

Background: PKA increases NMDA receptor responses and phosphorylates multiple residues on C-terminal domains (CTD).
Results: PKA inhibition reduced gating through GluN2B CTD and reduced Ca2+ permeability through GluN1 CTD.
Conclusion: PKA controls NMDA receptor gating and Ca2+ permeability through distinct sites.
Significance: Dissecting the complex modulatory effects of PKA on NMDA receptors helps delineate fundamental mechanisms of synaptic regulation.
Keywords: Calcium Signaling, Ionotropic Glutamate Receptor, Neurotransmitter Receptor, Protein Phosphorylation, Receptor Desensitization, cAMP Protein Kinase, Permeation Mechanism, Reaction Mechanism, Single-channel Kinetics
Abstract
Protein kinase A (PKA) enhances synaptic plasticity in the central nervous system by increasing NMDA receptor current amplitude and Ca2+ flux in an isoform-dependent yet poorly understood manner. PKA phosphorylates multiple residues on GluN1, GluN2A, and GluN2B subunits in vivo, but the functional significance of this multiplicity is unknown. We examined gating and permeation properties of recombinant NMDA receptor isoforms and of receptors with altered C-terminal domain (CTDs) prior to and after pharmacological inhibition of PKA. We found that PKA inhibition decreased GluN1/GluN2B but not GluN1/GluN2A gating; this effect was due to slower rates for receptor activation and resensitization and was mediated exclusively by the GluN2B CTD. In contrast, PKA inhibition reduced NMDA receptor-relative Ca2+ permeability (PCa/PNa) regardless of the GluN2 isoform and required the GluN1 CTD; this effect was due primarily to decreased unitary Ca2+ conductance, because neither Na+ conductance nor Ca2+-dependent block was altered substantially. Finally, we show that both the gating and permeation effects can be reproduced by changing the phosphorylation state of a single residue: GluN2B Ser-1166 and GluN1 Ser-897, respectively. We conclude that PKA effects on NMDA receptor gating and Ca2+ permeability rely on distinct phosphorylation sites located on the CTD of GluN2B and GluN1 subunits. This separate control of NMDA receptor properties by PKA may account for the specific effects of PKA on plasticity during synaptic development and may lead to drugs targeted to alter NMDA receptor gating or Ca2+ permeability.
Introduction
In central neurons, NMDA receptors mediate Ca2+ fluxes that are crucial to synaptic plasticity, synapse development, and excitotoxity (1–3). The mechanisms that control NMDA receptor Ca2+ currents are largely unknown, yet they impact these fundamental physiological and pathological processes in the brain and spinal cord. Relative to other Ca2+-permeable synaptic receptors, NMDA receptors produce large Ca2+ fluxes because they have characteristically slow gating, large unitary conductance, and high Ca2+ permeability (4–6). NMDA receptors are heterotetramers, containing two GluN1 (N1), which are expressed ubiquitously in central neurons, and two GluN2 (N2) subunits, of which the most abundant are N2A and N2B (7). Of the receptor regions known to influence NMDA receptor Ca2+ permeability, two are located on the obligatory N1 subunit; these consist of a group of charged residues, which line the external mouth of the pore, and of asparagine residues located deep within the membrane at the narrow constriction of the pore (8, 9). NMDA receptors have large intracellular C-terminal domains (CTD)2 that contain numerous phosphorylation substrates for a variety of kinases (10), which may control NMDA receptor Ca2+ signals as well.
Protein kinase A (PKA) phosphorylates NMDA receptors at numerous sites on N1 and N2 subunits (11, 12); however, the functional significance of this redundancy is unclear. In certain conditions, PKA can alter specifically the synaptic NMDA receptor Ca2+ signal without changing the magnitude of the excitatory postsynaptic current (EPSC) (13–15). Other reports indicate, however, that PKA activity can increase the amplitude of the NMDA receptor currents (13, 16–18) and prevent calcineurin-induced reduction in EPSCs (19). PKA inhibition has specific effects on N2A- and N2B-containing receptors; it decreases both Ca2+ influx and macroscopic current amplitude in N2B-containing receptors, but it reduces Ca2+ flux without changing the macroscopic traces of N2A-containing receptors (13). Consistent with these observations, PKA phosphorylation of NMDA receptors can alter the size and threshold of synaptic plasticity (13, 18, 20–22). The mechanisms by which PKA can change current amplitude and Ca2+ flux to influence neuronal currents and ultimately plasticity are unknown.
To investigate how PKA activity reshapes specific aspects of NMDA receptor currents, we used single-channel and macroscopic recordings from NMDA receptor isoforms and receptors carrying mutations and measured unitary gating kinetics, Ca2+ conductance, and Ca2+ permeability. We found that PKA activity influenced gating and Ca2+ permeability of NMDA receptors by separate mechanisms, and these effects were mediated by sites located on N2B and N1 subunits, respectively.
EXPERIMENTAL PROCEDURES
Cell Culture and Transfections
HEK293 cells (ATCC number CRL-1573) between passages 24 and 35 were transiently transfected with ∼3 μg of DNA, which included rat GluN1-1a (N1, U08261), GluN2 (N2), and GFP in a 1:1:0.8 ratio or GCamP6f (for patch clamp fluorometry experiments; from Arnd Pralle) in a 1:1:1 ratio, all expressed from pcDNA3.1(+). Wild-type subunits were rat GluN2A (N2A, M91561) or GluN2B (N2B, M91562). For some experiments, we used a chimeric mouse subunit, N2A2BCTD, which had residues 1–837 of N2A and 839–1483 of N2B (gift from Martha Constantine-Paton) (23). Additional mutations included C-terminal truncations as follows: N1stop, which terminates after Lys-838 and N2Astop, which terminates after Lys-844 (gifts from Gary Westbrook) (24, 25); and N2Bstop, which terminates at Gln-845, was generated previously (26). N1S897A and N1S897D were gifts from John Woodward (27). Additional mutations, N2BS1166A and N2BS1166D, were produced by site-directed mutagenesis (Qiagen) (28). All constructs were verified by full insert sequencing.
Electrophysiology
Cells were used 24–48 h post-transfection. For gating measurements, Na+-only currents were recorded from one-channel cell-attached patches using 12–24-megohm pipettes filled with extracellular solution containing (in mm) the following: 150 NaCl, 2.5 KCl, 1 EDTA, and 10 HEPBS at pH 8.0 with NaOH, with saturating concentrations of glutamate (1) and glycine (0.1), and applied pipette potential of +100 mV (Em approximately −110 mV). Unless otherwise indicated, cells were bathed in PBS. For paired recordings, basal activity was recorded for ∼15 min, after which 1 μm PKI (PKI(14–22)-amide myristoylated and dissolved in water, Enzo Biosciences, Farmingdale, NY) was added to the bath (29). For preincubation experiments, 1 μm PKI, 10 μm H-89, or 100 μm 8-Br-cAMP were added at least 10 min prior to recordings.
For single-channel recordings with extracellular Ca2+, the recording pipette contained (in mm) the following: 150 NaCl, 2.5 KCl, 10 HEPBS, 10 Tricine, 1 glutamate, and 0.1 glycine, at pH 8.0 with NaOH (total Na+, 158.5), with or without 2 or 10 CaCl2. These patches were held at −40 mV to reduce any residual block by trace Mg2+, while still maintaining a substantial inward driving force for Na+. To measure unitary Ca2+ currents, the recording pipette solution contained (in mm) the following: 75 CaCl2, 2.5 KCl, 10 Tricine, 10 HEPBS, 1 glutamate, 0.1 glycine, pH 8.0, with Tris-OH, and +100 mV applied potential. For all unitary current amplitude (i) measurements, cells were bathed in high-K+ solution containing (in mm) the following: 142 KCl, 5 NaCl, 1.8 CaCl2, 1.7 MgCl2, 10 HEPBS, pH 7.4, with KOH, to cancel the intrinsic resting membrane potential (Em, 0 mV). Current-voltage (i/V) relationships were determined from multichannel cell-attached recordings by isolating and analyzing clusters of single openings. Pipette potentials were between −10 and +100 mV and were applied in 10-mV steps that lasted at least 10 s and produced tens to hundreds of events per step. Reversal potentials and conductance (γ) were determined as the intercept and slope of linear fits to i/V data, respectively. Recordings with i/V fits that had R2 <0.95 were excluded. For all cell-attached recordings, currents were amplified and low-pass filtered at 10 kHz (Axopatch 200B); they were acquired directly into digital files by sampling at 40 kHz (NIDAQ board) with QuB software. Traces are displayed digitally filtered at 1 kHz.
Whole-cell currents were recorded with pipettes (2–5 megohms) filled with intracellular solution containing (in mm) the following: 140 CsCl, 2 MgCl2, 10 HEPES, 1 EGTA, pH 7.4, with CsOH. For some experiments, 1 μm myristoylated PKI(14–22) (myr-PKI) or PKI 6–22 (non-myr-PKI) was included in the pipette. Cells were held at −70 mV with the series resistance compensated >75%; data were sampled at 5 kHz, amplified and filtered at 2 kHz (AxoPatch200B), and acquired into digital files with pClamp 10.0 software. Using a pressurized solution exchange system (ALA Scientific, Farmingdale, NY), cells were continuously perfused with extracellular wash solution containing (in mm) the following: 150 NaCl, 2.5 KCl, 0.5 CaCl2, 10 HEPBS, 0.01 EDTA, 0.1 glycine, pH 7.4 or 8.0 (with NaOH), and were pulsed for 5 s with wash solution plus 1 mm glutamate.
Patch Clamp Fluorometry
NMDA receptor responses were elicited by applying glutamate-containing solutions (1 mm, 5 s), and the fast, ultrasensitive fluorescent protein GCamP6f was concurrently excited with 480 nm light (X-Cite 120LED, Lumen Dynamics) (30). Changes in fluorescence were detected with a photomultiplier tube module (Hamamatsu Photonics) driven by a low voltage power supply (Hamamatsu Photonics) held at 0.5 V. Fluorescence signals were sampled (5 kHz, Digidata 1440A), low-pass filtered (100 Hz), and stored as digital files (pClamp10 software, Molecular Devices).
Data Analysis and Statistics
Single-channel data were idealized with the SKM algorithm (31) using QuB software (University at Buffalo, Buffalo, NY), and the idealized events were fit by kinetic models with the MIL algorithm (QuB) as described previously (32, 33). Macroscopic traces (simulated or recorded) were analyzed in IgorPro 6.2 (WaveMetrics, Lake Oswego, OR).
Statistical significance of differences between data sets obtained before and after treatment was assessed with Student's paired t test for all values obtained on the same patch or with Student's unpaired t test when basal and +PKI data sets were obtained from separate cells. For the multiple comparisons, as in Fig. 1, a one-way analysis of variance was used with Tukey's post hoc analysis. Differences of means were considered significant for p < 0.05. For multiple comparisons done per one type of measurement (i.e. current amplitudes with different ionic conditions ±PKI, as in Fig. 5, Erev with different Ca2+ concentrations or mutations ±PKI preincubation, as in Figs. 6 and 7, and ΔF/F with different mutations ±PKI, as in Fig. 7), the significance threshold was adjusted according to the Bonferroni method. Accordingly, for two or three separate comparisons, the significance threshold was p < 0.025 or p < 0.0167, respectively.
FIGURE 1.

Pharmacological inhibition of PKA reduces N1/N2B receptor activity. A, left, whole-cell currents recorded in physiological extracellular solutions (2 mm Ca2+, pH 7.4) before (basal, black), after bath application of 1 μm myristoylated PKI(14–22) peptide (myr-PKI, red), and after removing the peptide (wash, blue). Right, summary of normalized current amplitudes (n = 4). B, top, whole-cell currents recorded in modified extracellular solutions (0.5 mm Ca2+, pH 8) at time 0 (black) or at 5 min after break-in (red) without (black) or with (red) PKI added to the pipette (intracellular) solution. Bottom, time dependence of peak current amplitudes for basal (n = 5), with myr-PKI (n = 6), and with non-myr-PKI (n = 5) peptides included in the pipette (intracellular) solution. C, cell-attached currents (inward Na+) recorded with modified pipette (extracellular) solutions (0 mm Ca2+, pH 8) without (basal) or after pretreating the cells with drugs: myr-PKI, H-89, or 8-Br-cAMP. D, summary of Po for basal (n = 11), myr-PKI (n = 5), H-89 (n = 6), and 8-Br-cAMP (n = 6). *, F(3,27) = 5.73, p < 0.05 with Tukey's post hoc analysis.
FIGURE 5.

PKI reduced unitary Ca2+ conductance. A, cell-attached currents (inward Ca2+ and Na+) recorded with 2 mm Ca2+ and 150 mm Na+ in the pipette solution, pH 8, from the same receptor before (basal, left) or after adding PKI to the bath (+PKI, right). B, time course of Po per min of recording for the receptor shown in A. Red bar indicates time of PKI application. C, summary of measured open probability (Po) before (gray) and after (red) adding PKI to the bath for both N1/N2A (triangles; n = 4) and N1/N2B (circles; n = 5) receptors. D, unitary currents were recorded from cell-attached patches of cells bathed in high (145 mm) K+ extracellular solution, with Na+ (150 mm) or Ca2+ (75 mm) as the principal permeating ion, before (basal, top) or after PKI preincubation (+PKI, bottom). Pipette potential is indicated for each panel. E, summary of unitary current amplitudes measured from experiments illustrated in D. The permeating ion is indicated along the corresponding bars; basal (gray): Na+, n = 22 and paired Ca2+ and K+ traces, n = 24; PKI preincubation (red), Na+, n = 23 and paired Ca2+ and K+ traces, n = 19. *, p < 0.016, unpaired Student's t test with Bonferroni correction.
FIGURE 6.

PKI reduced relative Ca2+ permeability. A, top, unitary cell-attached currents recorded from cells bathed in high (145 mm) external K+ at several pipette potentials with 0, 2, or 10 mm Ca2+ and 150 Na+ in the pipette (extracellular) solution, with or without PKI treatment. Bottom, summary of i/V dependences for each patch. Erev values were calculated as the extrapolations of linear fits to the data to 0 mV. B, summary of Erev values pooled for N1/N2A and N1/N2B receptors; means for each population are indicated; n (basal, +PKI): 0 Ca2+ (16, 13), 2 Ca2+ (14, 14), and 10 Ca2+ (6, 7); * marks statistically significant differences of means (p < 0.016, unpaired Student's t test with Bonferroni correction). C, evaluation of Ca2+ block as the degree of Ca2+-dependent reduction in unitary conductance (calculated as the slope of the i/V plots (A) with (red) or without (black) preincubation with PKI; S.E. values as smaller than symbols).
FIGURE 7.
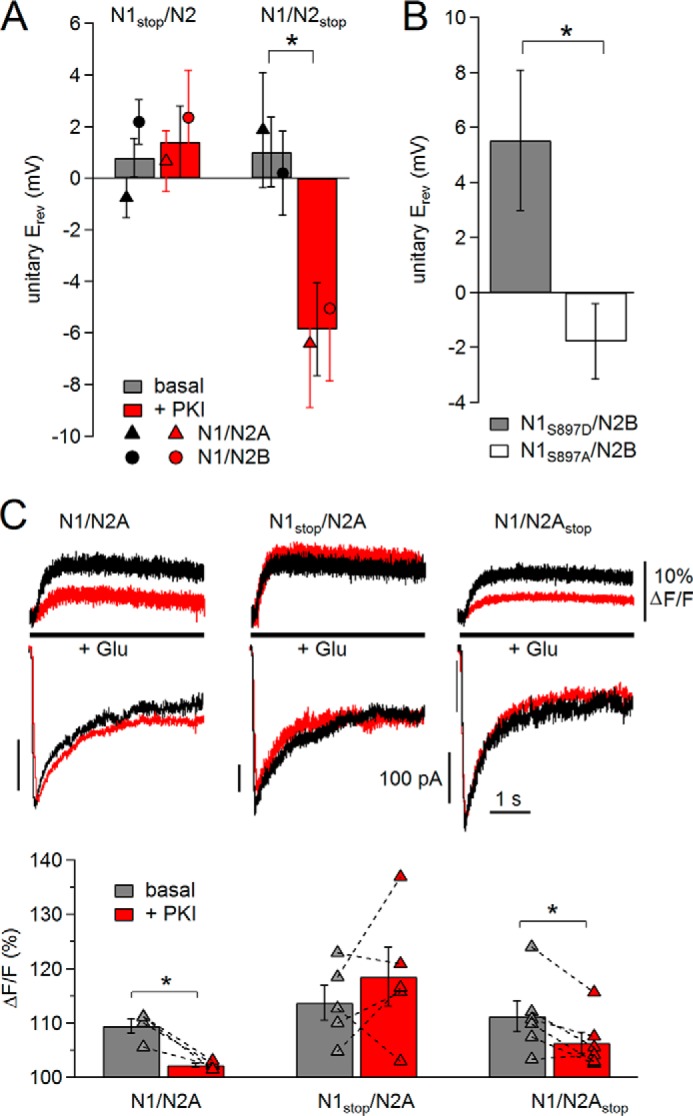
PKI reduction in Ca2+ permeability is mediated by N1 CTD, in part through Ser-897. A and B, summary of Erev values calculated for the conditions indicated; n (basal, +PKI): N1stop (19, 18); N2stop (18, 15); N1S897D (7), and N1S897A (8). * marks differences of means (p < 0.025, unpaired Student's t test with Bonferroni correction (A) or p < 0.05, unpaired Student's t test (B)). Measurements were made from cells bathed in high (145 mm) external K+ at several pipette potentials with 2 mm Ca2+ and 150 Na+ in the pipette (extracellular) solution from receptors containing N1stop or N2stop (A) and N1S897D or N1S897A subunits (B). C, top, simultaneous recordings of Ca2+-dependent increase in fluorescence (top) and whole-cell currents (bottom) from receptors containing wild-type (left), N1stop (middle), or N2Astop (right) subunits before (black) and after (red) PKI treatment. Bottom, summary of fluorescence data for basal (gray) and +PKI (red) conditions for receptors containing wild-type (n = 4), N1stop (n = 5), or N2Astop (n = 6) subunits. * marks differences of means (p < 0.016, paired Student's t test with Bonferroni correction).
RESULTS
PKA Inhibitors Reduced N1/N2B Receptor Current by Increasing Closed-channel Time
We used whole-cell current recordings to select conditions that allowed us to isolate and detect PKA-dependent changes in NMDA receptor signals. Protein kinase A inhibitor (PKI) is a naturally occurring peptide with high and specific inhibitory activity toward the catalytic subunit of PKA (34). A cell-permeable reagent, myristoylated 14–22 (myr-PKI) was found to modulate NMDA receptor properties in neurons and heterologous cells (13). Likewise, we observed that in physiological conditions (2 mm Ca2+, pH 7.4) bath application of myr-PKI reduced ∼3-fold the peak amplitude of glutamate-elicited whole-cell currents from HEK293 cells expressing N1/N2B receptors, and the currents recovered partially after removing the peptide (Fig. 1A, basal, −268 ± 55; +PKI, −81 ± 18 pA, p < 0.05).
Next, we asked whether bath-applied myr-PKI produced effects that were similar to those produced by intracellularly applied myr-PKI and whether this modulation also occurred in the modified conditions usually employed for NMDA receptor gating measurements, which require low concentrations of protons and divalent cations (Ca2+ and Mg2+) (35). We repeated whole-cell current recordings with modified extracellular solutions (0.5 mm Ca2+ and pH 8) and included myr-PKI in the recording pipette. We observed that peak current amplitudes recorded immediately after break-in were stable when recorded with regular intracellular solutions (basal) but declined within minutes when the intracellular solution contained myr-PKI or non-myristoylated PKI(6–22) (non-myr-PKI) (Fig. 1B). Based on these results, we concluded the following: PKA inhibitors reduced NMDA receptor currents independently of extracellular pH and Ca2+; myr-PKI was effective whether bath- or intracellularly applied; and myr-PKI (referred to as PKI throughout this report) had effects on NMDA receptor currents that were similar to those of the non-myristoylated peptide.
To investigate whether PKA modulates N1/N2B receptor gating, we recorded steady-state Na+-only currents from channels in cell-attached patches (0 mm Ca2+, 1 mm EDTA, and pH 8) (36) without (basal) or after pretreating the cells for ∼10 min with PKA inhibitors (PKI or H-89). This approach allowed us to focus on specific gating effects of PKA inhibitors because during cell-attached recordings receptors maintain native intracellular interactions and, in the absence of permeating Ca2+, Ca2+-dependent processes such as desensitization, inactivation, and run down are prevented (25, 37–40). Consistent with the macroscopic effects in Fig. 1, A and B, pretreating cells with PKI or H-89 reduced channel open probability (Po) ∼4-fold (basal, 0.19 ± 0.04; PKI, 0.05 ± 0.02; H-89, 0.02 ± 0.01; F(3,27) = 5.73, p < 0.05 with Tukey's post hoc analysis) (Fig. 1, C and D). Pretreating the cells with 8-Br-cAMP, a membrane-permeable cAMP analog, however, did not further increase channel activity over basal levels (Po, 8-Br-cAMP, 0.17 ± 0.04, p > 0.05 versus basal) (Fig. 1, C and D). Together, these results indicate that ongoing PKA activity is necessary to maintain high Po activity for N1/N2B receptors and that bath-applied PKI is an effective strategy to investigate the mechanisms by which PKA modulates NMDA receptor properties.
Next, we examined PKI effects on N1/N2B gating. We obtained a set of single-channel cell-attached recordings where we could compare the activity of each channel before (basal, ∼15 min) and after (+PKI, >15 min) adding PKI to the bath. Fig. 2A illustrates two continuous portions of current recorded from the same N1/N2B receptor before (left) and after (right) adding PKI (1 μm) to the bath. The bursting activity was visibly reduced within 5 min of adding PKI, and this reduction lasted for the remainder of the recording. To investigate the cause of this decline in activity, we examined how the single-channel kinetic parameters varied with time. For the receptor shown in Fig. 2A, unitary (Na+) current amplitude (i) and mean open time remained unchanged for the entire course of the recording, whereas Po decreased abruptly and was accompanied by an increase in mean closed times (MCT) (Fig. 2B). Relative to basal levels, PKI reduced Po ∼2.3-fold (basal, 0.19 ± 0.07; +PKI, 0.08 ± 0.03; n = 7, p < 0.05) with no detectable effects on unitary current or open durations (Table 1).
FIGURE 2.

PKA inhibition reduces N1/N2B receptor activity by lengthening closures. A, one-channel cell-attached currents (inward Na+) recorded with modified pipette (extracellular) solutions (0 mm Ca2+, pH 8) from the same receptor before (basal, left) or after adding to the bath myr-PKI 14–22 peptide (+PKI, right). B, time course of unitary current amplitude (i), Po, MOT, and MCT per min of recording for the receptor shown in A. Red bar indicates the time of PKI application. C, time dependence of activity (Po) per min recorded before (basal) or after adding water (+water, n = 14) or PKI (+PKI, n = 7) to the bath. D, summary of change in single-channel kinetic parameters after adding water (gray, n = 14) versus PKI (red, n = 7). *, p < 0.05, unpaired Student's t test.
TABLE 1.
NMDA receptor single-channel parameters before and after PKA inhibition
| GluN2 subunit | n | i | Po | MOT | MCT | Events × 106 |
|---|---|---|---|---|---|---|
| pA | ms | ms | ||||
| N1/N2B | ||||||
| Basal | 7 | 9.1 ± 0.5 | 0.19 ± 0.07 | 4.0 ± 0.5 | 65 ± 34 | 0.45 |
| +PKI | 7 | 8.9 ± 0.4 | 0.08 ± 0.03a | 3.2 ± 0.3 | 136 ± 67 | 1.42 |
| N1/N2A | ||||||
| Basal | 4 | 9.4 ± 0.6 | 0.48 ± 0.10 | 5.6 ± 0.6 | 8 ± 3 | 0.55 |
| +PKI | 4 | 9.3 ± 0.6 | 0.55 ± 0.06 | 6.4 ± 0.8 | 5 ± 1 | 1.49 |
| N1/N2A2BCTD | ||||||
| Basal | 5 | 9.2 ± 0.4 | 0.47 ± 0.14 | 6.4 ± 1.6 | 12 ± 5 | 0.44 |
| +PKI | 5 | 8.7 ± 0.3 | 0.33 ± 0.12 | 5.9 ± 1.3 | 28 ± 13 | 0.72 |
| N1/N2BS1166D | ||||||
| Basal | 6 | 9.7 ± 0.5 | 0.24 ± 0.06 | 4.6 ± 0.9 | 16 ± 2 | 0.57 |
| +PKI | 6 | 9.5 ± 0.6 | 0.08 ± 0.02 | 3.3 ± 0.6 | 43 ± 6 | 0.66 |
a Boldface entries indicate values that are significantly different relative to paired basal (p < 0.05).
As noted previously for N1/N2B receptors, we also observed substantial patch-to-patch variability in Po and even more so in MCT (35). For this reason, and to test whether the decrease in Po was PKI-specific, we obtained a separate set of recordings where instead of PKI we added solvent-only (+water) (Fig. 2C). None of the kinetic parameters examined changed significantly after adding water (Po, mean open time, MCT, i) (Fig. 2D). Relative to basal, however, MCT was increased significantly more in +PKI compared with +water (Fig. 2D), 2.7 ± 0.4-fold versus 1.5 ± 0.3-fold, p < 0.05. Together with the observation that PKI did not change current amplitude and open durations, these results strongly indicate that PKI decreased the Po of N1/N2B receptors largely by lengthening channel closed durations, and that ongoing phosphorylation by PKA is necessary to maintain basal N1/N2B activity at high levels.
PKI Caused N1/N2B Receptors to Accumulate in Desensitized States
To examine the effects of PKA inhibition on the open and closed event durations, we compiled duration histograms after separating for each record all events that occurred prior to (basal) or after treatment (+PKI) (Fig. 3A). As described in detail elsewhere, open distributions of neuronal as well as recombinant N1/2B receptors have two kinetic components within each kinetic mode, and up to four open components (E1 to E4) can be detected when a record contains three gating modes. In contrast, regardless of modes, closed distributions always have five components (E1 to E5), of which the longest, E4 and E5, represent desensitized components (35, 41). Consistent with our global open time analyses (MOT), the results showed that neither the number of open components nor their mean durations changed consistently after treatment (+PKI or +water), and all distributions had components with τ of ∼0.2, 3, and 6 ms, before and after PKI or water treatment (p > 0.05, data not shown). This result demonstrates that PKA phosphorylation did not influence the stability of open states or the modal gating kinetics of N1/N2B receptors. In contrast to this lack of effect on open events, closed event distributions revealed that PKI specifically increased the durations of the two desensitized components, τ4 and τ5, ∼2-fold with no influence on the shorter closed components (Fig. 3A, bottom). We conclude that this effect was PKI-specific because neither the exponential distributions of open nor those of closed durations were significantly different from basal conditions in the +water periods (data not shown).
FIGURE 3.

Kinetic mechanism of N1/N2B receptor inhibition by PKI. A, open (top) and closed (bottom) interval distributions for the currents recorded from the patch shown in Fig. 2A before (left, basal) and after PKI application (right, +PKI). Data (histograms) are overlaid with exponential components (E, lines) calculated by fitting multistate kinetic models to the one-channel data. Insets, time constants (τ, ms) and fractional areas (%). B, state models and predicted occupancies (C, closed; O, open) for receptors before (basal, top) or after (+PKI, bottom). All states represent fully liganded (glutamate and glycine) receptor conformations. Transition rates (s−1) were optimized from fits to individual single-channel records and are given as rounded mean values for all patches (n = 7). * indicates differences relative to basal (p < 0.05, paired Student's t test). C, left, macroscopic responses to glutamate (1 mm, 5 s) were simulated with the models in B and amplitudes in Fig. 2 (1000 channels) using published glutamate association and dissociation rates (35) (top) or were recorded experimentally as whole-cell currents (bottom, n = 6), without (black) or with extracellular PKI (red). Right, responses normalized to peak basal currents.
To further understand these observed changes in closed durations, we used a simplified kinetic model that was previously validated for N1/N2B receptors (35). This scheme consists of five closed states (C1 to C5) and thus accounts for all the closed components observed here (E1 to E5), and it combines all the openings into one aggregated open state (O). Fitting this scheme to our single-channel data obtained before or after treatment, we found that PKA inhibition slightly but significantly slowed the first two transitions along the opening pathway (basal versus +PKI) as follows: C3 → C2, 200 ± 40 s−1 versus 120 ± 30 s−1, and C2 → C1, 440 ± 30 s−1 versus 370 ± 30 s−1 (n = 7; p < 0.05); and it slowed at least ∼3-fold the recovery steps from the two desensitized states C5 and C4 as follows: C3 ← C5, 1.0 ± 0.3 s−1 versus 0.3 ± 0.1 s−1, and C2 ← C4, 26 ± 8 s−1 versus 5 ± 2 s−1 (Fig. 3B). In contrast, we observed no significant differences in any of the 10 rate constants after water treatment (data not shown). These changes in gating predict that PKA inhibition would cause active channels to accumulate in desensitized states, as illustrated by the occupancy plots in Fig. 3B where, following PKI treatment, fractional occupancy of open (O) and pre-open (C3, C2, and C1) states decreased and that of desensitized states (C5 and C4) increased from 0.45 to 0.75.
To determine how these gating changes observed for individual channels alter ensemble responses, we simulated with the kinetic schemes illustrated in Fig. 3B currents from 1000 channels. The kinetic models predicted that PKA inhibition would reduce the peak of the macroscopic response without significantly altering the time course of macroscopic desensitization (Fig. 3C; basal, τ = 495; +PKI, τ = 544 ms). Consistent with this prediction, PKI reduced the experimentally recorded whole-cell peak current amplitude ∼2-fold and did not change the decay time course (τbasal = 610 ± 300 ms, τPKI = 580 ± 200 ms, n = 5, p < 0.5). The model predicted slightly more inhibition of the steady-state current than observed experimentally, which may reflect additional differences due to distinct (cell-attached versus whole cell) recording configuration. In summary, we found that PKI treatment caused N1/N2B receptors to activate more slowly and substantially stabilized their desensitized states.
N2B CTD Mediates PKI Gating Effect, in Part through Ser-1166
We probed the molecular basis for the isoform specificity of PKA modulation of NMDA receptors by carrying out parallel experiments with N1/N2A receptors. In Fig. 4A, current traces recorded from a patch containing a single N1/N2A receptor are shown before and after PKI application. In contrast to our observations with N1/N2B receptors, the activity of N1/N2A receptors remained unchanged, even 45 min after PKI treatment (Table 1). N1/N2A receptors, however, became sensitive to PKI treatment when the N2A CTD was replaced with that of the N2B subunit (Fig. 4B and Table 1) (23). We reported previously that chimeric N1/N2A2BCTD receptors maintained high N1/N2A-like basal activity and displayed considerably lower kinetic variability compared with N1/N2B receptors (26, 41). In contrast to wild-type N2A-containing receptors, PKI treatment produced a significant decrease in Po for chimeric receptors, and this occurred through a similar mechanism as the effects of PKI on N2B-containing receptors (Fig. 4C). This result demonstrates that the C-terminal tail of N2B is sufficient to transfer gating sensitivity to PKI onto N1/N2A receptors and that in both receptor isoforms this CTD-mediated effect results from longer closed times.
FIGURE 4.

Gating effects of PKI are mediated by N2B CTD, in part through Ser-1166. A and B, one-channel cell-attached currents (inward Na+) recorded with modified pipette (extracellular) solutions (0 mm Ca2+, pH 8) from the same receptor before (basal, left) or after adding PKI (+PKI, right) from cells expressing N1/2A (A) or chimeric N1/N2A2BCTD (B) receptors. C, summary of changes induced by PKI to the kinetic parameters for N1/N2A (gray, n = 4) or N1/2A2BCTD (white, n = 5) relative to basal levels. D, one-channel activity recorded from phosphomimic (S1166D, left) and phosphodeficient (S1166A, right) receptors. E, reaction mechanisms for the conditions illustrated in D. F, reaction mechanisms with rates calculated from single-channel records of S1166D receptors before (left) or after PKI treatment (right). * indicates statistical significance of S1166A relative to S1166D (E, unpaired) and relative to basal (F, paired), p < 0.05, Student's t test.
The N2B CTD therefore must contain PKA phosphorylation sites that influence receptor gating not present in N2A CTD. This is true for N2B Ser-1166, which is a PKA target and mediates the effect of PKA on neuronal NMDA receptor signals. Specifically, a phosphodeficient or phosphomimic point mutation (S1166A or S1166D) had distinct gating activity in a manner consistent with the effects we describe here for pharmacological inhibition of PKA; relative to S1166D, S1166A had lower Po due to longer closed times (28). We therefore proceeded to delineate gating mechanisms for phosphodeficient (S1166A) and phosphomimic (S1166D) N1/N2B receptors. We found that these molecular perturbations affected precisely the same rate constants as we observed with pharmacological inhibition of PKA. Relative to the phosphomimic, the phosphodeficient mutation produced slower activation rates (S1166D versus S1166A):C2 → C1, 500 ± 40 s−1 versus 330 ± 30 s−1 (n = 16, 14; p < 0.05); and slowed resensitization (S1166D versus S1166A): C3 ← C5, 1.0 ± 0.1 s−1 versus 0.5 ± 0.1 s−1; and C2 ← C4: 21 ± 4 s−1 versus 6 ± 3 s−1 (n = 16, 14; p < 0.05) (Fig. 4E). Remarkably, this result demonstrates that the state of a single PKA phosphorylation site (N2BS1166) is sufficient to influence receptor gating in a manner identical to PKI. To test whether Ser-1166 was solely responsible for the effect of PKI on N1/N2B gating, we recorded currents from cell-attached patches containing one N1/N2BS1166D receptor before and after PKI treatment, and we compared the respective reaction mechanisms. PKI reduced the Po of N1/N2BS1166D ∼3-fold (basal, 0.24 ± 0.05; +PKI, 0.08 ± 0.02). The inhibitory mechanism was distinct; however, resensitization rates were not affected, and in addition to some activation rates that were slower, closing rates also increased (Fig. 4F). Thus, although Ser-1166 appears to be a major conduit for PKI effects on N1/N2B gating, other residues may also participate.
PKI Changed Ca2+ Permeability by Specifically Reducing Unitary Ca2+ Conductance
In the experiments described above, to surpass well documented experimental limitations, we measured gating by examining Na+-only currents. A critical aspect of NMDA receptor function, however, is permeability to both Na+ and Ca2+. In physiological conditions (2 mm Ca2+), Ca2+ ions represent a fraction (∼10–15%) of the passed current (8, 9). The gating changes we measured predict that PKI treatment reduced the total charge transfer through N1/N2B receptors ∼2.5-fold, and by extrapolation, if the modulation did not affect channel conductance or permeability characteristics, PKI would also, proportionally, reduce the size of the Ca2+ flux during each activation. Recent reports show, however, that PKA can change the fraction of NMDA receptor current carried by Ca2+ (13, 15, 28). Because we did not observe any changes in Na+ conductance, we initiated experiments that included Ca2+ in extracellular solutions, such that a part of, or all the inward current, was carried by Ca2+.
We first focused on PKA-specific effects of gating, this time in the presence of Ca2+, and we recorded unitary currents from cell-attached patches with 2 mm CaCl2 added to the pipette solution. In these conditions, the basal currents recorded from N1/N2B receptors had lower Po and lower unitary current amplitude (i) as compared with 0 Ca2+ (Fig. 5A). Often, the Po decreased even during the basal recording period, before adding PKI, and we detected no additional PKI-induced reduction in Po (Fig. 5B). Because this was true for N1/N2B and N1/N2A receptors, we combined results from both isoforms, and we report these for the aggregated N1/N2 pool: basal, 0.03 ± 0.01; +PKI, 0.03 ± 0.01; p > 0.05 (Fig. 5C). These results indicate that following NMDA receptor Ca2+ influx, receptors reside in states with much lower activity, and further gating reduction by PKI cannot be detected. Whether PKI effects on gating of N1/N2B are merely masked or are entirely occluded remains unresolved.
We turned our attention to the effects of PKI on the composition of currents passed by NMDA receptors. First, we determined directly whether PKI changed NMDA receptor Ca2+ conductance. For this, we measured unitary amplitudes of inward currents recorded from cell-attached patches bathed in high K+ solutions, when the pipette solution contained as the principal permeating ion Na+ (150 mm NaCl, +100 mV) or Ca2+ (75 mm CaCl2, +100 mV). In the Ca2+-only patches, we also measured the unitary amplitudes of outward currents (−100 mV), which were presumably carried by intracellular K+. We found that in these basal conditions for both N1/N2A and N1/N2B, Ca2+ currents were much smaller (∼1 pA) than Na+ currents (∼10 pA) for the same applied potential (+100 mV) (Fig. 5D). Importantly, PKI treatment reduced the inward Ca2+ current amplitude significantly (basal, 0.96 ± 0.15 pA; +PKI, 0.40 ± 0.04, p < 0.0167). In contrast, PKI did not change the unitary amplitudes of monovalent currents, whether measured as inward Na+ (basal, 9.4 ± 0.03 pA; +PKI, 9.0 ± 0.02, p > 0.01675) or outward K+ (basal, 1.29 ± 0.17 pA; +PKI, 1.17 ± 0.16, p < 0.0167) (Fig. 5E). This latter observation was consistent with a lack of effect of PKI on Na+ unitary conductance observed in the Na+-only experiments performed in PBS-bathed cells (Figs. 2, B and D, and 4C).
These results clearly demonstrate that PKI reduced specifically the NMDA receptor unitary conductance for Ca2+ but not Na+. This novel finding may represent the principal mechanism by which, in physiological conditions, where Ca2+ ions represent a relatively small fraction of the current, PKI produces an observable reduction in Ca2+ influx but a change in total (Na+ and Ca2+) current amplitude escapes detection. Similarly, we did not detect PKI-induced changes in unitary current amplitudes with 2 mm Ca2+ in the recording pipette (and PBS in the bath) (basal, 2.3 ± 0.3; +PKI, 2.5 ± 0.5) (Fig. 5A).
If this specific reduction in Ca2+ conductance by PKI is maintained when both Na+ and Ca2+ are present in the permeating solutions, PKI would also produce a marked decrease in the Ca2+ permeability relative to that of Na+ (PCa/PNa). Classically, relative permeability is inferred by examining the magnitude of the reversal potential (Erev) shift in bi-ionic solutions. We took this approach to examine the effects of PKI treatment on Erev of single-channel NMDA receptor currents and on the shifts in Erev produced by increasing extracellular Ca2+ concentrations. We calculated Erev as the intercept of linear regressions to experimentally measured i/V data (Fig. 6A). Under basal conditions and 0 Ca2+, both N1/N2A and N1/N2B currents reversed polarity at membrane potentials close to −5 mV (N1/N2, −4.4 ± 1.2 mV), and this value shifted progressively to more positive potentials with increasing extracellular Ca2+: ∼0 mV in 2 mm Ca2+ (0.6 ± 0.9 mV) and ∼8 mV in 10 mm Ca2+ (7.8 ± 2.3 mV) (Fig. 6B). These values are equivalent with a PCa/PNa value of ∼7, similar to values reported previously (8, 9). In contrast, after PKI pretreatment, the shifts in Erev produced by increasing extracellular Ca2+ were barely detectable, and the Erev retained negative values for all conditions: 0 Ca2+, −4.9 ± 1.7 mV; 2 Ca2+, −5.1 ± 1.2 mV, and 10 Ca2+, −2.2 ± 2.5 mV. These results are similar to those reported for hippocampal neurons treated with H-89 (28) and indicate that upon exposure to PKI, channel PCa/PNa decreased substantially.
It is well established that permeating Ca2+ ions produce a concentration-dependent decrease in overall unitary conductance. This phenomenon is referred to in the literature as “Ca block,” suggestive of a hypothesis where the overall reduction in current occurs because in bi-ionic solutions the slow-moving Ca2+ ions replace Na+ ions in the pore, practically preventing Na+ passage for periods too brief to be observed in single-channel traces (42). With this consideration in mind, although the PKI-dependent decrease in relative permeability (PCa/PNa) we measured is consistent with a mechanism where PKI specifically reduced Ca2+ (but not Na+) conductance (Fig. 5, D and E), it does not address whether extracellular Ca2+ ions, although rendered “impermeant” could still block the passage of Na+ ions (42). We addressed this question by examining the extent of Ca2+ block before and after PKI treatment. We calculated unitary conductance (γ) in 0, 2, and 10 mm Ca2+ as the slope of linear fits to the i/V data illustrated in Fig. 6A. We reasoned that if PKI treatment reduced Ca2+ conductance and also reduced block of Na+ currents by Ca2+, increasing extracellular Ca2+ concentrations should have minimal effect on conductance. Conversely, if PKI reduced Ca2+ conductance but also increased the block of Na+ currents, increasing extracellular Ca2+ should decrease γ more than in basal conditions. We found that the decrease in γ with increasing extracellular Ca2+ was not different in basal and +PKI conditions (Fig. 6C). Based on this result, we conclude that PKI did not restore unitary conductance to the high level observed in the absence of Ca2+ and therefore did not substantially alleviate block. Instead, PKI reduced NMDA receptor fractional Ca2+ current by specifically reducing channel unitary Ca2+ conductance.
N1 CTD Mediates PKI Permeability Effect, in Part through N1 Ser-897
The molecular determinants for the PKI-induced decrease in Ca2+ permeability must reside intracellularly, most likely on the C termini of N1 and/or N2 subunits. To test this hypothesis, we measured Erev of unitary currents in 2 mm external Ca2+ for receptors lacking the intracellular tail of either N1 (N1stop) or N2 subunits (N2stop) before and after treating the cells with PKI (Fig. 7A). We found that the CTD of N1, but not that of N2 subunits, was required for the PKI-dependent reduction in Ca2+ permeability. This result is consistent with the indistinguishable effects of PKI on permeation properties of N1/N2A and N1/N2B receptors and suggests that phosphorylation of one or more residues in the CTD of the N1 subunit is necessary to maintain the characteristically high Ca2+ permeability of NMDA receptors.
To test this hypothesis directly, we asked whether a change in the phosphorylation status of a single residue in the N1 CTD can affect the NMDA receptor Ca2+ permeability. We selected for these experiments N1 Ser-897, which is a validated PKA target (12). We measured Erev in 2 mm Ca2+ as a measure of Ca2+ permeability for receptors with phosphomimic Ser/Asp and phosphodeficient Ser/Ala substitutions at this site. We measured a significantly more positive Erev for the phosphomimic compared with phosphodeficient receptors, 5.5 ± 2.6 mV versus −1.8 ± 1.4 mV, respectively (n = 7, 8, p < 0.05) (Fig. 7B). This result demonstrates that phosphorylation of a single residue in the N1 CTD can increase substantially the receptor Ca2+ permeability. Still, both Ser/Asp and Ser/Ala mutants remained sensitive to PKI-induced reduction in their Ca2+ permeability (data not shown), suggesting that additional PKA targets have the ability to modify receptor Ca2+ permeability.
Finally, to determine directly whether PKI-dependent changes in Ca2+ conductance translate in reduced Ca2+ transport in nonequilibrium conditions, we used patch clamp fluorometry. We recorded simultaneously whole-cell currents and fluorescence from the genetically encoded GCamP6f protein, which is a fast and highly sensitive Ca2+ fluorophore (30). For these experiments, we selected cells expressing N2A-containing receptors, because we have already established that PKI did not alter gating kinetics (Figs. 4, A and C, and 5C), but it reduced Ca2+ conductance (Fig. 5, D and E) and relative Ca2+ permeability (Fig. 6B) of these receptors. We found that under basal conditions, glutamate (1 mm, 5 s) elicited currents that were similar in magnitude and time course for all three constructs tested (N1/N2A, N1stop/N2A, and N1/N2Astop), and PKI treatment did not produce observable changes in either of these responses (Fig. 7C).
Consistent with substantial Ca2+ permeability of N1/N2A receptors, we observed that under basal conditions glutamate applications produced ∼10% increase in fluorescence, and this signal was reduced ∼5-fold after PKI treatment (ΔF/F basal, 109.4 ± 1.3; +PKI, 102.3 ± 0.3, n = 4, p < 0.016). This reduction in Ca2+-dependent fluorescence was absent for receptors lacking the N1 CTD (ΔF/F basal, 113.7 ± 3.2, +PKI 118.6 ± 5.5, n = 5, p > 0.016), but was unchanged for receptors lacking the N2A CTD (ΔF/F basal, 111.2 ± 2.8; +PKI 106.3 ± 2.0, n = 6, p < 0.016) (Fig. 7C). This result provides additional evidence that the N1 CTD, but not the N2A CTD, mediates PKI-induced changes in NMDA receptor fractional Ca2+ current.
Together, the results described here demonstrate that ongoing PKA activity is required to maintain the characteristically high gating and high Ca2+ permeability of NMDA receptors. Importantly, these PKA actions are mediated by separate sites and have distinct mechanisms; specifically, sites on N2B CTD control gating kinetics by maintaining fast activation rates and destabilizing desensitized states, whereas sites on N1 CTD control receptors Ca2+ permeability by modulating specifically their unitary Ca2+ conductance.
DISCUSSION
To better understand how PKA activity modulates neuronal signaling through NMDA receptors, we examined how changes in cellular PKA activity impacted the biophysical properties of recombinant NMDA receptor isoforms. We used pharmacological inhibition of PKA and site-directed mutagenesis to investigate the mechanisms and the molecular determinants responsible for PKA modulation of NMDA receptor signals. We determined that PKA inhibitors, such as the highly specific naturally occurring peptide PKI or the ATP-site PKA antagonist H-89, reduced macroscopic NMDA receptor responses regardless of extracellular pH, and this effect occurred with bath-applied cell-permeable inhibitors (myr-PKI, H-89), as well as with intracellularly applied inhibitors (non-myr-PKI) (Fig. 1). In the remainder of the experiments reported here, we examined the effects of bath-applied myr-PKI on the gating and permeability properties of NMDA receptors.
Using paired cell-attached recordings of Na+ currents produced by the same N1/N2B receptor before and after bath-applied PKI, we determined that PKI had no effects on Na+ conductance, but relative to water-treated controls, it reduced channel Po ∼2.5-fold, solely by increasing the time spent in closed-channel conformations (Fig. 2D). A more detailed analysis of closed event distributions revealed that PKI reduced receptor gating by specifically doubling the duration of the two longest closed components (Fig. 3A). Reaction mechanisms derived by fitting kinetic models to these single-channel data explained the PKI-induced decrease in Po by increased occupancies of desensitized states, from 0.45 for wild-type N1/N2B receptor to 0.75 for PKI-modified receptors (Fig. 3B).
This result was not intuitive because PKI did not produce marked changes in the desensitization time course of whole-cell currents (Figs. 1, A and B, and 3C). The model, however, is consistent with this interpretation as illustrated by the simulated traces in Fig. 3C. The desensitization time course and extent of macroscopic NMDA receptor responses vary substantially with patch configuration (whole-cell or excised patch) and the extracellular ionic composition (Zn2+, Gly, and Ca2+) (43). Therefore, our approach, which employed recording activity from receptors that resided in nonperturbed membranes (cell-attached patches) and external solutions devoid of extracellular modulators (pH 8, 0 mm Ca2+, and EDTA), revealed the specific effect of PKI on NMDA receptor-desensitized states by extricating it from uncontrolled confounding factors. More precisely, we were able to identify a change in the rates for receptor recovery from desensitized states and rates toward opening as the main kinetic mechanism of PKI-induced reduction on channel Po (Fig. 3B).
As expected, a PKI-dependent effect was not apparent in the presence of steady-state Ca2+ influx through the recorded receptors (Fig. 5A). Previous groups found that NMDA receptor-mediated Ca2+ influx activates the Ca2+-dependent phosphatase calcineurin (PP2B), which targets NMDA receptors and reduces their activity (19, 40). The already much lower activity (Po, 0.03) we recorded basally when 2 mm Ca2+ was included in the pipette may have prevented us from detecting further PKI-dependent desensitization; alternatively, active calcineurin may have preempted the effect of PKI, possibly by acting on the same residues. Relevant to this issue, Raman et al. (19) have shown that PKA prevents calcineurin-dependent desensitization of synaptic NMDA receptor responses. The phosphatase responsible for the PKI-induced reduction in N1/N2B Po described here, however, is currently unknown.
We traced the molecular basis of PKI modulatory effects on N1/N2B gating to the CTD of the 2B subunit. Moreover, we found that when swapped for the native 2A CTD, the 2B CTD can transfer PKI gating sensitivity onto otherwise insensitive N1/N2A receptors (Fig. 4C). This result readily explains the isoform specificity of PKI-induced decrease in NMDA receptor currents (13). Notably, although the chimeric N1/N2A2B CTD retained characteristic N2A-like kinetics, the mechanism of the PKI-induced decrease in Po was similar: >2.5-fold increase in mean closed times. Even more striking, phosphodeficient and phosphomimic mutations of the N2B-specific PKA site, Ser-1166, altered the reaction mechanism by changing the same rates as PKI when acting on wild-type N1/N2B or chimeric N1/2A2B CTD channels (Figs. 3B and 4E). The fact that the S1166D mutant remained sensitive to PKI inhibition suggested that N2B CTD contains additional PKA targets, but these mediate distinct kinetic effects. Consistent with this view, S1166A reduces phosphorylation of N2B peptides by only ∼50% relative to wild type (28). Nevertheless, our result that perturbations of a single N2B CTD residue (Ser-1166), which is most likely remote from the gate, can substantially alter gating with the same kinetic mechanisms as PKI represents strong evidence that Ser-1166 is a principal target for PKI on N1/N2B receptors.
The lack of PKI effect on single-channel N1/N2A gating we observed here is consistent with previous work showing that PKA inhibition does not alter macroscopic N1/N2A currents (13). In contrast however, PKA phosphorylation of N1/N2A receptors lengthens NMDA receptor-mediated EPSCs in neurons (44), and the Ser/Ala modification of known targets on N2A CTD (Ser-900 or Ser-929) alters receptor gating in a manner similar to that described here for N1/N2BS1166A and PKI treatment of wild-type N1/N2B (45). Together, these results suggest that the phosphorylation status of N2A-containing receptors was not altered by PKI treatment. Thus, although the gating of both N1/N2A and N1/N2B isoforms is PKA-sensitive and the modulation may have the same kinetic mechanism, several other factors critical to the modulation are distinct between isoforms, such as the kinetics and specificities of the phosphatases involved and/or the kinetics and affinity of PKA for N2A and N2B sites.
A second major focus of our investigation was the PKI effect on NMDA receptor permeability. We used single-channel electrophysiology to measure in basal and +PKI conditions the following: unitary conductance for Na+ and Ca2+ currents; Ca2+-dependent shifts in Erev; and the extent of Ca2+ block in bi-ionic conditions. In addition, we used patch clamp fluorometry to simultaneously measure whole-cell current and increase in intracellular Ca2+ before and after treating cells with PKI. We found that PKA inhibition produced a substantial decrease of the unitary Ca2+ conductance but not of monovalent cation (Na+ or K+) conductance. Consistent with a decrease in Ca2+ but not Na+ permeability, PKI also decreased the PCa/PNa, evaluated as changes in Erev with increasing extracellular [Ca2+]. PKI did not substantially change the extent of Ca2+ block, an indication of interactions between co-permeant Na+ and Ca2+ ions, however. Also, because we could not detect a PKI-induced change in unitary conductance when both Na+ and Ca2+ were present at physiological concentrations (2 mm Ca2+ and 150 mm Na+), we conclude that PKI decreased the amount of Ca2+ but not that of Na+ in the total current. If anything, Na+ currents might increase slightly due to less Ca2+-dependent inactivation/desensitization processes in +PKI condition. This interpretation is consistent with the result that PKI decreased the Ca2+ flux by ∼50% but not the total whole-cell current. Based on these results, we suggest that the principal effect of PKI on NMDA receptor permeability is a specific decrease in unitary Ca2+ conductance. These results are the first description of a selective modulation of the Ca2+ unitary conductance of NMDA receptors and demonstrate that changes in PKA activity (PKI treatment) and also perturbations at N1 Ser-897 can produce this effect.
This work therefore describes two mechanisms whereby PKA can alter NMDA receptor-mediated neuronal Ca2+ influx as follows: PKA modulates total charge transfer via an N2B-mediated change in the stability of desensitized states, and it modulates the amount of Ca2+ present in the current passed through an N1 CTD-mediated mechanism. Importantly, the intramolecular separation of sites affecting gating and permeation allows PKA to exert global (through N1) and more local, isoform-specific (through N2) control on NMDA receptors. These new observations indicate that sensitivity to PKA and the effects of PKA on synaptic currents will depend on the types and relative abundance of the NMDA receptor isoforms expressed and also on the phosphatases that act to balance PKA activity. Additionally, PKA phosphorylation of NMDA receptors is regulated by neuronal and synaptic activity (19, 46–48) and a variety of neuromodulators (14, 15, 19). The mechanisms of regulation described here begin to unravel this complex control of synaptic activity and plasticity throughout development, in distinct brain regions, and following different patterns of neuronal activity.
Acknowledgments
We thank Dr. Gary Westbrook for providing CTD truncation constructs for GluN1 and GluN2A subunits, Dr. Martha Constantine-Paton for providing chimeras with swapped CTDs, Dr. John Woodward for providing the GluN1 Ser-897 mutations, and Dr. Arndt Pralle for cCAMP6f.
This work was supported, in whole or in part, by National Institutes of Health Grants R01052669 (to G. K. P.) and F32NS077622 (to T. K. A.).
- CTD
- C-terminal domain
- Tricine
- N-[2-hydroxy-1,1-bis(hydroxymethyl)ethyl]glycine
- MOT
- mean open time
- MCT
- mean closed time
- myr
- myristoylated
- PKI
- protein kinase A inhibitor
- 8-Br-cAMP
- 8-bromo-cAMP
- EPSC
- excitatory postsynaptic current.
REFERENCES
- 1. Shouval H. Z., Bear M. F., Cooper L. N. (2002) A unified model of NMDA receptor-dependent bidirectional synaptic plasticity. Proc. Natl. Acad. Sci. U.S.A. 99, 10831–10836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Choi D. W. (1994) Calcium and excitotoxic neuronal injury. Ann. N.Y. Acad. Sci. 747, 162–171 [DOI] [PubMed] [Google Scholar]
- 3. Bliss T. V., Collingridge G. L. (1993) A synaptic model of memory: long-term potentiation in the hippocampus. Nature 361, 31–39 [DOI] [PubMed] [Google Scholar]
- 4. Nowak L., Bregestovski P., Ascher P., Herbet A., Prochiantz A. (1984) Magnesium gates glutamate-activated channels in mouse central neurones. Nature 307, 462–465 [DOI] [PubMed] [Google Scholar]
- 5. MacDermott A. B., Mayer M. L., Westbrook G. L., Smith S. J., Barker J. L. (1986) NMDA-receptor activation increases cytoplasmic calcium concentration in cultured spinal cord neurones. Nature 321, 519–522 [DOI] [PubMed] [Google Scholar]
- 6. Lester R. A., Clements J. D., Westbrook G. L., Jahr C. E. (1990) Channel kinetics determine the time course of NMDA receptor-mediated synaptic currents. Nature 346, 565–567 [DOI] [PubMed] [Google Scholar]
- 7. Monyer H., Burnashev N., Laurie D. J., Sakmann B., Seeburg P. H. (1994) Developmental and regional expression in the rat brain and functional properties of four NMDA receptors. Neuron 12, 529–540 [DOI] [PubMed] [Google Scholar]
- 8. Burnashev N., Schoepfer R., Monyer H., Ruppersberg J. P., Günther W., Seeburg P. H., Sakmann B. (1992) Control by asparagine residues of calcium permeability and magnesium blockade in the NMDA receptor. Science 257, 1415–1419 [DOI] [PubMed] [Google Scholar]
- 9. Watanabe J., Beck C., Kuner T., Premkumar L. S., Wollmuth L. P. (2002) DRPEER: a motif in the extracellular vestibule conferring high Ca2+ flux rates in NMDA receptor channels. J. Neurosci. 22, 10209–10216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chen B. S., Roche K. W. (2007) Regulation of NMDA receptors by phosphorylation. Neuropharmacology 53, 362–368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Leonard A. S., Hell J. W. (1997) Cyclic AMP-dependent protein kinase and protein kinase C phosphorylate N-methyl-d-aspartate receptors at different sites. J. Biol. Chem. 272, 12107–12115 [DOI] [PubMed] [Google Scholar]
- 12. Tingley W. G., Ehlers M. D., Kameyama K., Doherty C., Ptak J. B., Riley C. T., Huganir R. L. (1997) Characterization of protein kinase A and protein kinase C phosphorylation of the N-methyl-d-aspartate receptor NR1 subunit using phosphorylation site-specific antibodies. J. Biol. Chem. 272, 5157–5166 [DOI] [PubMed] [Google Scholar]
- 13. Skeberdis V. A., Chevaleyre V., Lau C. G., Goldberg J. H., Pettit D. L., Suadicani S. O., Lin Y., Bennett M. V., Yuste R., Castillo P. E., Zukin R. S. (2006) Protein kinase A regulates calcium permeability of NMDA receptors. Nat. Neurosci. 9, 501–510 [DOI] [PubMed] [Google Scholar]
- 14. Chalifoux J. R., Carter A. G. (2010) GABAB receptors modulate NMDA receptor calcium signals in dendritic spines. Neuron 66, 101–113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Higley M. J., Sabatini B. L. (2010) Competitive regulation of synaptic Ca2+ influx by D2 dopamine and A2A adenosine receptors. Nat. Neurosci. 13, 958–966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Cerne R., Rusin K. I., Randić M. (1993) Enhancement of the N-methyl-d-aspartate response in spinal dorsal horn neurons by cAMP-dependent protein kinase. Neurosci. Lett. 161, 124–128 [DOI] [PubMed] [Google Scholar]
- 17. Westphal R. S., Tavalin S. J., Lin J. W., Alto N. M., Fraser I. D., Langeberg L. K., Sheng M., Scott J. D. (1999) Regulation of NMDA receptors by an associated phosphatase-kinase signaling complex. Science 285, 93–96 [DOI] [PubMed] [Google Scholar]
- 18. Bird G. C., Lash L. L., Han J. S., Zou X., Willis W. D., Neugebauer V. (2005) PKA-dependent enhanced NMDA receptor function in pain-related synaptic plasticity in amygdala neurons. J. Physiol. 5, 907–921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Raman I. M., Tong G., Jahr C. E. (1996) β-Adrenergic regulation of synaptic NMDA receptors by cAMP-dependent protein kinase. Neuron 16, 415–421 [DOI] [PubMed] [Google Scholar]
- 20. Sprengel R., Suchanek B., Amico C., Brusa R., Burnashev N., Rozov A., Hvalby O., Jensen V., Paulsen O., Andersen P., Kim J. J., Thompson R. F., Sun W., Webster L. C., Grant S. G., Eilers J., Konnerth A., Li J., McNamara J. O., Seeburg P. H. (1998) Importance of the intracellular domain of NR2 subunits for NMDA receptor function in vivo. Cell 92, 279–289 [DOI] [PubMed] [Google Scholar]
- 21. Lau C. G., Takeuchi K., Rodenas-Ruano A., Takayasu Y., Murphy J., Bennett M. V., Zukin R. S. (2009) Regulation of NMDA receptor Ca2+ signalling and synaptic plasticity. Biochem. Soc. Trans. 37, 1369–1374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Li B., Devidze N., Barengolts D., Prostak N., Sphicas E., Apicella A. J., Malinow R., Emamian E. S. (2009) NMDA receptor phosphorylation at a site affected in schizophrenia controls synaptic and behavioral plasticity. J. Neurosci. 29, 11965–11972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Foster K. A., McLaughlin N., Edbauer D., Phillips M., Bolton A., Constantine-Paton M., Sheng M. (2010) Distinct roles of NR2A and NR2B cytoplasmic tails in long-term potentiation. J. Neurosci. 30, 2676–2685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Krupp J. J., Vissel B., Thomas C. G., Heinemann S. F., Westbrook G. L. (2002) Calcineurin acts via the C terminus of NR2A to modulate desensitization of NMDA receptors. Neuropharmacology 42, 593–602 [DOI] [PubMed] [Google Scholar]
- 25. Krupp J. J., Vissel B., Thomas C. G., Heinemann S. F., Westbrook G. L. (1999) Interactions of calmodulin and α-actinin with the NR1 subunit modulate Ca2+-dependent inactivation of NMDA receptors. J. Neurosci. 19, 1165–1178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Maki B. A., Aman T. K., Amico-Ruvio S. A., Kussius C. L., Popescu G. K. (2012) C-terminal domains of N-methyl-d-aspartic acid receptor modulate unitary channel conductance and gating. J. Biol. Chem. 287, 36071–36080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Xu M., Smothers C. T., Woodward J. J. (2011) Effects of ethanol on phosphorylation site mutants of recombinant N-methyl-d-aspartate receptors. Alcohol 45, 373–380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Murphy J. A., Stein I. S., Lau C. G., Peixoto R. T., Aman T. K., Kaneko N., Aromolaran K., Saulnier J. L., Popescu G. K., Sabatini B. L., Hell J. W., Zukin R. S. (2014) Phosphorylation of Ser1166 on GluN2B by PKA is critical to synaptic NMDA receptor function and Ca2+ signaling in spines. J. Neurosci. 34, 869–879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Tash J. S., Welsh M. J., Means A. R. (1980) Protein inhibitor of cAMP-dependent protein kinase: production and characterization of antibodies and intracellular localization. Cell 21, 57–65 [DOI] [PubMed] [Google Scholar]
- 30. Chen T.-W., Wardill T. J., Sun Y., Pulver S. R., Renninger S. L., Baohan A., Schreiter E. R., Kerr R. A., Orger M. B., Jayaraman V., Looger L. L., Svoboda K., Kim D. S. (2013) Ultrasensitive fluorescent proteins for imaging neuronal activity. Nature 499, 295–300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Qin F., Auerbach A., Sachs F. (1996) Estimating single-channel kinetic parameters from idealized patch-clamp data containing missed events. Biophys. J. 70, 264–280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Qin F., Auerbach A., Sachs F. (1997) Maximum likelihood estimation of aggregated Markov processes. Proc. Biol. Sci. 264, 375–383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kussius C. L., Kaur N., Popescu G. K. (2009) Pregnanolone sulfate promotes desensitization of activated NMDA receptors. J. Neurosci. 29, 6819–6827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Dalton G. D., Dewey W. L. (2006) Protein kinase inhibitor peptide (PKI): a family of endogenous neuropeptides that modulate neuronal cAMP-dependent protein kinase function. Neuropeptides 40, 23–34 [DOI] [PubMed] [Google Scholar]
- 35. Amico-Ruvio S. A., Popescu G. K. (2010) Stationary gating of GluN1/GluN2B receptors in intact membrane patches. Biophys. J. 98, 1160–1169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Popescu G., Auerbach A. (2003) Modal gating of NMDA receptors and the shape of their synaptic response. Nat. Neurosci. 6, 476–483 [DOI] [PubMed] [Google Scholar]
- 37. Krupp J. J., Vissel B., Heinemann S. F., Westbrook G. L. (1996) Calcium-dependent inactivation of recombinant N-methyl-d-aspartate receptors is NR2 subunit specific. Mol. Pharmacol. 50, 1680–1688 [PubMed] [Google Scholar]
- 38. Legendre P., Rosenmund C., Westbrook G. L. (1993) Inactivation of NMDA channels in cultured hippocampal neurons by intracellular calcium. J. Neurosci. 13, 674–684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Rosenmund C., Westbrook G. L. (1993) Rundown of N-methyl-d-aspartate channels during whole-cell recording in rat hippocampal neurons: role of Ca2+ and ATP. J. Physiol. 470, 705–729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Tong G., Shepherd D., Jahr C. E. (1995) Synaptic desensitization of NMDA receptors by calcineurin. Science 267, 1510–1512 [DOI] [PubMed] [Google Scholar]
- 41. Borschel W. F., Myers J. M., Kasperek E. M., Smith T. P., Graziane N. M., Nowak L. M., Popescu G. K. (2012) Gating reaction mechanism of neuronal NMDA receptors. J. Neurophysiol. 108, 3105–3115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ascher P., Nowak L. (1988) The role of divalent cations in the N-methyl-d-aspartate responses of mouse central neurones in culture. J. Physiol. 399, 247–266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Amico-Ruvio S. A., Murthy S. E., Smith T. P., Popescu G. K. (2011) Zinc effects on NMDA receptor gating kinetics. Biophys. J. 100, 1910–1918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Townsend M., Liu Y., Constantine-Paton M. (2004) Retina-driven dephosphorylation of the NR2A subunit correlates with faster NMDA receptor kinetics at developing retinocollicular synapses. J. Neurosci. 24, 11098–11107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Maki B. A., Cole R., Popescu G. K. (2013) Two serine residues on GluN2A C-terminal tails control NMDA receptor current decay times. Channels 7, 126–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Sobczyk A., Svoboda K. (2007) Activity-dependent plasticity of the NMDA-receptor fractional Ca2+ current. Neuron 53, 17–24 [DOI] [PubMed] [Google Scholar]
- 47. Chatterjea D., Hamid E., Leonard J. P., Alford S. (2010) Phosphorylation-state-dependent regulation of NMDA receptor short-term plasticity modifies hippocampal dendritic Ca2+ transients. J. Neurophysiol. 104, 2203–2213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Gambrill A. C., Storey G. P., Barria A. (2011) Dynamic regulation of NMDA-Receptor transmission. J. Neurophysiol. 105, 162–171 [DOI] [PMC free article] [PubMed] [Google Scholar]