

Background: CD3 subunits are essential signaling components of the TCR.
Results: The membrane proximal CD3 CXXC motif is constitutively oxidized and critical for subunit conformation.
Conclusion: The CXXC intramolecular disulfide bond is an important structural feature of the CD3 subunits that couples extracellular activating events to intracellular signaling regulation.
Significance: Redox characterization provides insight into CD3 rigidifying elements in mechanotransduction.
Keywords: Disulfide, Mass Spectrometry (MS), Mechanotransduction, Nuclear Magnetic Resonance (NMR), Signaling, T-cell Receptor (TCR), CD3, Transmembrane
Abstract
The CD3ϵγ and CD3ϵδ heterodimers along with the CD3ζζ homodimer are the signaling components of the T cell receptor (TCR). These invariant dimers are non-covalently associated on the T cell plasma membrane with a clone-specific (i.e. clonotypic) αβ heterodimer that binds its cognate ligand, a complex between a particular antigenic peptide, and an MHC molecule (pMHC). These four TCR dimers exist in a 1:1:1:1 stoichiometry. At the junction between the extracellular and transmembrane domains of each mammalian CD3ϵ, CD3γ, and CD3δ subunit is a highly conserved CXXC motif previously found to be important for thymocyte and T cell activation. The redox state of each CXXC motif is presently unknown. Here we show using LC-MS and a biotin switch assay that these CXXC segments are constitutively oxidized on resting and activated T cells, consistent with their measured reduction potential. NMR chemical shift perturbation experiments comparing a native oxidized CD3δ CXXC-containing segment with that of a mutant SXXS-containing CD3δ segment in LPPG (1-palmitoyl-sn-glycero-3-phospho-(1′-rac-glycerol) (sodium salt)) micelles show extensive chemical shift differences in residues within the membrane-proximal motif as well as throughout the transmembrane and cytoplasmic domains as a result of the elimination of the native disulfide. Likewise, direct comparison of the native CD3δ segment in oxidizing and reducing conditions reveals numerous spectral differences. The oxidized CXXC maintains the structure within the membrane-proximal stalk region as well as that of its contiguous transmembrane and cytoplasmic domain, inclusive of the ITAM (immunoreceptor tyrosine-based activation motif) involved in signaling. These results suggest that preservation of the CD3 CXXC oxidized state may be essential for TCR mechanotransduction.
Introduction
The CD3ϵ, CD3γ, and CD3δ integral transmembrane proteins are components of both the pre-TCR2 and the TCR complexes and, as such, are required for proper T cell development and cognate recognition of antigen by mature T cells (1–5). The critical role of CD3 has been highlighted in cases of human CD3 deficiency (6–9). Each CD3 subunit contains extracellular, transmembrane (TM), and intracellular components that are necessary to mediate TCR function (10–12). These CD3 subunits interact to form CD3ϵγ and CD3ϵδ heterodimers that non-covalently associate with the TCRα and -β subunits on the surface of αβ T cells (for review, see Ref. 3). After assembly of these three heterodimeric components on the T cell membrane, the CD3ζζ homodimer associates, thereby forming the TCR complex. The TCRα and -β chains engage in a direct interaction with antigen upon recognition of surface-displayed peptide-MHC complexes (pMHC) on antigen-presenting cells but themselves lack any significant intracellular signaling motifs, being composed of ∼5–6-amino acid-long cytoplasmic tails (3, 13). Conversely, although the CD3 subunits themselves do not participate directly in pMHC antigen recognition, they are critical for relaying TCR-pMHC binding events intracellularly using their ITAM (immunoreceptor tyrosine-based activation motif)-containing, relatively lengthy (50–60-amino acid-long) cytoplasmic tails (11, 14, 15). The TM domain of each CD3 subunit is important for TCR complex assembly and is pivotal in linking extracellular binding events to intracellular signaling outcomes (16).
Just bordering the TM domains of CD3ϵ, CD3γ, and CD3δ is a highly conserved CXXC motif within this membrane proximal region of each subunit. The CD3 CXXC motif is important for CD3ϵγ and CD3ϵδ heterodimer association (17–20). Moreover, mutation of the cysteine residues in the CXXC motif has a significant effect on pre-TCR and αβ TCR development and signaling (19, 21). Although the physiological state of the CXXC motif has not been definitively characterized, it is known that the cysteines residues neither participate in intermolecular disulfide bonding interactions nor in metal ion coordination (19, 20). It has been postulated that the adjacent, closely spaced cysteines might form an intramolecular disulfide bond to provide structural stability to the membrane proximal region and TM domain (18). In this regard, CXXC motifs at the N terminus of α-helices have been observed to form intramolecular disulfide bonds that cap the helix and confer marked stability to the helical structure (22). If this were the case, then the contribution of a CD3 CXXC intramolecular disulfide bond at the N terminus of the TM domain could provide needed rigidity to the protein subunit to facilitate the recently described mechanosensing properties of the TCR (23–27). The force generated from TCR-pMHC binding interactions may be transmitted to the structurally stabilized CXXC motif in the membrane proximal region and capped TM segment for efficient transfer across the plasma membrane to initiate intracellular signaling cascades in response to extracellular antigen ligation under load (i.e. low piconewton force).
To investigate these possibilities we employed a combination of NMR, MS, and biochemical techniques to definitively characterize the native redox state of the CD3 CXXC motif on the surface of T cells. We can conclude from our results that each CXXC motif found in the CD3ϵ, CD3γ, and CD3δ subunits form highly stable intramolecular disulfide bonds. The CXXC disulfide bond remains constitutively oxidized even in the presence of a large excess of chemical reductant or when incubated with thioredoxin. Additionally, the oxidized state is preserved during T cell activation. Loss of the intramolecular disulfide bond either by reduction or mutation induced extensive conformational effects within the CXXC motif that were propagated throughout the TM domain and cytoplasmic tail when examined by NMR measurements using purified CD3 protein segments. The oxidation state of the CXXC is, therefore, coupled to the conformation of the signaling domain.
EXPERIMENTAL PROCEDURES
Reagents
Trypsin (sequencing grade) was purchased from Promega (Madison, WI), endoproteinase Lys-C (MS grade) was from Wako Chemicals USA (Richmond, VA), and endoproteinase Glu-C (sequencing grade was from Staphylococcus aureus V8) from Roche Applied Science. Peptide N-glycosidase F (proteomics grade), guanidine hydrochloride, ammonium bicarbonate, l-dithiothreitol (DTT), iodoacetamide, N-ethylmaleimide (NEM), and formic acid (optima LC/MS) were obtained from Sigma. LC-MS grade acetonitrile and water was purchased from Mallinckrodt Baker.
His-GB1-CXXC Peptide Constructs and Expression
The peptide constructs contained an N-terminal His tag, GB1 domain, a Met residue followed by the CXXC motif, and five residues C-terminal corresponding to each specific mouse CD3 subunit. The poly-Ala control peptide sequence used was CAACAAAAKAAAAKGY (22). Cloned constructs were isotopically expressed in the soluble fraction at yields of ∼5–10 mg/liter in M9 minimal media containing [15N]ammonium chloride (Cambridge Isotope Laboratories) as the sole nitrogen source. Expressed proteins were purified in PBS using cobalt metal affinity resin (Clontech) followed by S75 size exclusion chromatography.
CD3δ TMC-CXXC and CD3δ TMC-SXXS Constructs and Expression
Each construct contained an N-terminal His tag, GB1 domain, and a flexible linker containing a tobacco etch virus protease site followed by the native mouse CD3 sequences beginning at four residues N-terminal to the CXXC motif and extending to the C terminus of the cytoplasmic tail. Cloned constructs were isotopically expressed at yields of ∼10 mg/liter in M9 minimal media containing [15N]ammonium chloride as the sole nitrogen source. Expressed proteins were solubilized from the insoluble fraction using TBS containing 1% SDS. During solubilization of the CD3δ TMC-CXXC segment, the protein was oxidized with 5 mm GSSG and 2 mm S-methyl methanethiosulfonate, whereas CD3δ TMC-SXXS was oxidized with 2 mm only. The protein segments were purified by cobalt metal affinity resin, and then digested with tobacco etch virus protease in Tris buffer, pH 7.5, containing 4 mm dodecyl maltoside (Anatrace) and 4 mm DTT. After complete digestion, the samples were TCA-precipitated, washed with acetone, and dried thoroughly. The protein pellets were then resolubilized in TBS containing 5 mm SDS and reoxidized with GSSG and S-methyl methanethiosulfonate (CD3δ TMC CXXC) or S-methyl methanethiosulfonate alone (CD3δ TMC SXXS). Cleaved N-terminal tags were removed from the protein solution by cobalt metal affinity resin, and the CD3 segments were purified to homogeneity by size exclusion chromatography. After the final purification step, the protein segments were TCA-precipitated, washed with acetone, and dried thoroughly. The samples were then dissolved in 30 mm Tris buffer, pH 7.0, containing 100 mm LPPG, 10% D2O, and 0.02% NaN3 for NMR analysis. The CD3δ TMC CXXC sample used for completion of the backbone assignments was solubilized in [2H]palmitoyl lysophosphoglycerol (16:0) buffer (d31-LPPG was obtained from fbreagents.com, Cambridge, MA).
Jurkat Cell Culture and Reduction Experiments
Rex cells were previously enriched via repeated cell sorting of a high CD3-expressing population from Jurkat cell lines (28). Rex cells were cultured using DMEM media supplemented with 20% heat-inactivated fetal bovine serum, l-glutamine (2 mm), penicillin (100 units/ml), streptomycin (100 μg/ml), and 5 mm 2-mercaptoethanol. Cells were cultured in a 37 °C incubator with 5% CO2. Rex cells were maintained at a concentration of less than one million cells/ml of media during culture. Over a billion cells were grown from each seed culture, and a new stock was thawed for subsequent expansions. Approximately 5 × 108 cells were pooled for each control sample and tested sample condition, washed with 37 °C PBS, and resuspended in PBS at 37 °C for the reduction experiments. For samples investigating CD3 reduction by DTT, the control sample was treated with 6 mm NEM, and the reduced sample was treated with 2 mm DTT. Both samples were incubated at 25 °C for 10 min. After incubation, 6 mm NEM was added to the reduced sample, and then both samples underwent CD3 subunit purification. For samples investigating CD3 reduction by thioredoxin (Trx; Sigma, human recombinant), the control was treated with 100 μm DTT, and the reduced sample was treated with 100 μm DTT and 5 μg/ml Trx. Both samples were incubated at 25 °C for 15 min, and then 1.5 mm NEM was added to each sample before CD3 subunit purification.
PBMC Cell Culture and Activation
Leukopacks were obtained from the Jimmy Fund Kraft Family Blood Donor Center at the Dana-Farber Cancer Institute under an institute-approved board. PBMCs were isolated from leukopacks using Ficoll-Paque centrifugation and plated at one million cells/ml using DMEM media supplemented with 20% heat inactivated fetal bovine serum, l-glutamine (2 mm), penicillin (100 units/ml), and streptomycin (100 μg/ml). T cells in PBMCs were activated with anti-CD3 antibody (2AD2 (anti-CD3ϵ) (1:500 dilution) (29) or the combination of mitogenic anti-T112 and anti-T113 anti-CD2 antibodies (1:200 dilution) (30) and 1 μg/ml PMA). Antibodies were maintained as frozen ascitic fluid stocks at ∼1–2 mg/ml as previously described (29, 30). The surface of 2Ad2/PMA-activated PBMCs were monitored at 0, 1, 24, and 48 h with anti-CD3 (clone HIT3a), anti-CD4 (clone RPA-T4), and anti-CD8 (clone RPA-T8) in PBS buffer with 5% FBS, 0.1% sodium azide for 30 min. Samples were washed, fixed with 4% paraformaldehyde (Sigma), and analyzed using a FACsAria flow cytometer (BD Biosciences).
CD3 Purification from Jurkat Cells
Jurkat cells were centrifuged at 2000 rpm and washed twice with ice-cold PBS containing 1.5 mm NEM. The cells were first lysed in 20 mm Tris, pH 7.5, 1.5 mm NEM hypotonic buffer and centrifuged at 16,000 rpm, and the supernatant was removed. The cell pellets were resuspended in TBS lysis buffer containing 1% Triton X-100, 60 mm octyl glucoside, 1.5 mm NEM, 1 mm PMSF, and a Roche Applied Science protease inhibitor tablet. Lysis proceeded on ice for 15 min, and then the samples were centrifuged at 16,000 rpm. The cell lysate was then added to an anti-CD3ϵ or concanavalin A affinity purification column.
Anti-CD3ϵ Affinity Purification
Lysate was added to either an anti-CD3ϵ RW2–8C8 (31) or Leu4 (BD Biosciences) antibody column prepared at a concentration of 5 mg/ml of antibody and cross-linked to protein-G beads and incubated on the column overnight at 4 °C with rotation. After incubation, the lysate flowed through the column, the column was washed extensively with lysis buffer, and the CD3 subunits were eluted with 0.1 m glycine, pH 3.0, and neutralized immediately upon elution with Tris buffer. Column fractions were run on non-reducing SDS-PAGE gels and silver-stained. Those fractions containing bands in the 25- to 15-kDa region, corresponding to the molecular masses of the CD3 subunits, were pooled and concentrated to 0.2 ml using 10-kDa Millipore centrifugal filter unit. Samples of the concentrated eluate were run on non-reducing SDS-PAGE gels and Coomassie-stained. Bands in the 15–25-kDa region were excised and underwent MS analysis.
Concanavalin A Affinity Purification
Cell lysate was added to a 2-ml concanavalin A (ConA) column at ∼10 mg of ConA/ml of bead (GE Healthcare) and incubated overnight on the column at 4 °C with rotation. After incubation, the lysate flowed through the column, which was then washed with radioimmune precipitation assay buffer containing 1.5 mm NEM followed by TBS, pH 7.0, containing 1% Triton X-100, 1.5 mm NEM. CD3 was then eluted from the column with TBS, pH 7.0, containing 1% Triton X-100, 0.5 m methyl mannoside, and 1.5 mm NEM (CD3 samples treated with Trx or DTT post-lysis and purification lacked NEM from the second column wash step and elution). Samples from the biotin switch assay were directly run on a non-reducing SDS-PAGE gel, transferred to a PVDF membrane, and Western-blotted for anti-CD3ϵ (32) or streptavidin-HRP (Pierce) reactivity. Samples undergoing MS analysis were TCA-precipitated, washed with acetone, and dried. The samples were then resolubilized in SDS-PAGE gel running buffer containing NEM and separated by non-reducing SDS-PAGE. The gel was Coomassie-stained, and bands in the 15–25-kDa region were excised and underwent MS analysis.
Native In-gel Digestion
The gel bands were cut into 1-mm × 1-mm pieces, destained by acetonitrile, and dried in a SpeedVac (Labconco, Centrivap Cold Trap). The gel pieces were then rehydrated into the enzyme buffer, which was 12.5 ng/μl of each enzyme (Lys-C, trypsin, Glu-C, and peptide N-glycosidase F) in 25 mm ammonium bicarbonate at 4 °C for 30 min. Subsequently, the enzyme buffer was replaced by 25 mm ammonium bicarbonate and incubated at 37 °C overnight. Finally, the digest was extracted by acetonitrile and concentrated to almost complete dryness in the SpeedVac.
LC-MS
An Ultimate 3000 nano-LC pump (Dionex, Mountain View, CA) and a self-packed C18 column (Magic C18, 200 Å pore and 5-μm particle size, 75 μm inner diameter × 15 cm) (Michrom Bioresources, Auburn, CA) was coupled online to an LTQ-Orbitrap-ETD XL mass spectrometer (Thermo Fisher Scientific, San Jose, CA) through a nanospray ion source (New Objective, Woburn, MA). Mobile phase A (0.1% formic acid in water) and mobile phase B (0.1% formic acid in acetonitrile) were used for the gradient, which consisted of (i) 20 min at 2% B for sample loading 0.2 μl/min, (ii) linear from 2 to 5% B for 2 min, (iii) linear from 5 to 35% B for 60 min, (iv) linear from 35 to 90% B for 3 min, and finally (v) isocratic at 90% B for 5 min. The LTQ-Orbitrap-ETD XL mass spectrometer was operated initially in the data-dependent mode as follows: survey full-scan MS spectra (m/z 350–2000) were acquired in the Orbitrap with a mass resolution of 30,000 at m/z 400 (with an ion target value of 5 × 105 ions) followed by nine sequential MS2 scans using the LTQ mass spectrometer.
Data Analysis
The raw data were initially searched against the human CD3ϵ, -γ, and -δ sequence database in Proteome Discoverer 1.3 with a mass tolerance of ≤10 ppm for the precursor ions and ±0.8 Da of product ions with unspecific enzymatic digestion. The final confirmation of the peptide assignment was obtained by manual inspection to match the high abundant product ions with the precursor ion mass accuracy <5 ppm. The intrachain disulfide was assigned by the loss of a hydrogen (−1 Da) at each disulfide-involved cysteine.
CD3 Reduction Experiments after Cell Lysis and CD3 Purification
Concanavalin A-purified samples were divided into 200-μl aliquots, and 200 μm DTT was added to the control, Trx, and lipoic acid sample. Trx was added at a concentration of 10 μg/ml and lipoic acid (Sigma) at 10 μm to each respective sample. The DTT reduction sample was treated with 1 mm DTT. All samples were incubated at 25 °C for 30 min, and then 1 mm NEM-biotin (Pierce) was added to each sample. The samples were separated by SDS-PAGE, transferred to a PVDF membrane, and Western-blotted for anti-CD3ϵ using a rabbit anti-CD3ϵ heteroantisera (32) or streptavidin-HRP (Pierce) reactivity.
NMR Spectroscopy
NMR spectra were acquired on a Bruker 500 and 750 or Varian 700 MHz spectrometers equipped with a 5-mm cryogenic probe. 1H,15N heteronuclear single quantum correlation experiments (HSQC) NMR experiments for the His-GB1-CXXC peptides were performed at 298 K. Samples were in PBS buffer containing 2 mm GSSG, increasing amounts of GSH were titrated into the sample, and a spectrum was recorded at each titration point. Transverse relaxation optimization spectroscopy (TROSY)-enhanced 1H,15N HSQC spectra were collected on the CD3δ TMC-CXXC and CD3δ TMC-SXXS protein segments at 310 K. The standard array of three-dimensional TROSY-enhanced triple resonance backbone experiments was carried out on the CD3δ TMC-CXXC segment for completion of the backbone resonance assignments (18). The two indirect dimensions were non-uniformly sampled, with 12–15% of the two-dimensional grid acquired using a Poisson Gap sampling. The non-uniformly sampled data were reconstructed using istHMS software (33, 34). Linearly acquired data were processed with NMRPipe (35), and all the data were analyzed with NMRView (36) and CARA (37).
RESULTS
The Measured Equilibrium Reduction Potentials Are Consistent with the CD3 CXXC Motifs Being Oxidized on the Cell Surface
The ectodomain architecture of CD3ϵγ and relative position of the respective CXXC motifs, TM, and cytoplasmic segments are shown in Fig. 1. The equilibrium reduction potential of shortened segments of the CD3ϵ, CD3γ, and CD3δ subunits were determined to elucidate the physiologically relevant state of the CXXC motif on the surface of T cells. The CD3 CXXC-containing segments began with an N-terminal Met residue followed by the CXXC motif and extended five residues C-terminally and were produced in recombinant fusions with GB1 protein. After expression and purification, the standard reduction potentials (Eo′) were calculated by analysis of changes in HSQC cross-peak intensities (Fig. 2A), reflective of the amounts of reduced and oxidized CXXC peptide, in response to changes in GSH and GSSG composition of redox buffer (38). All CD3 equilibrium reduction potentials were found to be significantly more negative (Eo′ for CD3ϵ, -γ, and -δ are −0.211 V, −0.221 V, and −0.226 V, respectively, Fig. 2B) than the redox potential of the endoplasmic reticulum (Eo′ = −0.137 to −0.185 V) (39) or extracellular pools of cysteine/cystine, the predominant thiol pair in human plasma (−0.080V) (40). These results substantiate the hypothesis that the CXXC motifs are oxidized during folding in the endoplasmic reticulum and are, therefore, oxidized on the T cell surface. Previous data probing cell surface CD3γ using anti-CD3γ-antibody specific for its N terminus showed reactivity with WT CD3γ on the surface TCR complex but not of a SXXS motif variant of CD3γ in which both cysteines of CXXC were mutated to serine (19). Collectively, these data suggest that the normal, active state of CD3-CXXC in CD3γ, CD3δ, and CD3ϵ on the cellular surface is oxidized and that this state supports proper domain topology.
FIGURE 1.

Domain architecture of the CD3 subunits. A, the heterodimeric CD3ϵγ subunit complex is illustrated, where the CD3ϵ subunit is drawn in blue and the CD3γ subunit is in yellow. Select interdomain hydrogen bonds are shown on the G strands, in which the amide protons are drawn in gray and the carbonyl oxygen atoms are drawn in red. The cytoplasmic tails are illustrated vertically to depict receptor length, whereas physiologically, the tails may be associated with the inner leaflet of the plasma membrane. Each CD3 subunit contains an extracellular, transmembrane, and cytoplasmic domain. The CD3ϵγ extracellular domain structure was generated from the deposited PDB file 1XMW (18) using PyMOL (60). B, the partial mouse CD3 amino acid sequences are displayed and initiated at the membrane proximal region in each subunit. The TM domains are underlined. The boxed region drawn on each sequence highlights the CXXC motif. The sequence emphasized with a line above represents the peptides generated for measurement of the equilibrium reduction potentials. The CD3δ Cys to Ser mutations are denoted with red S labels above the native C labels in the sequence.
FIGURE 2.

Calculated equilibrium reduction potentials of the CD3 CXXC motifs. Peptides containing the CXXC regions of murine CD3ϵ, -γ, and ϵ as well as a poly-Ala-containing peptide as a control were produced in Escherichia coli as 15N isotopically labeled His6-GB1 fusion proteins. The peptides included an N-terminal Met residue, the CXXC motif, and five additional residues C-terminal. To aid in sample handling, the protein segments did not include the significantly hydrophobic TM residues. A, during the titration experiments, the GB1 peptides were equilibrated with 2 mm GSSG and varying amounts of GSH (0, 3, 5.1, 9, 15.6, 27, and 46 mm) were added and monitored using 1H,15N HSQC spectra. The chemical shift intensities of non-GB1 residues were monitored in a completely oxidizing (Ox) environment (blue) to a significantly reducing (Rd) environment (red). B, shown are curves fit to data determining the Keq of each CXXC. Each dot in the curve plots for the titration points represents an individual resonance monitored during the experiment. Standard reduction potentials were calculated from the equilibrium data for CD3γ, -δ, and -ϵ and determined to be −0.221 V, −0.226 V, and −0.211 V, respectively. The poly-Ala control reduction potential was −0.218 V, in agreement with the published value of −0.230 V of a longer, more helical peptide.
The CD3 CXXC Motif Forms an Intramolecular Disulfide Bond on the Surface of T Cells
To experimentally determine the oxidation status of the CD3 CXXC motif on the surface of resting and activated T cells directly, mAb affinity column-purified CD3 was examined by LC/MS analysis. Approximately 0.5 × 109 cells Jurkat REX T cells were harvested and lysed. Initially, the CD3 subunits were isolated from the cell lysate via the anti-CD3ϵ antibodies, Leu4 or RW2–8C8 (31). The results with each anti-CD3ϵ mAb were indistinguishable (data not shown). The Fig. 3A inset shows the region of the Coomassie-stained SDS-PAGE from which the CD3 components were excised. As described under “Experimental Procedures,” after multiprotease enzymatic digestions followed by LC/MS analysis, the relevant CXXC motif of the CD3γ, CD3δ, and CD3ϵ subunits was identified (Fig. 3, A–C). The CXXC peptides were each observed to be oxidized, having a mass 2 daltons lower than the calculated mass for the reduced form, confirming that the CD3 CXXC motifs are in the oxidized state, and forming an intramolecular disulfide bond (Fig. 3) irrespective of the anti-CD3 mAb used for the isolation. The results with CD3ϵ (Fig. 3A) are an exemplar, where the left trace shows the extracted ion chromatogram of the m/z (1+) peptide, and the insert shows the experimental monoisotonic m/z 825.26 (1+) containing an intrachain disulfide from the mass spectrum. The MS2 spectrum of the collision-induced dissociation to the right of the mass spectrum confirms the identity of this peptide.
FIGURE 3.

MS analysis of the oxidation status of the CXXC motif from CD3 subunits isolated from T cells. Antibody purified CD3 subunits isolated and concentrated samples from Jurkat T cells were separated by SDS-PAGE and Coomassie-stained. The region excised for MS analysis is illustrated. A, CD3ϵ; the identification of disulfide-linked VCENCME by mass spectrometry. The extracted ion chromatogram (XIC) of the peptide is shown from the C18 reversed phase elution with an inset showing the experimental m/z (1+) of the peptide. The experimental monoisotopic m/z 825.26 (1+) ion indicates an intrachain disulfide (theoretical monoisotopic m/z 827.27, 1+). The right figure shows the collision-induced dissociation-MS2 spectrum. The fragment ions confirm the identity of peptide VCENCME. B, CD3γ; the identification of disulfide-linked MCQNCIE by mass spectrometry. The extracted ion chromatogram of the peptide is shown from the C18 reversed phase elution with an inset showing the experimental m/z (1+) of the peptide. The experimental monoisotopic m/z 838.29 (1+) ion indicates an intrachain disulfide (theoretical monoisotopic m/z 840.30, 1+). The right figure shows the collision-induced dissociation-MS2 spectrum. The fragment ions confirm the identity of peptide MCQNCIE. C, CD3δ; the identification of disulfide-linked MCQSCVE by mass spectrometry. The extracted ion chromatogram of the peptide is shown from the C18 reversed phase elution with an inset showing the experimental m/z (1+) of the peptide. The experimental monoisotopic m/z 797.26 (1+) indicates an intrachain disulfide (theoretical monoisotopic m/z 799.28, 1+). The right figure shows the collision-induced dissociation-MS2 spectrum. The fragment ions confirm the identity of peptide MCQSCVE.
To remove the potential of antibody bias toward the selective purification of oxidized CD3 subunits, CD3 was also captured from cell lysates with a ConA-coupled resin. This lectin binds α-mannosyl-/α-glucosyl residues and demonstrates high affinity for oligomannose-type N-glycans and hybrid type N-glycans (41). As such, ConA affinity chromatography can be used to capture the CD3γ and CD3δ subunits that contain such N-linked glycans and the tightly non-covalently associated non-glycosylated CD3ϵ subunit. LC/MS analysis of ConA-purified CD3 subunits yielded comparable results to those shown in Fig. 3 and as summarized in Table 1. We conclude that the resting, native state of the CD3 CXXC motif of CD3γ, CD3δ, and CD3ϵ on the T cell surface is that of an intramolecular disulfide regardless of the isolation method.
TABLE 1.
Characterization of the CD3 oxidation state from purified protein and on the surface of intact T cells
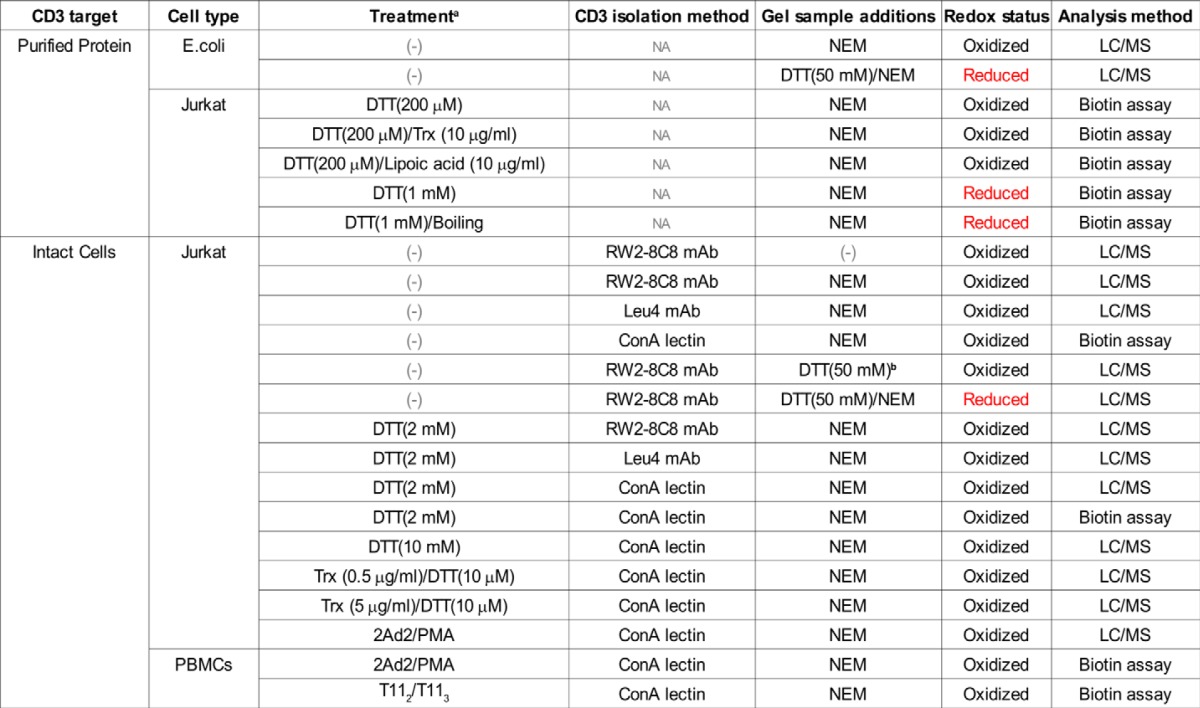
a Purified CD3 or intact cells were either not treated (−) or treated with potential reductant (DTT, Trx, lipoic acid, or mAb cellular activation (2Ad2/PMA or T112/T113) before analysis of the redox status.
b Despite the addition of DTT, the disulfide bond readily reforms on the SDS-PAGE gel without cysteine methylation via NEM.
The CD3 CXXC Motif Is Highly Resistant to Reduction
Next, the potential for modulation of the CXXC oxidation state on T cells was also explored. Initially, Jurkat T cells were incubated with either 2 or 10 mm DTT before lysis in the presence of NEM, isolation of CD3, and LC/MS analysis. However, in the presence of either concentration of DTT, the CXXC motif remained oxidized (Table 1). In fact, reduced CXXC peptides could only be detected by LC/MS after isolation from the cell surface into a detergent solution and subsequent treatment with DTT and NEM just before SDS-PAGE gel separation (Table 1). These results suggest that the CXXC motif may be occluded on the T cell surface, with reductant unable to access this region until CD3 is extracted from the cell membrane.
Biotin Switch Assay
In addition to MS analysis, reduction of CD3 by DTT was also monitored using a biotin-switch assay (for review, see Ref. 42). In this assay, reduced cysteine residues are modified by the addition of NEM-biotin and subsequently detected by streptavidin-HRP chemiluminescence after separation via SDS-PAGE and transfer to PVDF membrane. Although a notable increase in reduced proteins was observed in the DTT-treated samples in the 60–200-kDa range (Fig. 4A, left panel lanes), CD3 did not give rise to observable streptavidin-HRP signal and hence remained oxidized, consistent with the MS results (Fig. 3). Note that anti-CD3ϵ rabbit antisera Western blotting readily detected CD3 in the samples. The minor CD3ϵ Western blot signal observed at ∼40 kDa represents CD3ϵ-CD3ϵ misfolded intracellular proteins (39), further enhanced through rapid dimer formation after DTT treatment.
FIGURE 4.
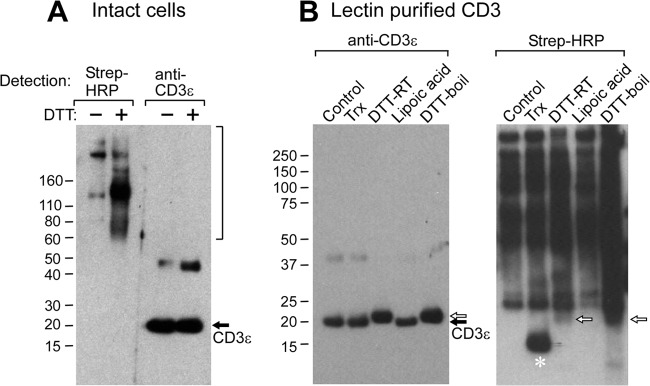
Analysis of the oxidation state in T cells by the biotin switch assay. A, 0.5 × 109 Jurkat REX cells were either untreated (−) or treated (+) with 2 mm DTT followed by biotin-NEM addition. Then CD3 was isolated from the cell lysate with ConA resin and subsequently separated by SDS-PAGE gel. Proteins were then transferred to PVDF membrane and blotted for streptavidin-HRP signal or for anti-CD3ϵ reactivity. The bracketed region from 60 kDa to >200 kDa represents multiple proteins that have been reduced and biotin-modified. The band migrating at ∼40 kDa represents a small fraction of CD3 ϵ dimers. B, CD3-containing samples were initially ConA purified from untreated Jurkat REX T cells, and then samples were either left untreated or treated with Trx, DTT at room temperature (DTT-RT), lipoic acid, or DTT and boiling before separation on SDS-PAGE gel. Proteins were then transferred to PVDF membrane and blotted for streptavidin-HRP signal or for anti-CD3ϵ reactivity. The band demarcated with an asterisk (*) represents exogenous Trx that had been added to the sample and became NEM-biotin-modified during the sample processing step. The reduced and biotin modified CD3 subunits are highlighted with a white arrow.
Thioredoxin Is Incapable of Reducing the CXXC Disulfide Bond
Although the small molecule reductant DTT did not reduce the CXXC motif on the cell surface, the ability of the CD3 subunits to undergo enzyme-mediated reduction was then tested by the addition of exogenous Trx to Jurkat T cells. Trx is known to be secreted into the extracellular space and found to regulate cellular functions (43–45). The cells were incubated with varying amounts of Trx and then lysed and purified as described above. MS analysis of Trx-treated cells showed that the CXXC motif remained oxidized under the conditions tested (Table 1). The ability of Trx to reduce the CD3 subunits after isolation from the cell membrane, where they are potentially more accessible to reduction, was analyzed by using the gel-based biotin switch assay. CD3 was purified from the cell lysates via ConA resin, then Trx was added with 10 μm DTT as an electron source for enzymatic-mediated reduction of the ConA-eluted material. After incubation, NEM-biotin was added to the samples, and samples were separated by SDS-PAGE gel and then analyzed for streptavidin-HRP signal. Regardless of whether CD3 was located on the T cell surface or isolated from the membrane, Trx at concentrations up to 10 μg/ml was not able to reduce the CXXC disulfide bond as observed by MS (Table 1) or the biotin-switch assay (Fig. 4B). The failure of Trx to reduce extracted CD3 may be the result of its 35-kDa size relative to the 154-Da size of DTT and henceforth limited accessibility to the CXXC disulfide bond despite possessing a somewhat structurally flexible active site (46). As shown in Fig. 4B, high concentrations of DTT or DTT/boiling were capable of reducing the CD3 subunits in the biotin switch assay when CD3 was extracted from the cell membrane. Evidence of such reduction is the appearance of streptavidin-HRP signal (white arrows Fig. 4B, right panel) and a mobility shift of CD3 on the SDS-PAGE gel in the Western blot analysis (white arrows Fig. 4B, left panel). The observed change in migration (Fig. 4B, left panel) is most likely due to reduction of the Ig domain Cys residues with or without reduction of the CXXC motif rather than of the Cys residues in the CXXC motif alone. The distal Ig disulfide bond will result in a much greater change in the hydrodynamic radius upon reduction than the membrane proximal disulfide bond in the CXXC motif, therefore, resulting in an observable up-shift on the gel. The lipid-soluble, small molecule reductant lipoic acid (47) at a concentration of 10 μm was unable to reduce the isolated CD3 subunits regardless of its significantly greater hydrophobic character than DTT and smaller size than Trx. It would appear that there may be a requirement for denaturation and/or reduction of the CD3 Ig-like domain disulfide to afford accessibility to the membrane proximal CXXC motif disulfide bond for reduction.
The CD3 Oxidation State Is Unaffected by T Cell Activation via the TCR Complex or CD2 Pathways
To determine if the oxidation state of CD3 could be modified by activation, T cells isolated from PBMCs were characterized in the resting and activated state by monitoring disulfide bond reduction using the biotin switch assay. T cells were activated via the CD3ϵ pathway using the anti-human CD3 2Ad2 murine IgM isotype antibody (29) for various times. Cell samples were removed after 0, 1, 24, and 48 h and treated with biotin-NEM. A decrease in the amount of CD3 was observed over the course of the activation (Fig. 5A, left panel). This result is expected, because activation via the TCR complex either with anti-CD3 mAb plus PMA or antigen results in loss of CD3 expression both by internalization and membrane shedding (29). The detectable CD3 remained oxidized in the resting and activated cells as tracked through streptavidin-HRP chemiluminescence (Fig. 5A, right panel). The CD3ϵ dimer, as observed in Fig. 4A and described previously (48), decreases at a slower rate. Perhaps this dimer represents an internal CD3ϵ-CD3ϵ pool lost more slowly. Fig. 5B shows that the CD3 surface expression on mature CD8 T cells by FACs analysis is rapidly lost upon 2Ad2 addition, going from a mean fluorescence intensity of 4000 to 200 in just 1 h. Although not shown, a similar pattern of CD3 loss was detected on CD4 T cells. Unlike CD3, other proteins co-purified by the ConA column do display an increase in reduction over the course of activation by this assay (Fig. 5A, right panel). This phenomenon has been previously reported in the literature (49).
FIGURE 5.
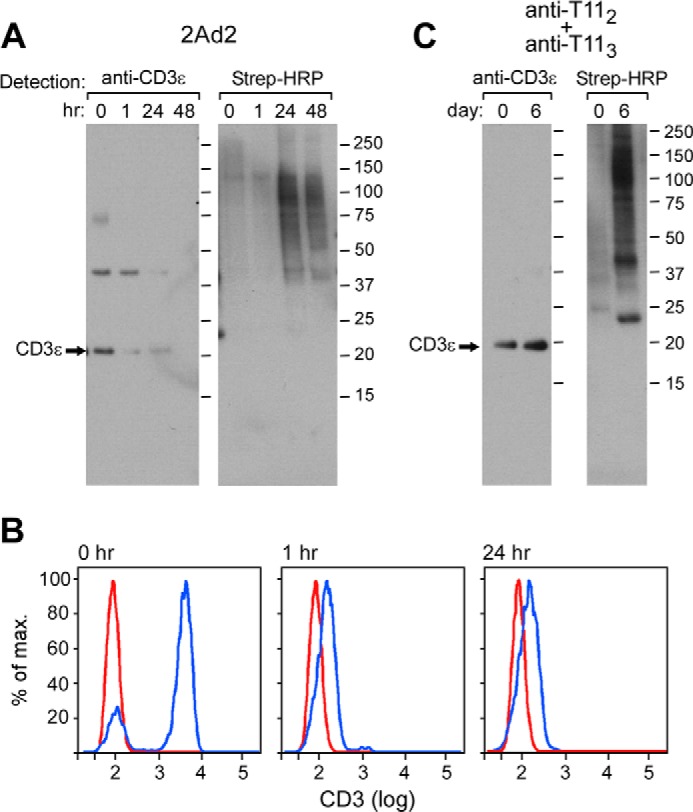
Conservation of the CXXC disulfide bond in both resting and activated T cells. A, 0.5 × 109 T cells isolated from PBMCs were either left untreated or treated with the activating antibody 2Ad2/PMA for 1–48 h. B, CD3 staining on the surface of 2Ad2/PMA-activated T cells was monitored at 0, 1, and 24 h with anti-CD3 FITC and anti-CD8 antigen-presenting cell. The unstained background control is depicted with the red line, and the anti-CD3 FITC staining gated on CD8 antigen-presenting cell-positive T cells is depicted with the blue line. C, 0.5 × 109 T cells isolated from PBMCs were either left untreated or treated with the activating anti-T112 and anti-T113 antibody combination for 6 days. In both A and C, CD3 was isolated from the cell lysate with Con A resin and then separated by SDS-PAGE gel. Proteins were then transferred to PVDF membrane and blotted for streptavidin-HRP signal or for anti-CD3ϵ reactivity.
Next, to bypass the CD3 pathway and the concomitant TCR down modulation, T cells were activated via the CD2 pathway using the combination of mitogenic anti-T112 and anti-T113 antibodies (30). Cell samples were removed after 6 days when cellular proliferation was already maximal (30) and treated with biotin-NEM. With this stimulation, the levels of CD3 remain constant over the activation period (Ref. 30 and data not shown). Nonetheless, biotin-modified CD3 (i.e. reduced CD3) was not detectable in the resting and activated T cells despite an overall observable increase in multiple reduced proteins that are ConA-co-purified from the cell samples (Fig. 5). The combined cellular results strongly support the notion that the CD3 CXXC motif is stably oxidized in both resting and activated T cells; furthermore, the disulfide bond is resistant to reduction via DTT, Trx, or T cell activation.
A Structural Role for the CXXC Motif in Maintenance of TM and Cytoplasmic Segment Conformations as Revealed by TROSY 1H,15N HSQC Spectra
The structural role of the CXXC motif was then investigated using two approaches. In the first approach, a double Cys to Ser mutant of the CD3δ segment (SXXS) was prepared and compared with the wild type CD3δ segment (CXXC) by NMR spectroscopy. Each construct began four residues N-terminal to the CXXC motif and extended to the C-terminal end of the cytoplasmic domain (Fig. 1). The protein segments were 15N isotopically labeled during recombinant expression, purified to homogeneity, and then incorporated into LPPG micelles for NMR analysis. The chemical shift dispersion in the TROSY 1H,15N HSQC spectrum of the CXXC construct was consistent with that of a membrane-spanning motif coupled with a partially structured soluble cytoplasmic tail (data not shown); resonances were spread within a relatively narrow range characteristic of either α helical or random coil secondary structure. Resonances associated with transmembrane residues (resonances 80–102) were uniformly less intense than those of the more mobile cytoplasmic region residues (resonances 103–151). Upon comparison of the TROSY 1H,15N HSQC spectra of the CXXC and SXXS segments, extensive chemical shift changes were readily observed in residues Cys-71, Thr-81, and Ile-91. Based on the partial backbone assignments, the largest chemical shift changes occurred in the N-terminal portion of the protein segment that includes the membrane-proximal region where the native CXXC motif resides (Fig. 6, resonances 67–78). Of significance, the chemical shift changes also extend C-terminally into the TM and cytoplasmic domains. Characterization of the spectral differences between the CXXC and SXXS protein segments suggests that the membrane proximal CXXC plays an important structural role in governing the conformation of the CD3 segment that includes the intracellular signaling domain (resonances 103–151).
FIGURE 6.
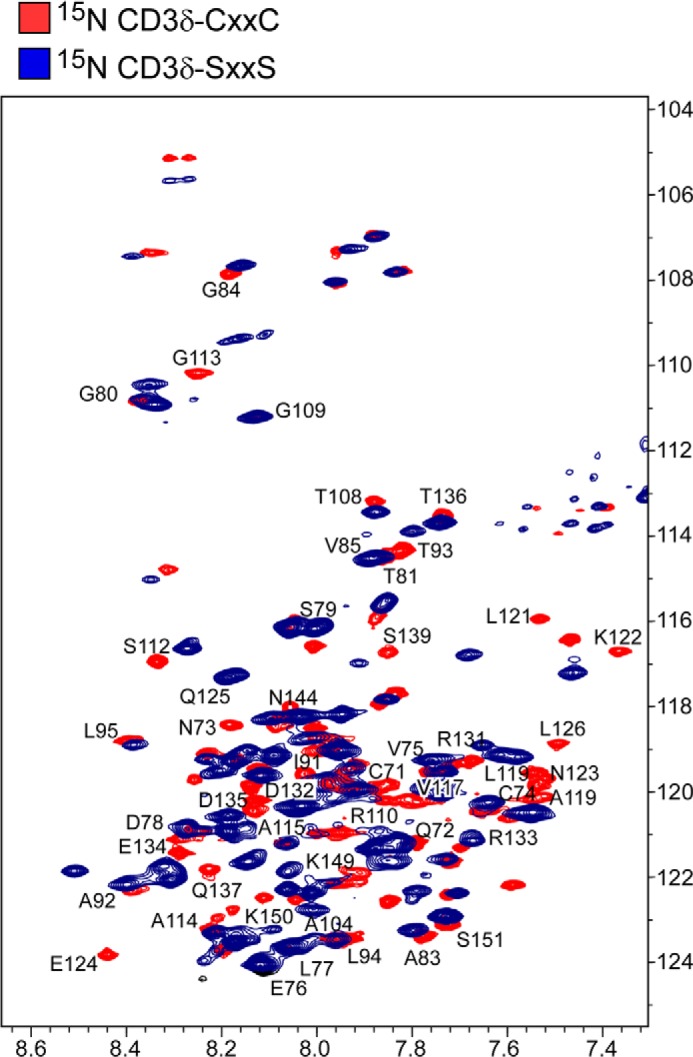
Structural alterations in CD3δ by SXXS mutation. 1H,15N TROSY HSQC spectrum of CD3δ-CXXC (blue) is overlaid with the TROSY HSQC spectrum of CD3δ-SXXS (red). Representative resonances are labeled in the membrane proximal, transmembrane, and cytoplasmic tail of CD3δ. Numbering corresponds to the full-length murine wild type CD3δ protein where the CXXC motif is at residues 71–74 (Fig. 1A).
In the second approach, chemical reduction of the native CXXC motif in the CD3 segment was also investigated by NMR spectroscopy. A titration experiment from 0 to 8 mm DTT was carried out using the wild type CD3δ segment described above. Consistent with the CXXC versus SXXS chemical shift comparison, considerable spectral differences were observed in CD3δ upon the addition of DTT, most notably in the N-terminal CXXC region (Fig. 7). As shown in Fig. 7A, comparison of 0- and 8-mm DTT specifically identified changes include Cys-71, Asn-73, and Asp-78. Chemical shift changes were also detected in the TM and cytoplasmic domains, i.e. Thr-81, Leu-95, Ala-119. Interestingly, Fig. 7B, which focuses on one region of the spectra with additional DTT amounts and incubation kinetics shown, indicates that the CXXC disulfide bond appears occluded, as was observed in the T cell experiments. Thus complete reduction of the protein segment was not observed until after a significant lapse of time (i.e. 12 h) even in the presence of 4-fold molar excess DTT (4 mm). Taken together, these results suggest that the highly favored, intramolecular disulfide bond in the CD3 CXXC motif appears protected from reduction in a membrane and membrane-like environment.
FIGURE 7.

Structural effects of disulfide bond reduction of the CD3Δa CXXC motif. A, 1H,15N TROSY HSQC spectrum of CD3δ-CXXC (red) overlaid with the TROSY HSQC spectrum of CD3δ in the presence of 8 mm DTT (blue). B, the boxed portion of the TROSY HSQC spectra of CD3δ in panel A is expanded for greater detail of illustrative residues over the course of the DTT titration experiment. Included in this view is a sample where 4 mm DTT was added without (yellow) or with (green) an additional 12 h of incubation before NMR analysis. The 0 mm (red) and 8 mm (blue) DTT conditions are also superimposed.
DISCUSSION
The CD3ϵ, CD3γ, and CD3δ membrane proximal CXXC motif is an important and highly conserved structural element within each of the CD3 subunits of all mammalian species. Based on the presented results, the CXXC intramolecular disulfide bond is likely formed during the folding process in the endoplasmic reticulum, and this oxidized state is then preserved on the surface of T cells. Loss of the disulfide bond either by mutation or reduction has a significant impact on the conformation of the protein segment in the vicinity of the CXXC motif and also impacts the positioning of the TM domain and cytoplasmic tail as shown for CD3δ. It is expected that CD3γ and CD3ϵ segments behave in a similar manner to CD3δ as evidenced by the similarity of their reduction potentials and behavior on the surface of T cells. The results herein thus bolster a model in which the CD3 CXXC motifs rigidify the structure of CD3 heterodimeric subunits within the extracellular to intracellular junction, potentially mechanically coupling the position and motion of the extracellular TCR complex to the intracellular signaling motifs. This rigidification adds to that of the squat ectodomains paired through extensive interface contacts and conjoint G strands (50).
Mutational studies of the Cys residues in the CD3γ and CD3ϵ CXXC motif have been shown to have significant physiological effects. For example, mutation of CD3γ Cys residues leads to structural alterations in both the CD3γ and CD3ϵ subunits when measured by conformationally specific antibody binding and also resulted in impairment of CD3ϵγ heterodimeric pairing (19). Cellular studies completed on CD3ϵ CXXC Cys mutants recently disclosed defects in TCR-dependent development and activation. These irregularities are presumably due to dysfunction induced by the CD3ϵ mutation in both CD3ϵγ and CD3ϵδ heterodimers (21). However, CD3γ mutation alone caused dysfunction of the pre-TCR as judged by attenuated DN3 and DN4 thymocyte transition and impairment in developmental progress to the double-positive stage without reduction in surface receptor expression (19). The CD3 CXXC Cys mutant subunits also appeared to affect TCR complex intersubunit interactions (51, 52), with observed diminished membrane proximal-mediated association of CD3ϵδ with TCRα in an in vitro translation experimental system (20). Although there appears to be some variability in the impact of the Cys mutations on subunit association and T cell functioning ascribed to each CD3 subunit, preservation of the CXXC motif is undoubtedly critical for proper TCR subunit positioning and cellular signaling.
The standard reduction potentials of −0.226 V, −0.211 V, and −0.221 V for CD3δ, CD3ϵ, and CD3γ CXXC motifs, respectively, are between the calculated values of the CXXC disulfide bonds found in PDI (−0.175 V) and thioredoxin (−0.270 V) (53). The CD3ϵ value is observed to be slightly less negative than CD3δ and CD3γ, which may be due to the presence of the more bulky aromatic side chain from a Tyr residue in the intervening sequence (Fig. 1). The reduction potentials for all three CD3 segments are considerably more negative than the redox potential of the endoplasmic reticulum, where redox-mediated folding occurs (39). The calculated values agree with the experimental data showing only oxidized CD3 CXXC motifs on the cell surface.
Interestingly, we observed herein an inability to reduce the CD3 CXXC motifs on the T cell surface after treatment with the chemical reductant, DTT, or the enzyme-mediated reductase, Trx. In contrast, in the soluble, non-membrane-associated peptide model system it was possible to reduce the disulfide bond with increasing amounts of reductant. On the other hand, a significant number of cell surface proteins did become more reduced after treatment with DTT or Trx in the intact cellular experiments despite CD3ϵ, CD3δ, and CD3γ remaining oxidized. A similar phenomenon was observed during the T cell activation experiments. The CD3 subunits remained oxidized whether the T cell was activated through the TCR or CD2 pathways; however, an overall increase in other reduced, co-purifying proteins was detected. Collectively, these data suggest that the CXXC region is not accessible to reduction in the TCR complex. Thus, although the peptide model system is optimal for determining the thermodynamic properties of the disulfide bond, it is problematic when considering the CD3 subunits embedded in a lipid environment. Given that CD3 CXXC disulfides were resistant to reduction by the high concentrations of DTT present in the cellular experiments, beyond which cellular viability was compromised, it is unlikely that an environment exists in which these motifs are reduced at the surface of living cells in vivo. Hence, we conclude that CD3 CXXC motifs are not redox switches linked to T cell activation.
The completeness of the oxidized status and the difficulty in reducing the CXXC motifs in a membrane-like environment may be a result of one or more factors. First, there may be a contribution to the stability of the disulfide through thermodynamic coupling to the formation of the transmembrane helix as has been reported in model studies on soluble helices (22, 54). Indeed, the reduction potentials of the CD3 motifs may be significantly more negative than those determined here, as is the value for the poly(A) helix determined by Iqbalsyah et al. (22) compared with that shown here (−0.230 V versus −0.218 V). Secondly, there may be some occlusion of the site due to its location within the CD3 molecule itself and/or due to ectodomain quaternary associations within the TCR complex (50). Although this is not a factor in experiments studying the isolated CD3δ fragment, the position of the CXXC motif proximal to the central G-strands of the CD3 heterodimers means that it is likely that significant steric hindrance would be encountered by an attacking thiolate anion, which must approach in line with the existing disulfide bond in order for thiol-disulfide exchange to occur. Similarly, the CXXC motif may be partially occluded by the lipid bilayer itself, with the transmembrane helix retaining sufficient rigidity to prevent access by an incoming nucleophile to the disulfide bond.
In conjunction with cellular defects, substantial structural alterations are directly observed by NMR with mutation of the Cys to Ser residues in the CD3δ CXXC motif-containing protein segment. The extensive chemical shift changes observed in CD3δ-CXXC relative to CD3δ-SXXS that occur throughout the protein segment are consistent with the broad structural effects of CXXC mutation impacting conformationally specific antibody detection observed previously with CD3γ (19). Moreover, the large structural changes are suggestive of the importance of the CXXC intramolecular disulfide bond on maintaining the structural rigidity of the membrane proximal region and TM domain, which also appears to be coupled to the positioning of the cytoplasmic tail. One may then infer that the tightly paired and associated G-strands N-terminal to the membrane proximal region in the CD3 extracellular heterodimer (Fig. 1) will be similarly structurally perturbed with loss of the CXXC disulfide bond, thereby linking the mechanosensor function of the αβ TCR to the redox state of the CD3 subunits as described below.
Additionally, loss of the intramolecular CXXC disulfide bond through reduction by DTT results in significant conformational changes in the protein segment as was observed with mutation. It is noteworthy that the ability to reduce the CD3δ CXXC disulfide bond in a membrane-like environment in the presence of a large molar excess of DTT was not immediate (Fig. 7). This is in agreement with the inability to reduce the CXXC motif in the CD3 subunits displayed on the surface of T cells and provides further evidence that the CXXC is not readily accessible for reduction in a lipid environment. Therefore, given the likely close proximity of the CXXC motif to the plasma membrane in the native state, mutation or reduction would then affect the disposition of the CD3 segments on the plasma membrane consistent with data on anti-CD3γ ectodomain antibody binding (19). Significant cell surface positional changes would most certainly correlate to conformational changes throughout the TM domain and into the cytoplasmic tail, therefore in agreement with the observed conformational changes in the CD3δ segment induced through mutation or reduction monitored by NMR.
CXXC motifs present at the N terminus of helical peptides have a propensity to form a helical cap and contribute to a measurable increase in helical stability (22, 54). Thus, the intramolecular disulfide bond formed by the CD3 CXXC motif may function to stabilize the TM domain helix and further rigidify the membrane proximal region either by formation of a helical cap or other structural element constrained by the disulfide bond. This rigidity will contribute to the overall CD3 heterodimeric stability within the TCR and contributes to the rigid CD3 heterodimer connectivity to the T cell membrane. The TCR will then become more responsive to force induced pMHC binding events and thereby be better equipped to convert binding energy into intracellular signaling cascade activation (3).
TCR-dependent recognition results from the cross-junctional TCR-pMHC binding at the T cell-APC interface, i.e. a two-dimensional binding interface, that is undoubtedly subjected to mechanical forces. As noted previously, the TCR complex is composed of an immunoglobulin Fab-like αβ heterodimer and the non-covalently associated CD3 signaling components. The juxtaposition of the squat and rigid heterodimeric CD3 structures on short stalks that flank the taller αβ heterodimer, itself tethered to the T-cell membrane by long linkers, suggested a TCR-based signal transduction mechanism initiated by mechanical triggering of the extracellular domains (21). Dynamic mechanosensing might occur during T-cell immune surveillance as well as accompanying molecular rearrangement at the immunological synapse post-cessation of cell scanning via outside-in and inside-out force, respectively. This force-driven signaling notion has recently been confirmed by optical tweezer experiments (25) and extended by biomembrane force probe studies demonstrating that force prolongs TCR-pMHC bond lifetime for agonist peptides (55). Given that the TCR functions as a mechanosensor to communicate the pMHC-specific information from the ligand-binding site on the αβ TCR ectodomains through the plasma membrane to the phosphorylation sites of the cytosolic tails of the CD3 subunits via a mechanical process, its architecture is finely tuned to detect, amplify, and transduce piconewton-scale forces and nanometer-scale mechanical events through its structural features. In this regard, the uniquely elongated TCRβ constant domain FG loop can push on the CD3ϵγ ectodomain during mechanical movement as described (4, 21). It now seems clear that the rigidity imparted by the CXXC motifs of the CD3 heterodimers will efficiently facilitate signaling to and through the membrane, on the one hand, and offer a registered vertical alignment on the membrane to optimize signaling in a coordinated fashion, on the other.
An increasing number of receptors appear to work via mechanotransduction; that is, the explicit ability of force to induce a biochemical signal (for review, see Refs. 56 and 57). The von Willebrand factor protein provides one example of a structure highly reinforced through the presence of disulfide bonds (58, 59). Von Willebrand factor contains a number of disulfide bonds that contribute to the global stability of the protein and a C-terminal domain containing a cystine knot motif. The intertwined Cys residues respond to a pulling force under sheer stress that then leads to conformational changes in von Willebrand factor protein binding, ultimately regulating blood coagulation responses (58, 59).
In sum, the CD3 CXXC motifs form highly stable intramolecular disulfide bonds on the surface of T cells that appear to be critical in maintaining the conformation of the CD3 subunits, TCR intersubunit interactions, and intracellular signaling responses to extracellular TCR-pMHC binding interactions. It is clear that as our understanding of biology becomes more precise, then the fine structural details underpinning the extraordinary sensitivity and specificity of receptor function will be revealed.
Acknowledgments
We gratefully acknowledge Tara Mayo and Jaewon Choi for technical assistance with Jurkat cell culture. We thank Dr. Cox Terhorst for technical discussion handling ConA purified CD3, Maris Handley for flow cytometry, and Dr. Sven Hyberts for non-uniformly sampled NMR data processing.
This work was supported, in whole or in part, by National Institutes of Health Grants AI100643-01 (to E. L. R.), GM047467 (to E. L. R. and G. W.), AI375581, P41-EB002026, and S10-RR029236 (to G. W.) and GM15847 (to B. L. K.).
- TCR
- T cell receptor
- TM
- transmembrane
- pMHC
- peptide-MHC complex
- NEM
- N-ethylmaleimide
- Trx
- thioredoxin
- HSQC
- heteronuclear single quantum correlation experiment
- TROSY
- transverse relaxation optimization spectroscopy
- ConA
- concanavalin A
- PMA
- phorbol 12-myristate 13-acetate
- PBMC
- peripheral blood mononuclear cell
- MMTS
- S-methyl methanethiosulfonate
- LPPG
- 1-palmitoyl-sn-glycero-3-phospho-(1′-rac-glycerol) (sodium salt)
- TMC
- transmembrane-cytoplasmic domain.
REFERENCES
- 1. Dave V. P., Cao Z., Browne C., Alarcon B., Fernandez-Miguel G., Lafaille J., de la Hera A., Tonegawa S., Kappes D. J. (1997) CD3δ deficiency arrests development of the αβ but not the γδ T cell lineage. EMBO J. 16, 1360–1370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Haks M. C., Krimpenfort P., Borst J., Kruisbeek A. M. (1998) The CD3γ chain is essential for development of both the TCRαβ and TCRγδ lineages. EMBO J. 17, 1871–1882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kim S. T., Shin Y., Brazin K., Mallis R. J., Sun Z. Y., Wagner G., Lang M. J., Reinherz E. L. (2012) TCR mechanobiology: torques and tunable structures linked to early T cell signaling. Front. Immunol. 3, 76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Malissen M., Gillet A., Ardouin L., Bouvier G., Trucy J., Ferrier P., Vivier E., Malissen B. (1995) Altered T cell development in mice with a targeted mutation of the CD3-ϵ gene. EMBO J. 14, 4641–4653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Smith-Garvin J. E., Koretzky G. A., Jordan M. S. (2009) T cell activation. Annu. Rev. Immunol. 27, 591–619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Arnaiz-Villena A., Timon M., Rodriguez-Gallego C., Iglesias-Casarrubios P., Pacheco A., Regueiro J. R. (1993) T lymphocyte signalling defects and immunodeficiency due to the lack of CD3γ. Immunodeficiency 4, 121–129 [PubMed] [Google Scholar]
- 7. de Saint Basile G., Geissmann F., Flori E., Uring-Lambert B., Soudais C., Cavazzana-Calvo M., Durandy A., Jabado N., Fischer A., Le Deist F. (2004) Severe combined immunodeficiency caused by deficiency in either the δ or the ϵ subunit of CD3. J. Clin. Invest. 114, 1512–1517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gil J., Busto E. M., Garcillán B., Chean C., García-Rodríguez M. C., Díaz-Alderete A., Navarro J., Reiné J., Mencía A., Gurbindo D., Beléndez C., Gordillo I., Duchniewicz M., Höhne K., García-Sánchez F., Fernández-Cruz E., López-Granados E., Schamel W. W., Moreno-Pelayo M. A., Recio M. J., Regueiro J. R. (2011) A leaky mutation in CD3D differentially affects αβ and γδ T cells and leads to a Tαβ-Tγδ+B+NK+ human SCID. J. Clin. Invest. 121, 3872–3876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Tokgoz H., Caliskan U., Keles S., Reisli I., Guiu I. S., Morgan N. V. (2013) Variable presentation of primary immune deficiency: two cases with CD3 γ deficiency presenting with only autoimmunity. Pediatr. Allergy Immunol. 24, 257–262 [DOI] [PubMed] [Google Scholar]
- 10. Blumberg R. S., Alarcon B., Sancho J., McDermott F. V., Lopez P., Breitmeyer J., Terhorst C. (1990) Assembly and function of the T cell antigen receptor. Requirement of either the lysine or arginine residues in the transmembrane region of the α chain. J. Biol. Chem. 265, 14036–14043 [PubMed] [Google Scholar]
- 11. Letourneur F., Klausner R. D. (1992) Activation of T cells by a tyrosine kinase activation domain in the cytoplasmic tail of CD3ϵ. Science 255, 79–82 [DOI] [PubMed] [Google Scholar]
- 12. Manolios N., Letourneur F., Bonifacino J. S., Klausner R. D. (1991) Pairwise, cooperative, and inhibitory interactions describe the assembly and probable structure of the T-cell antigen receptor. EMBO J. 10, 1643–1651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kuhns M. S., Davis M. M. (2012) TCR signaling emerges from the sum of many parts. Front. Immunol. 3, 159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Reth M. (1989) Antigen receptor tail clue. Nature 338, 383–384 [PubMed] [Google Scholar]
- 15. van der Merwe P. A., Dushek O. (2011) Mechanisms for T cell receptor triggering. Nat. Rev. Immunol. 11, 47–55 [DOI] [PubMed] [Google Scholar]
- 16. Call M. E., Wucherpfennig K. W. (2005) The T cell receptor: critical role of the membrane environment in receptor assembly and function. Annu Rev. Immunol. 23, 101–125 [DOI] [PubMed] [Google Scholar]
- 17. Borroto A., Mallabiabarrena A., Albar J. P., Martínez-A C., Alarcón B. (1998) Characterization of the region involved in CD3 pairwise interactions within the T cell receptor complex. J. Biol. Chem. 273, 12807–12816 [DOI] [PubMed] [Google Scholar]
- 18. Sun Z.-Y. J., Kim K. S., Wagner G., Reinherz E. L. (2001) Mechanisms contributing to T cell receptor signaling and assembly revealed by the solution structure of an ectodomain fragment of the CD3ϵγ heterodimer. Cell 105, 913–923 [DOI] [PubMed] [Google Scholar]
- 19. Touma M., Sun Z. Y., Clayton L. K., Marissen W. E., Kruisbeek A. M., Wagner G., Reinherz E. L. (2007) Importance of the CD3γ ectodomain terminal β-strand and membrane proximal stalk in thymic development and receptor assembly. J. Immunol. 178, 3668–3679 [DOI] [PubMed] [Google Scholar]
- 20. Xu C., Call M. E., Wucherpfennig K. W. (2006) A membrane-proximal tetracysteine motif contributes to assembly of CD3δϵ and CD3γϵ dimers with the T cell receptor. J. Biol. Chem. 281, 36977–36984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wang Y., Becker D., Vass T., White J., Marrack P., Kappler J. W. (2009) A conserved CXXC motif in CD3ϵ is critical for T cell development and TCR signaling. PLoS Biol. 7, e1000253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Iqbalsyah T. M., Moutevelis E., Warwicker J., Errington N., Doig A. J. (2006) The CXXC motif at the N terminus of an α-helical peptide. Protein Sci. 15, 1945–1950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Husson J., Chemin K., Bohineust A., Hivroz C., Henry N. (2011) Force generation upon T cell receptor engagement. PLoS ONE 6, e19680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Judokusumo E., Tabdanov E., Kumari S., Dustin M. L., Kam L. C. (2012) Mechanosensing in T lymphocyte activation. Biophys. J. 102, L5–L7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kim S. T., Takeuchi K., Sun Z. Y., Touma M., Castro C. E., Fahmy A., Lang M. J., Wagner G., Reinherz E. L. (2009) The αβ T cell receptor is an anisotropic mechanosensor. J. Biol. Chem. 284, 31028–31037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Li Y. C., Chen B. M., Wu P. C., Cheng T. L., Kao L. S., Tao M. H., Lieber A., Roffler S. R. (2010) Cutting edge: mechanical forces acting on T cells immobilized via the TCR complex can trigger TCR signaling. J. Immunol. 184, 5959–5963 [DOI] [PubMed] [Google Scholar]
- 27. Ma Z., Finkel T. H. (2010) T cell receptor triggering by force. Trends Immunol. 31, 1–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Acuto O., Hussey R. E., Fitzgerald K. A., Protentis J. P., Meuer S. C., Schlossman S. F., Reinherz E. L. (1983) The human T cell receptor: appearance in ontogeny and biochemical relationship of α and β subunits on IL-2-dependent clones and T cell tumors. Cell 34, 717–726 [DOI] [PubMed] [Google Scholar]
- 29. Reinherz E. L., Meuer S., Fitzgerald K. A., Hussey R. E., Levine H., Schlossman S. F. (1982) Antigen recognition by human T lymphocytes is linked to surface expression of the T3 molecular complex. Cell 30, 735–743 [DOI] [PubMed] [Google Scholar]
- 30. Meuer S. C., Hussey R. E., Fabbi M., Fox D., Acuto O., Fitzgerald K. A., Hodgdon J. C., Protentis J. P., Schlossman S. F., Reinherz E. L. (1984) An alternative pathway of T-cell activation: a functional role for the 50 kD T11 sheep erythrocyte receptor protein. Cell 36, 897–906 [DOI] [PubMed] [Google Scholar]
- 31. Meuer S. C., Cooper D. A., Hodgdon J. C., Hussey R. E., Fitzgerald K. A., Schlossman S. F., Reinherz E. L. (1983) Identification of the receptor for antigen and major histocompatibility complex on human inducer T lymphocytes. Science 222, 1239–1242 [DOI] [PubMed] [Google Scholar]
- 32. Koning F., Maloy W. L., Coligan J. E. (1990) The implications of subunit interactions for the structure of the T cell receptor-CD3 complex. Eur. J. Immunol. 20, 299–305 [DOI] [PubMed] [Google Scholar]
- 33. Hyberts S. G., Milbradt A. G., Wagner A. B., Arthanari H., Wagner G. (2012) Application of iterative soft thresholding for fast reconstruction of NMR data non-uniformly sampled with multidimensional Poisson Gap scheduling. J. Biomol. NMR 52, 315–327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hyberts S. G., Takeuchi K., Wagner G. (2010) Poisson-gap sampling and forward maximum entropy reconstruction for enhancing the resolution and sensitivity of protein NMR data. J. Am. Chem. Soc. 132, 2145–2147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Delaglio F., Grzesiek S., Vuister G. W., Zhu G., Pfeifer J., Bax A. (1995) NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6, 277–293 [DOI] [PubMed] [Google Scholar]
- 36. Johnson B. A., Blevins R. A. (1994) NMRView: a computer program for the visualization and analysis of NMR data. J. Biomol. NMR 4, 603–614 [DOI] [PubMed] [Google Scholar]
- 37. Keller R. (2004) Computer aided resonance assignment tutorial. Cantina, Zurich, Switzerland [Google Scholar]
- 38. Piotukh K., Kosslick D., Zimmermann J., Krause E., Freund C. (2007) Reversible disulfide bond formation of intracellular proteins probed by NMR spectroscopy. Free Radic. Biol. Med. 43, 1263–1270 [DOI] [PubMed] [Google Scholar]
- 39. Hwang C., Sinskey A. J., Lodish H. F. (1992) Oxidized redox state of glutathione in the endoplasmic reticulum. Science 257, 1496–1502 [DOI] [PubMed] [Google Scholar]
- 40. Jones D. P., Carlson J. L., Mody V. C., Cai J., Lynn M. J., Sternberg P. (2000) Redox state of glutathione in human plasma. Free Radic. Biol. Med. 28, 625–635 [DOI] [PubMed] [Google Scholar]
- 41. Varki A. (2009) Essentials of Glycobiology, 2nd ed., Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY: [PubMed] [Google Scholar]
- 42. Forrester M. T., Foster M. W., Benhar M., Stamler J. S. (2009) Detection of protein S-nitrosylation with the biotin-switch technique. Free Radic. Biol. Med. 46, 119–126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Miyamoto S., Sakamoto T., Soejima H., Shimomura H., Kajiwara I., Kojima S., Hokamaki J., Sugiyama S., Yoshimura M., Ozaki Y., Nakamura H., Yodoi J., Ogawa H. (2003) Plasma thioredoxin levels and platelet aggregability in patients with acute myocardial infarction. Am. Heart. J. 146, 465–471 [DOI] [PubMed] [Google Scholar]
- 44. Son A., Kato N., Horibe T., Matsuo Y., Mochizuki M., Mitsui A., Kawakami K., Nakamura H., Yodoi J. (2009)) Direct association of thioredoxin-1 (Trx) with macrophage migration inhibitory factor (MIF): regulatory role of Trx on MIF internalization and signaling. Antioxid. Redox Signal. 11, 2595–2605 [DOI] [PubMed] [Google Scholar]
- 45. Xu S.-Z., Sukumar P., Zeng F., Li J., Jairaman A., English A., Naylor J., Ciurtin C., Majeed Y., Milligan C. J., Bahnasi Y. M., Al-Shawaf E., Porter K. E., Jiang L.-H., Emery P., Sivaprasadarao A., Beech D. J. (2008) TRPC channel activation by extracellular thioredoxin. Nature 451, 69–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Lillig C. H., Holmgren A. (2007) Thioredoxin and related molecules: from biology to health and disease. Antioxid. Redox Signal. 9, 25–47 [DOI] [PubMed] [Google Scholar]
- 47. Ohmori Hnt, Yamauchi T., Yamamoto I. (1986) Augmentation of the antibody response by lipoic acid in mice. I. Analysis of the mode of action in an in vitro cultures system. Jpn. J. Pharmacol. 42, 135–140 [DOI] [PubMed] [Google Scholar]
- 48. Jin Y. J., Koyasu S., Moingeon P., Steinbrich R., Tarr G. E., Reinherz E. L. (1990) A fraction of CD3 ϵ subunits exists as disulfide-linked dimers in both human and murine T lymphocytes. J. Biol. Chem. 265, 15850–15853 [PubMed] [Google Scholar]
- 49. Metcalfe C., Cresswell P., Ciaccia L., Thomas B., Barclay A. N. (2011) Labile disulfide bonds are common at the leucocyte cell surface. Open Biol. 1, 110010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Sun Z.-Y. J., Kim S. T., Kim I. C., Fahmy A., Reinherz E. L., Wagner G. (2004) Solution structure of the CD3ϵΔ ectodomain and comparison with CD3ϵγ as a basis for modeling T cell receptor topology and signaling. Proc. Natl. Acad. Sci. U.S.A. 101, 16867–16872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Manolios N., Bonifacino J. S., Klausner R. D. (1990) Transmembrane helical interactions and the assembly of the T cell receptor complex. Science 249, 274–277 [DOI] [PubMed] [Google Scholar]
- 52. Cosson P., Lankford S. P., Bonifacino J. S., Klausner R. D. (1991) Membrane protein association by potential intramembrane charge pairs. Nature 351, 414–416 [DOI] [PubMed] [Google Scholar]
- 53. Aslund F., Berndt K. D., Holmgren A. (1997) Redox potentials of glutaredoxins and other thiol-disulfide oxidoreductases of the thioredoxin superfamily determined by direct protein-protein redox equilibria. J. Biol. Chem. 272, 30780–30786 [DOI] [PubMed] [Google Scholar]
- 54. Indu S., Kumar S. T., Thakurela S., Gupta M., Bhaskara R. M., Ramakrishnan C., Varadarajan R. (2010) Disulfide conformation and design at helix N termini. Proteins 78, 1228–1242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Liu B., Chen W., Evavold B. D., Zhu C. (2014) Accumulation of dynamic catch bonds between TCR and agonist peptide-MHC triggers T cell signaling. Cell 157, 357–368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Chen W., Zhu C. (2013) Mechanical regulation of T-cell functions. Immunol. Rev. 256, 160–176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Choi Y. I., Duke-Cohan J. S., Chen W., Liu B., Rossy J., Tabarin T., Ju L., Gui J., Gaus K., Zhu C., Reinherz E. L. (2014) Dynamic control of β1 integrin adhesion by the plexinD1-sema3E axis. Proc. Natl. Acad. Sci. U.S.A. 111, 379–384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Zhou Y. F., Eng E. T., Zhu J., Lu C., Walz T., Springer T. A. (2012) Sequence and structure relationships within von Willebrand factor. Blood 120, 449–458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Zhou Y. F., Springer T. A. (2014) Highly reinforced structure of a C-terminal dimerization domain in von Willebrand factor. Blood 123, 1785–1793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. DeLano W. L. (2010) The PyMOL Molecular Graphics System, Version 1.3r1, Schrodinger, LLC, New York [Google Scholar]