was recently examined with density functional theory (DFT) calculations (11). Kim et al. (11) predicted that AA9 LPMOs employ a Cu(II)-oxyl reactive oxygen species for hydrogen abstraction from the substrate, followed by an oxygen-rebound mechanism for substrate hydroxylation. This step will be followed by an elimination reaction, resulting in glycosidic bond cleavage. To activate the LPMO catalytic cycle, the initial dioxygen binding was hypothesized to require reduction of Cu(II) oxidation state of the enzyme to a Cu(I) state, likely mediated by an enzymatic or small molecule reducing agent.
Until recently, there has been a dearth of structural data for metal binding in AA10 LPMOs compared with fungal AA9 LPMOs. Recently, Hemsworth et al. (27) reported the structure of Bacillus amyloliquefaciens CBM33 (BamCBM33), with unknown catalytic activity, binding Cu(I). It was shown that BamCBM33 is stabilized in the presence of copper and that the active site of BamCBM33 with a Cu(I) ion adopts a T-shaped geometry (PDB codes 2YOX and 2YOY). A Cu(II) form of BamCBM33 was not crystallized, but x-ray absorption near edge structure (XANES) and EPR spectroscopic methods were used to demonstrate that the enzyme was readily photoreduced during crystallization from the Cu(II) form to a Cu(I) state (27).
In this study, we investigate the active site of EfaCBM33 by progressively photoreducing the catalytic copper from Cu(II) to Cu(I) in the x-ray beam using a data collection minimizing the x-ray dose that is deposited in the sample. During photoreduction, we determine successive structural states by collecting x-ray diffraction data sets on the same crystal. By comparing the structures to known Cu(I) and Cu(II) analogues found in the Cambridge Structural Database (CSD) (31), we ascertain that the obtained structures of the EfaCBM33A unambiguously describe varying oxidation states ranging from Cu(II) to Cu(I). Lastly, we conduct quantum mechanical calculations on an active site model of the Cu(II) and Cu(I) forms of EfaCBM33, which suggest that the electronic structure of the active site remains quite similar as measured by atomic charges. Because the initial reduction of Cu(II) is likely a requirement for LPMO activity, these results offer a structural and electronic picture of how LPMO active sites are preactivated for oxygen binding and subsequent catalysis.
EXPERIMENTAL PROCEDURES
Protein Preparation and Crystallization of EfaCBM33A
EfaCBM33A was expressed and purified as previously described (24). The protein was incubated with 1 mm CuSO4 for 0.5 h. After soaking, the protein solution was desalted using a 10 DG column (Bio-Rad) and concentrated to 25 mg/ml prior to crystallization. EfaCBM33A crystals were obtained in 20% (w/v) PEG 8000 and 0.1 m HEPES, pH 7.5, by the sitting drop vapor diffusion method as previously described (24). Rod-shaped crystals grew to an approximate size of 400 × 53 × 40 μm after 2 days of incubation at 20 °C. The crystal used for data collection was soaked in the crystallization solution with the addition of 20% PEG 400 as a cryoprotectant for ∼10 s prior to being plunged into liquid nitrogen.
Diffraction Data Collection and Structure Solution
X-ray diffraction experiments were performed at Beamline ID14-EH1 at the European Synchrotron Radiation Facility, Grenoble, France. Six diffraction data sets were collected using the same single crystal. By utilizing a rod-shaped crystal, monitoring the evolution of UV-visible absorption spectra with x-ray dose (32), and a strategy for helical data collection (33), the radiation dose was minimized, and data of a minimally photoreduced state of EfaCBM33 could be collected (34). A helical data collection consists of defining two points on the crystal along the rotation axis of the goniometer. Although the crystal is rotated over a total 97° angular wedge by 1° steps, it is automatically translated along the rotation axis in between two consecutive rotation steps, thus presenting a fresh part of the crystal to the beam for each diffraction frame. Eventually, the x-ray dose deposited in the sample will approximately be d/w smaller than that deposited with a standard data collection protocol, where d is the horizontal distance between the two points, and w is the horizontal width of the beam. Two points on a limited region (320 × 53 × 40 μm) of the crystal were set up as start and end points for data collection with a 50 × 100-μm x-ray beam. A different exposure dose per image was used for some data sets, as shown in Table 1. Collecting subsequent data sets by this method allowed for the analysis of the effects of photoreduction on the active site copper with minimal systematic errors, because all data sets were collected from multiple and subsequent exposures of the same crystal volume.
TABLE 1.
X-ray data collection and processing, structure refinement, and final model statistics
| PDB code | 4ALC | 4ALE | 4ALR | 4ALQ | 4ALS | 4ALT |
|---|---|---|---|---|---|---|
| Data quality | ||||||
| Space group | P212121 | P212121 | P212121 | P212121 | P212121 | P212121 |
| Cell dimensions (Å) | ||||||
| a | 43.42 | 43.42 | 43.42 | 43.46 | 43.44 | 43.40 |
| b | 48.56 | 48.57 | 48.58 | 48.62 | 48.61 | 48.57 |
| c | 68.45 | 68.46 | 68.46 | 68.51 | 68.50 | 68.43 |
| Data collections statistics | ||||||
| Wavelength (Å) | 0.9334 | 0.9334 | 0.9334 | 0.9334 | 0.9334 | 0.9334 |
| Resolution range (Å) | 48.56–1.49 | 48.57–1.48 | 39.68–1.49 | 48.68–1.48 | 48.61–1.47 | 39.60–1.49 |
| No. of unique reflections | 23,919 (3317) | 24,395 (3216) | 23,936 (3324) | 24,358 (3235) | 24,138 (3119) | 23,899 (3307) |
| Multiplicity | 3.4 (3.0) | 3.4 (2.9) | 3.4 (3.0) | 3.3 (2.9) | 2.9 (2.3) | 3.4 (3.1) |
| Completeness | 98.6 (96.1) | 97.8 (90.5) | 98.6 (96.1) | 97.8 (91.2) | 96.6 (87.9) | 98.6 (95.9) |
| Rmergea | 0.094 (0.477)b | 0.096 (0.503) | 0.094 (0.484) | 0.067 (0.237) | 0.056 (0.142) | 0.100 (0.564) |
| Exposure time/frame (s) | 1 | 1 | 1 | 3 | 6 | 1 |
| Total exposure time (s) | 97 | 194 | 485 | 776 | 1358 | 1455 |
| Dose (grays) | 8.27 × 104 | 1.65 × 105 | 2.47 × 105 | 4.94 × 105 | 9.86 × 105 | 1.07 × 106 |
| Refinement | ||||||
| Resolution range (Å) | 39.60–1.49 | 39.61–1.48 | 39.62–1.49 | 39.65–1.48 | 39.64–1.47 | 39.60–1.49 |
| Observations | 80,452 (10080) | 81,915 (9417) | 80,300 (10,018) | 81,296 (9301) | 69,202 (7187) | 80,875 (10,226) |
| No. of reflections used (working set) | 22,667 | 23,130 | 22,679 | 23,087 | 22,872 | 22,647 |
| Rwork (%) | 16.1 | 16.1 | 16.1 | 15.6 | 15.6 | 16.1 |
| Rfree (%) | 18.8 | 18.4 | 18.8 | 18.3 | 19.6 | 18.8 |
| No. of residues | 167 | 167 | 167 | 167 | 167 | 167 |
| No. of water molecules | 289 | 286 | 282 | 281 | 292 | 280 |
| No. of copper atoms | 1 | 1 | 1 | 1 | 1 | 1 |
| RMSD bond lengths (Å) | 0.008 | 0.008 | 0.008 | 0.008 | 0.008 | 0.008 |
| RMSD angles (°) | 1.230 | 1.229 | 1.230 | 1.210 | 1.221 | 1.242 |
| Ramachandranc | ||||||
| In favored regions (%) | 100 | 100 | 100 | 100 | 100 | 100 |
| Outliers (%) | 0 | 0 | 0 | 0 | 0 | 0 |
a Rmerge = ΣhklΣi|I-i(hkl) − <I(hkl)>|/ΣhklΣi Ii(hkl).
b Values for the highest resolution shell are given in parentheses.
c Calculated using a strict boundary Ramachandran definition given by Kleywegt and Jones (41).
All data sets were indexed and integrated using the program XDS (35) and scaled with the CCP4 program suite version 6.2.0 (36). The structures of EfaCBM33A were solved by molecular replacement using CBP21 (22) (PDB code 2BEM) as the starting model. Model building and maximum-likelihood refinement were performed with iterative cycles of model building in COOT version 0.6.2 (37), by inspection of 2mFo-DFc and mFo-DFc σA-weighted maps, and model refinement in Refmac5 version 5.6.0117 (38). The bound copper ion was modeled in at a final stage of the refinement. PyMOL 1.5 (39) was used for analysis of the structures and figure preparations. LSQMAN (40) was used for structural alignments, and Ramachandran statistics were determined using MOLEMAN2 (41). Omit maps were calculated using Phenix 1.8.1 (42). Dose calculations for the exposed crystal regions were performed using RADDOSE version 2 (43, 44).
Crystallographic Database Search
A search of the Cambridge Structural Database was performed using ConQuest 1.14. Detailed search parameters are described below in the text.
Quantum Mechanical Calculations
Quantum mechanical calculations based on DFT were performed using Gaussian09 (45) on an active site model (ASM) of EfaCBM33, which includes His29, Glu64, Ala112, His114, Trp176, Ile178, Phe185, and the copper ion. The crystallographic water molecules that coordinate the copper ion were also included where appropriate. Smaller and larger ASMs were considered (data not shown), and this model was found to provide the optimal balance between reproducing the crystal structure and being computationally tractable. Additionally, as reported under “Results,” the RMSDs of the resulting models of both the Cu(II) and Cu(I) states were below 0.4 Å, a value that is well within the range considered to represent sufficient accuracy in cluster models of enzyme active sites (46). All geometry optimizations were conducted with the local meta-GGA M06-L functional (47, 48) and the 6–31G(d) basis set for all atoms. M06-L was chosen because of improved accuracy for the dispersive and mixed binding complexes. The local density function is computationally efficient for optimization of large structures, employing thousands of basis functions. All α- and β-carbons were fixed during optimizations. All systems were treated with the conductor-like polarizable continuum model (49, 50) using diethyl ether solvation (ϵ = 4 for the protein environment) (51). We computed the harmonic vibrational frequencies for all optimized structures to confirm that they are minima, possessing zero imaginary frequencies. Atomic charges were calculated using natural population analysis from NBO 6.0.
RESULTS
Overall Structure of EfaCBM33A in Complex with Copper
The EfaCBM33A with a bound copper atom was crystallized in space group P212121 with cell dimensions of 43.4 × 48.6 × 68.5 Å, one protein molecule per asymmetric unit, and a Vm (Matthews coefficient) (53) of 1.97 Å3/Da including all Cα atoms in the structure. We present six structures of EfaCBM33A along the process of x-ray induced photoreduction, all refined at 1.5 Å and final R and Rfree values of 15.6–16.1% and 18.3–19.6%, respectively. The data collection and refinement statistics are summarized in Table 1. In all the structure models, there is clear electron density for all the 169 amino acid residues, ∼285 water molecules, and 1 copper atom bound to the protein. Negligible pairwise RMSD values of 0.03–0.04 Å over all protein atoms show that the structures are essentially identical. The primary differences are found in the coordination geometry of the copper ion as a function of the x-ray dose. With the exception of the active site, the structure of the copper-bound EfaCBM33A herein is very similar to the previously published apo form without copper (PDB code 4A02 (24); 0.54 Å RMSD on Cα atoms).
The overall structure and the active site of EfaCBM33A, as observed in the structure determined from the data set obtained after the lowest radiation dose (PDB code 4ALC) exhibits a trigonal bipyramidal (tbp) structure coordinated by two conserved histidine residues (the histidine brace) and two water molecules (Fig. 1, A–C). In this configuration, the N-terminal histidine (His29) forms a bidentate coordination to the copper ion wherein the backbone N atom occupies one of the three equatorial coordination positions, and the side chain Nδ atom occupies one axial position. The other axial position is occupied by the Nϵ atom in the His114 residue. The remaining equatorial positions are occupied by two water molecules. Three additional residues conserved in AA10 LPMOs are shown in Fig. 1C, Glu64, Ala112, and Phe185. Ala112 is not conserved in AA9 LPMOs and is thought to play a role in the potential mechanistic differences between AA9 and AA10 LPMOs (27, 54). Phe185 is located in a similar position to a conserved tyrosine in AA9 LPMOs, which in the latter case therein imparts an octahedral coordination state around the Cu(II) ion (7, 13, 28, 55, 56). In AA9 LPMOs, the Glu64 residue is replaced by a conserved glutamine residue. The 4ALC data set shows two spherical electron densities (1.93 and 1.91 e/Å3, respectively) at 2.21 and 2.19 Å from the copper ion, which were modeled and refined as water molecules and are shown as red spheres in Fig. 1C. For the water molecules bound to copper, there are no other stabilizing interactions with the enzyme. Thus, the positions of the water molecules are primarily dictated by coordination to the copper ion.
FIGURE 1.

Overall structure of EfaCBM33. A and B, side (A) and bottom (B) views of the crystal structure of Cu(II)-bound EfaCBM33A with a cartoon and transparent surface model in gray (PDB code 4ALC; the structure with the lowest radiation dose). The active center is highlighted in yellow, and the two residues making up the histidine brace are shown in stick format. The copper atom is shown as a brown sphere, and two water molecules coordinated to the copper are shown as red spheres. C, the active site in the oxidized (Cu(II)) form of EfaCBM33 (PDB code 4ALC). Note that there are not other stabilizing interactions between the two coordinating water molecules and the enzyme. D, the active site in BamCBM33 binding Cu(I) (PDB code 2YOX) (27). Distances to the copper ion are provided in Å.
The only other known AA10 structure with a copper ion bound reported to date is BamCBM33 from Hemsworth et al. (27), wherein all copper ions were photoreduced to a Cu(I) oxidation state. The BamCBM33 enzyme active site is illustrated in Fig. 1D. The coordination geometry therein is in a T-shaped (Tsh) geometry with no water molecules coordinated to the copper ion. The corresponding protein-copper interactions retain the structure of the histidine brace. The difference in observed geometry between the 4ALC structure and the BamCBM33 structure indicates a difference in copper oxidation state, as described in detail further below.
Structural Changes Induced by X-ray Photoreduction
The structural changes caused by the increase in x-ray dosage during photoreduction were limited to the local environment of the copper ion (Fig. 2). Omit map analysis of the copper-coordinated water molecules shows a continuous decay of electron density correlated with x-ray exposure, and at ∼1 megagray accumulated radiation (4ALT), both water molecules are completely lost. The electron density for the water molecule closest to Ala112 is retained slightly longer than the other. The decay of the electron density for the two water molecules coordinated to the copper represents a change in the fraction of Cu(II) to Cu(I) populations between the six structures, as a result of the accumulated radiation dose of the exposed crystal region used for data collection. The structures obtained at higher doses of x-ray radiation reveal a continuous shift in the copper coordination configuration from tbp coordination to Tsh geometry in the structures that lack the copper-bound water molecules (Fig. 2).
FIGURE 2.
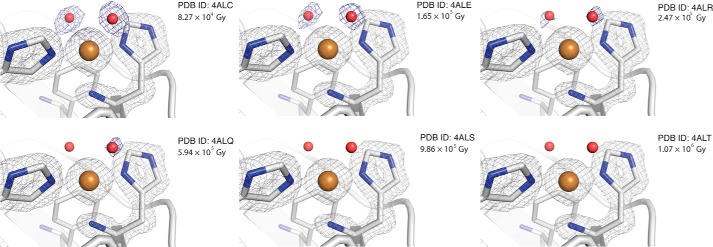
Close up view of the catalytic centers of EfaCBM33A at different levels of x-ray exposure. The blue omit-calculated 2mFo − Fc maps (52) are contoured at 0.87 e/Å3, the gray 2Fo − Fc maps are contoured at 0.89 e/Å3. The red oxygen-atom coordinates are taken from the 4ALC structure, are placed throughout the series for reference, and were not used in the calculation of the 2mFo − Fc omit maps. The structural images are labeled by the PDB codes and the dose of x-ray exposure.
LPMO Copper Oxidation State Determination by Analogy to Small Molecule Copper Complexes
To monitor the reduction of the copper ion bound to EfaCBM33A, UV-visible microspectrophotometry was used to record spectral changes of the crystal during x-ray exposure. However, the high background noise and low copper ion concentration in the sample prevented successful application of this method. Thus, to ascertain whether the x-ray-induced changes in the conformation of the copper site are indicative of an actual reduction of the copper ion from Cu(II) to Cu(I), the CSD was searched for relevant copper structures (31). The copper coordination in the retrieved structures was then compared with the initial and final EfaCBM33A structures.
The most obvious structural change upon x-ray exposure is that the two water molecules coordinating to the copper ion gradually disappear, as shown in Fig. 2. This demonstrates that the coordination number for the copper ion drops from five to three; the conformation of the copper site changes from a five-coordinated tbp structure to a three-coordinated Tsh geometry (Fig. 3). Gradual disappearance of electron density upon increasing the x-ray dosage was not observed for any other water molecule in the structure, suggesting that the effects seen for the copper-bound waters relates to a change in the copper ion. Among more than 40,000 copper structures in the CSD (31), nearly half fit our initial search criteria (1 ≤ coordination number ≤ 8; only nitrogen and/or oxygen as coordinating atoms), including 9,727 five-coordinate and 564 three-coordinate structures (data not shown). Limiting the search to only include those with histidine-like coordination surroundings and excluding strained structures left 10 five-coordinate structures, all Cu(II) (Table 2), and 24 three-coordinate structures, all Cu(I) (Table 3). Details regarding their Cu–N/O bonds, bond angles, torsion angles, and the resulting overall geometry of the copper site are shown in Tables 2 and 3, respectively.
FIGURE 3.
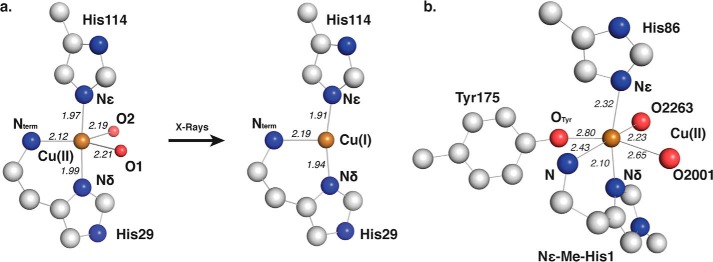
Copper coordination in EfaCBM33A and TauGH61A. Important residues, atoms, and coordination distances to the copper ion in Å. are indicated where appropriate. a, the copper binding site of EfaCBM33A (PDB code 4ALC) displays a trigonal bipyramidal (tbp) coordination of copper and after x-ray exposure adopts a T-shaped (Tsh) configuration (PDB code 4ALT). b, an octahedral Cu(II) coordination in the GH61A from T. aurantiacus (PDB code 2YET). In most AA10 LPMOs, including EfaCBM33A, the tyrosine residue labeled Tyr175 is replaced by phenylalanine.
TABLE 2.
Copper structures with similar coordination as EfCBM33A subjected to the lowest radiation dosage (4ALC)
Structures found in the CSD criteria described in the text listed by their code, oxidation state, bond lengths [Cu–N, unless noted], bond angle distribution, and configuration. All structures include a five-coordinated copper(II) ion, including NIDHOY (which is mislabelled in the CSD). One structure shows a copper configuration that is very similar to the configuration in 4ALC: ZUBHOT (tbp configuration with two equatorial oxygen atoms with the longest bond distances to the oxygen from the copper ion), whereas CISHIW also has the same general layout (highlighted in peach). When comparing individual bond distances, it should be noted that both ZUBHOT and CISHIW coordinate to one anionic species each, ClO4− and NCS−, respectively. sqpy, square pyramidal; tbp, trigonal bipyramidal; dist., distorted. The 4ALC structural details are included for reference at the end of the table (highlighted in turquoise).
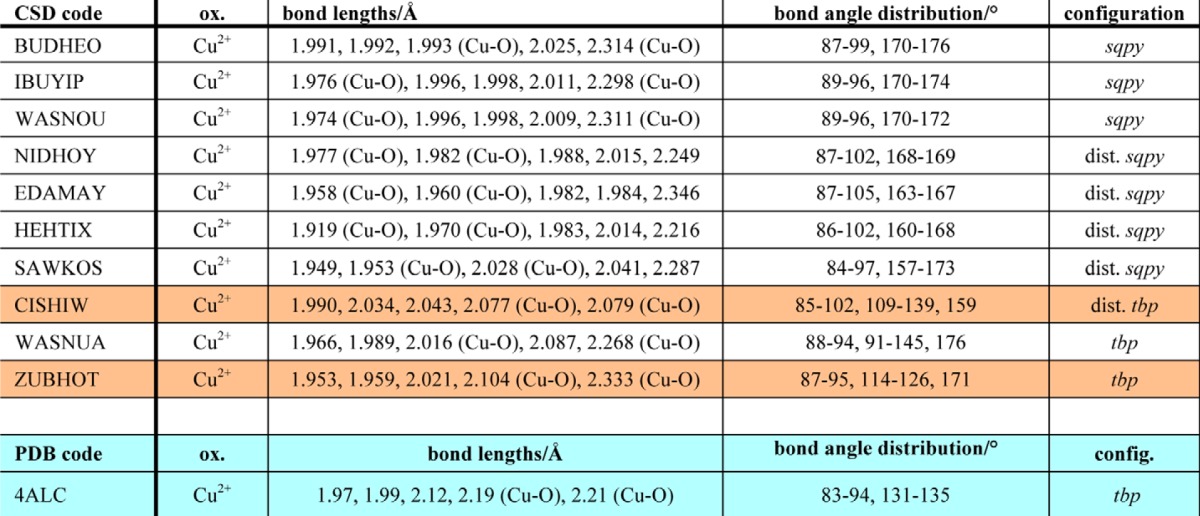
TABLE 3.
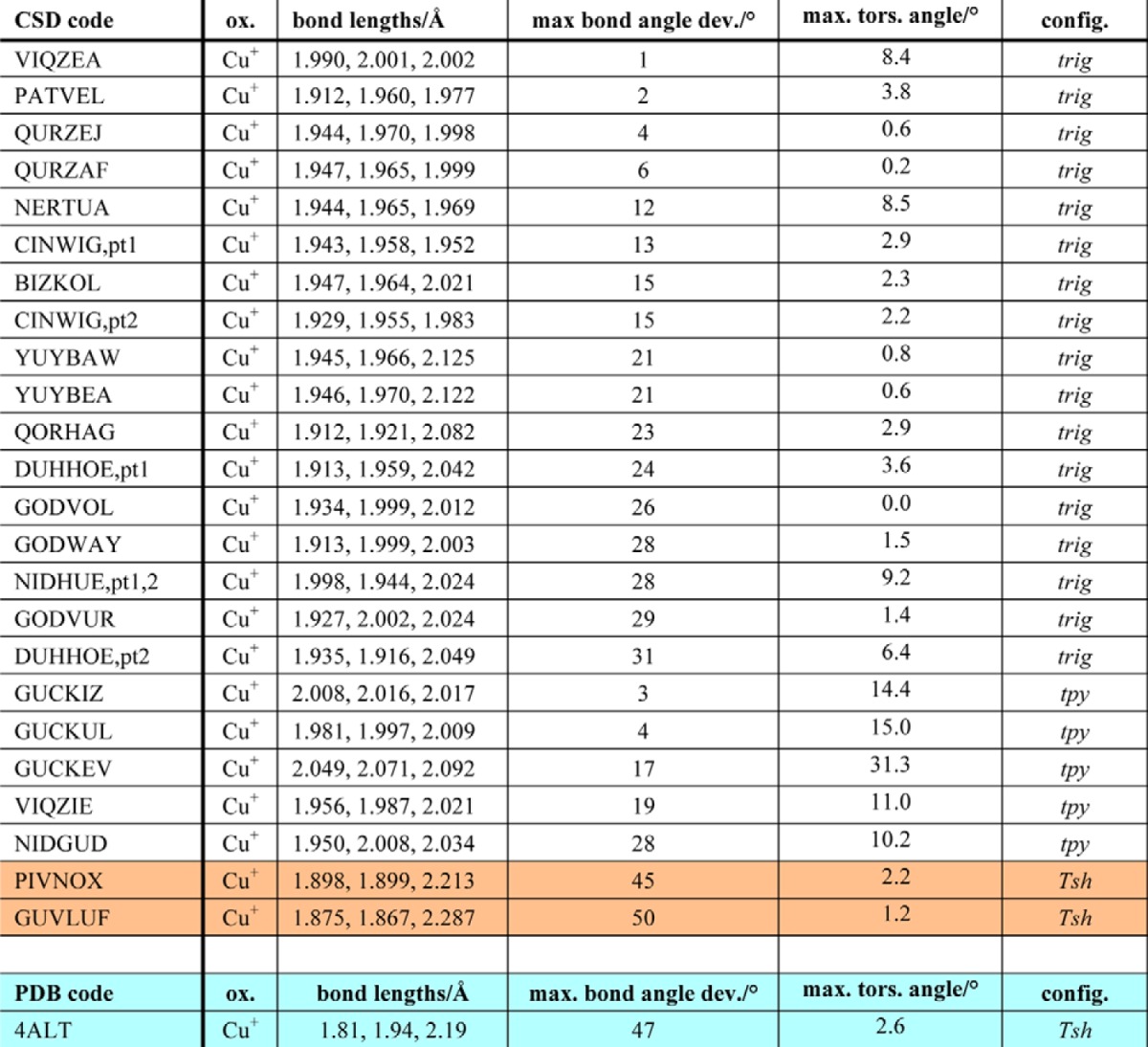
Copper structures with similar coordination as EfCBM33A after being subjected to radiation
Copper structures with similar coordination as EfCBM33A after being subjected to radiation, using the criterion described in the text as listed in CSD by their code, oxidation state, bond lengths, maximum N–Cu–N bond angle deviation (from 120°), maximum Cu–N–N–N torsion angle, and assigned configuration. All structures include three-coordinate copper(I) ions. One structures has nearly identical configuration to the irradiated form of EfCBM33A (4ALT): PIVNOX (Tsh, uneven bond distribution), whereas GUVLUF also has the same general layout (highlighted in peach). trig, trigonal; tpy, trigonal pyramidal; Tsh, T-shaped. pt1 and pt2 denote part 1 and part 2 of the same reported structure, denoting crystallographically independent atoms. The 4ALT structural details are included for reference at the end of the table (highlighted in turquoise).
The EfaCBM33 structures have two axial copper-nitrogen (Cu–Nax and Cu–Nax′) bond distances, which both decrease by 0.05 Å going from tbp to Tsh, whereas the equatorial copper-nitrogen (Cu–Neq) bond distance becomes 0.075 Å longer. Additionally, the nearly linear Nax–Cu–Nax′ angle in tbp, 176.2°, bends a bit off-axis in Tsh, 167.5°, whereas the Neq–Cu–Nax and Neq–Cu–Nax′ angles increase by 5.6 and 3.1°, respectively. The five-coordinated form is almost identical to the Cu(II) structure reported by Casella et al. (57) (CSD code ZUBHOT), whereas the three-coordinated counterpart most closely mimics the Tsh copper(I) structure reported by Sorrell et al. (58) (CSD code PIVNOX). A CSD search for five-coordinated, dihydrate copper structures also coordinated by three nitrogen atoms, returned seven structures—all Cu(II)—with several tbp examples similar to the hydrated version of EfaCBM33A seen in 4ALC (Table 4). Taken together, these observations show that the structural changes observed upon irradiation of EfaCBM33A reflect photoreduction of Cu(II) to Cu(I).
TABLE 4.
Five-coordinated, dihydrate copper structures in the CSD
Entries selected that exhibit two coordinating water molecules are listed by their CSD code, oxidation state, bond distances, and overall configuration. Notably, the mean Cu–N bond distance is shorter than the mean Cu–O distance. Two copper sites exhibit sqpy configuration (highlighted in red), but most are in a tbp configuration, as also observed in 4ALC. sqpy, square pyramidal; tbp, trigonal bipyramidal; dist., distorted; ax, axial; eq, equatorial.
Quantum Mechanical Calculations of the LPMO Active Site
The structures presented above enable DFT calculations to quantify how the electronic structure of the active site changes upon reduction. The active sites of 4ALC, the Cu(II) structure, and 4ALT, the Cu(I) structure, were both examined with the M06-L functional and the 6–31G(d) basis set, by employing an ASM representation of the system. Quantum mechanical geometry optimizations were conducted with a range of ASMs. The model consisting of the residues His29, Glu64, Ala112, His114, Trp176, Ile178, and Phe185 was found to yield the smallest RMSD values for a size that was still computationally tractable with a full quantum mechanical treatment of the ASM in both structures (Table 5; RMSDs of 0.37 and 0.32 Å for 4ALC and 4ALT, respectively). Fig. 4 shows comparisons between the crystal structures and the quantum mechanically optimized ASMs. All computed distances between coordinating nitrogen atoms and the copper differ from the crystallographically observed distances by less than 0.07 Å, which is well within the resolution of the structure (Table 5).
TABLE 5.
Structural and electronic parameters for the ASM (active site model) of 4ALC and 4ALT
NA, not applicable.
| 4ALC | 4ALCopta | 4ALT | 4ALTopta | |
|---|---|---|---|---|
| Cu-Nδ (His29) (Å) | 1.990 | 1.983 | 1.939 | 1.977 |
| Cu-N (His29) (Å) | 2.118 | 2.065 | 2.193 | 2.177 |
| Cu-Nϵ (His114) (Å) | 1.966 | 1.994 | 1.913 | 1.982 |
| Cu–O1 distances (Å) | 2.207 | 2.220 | NA | NA |
| Cu–O2 distances (Å) | 2.189 | 2.116 | NA | NA |
| Nδ (His29) chargeb | NA | −0.68 | NA | −0.71 |
| N (His29) chargeb | NA | −0.98 | NA | −1.01 |
| Nϵ (His114) chargeb | NA | −0.67 | NA | −0.68 |
| RMSD (Å)c | NA | 0.37 | NA | 0.32 |
| Coordination geometry | tbp | tbp | Tsh | Tsh |
a The “opt” subscript denotes the geometry-optimized structures.
b NPA charges (e).
c RMSD was calculated only with heavy atoms of the respective structures.
FIGURE 4.
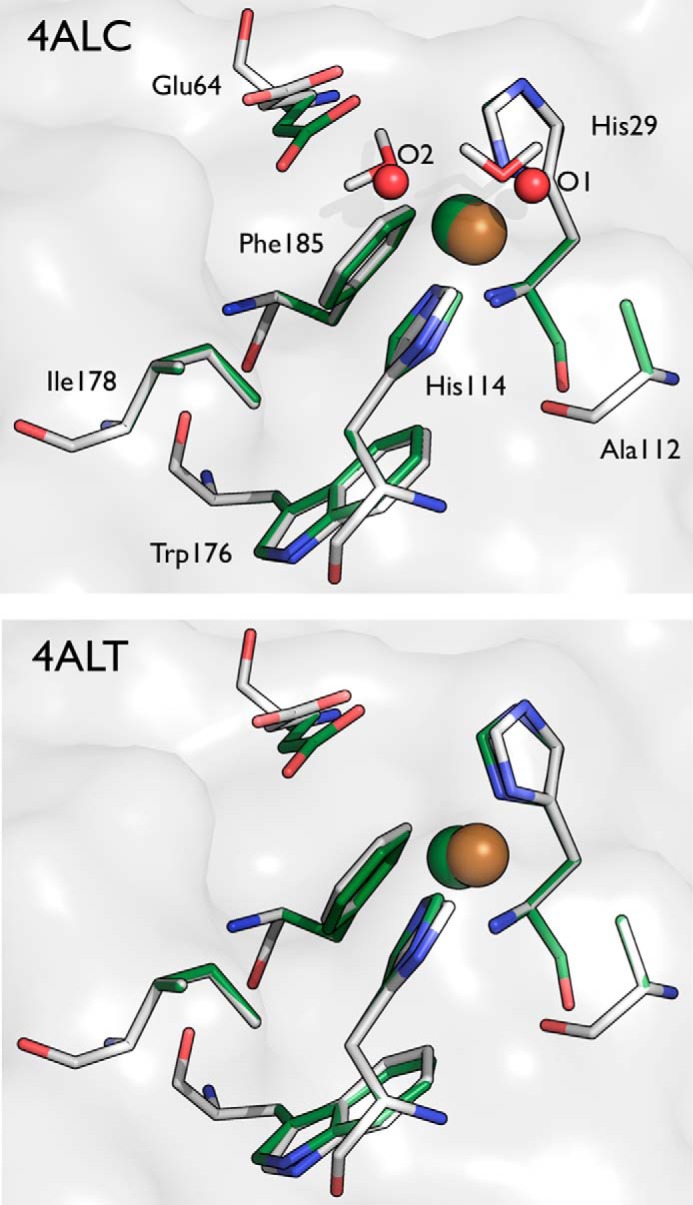
Comparison of the crystallographically determined active sites of 4ALC (top panel) and 4ALT (bottom panel) with quantum mechanically optimized active site models. The residues in gray (carbon), blue (nitrogen), and red (oxygen) represent the crystal structures, and the residues shown in green represent the geometry optimized structures from the DFT calculations. The copper is colored gold and green, respectively. The water molecules from the crystal structure are shown as red spheres in 4ALC, and the optimized water molecules are shown in stick format.
Subsequent to the geometry optimizations, natural population analysis was conducted to examine the charge distributions for both states. As shown in Table 5, the copper ion charges in the oxidized and reduced states of the active site are +1.48 and +0.99, respectively. These values agree well with the formal oxidation states of Cu(II) and Cu(I) and also agree remarkably well with the charges found in both the formal Cu(II) and Cu(I) oxidation states of +1.48 and +0.92, respectively, in an AA9 LPMO with a similar ASM approach (11). Interestingly, the charge distribution of the coordinating histidine residues does not show a significant change, despite the substantial change in the copper ion oxidation state. This result suggests that the LPMO active site is able to readily accommodate both oxidation states of copper with little overall change in the charge distribution in the enzyme.
DISCUSSION
The present study presents the second structure of a CBM33 with copper bound and the first structure of a CBM33 with a Cu(II) ion. Using a data collection strategy allowing for the structure determination of LPMO structures in both copper oxidation states, we were able to visualize structural and copper coordination changes associated with reduction. This experimental methodology is quite generalizable and can be used to capture the electronic and structural transitions in metalloenzyme reduction at advanced light sources.
It has been proposed that an electron from cellobiose dehydrogenase or from a small molecule reducing agent such as ascorbic acid can reduce the LPMO copper ion to a formal oxidation state of Cu(I) (10), prior to binding of dioxygen. This order of events is in accordance with the notion that molecular oxygen tends to bind copper proteins when the metal ion is in the reduced monovalent state (59). Subsequent to dioxygen binding, the catalytic cycle is initiated, which results in substrate hydroxylation, followed by elimination to cleave the glycosidic linkage (10, 11). This general mechanism will incorporate a single oxygen atom from molecular oxygen into the products. The elimination product can then undergo a hydrolysis reaction, which will incorporate an oxygen atom from water, as demonstrated in the mass spectrometry experiments performed by Vaaje-Kolstad et al. (6) with CBP21 and 18O-containing reagents. Interestingly, the series of structures described in the present study show very little structural variation in the conformation of the copper site, despite the change in the copper coordination state induced by x-ray photoreduction. This result highlights that the LPMO catalytic center is preorganized to readily accommodate both oxidation states of copper.
Building on the present results, including the use of the CSD to “annotate” copper site configurations, we conducted a survey of previously reported LPMO structures in terms of their copper oxidation state, the results of which are reported in Table 6. The structures that are annotated as having a Cu(II) metal, NcrPMO-2, NcrPMO-3, and TauGH61A, all exhibit an octahedral six-coordinated octahedral binding motif, which is compatible with the copper being Cu(II). Although the variation in bond lengths and angles is quite high between noncrystallographic symmetry-related molecules in some structures, as well as between the different structures, it is reasonable to conclude that all these published structures represent oxidized Cu(II), in accordance with annotation in the PDB. It is interesting to note that known AA9 LPMO structures with copper contain Cu(II), even though specific precautions to prevent x-ray photoreduction do not seem to have been taken. Under standard x-ray conditions, photoreduction of copper bound to CBM33s readily takes place (27), which could indicate a difference between the AA9 and AA10 LPMO copper sites, possibly caused by the extra coordinating tyrosine in AA9 LPMOs (Fig. 3b). Indeed, based on observed structural and EPR-spectrum differences, Hemsworth et al. (27) have suggested that the oxidative chemistries catalyzed by these enzyme families may differ. Further work is needed to substantiate this hypothesis. Regardless, considering the large overall similarity of the copper sites (Fig. 3), including the histidine brace, it seems reasonable to hypothesize that both enzyme types employ similar catalytic activation steps for reduction of the copper atom to prime the active site for binding molecular oxygen. Thus, it is likely that the structural and electronic insight obtained here for an AA10 LPMO will be relatively similar for an AA9 LPMO enzyme.
TABLE 6.
LPMO active site geometries and the likely oxidation state of the metal ion based on the coordination number and molecular geometry
| Protein | PDB ID | Chain ID | Metal | Geometrya | CN |
|---|---|---|---|---|---|
| EfaCBM33 | 4ALC | A | Cu2+ | tbp | 5 |
| EfaCBM33 | 4ALT | A | Cu+ | Tsh | 3 |
| SmaCBP21 | 2BEM | A | |||
| 2BEM | B | ||||
| 2BEM | C | Na+ | sqpl | 4 | |
| BamCBM33 | 2YOX | A | Cu+ | Tsh | 3 |
| B | Cu+ | Tsh | 3 | ||
| 2YOY | A | Cu+ | Tsh | 3 | |
| B | Cu+ | Tsh | 3 | ||
| PchGH61D | 4B5Q | A | Cu2+ | Oh | 6b |
| B | Cu2+ | Oh | 6b | ||
| NcrPMO-2 | 4EIR | A | Cu2+ | Oh | 6 |
| B | Cu2+ | Oh | 6 | ||
| NcrPMO-3 | 4EIS | A | Cu2+ | Oh | 6c |
| B | Cu2+ | sqpy | 5 | ||
| TteGH61E-1 | 3EII | A | Zn2+ | Oh | 6 |
| B | Zn2+ | Oh | 6 | ||
| C | Zn2+ | Oh | 6 | ||
| D | Zn2+ | sqpy | 5 | ||
| TteGH61E-2 | 3EJA | A | Mg2+ | sqpy | 6d |
| B | Mg2+ | tbp | 5e | ||
| C | Mg2+ | sqpy | 5 | ||
| D | Mg2+ | sqpy | 5 | ||
| HjeGH61B | 2VTC | A | Ni2+ | Oh | 6f |
| B | Ni2+ | Oh | 6 | ||
| TauGH61A | 2YETg | A | Cu2+ | Oh | 6 |
| B | Cu2+ | Oh | 6 | ||
| TauGH61A | 3ZUDh | A | Cu2+ | ||
| AoAA11 | 4MAH | A | Zn2+ | Tsh | 3 |
| 4MAI | A | Cu2+ | Tsh | 3 |
a tbp, trigonal bipyrimidal; Tsh, T-shaped; Oh, octahedral; sqpl, square planar; sqpy, square pyramidal.
b A glycerol molecule coordinates the two positions around the copper ion; it is not modeled as such.
c Long distances to O 3.6 Å and superoxide 3.44 Å.
d Coordinates an SO42− at 4.1 Å.
e Coordinates an SO42− at 4.1 Å.
f One oxygen ligand at 1.21 Å.
g Copper ions modeled with 20% occupancy.
h Copper modeled in dual conformations, partly surrounded by unmodeled density and a glycerol.
Lastly, the DFT calculations employed here reveal that primarily a coordination number change with only very minor geometry changes in the coordinating atoms is required for binding to the copper ion as it goes from a Cu(II) state to Cu(I). As measured by the atomic charges, very little change occurs in the surrounding protein residues electronically. This result is similar to that found for an AA9 LPMO in our recent mechanistic study (11). Therein, we computed the partial charges of the Thermoauscus aurantiacus AA9 LPMO (TauGH61A) upon copper reduction from Cu(II) to Cu(I) as the step before oxygen binding. This calculation showed that the partial charges of the coordinating atoms only very slightly change upon reduction and concomitant removal of the coordinating water molecules, which in an AA9 LPMO changes the copper coordination from distorted octahedral to tetrahedral coordination (11). Taken together, these results suggest that both fungal and nonfungal LPMO active sites are quite plastic and can readily bind both states of copper. Moreover, the development of a robust ASM for AA10 LPMOs will likely enable the study of the complete reaction mechanism of this family of LPMOs using a cluster model or theozyme approach, similar to that done for AA9 LPMOs (11).
CONCLUSIONS
In this study, we present a crystallographic and computational study of the effects of copper reduction in EfaCBM33, using the structures of well characterized small molecule copper complexes from the CSD to assign the oxidation state the copper ion. X-ray photoreduction causes clear changes in the active site of EfaCBM33, namely the loss of the coordinating water molecules. By correlating the structural data with the CSD, the two forms of EfaCBM33A were assigned as a Cu(II) and Cu(I) state with a trigonal bipyramidal and T-shaped geometry, respectively. DFT calculations reveal only minor changes in the atomic charges required for binding to either oxidation state of the copper ion, similar to what was found in a theoretical study for an AA9 LPMO (11). This study provides the first experimental data set to provide insight in the reductive step that activates an LPMO for catalysis.
Acknowledgment
We acknowledge the European Synchrotron Radiation Facility for beamtime.
This work was supported by the Faculty for Natural Resources and Agriculture, Swedish University of Agricultural Sciences, the research program MicroDrivE-Microbially Derived Energy, U. S. Department of Energy BioEnergy Technologies Office and National Science Foundation XSEDE Grant MCB090159 (to G. T. B. and S. K.) (through the Texas Advanced Computing Center), and Grants 214138 and 214613 from the Norwegian Research Council (to G. V.-K. and V. G. H. E.).
- GH
- glycoside hydrolase
- LPMO
- lytic polysaccharide monooxygenases
- RMSD
- root mean square deviation
- PDB
- Protein Data Bank
- CSD
- Cambridge Structural Database
- CBM
- carbohydrate-binding module
- AA
- Auxiliary Activity
- DFT
- density functional theory
- ASM
- active site model.
REFERENCES
- 1. Chundawat S. P., Beckham G. T., Himmel M. E., Dale B. E. (2011) Deconstruction of lignocellulosic biomass to fuels and chemicals. Annu. Rev. Chem. Biomol. Eng. 2, 121–145 [DOI] [PubMed] [Google Scholar]
- 2. Himmel M. E., Ding S. Y., Johnson D. K., Adney W. S., Nimlos M. R., Brady J. W., Foust T. D. (2007) Biomass recalcitrance: engineering plants and enzymes for biofuels production. Science 315, 804–807 [DOI] [PubMed] [Google Scholar]
- 3. Eijsink V. G., Vaaje-Kolstad G., Vårum K. M., Horn S. J. (2008) Towards new enzymes for biofuels: lessons from chitinase research. Trends Biotechnol. 26, 228–235 [DOI] [PubMed] [Google Scholar]
- 4. Cantarel B. L., Coutinho P. M., Rancurel C., Bernard T., Lombard V., Henrissat B. (2009) The Carbohydrate-Active EnZymes database (CAZy): an expert resource for Glycogenomics. Nucleic Acids Res. 37, D233–D238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Levasseur A., Drula E., Lombard V., Coutinho P. M., Henrissat B. (2013) Expansion of the enzymatic repertoire of the CAZy database to integrate auxiliary redox enzymes. Biotechnol. Biofuels 6, 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Vaaje-Kolstad G., Westereng B., Horn S. J., Liu Z., Zhai H., Sørlie M., Eijsink V. G. (2010) An oxidative enzyme boosting the enzymatic conversion of recalcitrant polysaccharides. Science 330, 219–222 [DOI] [PubMed] [Google Scholar]
- 7. Quinlan R. J., Sweeney M. D., Lo Leggio L., Otten H., Poulsen J. C., Johansen K. S., Krogh K. B., Jørgensen C. I., Tovborg M., Anthonsen A., Tryfona T., Walter C. P., Dupree P., Xu F., Davies G. J., Walton P. H. (2011) Insights into the oxidative degradation of cellulose by a copper metalloenzyme that exploits biomass components. Proc. Natl. Acad. Sci. U.S.A. 108, 15079–15084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Forsberg Z., Vaaje-Kolstad G., Westereng B., Bunaes A. C., Stenstrøm Y., MacKenzie A., Sørlie M., Horn S. J., Eijsink V. G. (2011) Cleavage of cellulose by a CBM33 protein. Protein Sci. 20, 1479–1483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Westereng B., Ishida T., Vaaje-Kolstad G., Wu M., Eijsink V. G., Igarashi K., Samejima M., Ståhlberg J., Horn S. J., Sandgren M. (2011) The putative endoglucanase PcGH61D from Phanerochaete chrysosporium is a metal-dependent oxidative enzyme that cleaves cellulose. PLoS One 6, e27807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Phillips C. M., Beeson W. T., Cate J. H., Marletta M. A. (2011) Cellobiose dehydrogenase and a copper-dependent polysaccharide monooxygenase potentiate cellulose degradation by Neurospora crassa. ACS Chem. Biol. 6, 1399–1406 [DOI] [PubMed] [Google Scholar]
- 11. Kim S., Ståhlberg J., Sandgren M., Paton R. S., Beckham G. T. (2014) Quantum mechanical calculations suggest that lytic polysaccharide monooxygenases use a copper-oxyl, oxygen-rebound mechanism. Proc. Natl. Acad. Sci. U.S.A. 111, 149–154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hemsworth G. R., Henrissat B., Davies G. J., Walton P. H. (2014) Discovery and characterization of a new family of lytic polysaccharide monooxygenases. Nat. Chem. Biol. 10, 122–126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Harris P. V., Welner D., McFarland K. C., Re E., NavarroPoulsen J. C., Brown K., Salbo R., Ding H., Vlasenko E., Merino S., Xu F., Cherry J., Larsen S., Lo Leggio L. (2010) Stimulation of lignocellulosic biomass hydrolysis by proteins of glycoside hydrolase family 61: structure and function of a large, enigmatic family. Biochemistry 49, 3305–3316 [DOI] [PubMed] [Google Scholar]
- 14. Horn S. J., Vaaje-Kolstad G., Westereng B., Eijsink V. G. (2012) Novel enzymes for the degradation of cellulose. Biotechnol. Biofuels 5, 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Merino S., Cherry J. (2007) Progress and challenges in enzyme development for biomass utilization. In Biofuels (Olsson L., ed) pp. 95–120, Springer, Berlin: [DOI] [PubMed] [Google Scholar]
- 16. Beckham G. T., Crowley M. F. (2011) Examination of the α-chitin structure and decrystallization thermodynamics at the nanoscale. J. Phys. Chem. B 115, 4516–4522 [DOI] [PubMed] [Google Scholar]
- 17. Beckham G. T., Matthews J. F., Peters B., Bomble Y. J., Himmel M. E., Crowley M. F. (2011) Molecular-level origins of biomass recalcitrance: decrystallization free energies for four common cellulose polymorphs. J. Phys. Chem. B 115, 4118–4127 [DOI] [PubMed] [Google Scholar]
- 18. Payne C. M., Himmel M. E., Crowley M. F., Beckham G. T. (2011) Decrystallization of oligosaccharides from the cellulose Iβ surface with molecular simulation. J. Phys. Chem. Lett. 2, 1546–1550 [Google Scholar]
- 19. Aachmann F. L., Sørlie M., Skjåk-Braek G., Eijsink V. G., Vaaje-Kolstad G. (2012) NMR structure of a lytic polysaccharide monooxygenase provides insight into copper binding, protein dynamics, and substrate interactions. Proc. Natl. Acad. Sci. U.S.A. 109, 18779–18784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Isaksen T., Westereng B., Aachmann F. L., Agger J. W., Kracher D., Kittl R., Ludwig R., Haltrich D., Eijsink V. G., Horn S. J. (2014) A C4-oxidizing lytic polysaccharide monooxygenase cleaving both cellulose and cello-oligosaccharides. J. Biol. Chem. 289, 2632–2642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Vaaje-Kolstad G., Horn S. J., van Aalten D. M., Synstad B., Eijsink V. G. (2005) The non-catalytic chitin-binding protein CBP21 from Serratia marcescens is essential for chitin degradation. J. Biol. Chem. 280, 28492–28497 [DOI] [PubMed] [Google Scholar]
- 22. Vaaje-Kolstad G., Houston D. R., Riemen A. H., Eijsink V. G., van Aalten D. M. (2005) Crystal structure and binding properties of the Serratia marcescens chitin-binding protein CBP21. J. Biol. Chem. 280, 11313–11319 [DOI] [PubMed] [Google Scholar]
- 23. Bøhle L. A., Mathiesen G., Vaaje-Kolstad G., Eijsink V. G. (2011) An endo-ss-N-acetylglucosaminidase from Enterococcus faecalis V583 responsible for the hydrolysis of high-mannose and hybrid-type N-linked glycans. FEMS Microbiol. Lett. 325, 123–129 [DOI] [PubMed] [Google Scholar]
- 24. Vaaje-Kolstad G., Bøhle L. A., Gåseidnes S., Dalhus B., Bjørås M., Mathiesen G., Eijsink V. G. (2012) Characterization of the chitinolytic machinery of Enterococcus faecalis V583 and high-resolution structure of its oxidative CBM33 enzyme. J. Mol. Biol. 416, 239–254 [DOI] [PubMed] [Google Scholar]
- 25. Vebø H. C., Snipen L., Nes I. F., Brede D. A. (2009) The transcriptome of the nosocomial pathogen Enterococcus faecalis V583 reveals adaptive responses to growth in blood. PLoS One 4, e7660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Vebø H. C., Solheim M., Snipen L., Nes I. F., Brede D. A. (2010) Comparative genomic analysis of pathogenic and probiotic Enterococcus faecalis isolates, and their transcriptional responses to growth in human urine. PLoS One 5, e12489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hemsworth G. R., Taylor E. J., Kim R. Q., Gregory R. C., Lewis S. J., Turkenburg J. P., Parkin A., Davies G. J., Walton P. H. (2013) The copper active site of CBM33 polysaccharide oxygenases. J. Am. Chem. Soc. 135, 6069–6077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wu M., Beckham G. T., Larsson A. M., Ishida T., Kim S., Payne C. M., Himmel M. E., Crowley M. F., Horn S. J., Westereng B., Igarashi K., Samejima M., Ståhlberg J., Eijsink V. G., Sandgren M. (2013) Crystal structure and computational characterization of the lytic polysaccharide monooxygenase GH61D from the basidiomycota fungus Phanerochaete chrysosporium. J. Biol. Chem. 288, 12828–12839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Beeson W. T., Phillips C. M., Cate J. H., Marletta M. A. (2012) Oxidative cleavage of cellulose by fungal copper-dependent polysaccharide monooxygenases. J. Am. Chem. Soc. 134, 890–892 [DOI] [PubMed] [Google Scholar]
- 30. Vu V. V., Beeson W. T., Phillips C. M., Cate J. H., Marletta M. A. (2014) Determinants of regioselective hydroxylation in the fungal polysaccharide monooxygenases. J. Am. Chem. Soc. 136, 562–565 [DOI] [PubMed] [Google Scholar]
- 31. Allen F. H. (2002) The Cambridge Structural Database: a quarter of a million crystal structures and rising. Acta Crystallogr. B 58, 380–388 [DOI] [PubMed] [Google Scholar]
- 32. McGeehan J., Ravelli R. B., Murray J. W., Owen R. L., Cipriani F., McSweeney S., Weik M., Garman E. F. (2009) Colouring cryo-cooled crystals: online microspectrophotometry. J. Synchrotron Radiat. 16, 163–172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Flot D., Mairs T., Giraud T., Guijarro M., Lesourd M., Rey V., van Brussel D., Morawe C., Borel C., Hignette O., Chavanne J., Nurizzo D., McSweeney S., Mitchell E. (2010) The ID23–2 structural biology microfocus beamline at the ESRF. J. Synchrotron Radiat. 17, 107–118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Adam V., Royant A., Nivière V., Molina-Heredia F. P., Bourgeois D. (2004) Structure of superoxide reductase bound to ferrocyanide and active site expansion upon x-ray-induced photo-reduction. Structure 12, 1729–1740 [DOI] [PubMed] [Google Scholar]
- 35. Kabsch W. (2010) XDS. Acta Crystallogr. D Biol. Crystallogr. 66, 125–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bailey S. (1994) The Ccp4 Suite: programs for protein crystallography. Acta Crystallogr. D 50, 760–763 [DOI] [PubMed] [Google Scholar]
- 37. Emsley P., Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 38. Murshudov G. N., Vagin A. A., Dodson E. J. (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D 53, 240–255 [DOI] [PubMed] [Google Scholar]
- 39. DeLano W. L. (2002) The PyMOL Molecular Graphics System, DeLano Scientific, San Carlos, CA [Google Scholar]
- 40. Kleywegt G. J. (1996) Use of non-crystallographic symmetry in protein structure refinement. Acta Crystallogr. D Biol. Crystallogr. 52, 842–857 [DOI] [PubMed] [Google Scholar]
- 41. Kleywegt G. J., Jones T. A. (1996) Phi/psi-chology: Ramachandran revisited. Structure 4, 1395–1400 [DOI] [PubMed] [Google Scholar]
- 42. Echols N., Grosse-Kunstleve R. W., Afonine P. V., Bunkóczi G., Chen V. B., Headd J. J., McCoy A. J., Moriarty N. W., Read R. J., Richardson D. C., Richardson J. S., Terwilliger T. C., Adams P. D. (2012) Graphical tools for macromolecular crystallography in PHENIX. J. Appl. Crystallogr. 45, 581–586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Murray J. W., Garman E. F., Ravelli R. B. (2004) X-ray absorption by macromolecular crystals: the effects of wavelength and crystal composition on absorbed dose. J. Appl. Crystallogr. 37, 513–522 [Google Scholar]
- 44. Paithankar K. S., Owen R. L., Garman E. F. (2009) Absorbed dose calculations for macromolecular crystals: improvements to RADDOSE. J. Synchrotron Radiat. 16, 152–162 [DOI] [PubMed] [Google Scholar]
- 45. Frisch M. J., Trucks G. W., Schlegel H. B., Scuseria G. E., Robb M. A., Cheeseman J. R., Scalmani G., Barone V., Mennucci B., Petersson G. A. (2010) Gaussian 09, revision B. 01 Gaussian, Inc., Wallingford, CT [Google Scholar]
- 46. Dechancie J., Clemente F. R., Smith A. J., Gunaydin H., Zhao Y.-L., Zhang X., Houk K. N. (2007) How similar are enzyme active site geometries derived from quantum mechanical theozymes to crystal structures of enzyme-inhibitor complexes? Implications for enzyme design. Protein Sci. 16, 1851–1866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Zhao Y., Truhlar D. (2008) The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Account 120, 215–241 [Google Scholar]
- 48. Zhao Y., Truhlar D. G. (2008) Density functionals with broad applicability in chemistry. Acc. Chem. Res. 41, 157–167 [DOI] [PubMed] [Google Scholar]
- 49. Barone V., Cossi M. (1998) Quantum calculation of molecular energies and energy gradients in solution by a conductor solvent model. J. Phys. Chem. A 102, 1995–2001 [Google Scholar]
- 50. Cossi M., Rega N., Scalmani G., Barone V. (2003) Energies, structures, and electronic properties of molecules in solution with the C-PCM solvation model. J. Comput. Chem. 24, 669–681 [DOI] [PubMed] [Google Scholar]
- 51. Liao R.-Z., Thiel W. (2013) On the effect of varying constraints in the quantum mechanics only modeling of enzymatic reactions: the case of acetylene hydratase. J. Phys. Chem. B 117, 3954–3961 [DOI] [PubMed] [Google Scholar]
- 52. Terwilliger T. C., Grosse-Kunstleve R. W., Afonine P. V., Moriarty N. W., Adams P. D., Read R. J., Zwart P. H., Hung L. W. (2008) Iterative-build OMIT maps: map improvement by iterative model building and refinement without model bias. Acta Crystallogr. D Biol. Crystallogr. 64, 515–524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Matthews B. W. (1968) Solvent content of protein crystals. J. Mol. Biol. 33, 491–497 [DOI] [PubMed] [Google Scholar]
- 54. Hemsworth G. R., Davies G. J., Walton P. H. (2013) Recent insights into copper-containing lytic polysaccharide mono-oxygenases. Curr. Opin. Struct. Biol. 23, 660–668 [DOI] [PubMed] [Google Scholar]
- 55. Karkehabadi S., Hansson H., Kim S., Piens K., Mitchinson C., Sandgren M. (2008) The first structure of a glycoside hydrolase family 61 member, Cel61B from Hypocrea jecorina, at 1.6 angstrom resolution. J. Mol. Biol. 383, 144–154 [DOI] [PubMed] [Google Scholar]
- 56. Li X., Beeson W. T., 4th, Phillips C. M., Marletta M. A., Cate J. H. (2012) Structural basis for substrate targeting and catalysis by fungal polysaccharide monooxygenases. Structure 20, 1051–1061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Casella L., Carugo O., Gullotti M., Doldi S., Frassoni M. (1996) Synthesis, structure, and reactivity of model complexes of copper nitrite reductase. Inorg. Chem. 35, 1101–1113 [DOI] [PubMed] [Google Scholar]
- 58. Sorrell T. N., Garrity M. L., Richards J. L., White P. S. (1994) Synthesis, structural characterization and dioxygen reactivity of imidazole-ligated Cu (I) complexes. Inorg. Chim. Acta 218, 103–108 [Google Scholar]
- 59. Que L., Jr., Tolman W. B. (2008) Biologically inspired oxidation catalysis. Nature 455, 333–340 [DOI] [PubMed] [Google Scholar]