

Background: The molecular mechanisms underlying activation of CRF1 receptor (CRF1R) were elusive.
Results: We determined specific residues in the transmembrane domains (TMs) of CRF1R that are critical for receptor activation.
Conclusion: A possible “transmission switch” involving TM interactions is important for CRF1R activation.
Significance: This knowledge may aid in the development of nonpeptide CRF1R antagonists for use in stress-related disorders.
Keywords: 7-Helix Receptor, G Protein-coupled Receptor (GPCR), Protein Drug Interaction, Protein Structure, Receptor Structure-Function, Corticotropin-releasing Factor (CRF), Type 1 CRF Receptor (CRF1R)
Abstract
The corticotropin-releasing factor (CRF) type 1 receptor (CRF1R) for the 41-amino acid peptide CRF is a class B G protein-coupled receptor, which plays a key role in the response of our body to stressful stimuli and the maintenance of homeostasis by regulating neural and endocrine functions. CRF and related peptides, such as sauvagine, bind to the extracellular regions of CRF1R and activate the receptor. In contrast, small nonpeptide antagonists, which are effective against stress-related disorders, such as depression and anxiety, have been proposed to interact with the helical transmembrane domains (TMs) of CRF1R and allosterically antagonize peptide binding and receptor activation. Here, we aimed to elucidate the role of the third TM (TM3) in the molecular mechanisms underlying activation of CRF1R. TM3 was selected because its tilted orientation, relative to the membrane, allows its residues to establish key interactions with ligands, other TM helices, and the G protein. Using a combination of pharmacological, biochemical, and computational approaches, we found that Phe-2033.40 and Gly-2103.47 in TM3 play an important role in receptor activation. Our experimental findings also suggest that Phe-2033.40 interacts with nonpeptide antagonists.
Introduction
G protein-coupled receptors (GPCRs)3 are essential for life as they regulate vital physiological functions in almost every eukaryotic organism, including fungi and plants (1). They accomplish this by triggering a large number of cellular signaling cascades, through their cognate heterotrimeric G proteins, as a result of their interaction with a vast number of chemically divergent molecules ranging from light to large polypeptides (1, 2).
Based on sequence similarity methods, the superfamily of GPCRs is classified into four families or classes (A, B, C, and smoothened), which display little sequence similarity and do not share common structural/functional motifs (3). Class A (or rhodopsin-like) is the largest family of GPCRs. Most of our understanding regarding the structure and function of GPCRs has been achieved in this family (4). Very recently the crystal structures of the type 1 receptor (CRF1R) for the 41-amino acid peptide, corticotropin-releasing factor (CRF), and the glucagon receptor (GCGR), which belong to class B GPCRs, have been published (5, 6). Notably, despite their overall low sequence identity, structures from different families share a common molecular architecture and activate a similar pool of intracellular signaling molecules, the G proteins. This architecture is characterized by the presence of seven α-helical transmembrane domains (TMs) connected to each other by three extracellular (EL) and three intracellular loops. Moreover, all GPCRs possess an extracellular N-terminal region (N-domain) and a cytoplasmic C terminus, containing an α-helix (Hx8) oriented parallel to the cell membrane. These common features lead to the biologically important hypothesis that GPCR activation utilizes conserved structural elements leading to production of a biological effect.
Although the crystal structure of a receptor provides the ultimate information for its structure, it represents a single snapshot of one conformational state. Specifically, the crystal structures of the apo-state of GCGR and the CRF1R in complex with the nonpeptide CRF antagonist CP-376395 represent the inactive states of truncated receptors, lacking their extracellular N-domain (5, 6). Despite the fact that these truncated forms of class B GPCRs bound small nonpeptide ligands with similar affinities with those of the full-length receptors, they are not able to bind peptides and be activated because they lack the functionally indispensable N-domain (7, 8). Thus, although crystallization of CRF1R or GCGR provided invaluable structural information for an inactive state of these receptors, the molecular mechanisms underlying receptor activation that convert ligand binding to a biological effect are still elusive.
In this study, we aimed to elucidate the molecular mechanisms underlying the activation of CRF1R and to validate the hypothesis that activation of class A and B GPCRs utilizes conserved structural elements. We accomplished this by using a combination of different experimental approaches and by comparing the available results on the role of the third TM (TM3) in the structure-function of class A with the much less studied class B GPCRs. TM3 is a structural and functional hub as suggested by the comparison of class A crystal structures, in their inactive and active states (9). The residues in TM3 are functionally important by mediating interactions with the extracellular ligand, as well as by forming key inter-TM interactions that define the GPCR-fold, a conserved disulfide bridge with the second EL (EL2), and an important interface for G protein binding. We focused on the CRF1R, which plays a key role in the response of our body to stressful stimuli and the maintenance of homeostasis, by regulating neural and endocrine functions (6, 10). Moreover, the TM3 of CRF1R has been shown to be involved in the binding of small ligand antagonists that are effective against stress-related disorders, such as depression and anxiety (6, 10).
EXPERIMENTAL PROCEDURES
Structural Alignment
All crystallized GPCRs (including 21 receptors, 18 of which belong to family A, two to family B, and one to family F) were structurally superimposed using MultiProt based on their Cβ atoms (11). This permitted us to obtain a structurally based sequence alignment, which was later revised using Jalview (12).
Model of the Inactive CRF1R
To study the TM3-TM5 interface of the unliganded state of CRF1R, a homology model based on the crystal structure of GCGR (Protein Data Bank code 4L6R) was built using MODELLER (5, 13). The primary sequence of human CRF1R was used (UniProt accession code P34998) in which the G2103.47C mutation was inserted. The highly conserved residues-motifs between family B (L1.42 in TM1, HXNL in TM2, W3.42 in TM3, GWGXP in TM4, N5.53 in TM5, PLLG in TM6, and G7.46 in TM7), were used as reference points in TM sequence alignments (supplemental Fig. S1). The N terminus of the receptor was disregarded. Residues are identified by the general numbering scheme of Ballesteros and Weinstein that allows easy comparison among residues in the 7TM segments of receptors belonging to different families.
Site-directed Mutagenesis
The cDNA sequences encoding wild-type CRF1R (WT) or ΔCys CRF1R(ΔCys) were subcloned into the bicistronic expression vector pcin4, thereby creating the vectors pcin4-WT or pcin4-ΔCys, respectively, as described previously (14). ΔCys is a mutant CRF1R, which is insensitive to sulfhydryl-specific reagents and has similar functional properties with those of wild-type receptor (14). Mutations were generated by the polymerase chain reaction-mediated mutagenesis, using Pfu polymerase (MBI Fermentas, Hanover, MD) and mutagenic oligonucleotides encoding the desired amino acid substitution. The polymerase chain reaction generated DNA fragments containing Cys, Trp, Ala, Ile, or Lys mutations. The fragments containing the Cys mutations were subcloned into the pcin4-ΔCys plasmid (creating the pcin4-substituted Cys mutant plasmids), whereas those containing the other mutations were subcloned into the pcin4-WT plasmid (creating the pcin4-X mutant plasmids). The mutations were confirmed by DNA sequencing.
Cell Culture, Transfection, and Harvesting
Human embryonic kidney (HEK) 293 cells were grown in Dulbecco's modified Eagle's medium/F-12 (1:1) containing 3.15 g/liter glucose and 10% bovine calf serum at 37 °C and 5% CO2. Sixty-millimeter dishes of HEK293 cells at 80–90% confluence were transfected with 2–3 μg of pcin4-WT (WT), pcin4-ΔCys (ΔCys), pcin4-X mutant (X mutants), or pcin4-substituted Cys mutant (substituted Cys mutants) plasmids using 9 μl of Lipofectamine and 2.5 ml of Opti-MEM (both from Invitrogen). To generate stably transfected pools of cells expressing the receptors 5–12 h after transfection, the medium was replaced by Dulbecco's modified Eagle's medium/F-12 (1:1) containing 3.15 g/liter glucose, 10% bovine calf serum (Hyclone Laboratories, Logan, UT), and 700 μg/ml G418 (Geneticin), an antibiotic (Invitrogen). The antibiotic was added to select a stably transfected pool of cells. Cells expressing WT, ΔCys, or mutants, at 100% confluence in 60- or 100-mm dishes, were washed with phosphate-buffered saline (PBS) (4.3 mm Na2HPO4·7H2O, 1.4 mm KH2PO4, 137 mm NaCl, and 2.7 mm KCl, pH 7.3–7.4, at 37 °C), briefly treated with PBS containing 2 mm EDTA (PBS/EDTA), and then dissociated in PBS/EDTA. Cells suspensions were centrifuged at 50 × g for 2 min at room temperature, and the pellets were resuspended in 1 ml of buffer M (25 mm HEPES containing 5.4 mm KCl, 140 mm NaCl, and 2 mm EDTA, pH 7.2, at 22–25 °C) for treatment with methanethiosulfonate reagents or in 1.5 ml of buffer H (20 mm HEPES, containing 10 mm MgCl2, 2 mm EGTA, 0.2 mg/ml bacitracin, and 0.93 μg/ml aprotinin, pH 7.2, at 4 °C) for binding assays.
125I-Tyr0-Sauvagine Binding
For radioligand binding assays, cell suspensions (1.5 ml) in buffer H were homogenized using an Ultra-Turrax T25 homogenizer (IKA Janke and Kunkel, Staufen, Germany) at setting ∼20 for 10–15 s, at 4 °C. The homogenates were centrifuged at 16,000 × g for 10 min at 4 °C, and the membrane pellets were resuspended in 1 ml of buffer B (buffer H containing 0.1% bovine serum albumin, pH 7.2, at 20 °C). The membrane suspensions were diluted in buffer B and used for homologous or heterologous competition binding studies as described previously (15). In brief, aliquots of diluted membrane suspensions (50 μl) were added into low retention tubes (Kisker-Biotech, Steinfurt, Germany), containing buffer B and 20–50 pm 125I-Tyr0-sauvagine with or without increasing concentrations of Tyr0-sauvagine (homologous competition binding), sauvagine, astressin, or antalarmin (heterologous competition binding) (American Peptide Co., Sunnyvale, CA). The mixtures were incubated at 20–21 °C for 120 min and then filtered using a Brandel cell harvester through Whatman 934AH glass fiber filters presoaked for 1 h in 0.3% polyethyleneimine at 4 °C. The filters were washed three times with 0.5 ml of ice-cold PBS, pH 7.1, containing 0.01% Triton X-100. Filters were assessed for radioactivity in a gamma counter (1275 minigamma, 80% efficiency; LKB Wallac, Chalfont St. Giles, Buckinghamshire, UK). The amount of membrane used was adjusted to ensure that the specific binding was always equal to or less than 10% of the total concentration of the added radioligand. Specific 125I-Tyr0-sauvagine binding was defined as total binding less nonspecific binding in the presence of 1000 nm sauvagine or antalarmin. Data for competition binding were analyzed by nonlinear regression analysis, using Prism 4.0 (GraphPad Software, San Diego). IC50 values were obtained by fitting the data from competition studies to a one-site competition model. The logKi values for astressin and antalarmin and the logKD values for 125I-Tyr0-sauvagine binding were determined from heterologous and homologous competition data, respectively, as described previously using Prism 4.0 (15).
Reactions with MTSEA
For treatment with MTSEA, aliquots (0.1 ml) of suspensions (in buffer M) of cells expressing ΔCys or substituted Cys mutants were incubated without or with 2.5 mm MTSEA for 15 s at 22–25 °C. Cell suspensions were then diluted 140-fold in buffer PBS/EDTA, pH 7.1, at 22–25 °C containing 10 mm MgCl2 and centrifuged at 250 × g for 10 min at 22–25 °C, and the pellets were resuspended in 1.5 ml of buffer M containing 10 mm MgCl2. Cell suspensions were centrifuged at 500 × g for 5 min at 22–25 °C, and the pellets were homogenized in 1.5 ml of buffer H, as described above. The homogenates were centrifuged at 16,000 × g for 10 min at 4 °C, and the membrane pellets were resuspended in 1 ml of buffer B (buffer H containing 0.1% bovine serum albumin, pH 7.2, at 20 °C). The membrane suspensions were used to assay for 125I-Tyr0-sauvagine binding as described above. Inhibition of binding was calculated as 1 − (specific binding after MTSEA/specific binding without MTSEA).
Protection experiments were performed by preincubation of aliquots of cell suspensions with astressin (1 μm), Tyr0-sauvagine (1 μm), or α-helical (9–41) CRF (1 μm) for 60 min at 20 °C in buffer M. Subsequently, the mixtures (0.1 ml) were treated with 2.5 mm MTSEA as described above. Cells were treated and homogenized as described above, and homogenates were used to assay for 125I-Tyr0-sauvagine binding.
cAMP Accumulation Assays
HEK293 cells stably expressing WT, ΔCys, or mutants were plated in 96-well cell culture plates (pretreated with 0.1 mg/ml poly-l-lysine). After incubation overnight at 37 °C in 5% CO2, the cells were 95–100% confluent. The medium was removed, and 100 μl of assay buffer (25 mm HEPES, pH 7.4, 2 mm choline, 288 mm sucrose, 0.9 mm CaCl2, 0.5 mm MgCl2, and 1 mm 3-isobutyl-1-methylxanthine) was added. After 1 h of incubation at 37 °C, more assay buffer without (basal levels) or with increasing concentrations of sauvagine in the presence or absence of 0.3 μm astressin or 1–2 μm antalarmin was added to a total volume of 200 μl, and the incubation was continued for 30 min at 37 °C. At the end of the incubation, the assay buffer was removed. The cells were placed on ice and lysed with 3% trichloroacetic acid. Lysates were incubated on ice for 30–60 min and stored at −20 °C. After 1–5 days, frozen lysates were thawed and centrifuged at 1800 × g for 10 min at 4 °C, and the supernatants were neutralized with 2 n NaOH. Quantification of cAMP in the neutralized supernatants was performed using a competitive binding assay as described previously (15). In brief, supernatants were transferred to polypropylene mini-tubes (20 μl/tube) containing buffer A (100 mm Tris-HCl, pH 7.4, 100 mm NaCl, and 5 mm EDTA) with 1–1.5 nm [2,8-3H]cAMP (PerkinElmer Life Sciences). Subsequently, cAMP-binding protein (∼100 mg of crude bovine adrenal cortex extract in 500 ml of buffer A) was added to each tube. After incubation on ice for 3 h, the mixtures were filtered through Whatman 934AH glass fiber filters as described for radioligand binding assays, using buffer C (120 mm NaCl and 10 mm Tris HCl, pH 7.4, at 4 °C) as washing buffer. The amount of cAMP in each sample (one-tenth of a well) was determined by comparison with a standard curve of known concentrations of unlabeled cAMP (1–100 pmol/tube). The logEC50 values were obtained by fitting the data to a one-site sigmoidal model using nonlinear regression analysis (Prism 4.0).
RESULTS
Structure-based Alignment of GPCRs with Known Structure
Fig. 1A shows the superimposition of the helical TMs of representative members of the currently available crystal structures of GPCRs in the inactive conformation. The structure of the cytoplasmic part of TMs is highly conserved, whereas the structure of their extracellular part is more divergent. This suggests that each receptor family has adjusted specific structural characteristics for selective ligand binding that triggers a conserved set of conformational rearrangements of the helices near the G protein binding domains. The publication of the structures of the ligand-free opsin, the β2-adrenergic receptor (β2-AR) bound to agonists and the β2-AR bound to Gs have shown the intracellular structural changes associated with activation of class A GPCRs (16–18). To compare these mechanisms with class B GPCRs, Fig. 1B shows a structure-based sequence alignment of all available crystal structures of GPCRs, and supplemental Fig. S1 shows the amino acid conservation, at structurally homologous positions, within classes A and B. This alignment compares the highly conserved amino acids in class A, used by Ballesteros and Weinstein to define a general numbering scheme for class A GPCRs, with class B (19). Class B includes both the adhesion and secretin subfamilies, as the latter is a descendant from the adhesion subfamily (3). In particular, TM3 contains the conserved Cys3.25 engaged in a disulfide bond with a conserved Cys in EL2. The positional equivalent of the highly conserved (D/E)R3.50Y motif in class A corresponds to (Y/H)L3.50(Y/H) in class B. Because of the spatial conservation of the TM amino acids between classes A and B, the Ballesteros-Weinstein nomenclature is used throughout.
FIGURE 1.

Structural alignment of GPCR families with known structure. A, comparison of the TM domains plus Hx8 of the currently available crystal structures in the inactive conformation. Rhodopsin (Protein Data Bank codes 1U19), β2-adrenergic (2RH1), dopamine D3 (3PBL), M3 muscarinic (4DAJ), μ-opioid (4DKL), and histamine H1 (3RZE) receptors are shown as representative structures of class A GPCRs. The TM structures of class A, B (CRF1 (4K5Y) and GCG (4L6R)), and smoothened (4JKV) receptors are shown as white, blue, and orange ribbons, respectively. The highly conserved amino acids in each helix, used by Ballesteros and Weinstein to define a general numbering scheme for class A GPCRs, are shown as gray sticks (19). The homologous amino acids, at these conserved class A positions, for the class F smoothened receptor are shown in orange and for class B CRF1 and GCG receptors in blue. B, sequence alignment derived from the structural superimposition between GPCRs with known crystal structures.
Use of the Substituted Cysteine Accessibility Method (SCAM) to Identify Amino Acids of TM3 Forming the Water-accessible Binding Site Crevice
To identify the TM3 residues that are located on the water-accessible binding site crevice of the inactive apo-state of the full-length CRF1R, we mutated each amino acid, one at a time, to Cys (engineered Cys) and determined their accessibilities by applying SCAM (20). SCAM is based on the ability of the positively charged MTSEA, to react with the free sulfhydryl group of a cysteine (supplemental Fig. S2). We detected the MTSEA reaction by determining its ability to inhibit the binding of the radiolabeled CRF analog, 125I-Tyr0-sauvagine, to the receptor. We used an MTSEA-insensitive CRF1R mutant (ΔCys), which has a wild-type pharmacological profile and has been created by replacing five endogenous Cys by Ser (14). Among the 22 TM3 engineered Cys (ΔCys + R1893.26C to ΔCys + G2103.47C), those at positions 1893.26, 1923.29, 1933.30, 1953.32, 1963.33, and 1993.36 reacted with MTSEA because the reaction significantly reduced the binding of 125I-Tyr0-sauvagine to these Cys-substituted mutants (Fig. 2A). Given that in the absence of MTSEA, the R1893.26C, T1923.29C, A1933.30C, Y1953.32C, N1963.33C, and H1993.36C substitutions did not significantly alter 125I-Tyr0-sauvagine affinity (Fig. 2B) and thus the overall conformation of receptor, we infer that these TM3 residues are located on the binding site crevice of CRF1R. This is consistent with the recent crystal structures of CRF1R and GCGR (5, 6). Similarly, the ΔCys + F2033.40C mutant reacted with MTSEA, because the reaction significantly altered 125I-Tyr0-sauvagine binding. Surprisingly, in contrast to the other positive TM3 Cys mutants, MTSEA reaction with ΔCys + F2033.40C increased binding (Fig. 2A). Notably, the potentiation of radioligand binding to ΔCys + F2033.40C by MTSEA was significantly decreased after pretreatment of cells expressing this mutant with 1 μm astressin, but not with α-helical (9–41) CRF or Tyr0-sauvagine (at concentrations of 1μm) (Fig. 3A). We infer that F2033.40 is also located on the binding site crevice of CRF1R, given that this TM3 residue interacts with small nonpeptide antagonists, such as antalarmin, as suggested by our results (Fig. 3B) and by the crystal structure of CRF1R (6). The hydrophobic antalarmin, even at the high concentration of 1 μm, failed to antagonize sauvagine binding to ΔCys after mutation of the hydrophobic F2033.40 to the hydrophilic Cys, regardless of the presence of MTSEA (Fig. 3B). In marked contrast, the binding affinity of astressin was not affected by the F2033.40C mutation even in the presence of MTSEA (Fig. 3C). Astressin is a CRF peptide antagonist that is less sensitive to receptor activation-associated conformational changes than agonists, and it interacts with the extracellular N-domain of CRF1R (8, 15).
FIGURE 2.

Structural and functional characterization of the TM3 of CRF1R. A, inhibition of specific 125I-Tyr0-sauvagine binding to MTSEA-insensitive CRF1R mutant (ΔCys) or its substituted Cys mutants after their reaction with MTSEA is represented by bars, which indicate the mean ± S.E. values from 3 to 32 independent experiments. Negative inhibition means potentiation of radioligand binding after MTSEA reaction. Solid bars with asterisks indicate substituted Cys mutants for which inhibition/potentiation was significantly different from ΔCys (p < 0.05; one way ANOVA). B, binding affinities (−logKD) of 125I-Tyr0-sauvagine for the ΔCys CRF1R (ΔCys) and its substituted Cys mutants (ΔCys + R1893.26C to ΔCys + G2103.47C) are represented as bars, which indicate the mean ± S.E. values from 3 to 7 independent experiments. Solid bars with asterisks indicate substituted Cys mutants for which 125I-Tyr0-sauvagine binding affinity was significantly different from that for ΔCys (p < 0.05; one way ANOVA).
FIGURE 3.

Effect of F2033.40C mutation on the binding properties of ΔCys CRF1R. A, potentiation of 125I-Tyr0-sauvagine binding to ΔCys + F2033.40C by MTSEA after pretreatment of cells expressing this receptor without or with 1 μm astressin, 1 μm Tyr0-sauvagine, or 1 μm α-helical (9–41) CRF (α-hel-CRF). The bars indicate the mean ± S.E. values from 2 to 7 independent experiments. Hatched bar with asterisk indicates that MTSEA potentiation of binding to ΔCys + F2033.40C was significantly inhibited after pretreatment of receptor with ligand (p < 0.05; one-way ANOVA). B–D, competition binding isotherms of antalarmin (B), astressin (C), and Tyr0-sauvagine (D) at the ΔCys CRF1R, its substituted Cys mutant, ΔCys + F2033.40C, and the MTSEA-treated ΔCys + F2033.40C. The means ± S.E. (duplicate determination) are shown from a representative experiment performed 3–6 times with similar results. The affinities (−logKi) of antalarmin, determined from these experiments, are 7.32 ± 0.05, <5.00 and <5.00, for ΔCys, ΔCys + F2033.40C, and MTSEA-treated ΔCys + F2033.40C, respectively. The affinities (−logKD) of 125I-Tyr0-sauvagine, are given in Table 1. The affinities (−logKi) of astressin are 8.00 ± 0.10, 7.58 ± 0.19, and 7.54 ± 0.27, for ΔCys, ΔCys + F2033.40C, and MTSEA-treated ΔCys+F2033.40C, respectively.
Quantification of the binding affinity of 125I-Tyr0-sauvagine for the ΔCys CRF1R indicated that the F2033.40C mutation decreased the affinity of receptor 18-fold (Figs. 2B and 3D and Table 1). Notably, MTSEA restored the low affinity of ΔCys + F2033.40C to normal levels (Figs. 2B and 3D and Table 1). Substitution of F2033.40 by Lys (F2033.40K) did not significantly modify the binding of 125I-Tyr0-sauvagine relative to wild-type receptor (Fig. 4A), in a manner similar to the effect of the MTSEA reaction that adds a Lys-like side chain to F2033.40C (Fig. 3D). In contrast, the F2033.40K mutation abolished antalarmin binding (Fig. 4B), as in the F2033.40C mutation (Fig. 3B). The binding affinity of astressin was not affected by the F2033.40K mutation (Fig. 4C), similar to the F2033.40C mutation, which did not affect astressin affinity even in the presence of MTSEA (Fig. 3C). The effects of the Cys mutation of F2033.40 and its modification by MTSEA on the binding of the peptide agonist Tyr0-sauvagine, but not on the peptide antagonist astressin, suggest that F2033.40 influences receptor activation.
TABLE 1.
Effect of MTSEA reaction on the binding properties of the substituted Cys mutants
The affinities of Tyr0-sauvagine (−logKD) for the substituted Cys mutants before and after their reaction with MTSEA were determined from homologous competitive binding experiments, using as radioligand the 125I-Tyr0-sauvagine. The mean ± S.E. values were obtained from 2 to 7 independent experiments with similar results.
| −logKD ± S.E. | + MTSEA | |
|---|---|---|
| ΔCys + R189C | 8.38 ± 0.11 | 8.47 ± 0.40 |
| ΔCys + T192C | 8.50 ± 0.24 | 9.03 ± 0.29 |
| ΔCys + A193C | 8.77 ± 0.12 | 8.98 ± 0.05 |
| ΔCys + Y195C | 8.94 ± 0.23 | 9.51 ± 0.21 |
| ΔCys + N196C | 8.85 ± 0.21 | 9.33 ± 0.17 |
| ΔCys + H199C | 8.61 ± 0.12 | 8.94 ± 0.15 |
| ΔCys + F203C | 7.33 ± 0.20 | 8.80 ± 0.33 |
FIGURE 4.
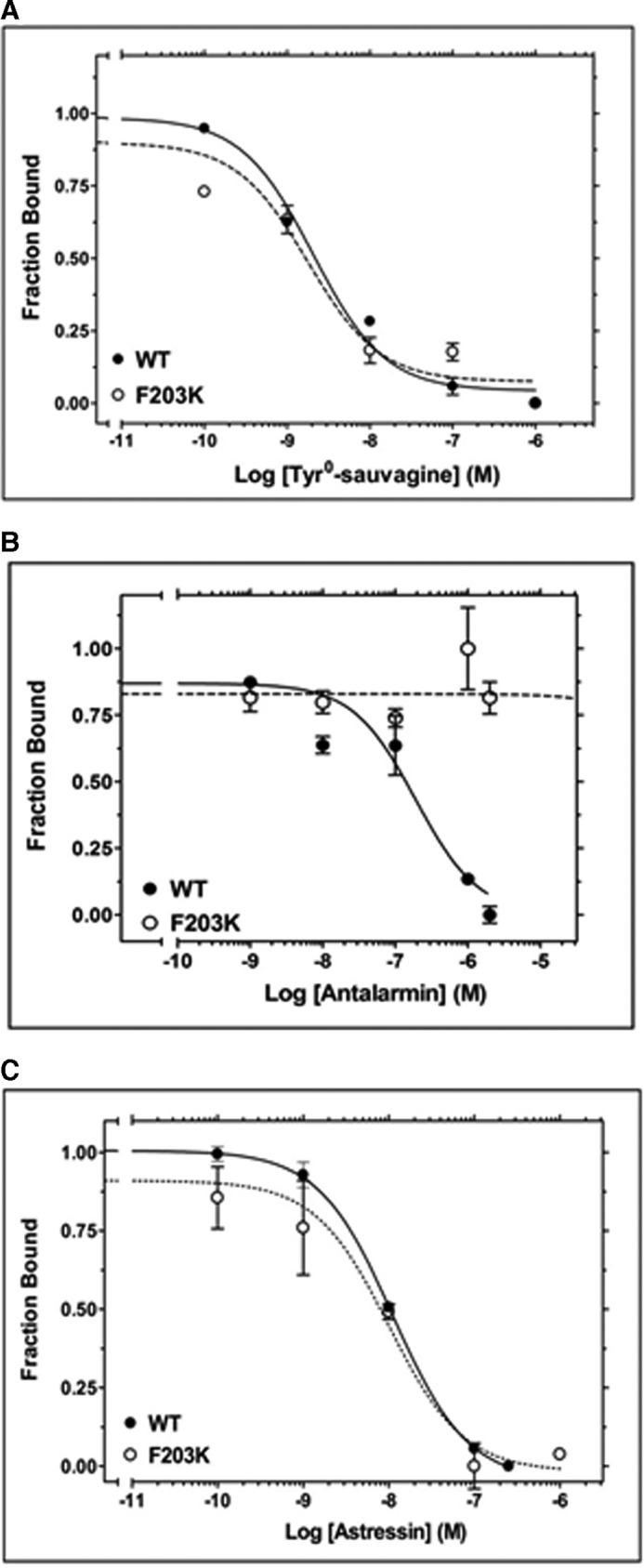
Effect of F2033.40K mutation on ligand binding. A, competition binding isotherms of Tyr0-sauvagine at the WT CRF1R and its F203K mutant. The means and S.E. (duplicate determination) are shown from a representative experiment performed 3–4 times with similar results. The affinities (−logKD) of 125I-Tyr0-sauvagine, determined from these experiments, are 8.38 ± 0.20 and 8.51 ± 0.21 for WT and F203K, respectively. B, competition binding isotherms of antalarmin at the F203K mutant and WT CRF1R. The means and S.E. (duplicate determination) are shown from a representative experiment performed 4–5 times with similar results. The affinities (−logKi) of antalarmin, determined from these experiments, are 7.62 ± 0.13 and <5.00 for WT and F203K, respectively. C, competition binding isotherms of astressin at the 203K mutant and WT CRF1R. The means and S.E. (duplicate determination) are shown from a representative experiment performed 3–6 times with similar results. The affinities (−logKi) of astressin, determined from these experiments, are 8.02 ± 0.07 and 8.02 ± 0.19 for WT and F203K, respectively.
Similar effects were observed in the binding of sauvagine, which lacks the N-terminal Tyr0 (Fig. 5A), to the F2033.40C mutant receptor. Sauvagine bound ΔCys + F2033.40C with low affinity, which was largely increased upon reaction with MTSEA (Fig. 5B).
FIGURE 5.

Effect of F2033.40C mutation on sauvagine binding. A, amino acid sequences of sauvagine and Tyr0-sauvagine. The amino acids in gray boxes interact with the second extracellular loop of CRF1R (15). B, competition binding isotherms of sauvagine at the ΔCys + F203C, before and after its reaction with MTSEA. The means and S.E. (duplicate determination) are shown from a representative experiment performed four times with similar results. The affinities (−logKi) of sauvagine, determined from these experiments, are 7.37 ± 0.11 and 8.20 ± 0.15, for the ΔCys + F203C and the MTSEA-treated ΔCys + F203C, respectively.
Role of F2033.40 in the Activation of the CRF1R
A GCGR-based homology model of the CRF1R used to depict the unliganded basal state shows a network of hydrophobic-aromatic interactions between TM3 and -6 (Fig. 6A), which is similar to the class A network (Fig. 6B) (17, 21). In this network of interactions, the hydrophobic-aromatic residues at positions 3.40 (Phe 77% and Tyr 20%) and 6.44 (Leu 65% and Phe 22%) are highly conserved in class B GPCRs (supplemental Fig. S1).
FIGURE 6.
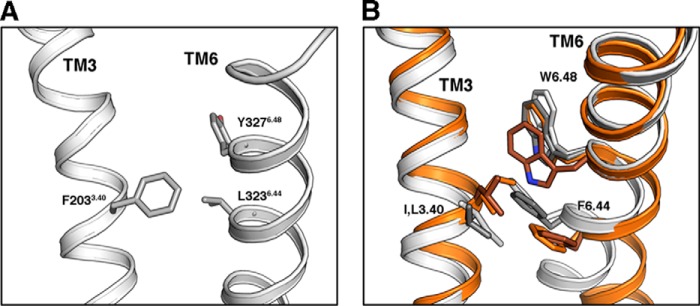
Role of F2033.40 in CRF1R activation. A, GCGR-based homology model of CRF1R, used to depict the ligand-free basal state. Residues F2033.40, L3236.44, and Y3276.48 are shown, whose homologous amino acids in class A GPCRs play a key role in receptor activation. B, comparison of the amino acids at positions 3.40, 6.44, and 6.48 in the inactive (white) and active (orange) β2-AR and the inactive (gray) and active (brown) rhodopsin.
The important functional role of F2033.40 of CRF1R is supported by the fact that its mutation to Trp or Ile does not modify the binding of 125I-Tyr0-sauvagine relative to WT receptor (Fig. 7A and Table 2). Importantly, removing the hydrophobic and bulky side chain at position 203 by mutating F2033.40 to Ala, reduced the affinity of the radiolabeled agonist 27-fold (Fig. 7A and Table 2). In contrast, the affinity of the antagonist astressin was not affected by these mutations (Fig. 7B and Table 2). The different effects of these mutations on peptide agonist versus antagonist binding further support that modifications at position 2033.40 of CRF1R affect the ability of the receptor to adopt the active conformation, which binds with high affinity agonists such as 125I-Tyr0-sauvagine. Antalarmin binding to CRF1R was abolished by the F2033.40A mutation, whereas F2033.40I and F2033.40W substitutions slightly modified and increased its binding affinity, respectively (Fig. 7C and Table 2).
FIGURE 7.

Interaction of antalarmin with F2033.40 of CRF1R. A–C, competition binding isotherms of Tyr0-sauvagine (A), astressin (B), and antalarmin (C) at the WT CRF1R and its F2033.40A, F2033.40I, and F2033.40W mutants. The means and S.E. (duplicate determination) are shown from a representative experiment performed 3–6 times with similar results. The affinities of 125I-Tyr0-sauvagine (−logKD), astressin (−logKi), and antalarmin (logKi)), determined from these experiments, are given in Table 2. D–G, stimulation of cAMP accumulation in HEK293 cells expressing WT (D), F2033.40W (E), F2033.40I (F), or F2033.40A (G) by sauvagine in the absence or presence of 0.3 μm astressin or 1–2 μm antalarmin. The means and S.E. (duplicate determination) are shown from a representative experiment performed 3–10 times with similar results. The potencies (−logEC50) of sauvagine without or with antalarmin or astressin, determined from these experiments, are given in Table 2. H, molecular model of the complex between antalarmin and CRF1R. The ethyl groups of the ligand interact with F2033.40, the aromatic ring interacts with M2063.43 and N2835.54, and the methyl groups of the terminal aromatic ring are located in a hydrophobic pocket of the receptor.
TABLE 2.
Effect of F2033.40 mutations on binding and signaling properties of CRF1R
Top, affinities of Tyr0-sauvagine (−logKD), astressin (−logKi), or antalarmin (−logKi) for WT CRF1R or F2033.40 mutants were determined from homologous or heterologous competitive binding experiments, using as radioligand the 125I-Tyr0-sauvagine. The mean ± S.E. values were obtained from 3 to 6 independent experiments with similar results. Bottom, potencies of sauvagine (−logEC50) for WT CRF1R or F2033.40 mutants were determined in cAMP accumulation experiments, in the absence or presence of 0.3 μm astressin or 1–2 μm antalarmin. The mean ± S.E. values were obtained from 3 to 10 independent experiments with similar results.
| WT | F203A | F203I | F203W | |
|---|---|---|---|---|
| −logKD or −logKi | ||||
| Tyr0-sauvagine | 8.38 ± 0.20 | 6.95 ± 0.11 | 8.40 ± 0.16 | 8.40 ± 0.37 |
| Astressin | 8.02 ± 0.07 | 8.03 ± 0.18 | 7.84 ± 0.18 | 7.74 ± 0.07 |
| Antalarmin | 7.62 ± 0.13 | <5.00 | 6.92 ± 0.15 | 9.54 ± 0.27 |
| −logEC50 | ||||
| Sauvagine | 9.90 ± 0.19 | 8.52 ± 0.23 | 9.79 ± 0.40 | 9.60 ± 0.06 |
| Sauvagine + astressin | 8.72 ± 0.21 | 7.40 ± 0.21 | 7.98 ± 0.30 | 8.05 ± 0.43 |
| Sauvagine + antalarmin | 8.38 ± 0.35 | 8.00 ± 0.29 | 6.74 ± 0.23 | 7.33 ± 0.69 |
To provide additional experimental evidence for the important role of F2033.40 in receptor activation, we determined the ability of sauvagine to stimulate cAMP accumulation in HEK293 cells before and after F2033.40 mutations. Clearly, F2033.40W and F2033.40I mutations did not affect the ability of sauvagine to activate CRF1R (Fig. 7, D–F, and Table 2). Specifically, F2033.40W and F2033.40I mutations reduced the potencies of sauvagine (−logEC50) only 2- and 1.3-fold, respectively. In contrast, the F2033.40A mutation had the greatest effect on sauvagine potency (Fig. 7G and Table 2), by decreasing it 24-fold. The potency of sauvagine (−logEC50) to stimulate cAMP accumulation was reduced by astressin or antalarmin by 15- or 33-fold, respectively, for WT receptor, 35- or 186-fold for the F2033.40W mutant, and 65- or 1117-fold for the F2033.40I mutant (Fig. 7, D–F, and Table 2).
Antalarmin Antagonizes Peptide Binding and Receptor Activation by Interacting with F2033.40
The importance of the aromatic ring at position 2033.40 in antalarmin binding to CRF1R has already been seen in different experiments (e.g. Figs. 3B, 4B, and 7C and Table 2). Moreover, the potency of sauvagine to stimulate cAMP accumulation greatly depended on the nature of the substituted residue at the 3.40 position (Fig. 7, D–G, and Table 2). Remarkably, antalarmin, even at a very high concentration (2 μm), was unable to inhibit sauvagine potency for F2033.40A, in agreement with the inability of this nonpeptide ligand to bind to F2033.40A (Fig. 7G and Table 2). In marked contrast, the peptide antagonist astressin decreased the potency of sauvagine to stimulate cAMP (Fig. 7G and Table 2). Fig. 7H shows a model of the complex between antalarmin and CRF1R based on the crystal structure of the similar CP-376395 ligand and CRF1R (6). F2033.40 interacts with the ethyl group of the ligand and restricts the binding site along with Y3276.48.
Gly2103.47 Plays an Important Role in the Activation of the CRF1R
The binding affinity of 125I-Tyr0-sauvagine for ΔCys + G2103.47C receptor was 129 times lower than that for ΔCys, whereas this mutation did not affect the binding affinity of astressin (Figs. 2B and 8A). Similarly, the potency of sauvagine to stimulate cAMP accumulation was decreased 121 times due to the G2103.47C mutation (Fig. 8B). The G2103.47A mutation had a significantly smaller impact on the affinity and potency of sauvagine compared with that of G2103.47C substitution. In specific the binding affinity of 125I-Tyr0-sauvagine and the potency of sauvagine for the ΔCys were only reduced 10- and 3.5-fold, respectively, by the G2103.47A mutation (Fig. 8, A and B). In addition, similar to G2103.47C mutation, the G2103.47A substitution did not affect the affinity of astressin (Fig. 8A). Fig. 8C shows that G2103.47 is pointing toward N2835.54 in TM5 (88% conserved in the class B sequences), which is forming an inter-helical hydrogen bond with the backbone oxygen of the amino acid at position 3.43 (Met-206). Mutation of G2103.47C, but not of G2103.47A, adds a polar side chain in the key interface between TM3 and -5 that interacts with N2835.54 (Fig. 8D). The publication of the crystal structure of the ligand-free opsin showed that during the process of receptor activation, the intracellular part of TM6 tilts outward and TM5 comes close to TM6 (16). Thus, we propose that the additional constraint between N2835.54 and C2103.47 that replaced G2103.47 restrains TM5 in the inactive conformation that makes difficult the activation of the mutant CRF1R by the extracellular ligand. As a consequence, only small, nonpolar amino acids are found at position 3.47 in class B GPCRs (Gly 57% and Ala 39%).
FIGURE 8.

Effect of G2103.47C and G2103.47A mutations on the binding and signaling properties of ΔCys CRF1R. A, binding affinities of astressin (−logKi) and 125I-Tyr0-sauvagine (−logKD) for ΔCys and its substituted Cys mutants, ΔCys + G2103.47C and ΔCys + G2103.47A, are represented as bars, which indicate the mean ± S.E. values from 3 to 4 independent experiments. The asterisk and pound indicate that mutations significantly alter 125I-Tyr0-sauvagine binding affinity compared with that of ΔCys and ΔCys + G2103.47C, respectively (p < 0.05; one-way ANOVA). B, stimulation of cAMP accumulation in HEK293 cells expressing ΔCys, ΔCys + G2103.47C, or ΔCys + G2103.47A by sauvagine. The means and S.E. (duplicate determination) are shown from a representative experiment performed 4–11 times with similar results. The potencies (−logEC50) of sauvagine for the ΔCys, ΔCys + G2103.47C, and ΔCys + G2103.47A, determined from these experiments, are 8.68 ± 0.14, 6.60 ± 0.13, and 8.15 ± 0.06, respectively. C and D, molecular model of the interface between TM3 and -5 of CRF1R, constructed from the structure of the ligand-free GCGR. C, N2835.54 in TM5 is forming an inter-helical hydrogen bond with the backbone of M2063.43. D, mutation of G2103.47 to Cys adds a polar side chain that interacts with N2835.54.
DISCUSSION
By using SCAM, we identified the TM3 residues of the apo-inactive state of the full-length CRF1R that are exposed in the water-accessible binding site crevice of the receptor. Seven among the 22 TM3 residues (R1893.26, T1923.29, A1933.30, Y1953.32, N1963.33, H1993.36, and F2033.40) were found to be located on the surface of the binding site crevice, whereas residues that are located deeper in TM3 than F2033.40 were inaccessible. These results suggest that the TM3 of CRF1R in its unliganded state is positioned such that half of this helix participates in the formation of a large cavity, whereas the other half is tightly packed with the other TMs. This prediction is compatible with the crystal structures of the solubilized, N-terminally truncated inactive states of both unliganded GCGR and CRF1R complexed with CP-376395 (5, 6). The identified large V-shape cavity of the CRF1R is capable of accommodating the large peptides that bind class B GPCRs.
Among the Cys substitutions of TM3 residues, the G2103.47C mutation had the largest impact on the high affinity of 125I-Tyr0-sauvagine binding. Specifically, substitution of G2103.47 to Cys decreased the binding affinity of 125I-Tyr0-sauvagine 129-fold. Similarly, the G2103.47C mutation decreased the potency of sauvagine 121-fold. These results suggest that mutation of G2103.47 to Cys largely decreased the ability of receptor to adopt its active state. G2103.47 is located one helical turn below residue M2063.43, whose backbone carbonyl group forms a hydrogen bond interaction with N2835.54 of TM5 (Fig. 8C), in the crystal structures of GCGR and CRF1R (5, 6). Our computational model shows that the –SH group of C2103.47 in the G2103.47C mutation forms a hydrogen bond with the side chain oxygen atom of N2835.54 (Fig. 8D). In contrast, mutation of G2103.47 to Ala, which cannot form this hydrogen bond, had a much smaller impact on the affinity and potency of sauvagine. Thus, strengthening the interface between TM3 and -5 further stabilizes the inactive state of receptor, thus rendering it unable to be activated by agonists to the same degree as the wild-type receptor. This leads to the speculation that the movement of TM3 and TM5 that occurs during the process of class A receptor activation might also occur in class B GPCRs (16). Similarly, the mechanism of receptor inactivation by the nonpeptide antagonist antalarmin could be partly due to the reinforcement of this interface between TM3 and -5 (Fig. 7H). The pyrrolo-pyrimidine group of antalarmin forms a hydrogen bond with N2835.54 and a hydrophobic interaction with M2063.43, whereas its ethyl group forms an aliphatic-aromatic interaction with F2033.40 (Fig. 7H).
Interestingly, the central part of the TM3-TM5 interface has also been suggested to play an important role in the activation of class A GPCRs (17, 21). Specifically, in the crystal structure of the inactive state of β2-AR, the relative positions of TM3, -5, and -6 are stabilized by interactions between a cluster of conserved hydrophobic and aromatic residues P2115.50, I1213.40, and F2826.44. Comparison of the agonist-bound active structure of the β2-adrenergic receptor with its inactive structure shows an activation-associated rearrangement (“transmission switch”) of I1213.40, P2115.50, and F2826.44 (Fig. 6B) (9, 17, 22). In specific, agonist binding triggers an anticlockwise rotation, viewed from the extracellular part, of I1213.40 and F2826.44 facilitating the outward movement of TM6 for β2-AR activation (Fig. 6B) (17). In addition to this rearrangement, the crystal structure of metarhodopsin II (23) also shows the shift of W6.48 toward TM5, relative to the inactive structure (Fig. 6B). Class B GPCRs also contain a conserved Pro residue (present in 87% of the sequences) at position 5.45 which, in the crystal structures of GCGR and CRF1R, does not cause the characteristic opening of the helix as observed at P5.50 in class A receptors. However, F2033.40 (Phe 77% and Tyr 20% in class B sequences) and L3236.44 (Leu 65% and Phe 22%) of CRF1R, which correspond to I1213.40 (Ile 40%, Val 25%, and Leu 11% in class A sequences), and F2826.44 (Phe 81% and Tyr 7%) in β2-AR (supplemental Fig. S1), respectively, form an aromatic-aliphatic interaction in the model of the apo, unliganded, and inactive state of CRF1R (Fig. 6). Importantly, removal of the aromatic F2033.40 side chain (mutation to Ala, F2033.40A) of CRF1R decreased the ability of receptor to adopt its active state, which is responsible for the high affinity binding of 125I-Tyr0-sauvagine and the high potency of sauvagine. In contrast, conservation of the aromaticity at this position by mutating F2033.40 to Trp (F2033.40W) or adding the bulky and aliphatic Ile side chain (F2033.40I), to mimic the β2-adrenergic sequence, did not considerably affect the pharmacological properties of the receptor. These results led us to speculate that class B GPCRs might also contain a “transmission switch” from TM3 toward TM6 that finally triggers the outward movement of the cytoplasmic end of TM6 for receptor activation and G protein binding.
Consistent with the notion that the presence of a bulky aromatic/aliphatic side chain at position 2033.40 is important for CRF1R activation, introduction of a smaller and hydrophilic Cys side chain (F2033.40C) also decreased the high affinity binding of 125I-Tyr0-sauvagine. Interestingly, reaction of C2033.40 of the mutant receptor with the MTSEA restored the binding affinity of radiolabeled agonist. Moreover, substitution of F2033.40 with Lys (F2033.40K) did not alter the high affinity binding of 125I-Tyr0-sauvagine relative to the wild-type receptor. Thus, addition of the positively charged side chain of K2033.40 or MTSEA-C2033.40, within the hydrophobic environment of the 2033.40 locus, probably forms a new set of interactions with nearby amino acids that facilitate receptor activation. Repositioning of the 3.40 residue and formation of a new set of interactions during receptor activation has also been suggested in class A GPCRs (17).
Similar to the modification of F2033.40 in CRF1R, Ala mutation of the homologous residue in the class B glucose-dependent insulinotropic polypeptide receptor has been shown to decrease the ability of peptides to activate the receptor (24). In contrast, Ala mutation of this residue in GCGR did not affect receptor activation by glucagon (5). Likewise, mutation of the corresponding residue in class A GPCRs had a different impact in receptor activation. For example, Ala mutation of the amino acid at position 3.40 decreased the basal and agonist-induced activation of the histamine H1 receptor (21). In marked contrast Ala mutation of the homologous residue in the muscarinic receptor constitutively activated the receptor and increased the binding affinity and signaling efficacy of agonists (25). Moreover, mutation of the corresponding residue in the thyrotropin-releasing hormone receptor did not change its pharmacological profile (26). Thus, members from different families of GPCRs could share common structural elements involved in their mechanism of activation, however without this pattern of conservation serving as an exclusive feature of all receptors belonging to these families. Furthermore, different ligands also trigger different activation mechanisms at a given receptor (27). These findings might provide the basis for a novel classification of GPCRs according to their common mechanisms of activation.
The effects of F2033.40A and F2033.40C mutations on 125I-Tyr0-sauvagine binding are not likely to be due to a possible loss of interaction between the Tyr0 of sauvagine and the aromatic group of F2033.40 because these mutations had similar impact on the binding of sauvagine, which lacks Tyr0. Similarly, the first four N-terminal residues of CRF peptides are not important for their biological activity (28). Likewise the aliphatic residues Ile-5 and Ile-7 of sauvagine are not likely to interact with F2033.40 because the introduction of a positive charge at position 2033.40 after F2033.40K mutation did not affect the high affinity binding 125I-Tyr0-sauvagine. The hydrophilic Ser-6 of sauvagine is also not compatible with the hydrophobic F2033.40 of CRF1R. In addition, sauvagine residues 8–11 do not enter the binding site crevice of CRF1R to reach F2033.40, because they interact with the EL2 of receptor (15). Thus, F2033.40 must play an allosteric role in peptide binding and peptide-mediated activation of CRF1R. The important conformational role of F2033.40 is further supported by the different effects of various ligands on MTSEA reactivity at this residue. The agonist Tyr0-sauvagine and the α-helical (9–41) CRF, which is an antagonist with partial agonist properties (29), were unable to protect F2033.40C from MTSEA modification. In marked contrast, the antagonist astressin significantly decreased the ability of MTSEA to potentiate 125I-Tyr0-sauvagine binding to F2033.40C. The different functional role of various CRF ligands is in agreement with previous studies. Astressin binding to CRF1R has been shown to be potentiated by the GTP analog, Gpp(NH)p, whereas that of the agonist CRF was decreased (30).
Interestingly, F2033.40 not only plays a key role in CRF1R activation but also is the most important contact site of the nonpeptide antagonist, antalarmin. Removing its side chain by mutating F2033.40 to Ala or replacing it with the hydrophilic Cys or the positively charged Lys abolished the binding of the hydrophobic nonpeptide antagonist, antalarmin, as well as its ability to decrease the potency of sauvagine for the CRF1R. Similar to our findings, the crystal structure of CRF1R has shown that F2033.40 interacts with CP-376395 (6). Thus, F2033.40 plays a common role in the binding of different nonpeptide CRF1R antagonists, and it is speculated that this interaction allosterically inhibits peptide binding and receptor activation.
Conclusively, TM3 plays a key role in the activation of the class B CRF1R, as in class A GPCRs. Remarkably, as in CRF1R, mutations at position 3.40 of the TM3 of class A, rhodopsin receptor, severely affect the receptor's function, being related to a decreased time in the meta II active state of receptor, poor retinal binding, reduced transducin activation, and retinitis pigmentosa (31). Moreover, the 3.40 residue of CRF1R is not just a mere contact site of nonpeptide antagonists, but possibly serves as a molecular determinant of allosteric antagonism for peptide binding and receptor activation by nonpeptide antagonists. This finding is not only of fundamental biological interest, but it also holds great potential for enhancing human health, given that small nonpeptide CRF1R antagonists have been shown to be effective against stress-related disorders, such as anxiety and depression (10).
Supplementary Material
Acknowledgment
We are grateful to Dr. George Chrousos for the gift of antalarmin.
This work was supported in part by a National Alliance for Research on Schizophrenia and Depression grant (to G. L.), a grant from the Greek Ministry of Health (to G. L.), and Grant SAF2013-48271-C2-2-R from Ministerio de Ciencia e Innovacion (to L. P.).

This article contains supplemental Figs. S1 and S2.
- GPCR
- G protein-coupled receptor
- CRF1R
- type 1 receptor for the corticotropin-releasing factor
- CRF
- corticotropin-releasing factor
- GCGR
- glucagon receptor
- SCAM
- substituted cysteine accessibility method
- MTSEA
- methanothiosulfonate ethylammonium
- TM
- transmembrane domain
- EL
- extracellular loop
- N-domain
- extracellular N-terminal region
- β2-AR
- β2-adrenergic receptor
- GppNHp
- guanosine 5′-[β,γ-imido]triphosphate
- ANOVA
- analysis of variance.
REFERENCES
- 1. Bockaert J., Pin J. P. (1999) Molecular tinkering of G protein-coupled receptors: an evolutionary success. EMBO J. 18, 1723–1729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Liapakis G., George L., Cordomí A., Arnau C., Pardo L., Leonardo P. (2012) The G protein coupled receptor family: actors with many faces. Curr. Pharm. Des. 18, 175–185 [DOI] [PubMed] [Google Scholar]
- 3. Nordström K. J., Lagerström M. C., Wallér L. M., Fredriksson R., Schiöth H. B. (2009) The secretin GPCRs descended from the family of adhesion GPCRs. Mol. Biol. Evol. 26, 71–84 [DOI] [PubMed] [Google Scholar]
- 4. Katritch V., Cherezov V., Stevens R. C. (2013) Structure-function of the G protein-coupled receptor superfamily. Annu. Rev. Pharmacol. Toxicol. 53, 531–556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Siu F. Y., He M., de Graaf C., Han G. W., Yang D., Zhang Z., Zhou C., Xu Q., Wacker D., Joseph J. S., Liu W., Lau J., Cherezov V., Katritch V., Wang M. W., Stevens R. C. (2013) Structure of the human glucagon class B G protein-coupled receptor. Nature 499, 444–449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hollenstein K., Kean J., Bortolato A., Cheng R. K., Doré A. S., Jazayeri A., Cooke R. M., Weir M., Marshall F. H. (2013) Structure of class B GPCR corticotropin-releasing factor receptor 1. Nature 499, 438–443 [DOI] [PubMed] [Google Scholar]
- 7. Hoare S. R. (2005) Mechanisms of peptide and nonpeptide ligand binding to Class B G protein-coupled receptors. Drug. Discov. Today 10, 417–427 [DOI] [PubMed] [Google Scholar]
- 8. Perrin M. H., Sutton S., Bain D. L., Berggren W. T., Vale W. W. (1998) The first extracellular domain of corticotropin releasing factor-R1 contains major binding determinants for urocortin and astressin. Endocrinology 139, 566–570 [DOI] [PubMed] [Google Scholar]
- 9. Venkatakrishnan A. J., Deupi X., Lebon G., Tate C. G., Schertler G. F., Babu M. M. (2013) Molecular signatures of G protein-coupled receptors. Nature 494, 185–194 [DOI] [PubMed] [Google Scholar]
- 10. Liapakis G., Venihaki M., Margioris A., Grigoriadis D., Gkountelias K. (2011) Members of CRF family and their receptors: from past to future. Curr. Med. Chem. 18, 2583–2600 [DOI] [PubMed] [Google Scholar]
- 11. Shatsky M., Nussinov R., Wolfson H. J. (2004) A method for simultaneous alignment of multiple protein structures. Proteins 56, 143–156 [DOI] [PubMed] [Google Scholar]
- 12. Waterhouse A. M., Procter J. B., Martin D. M., Clamp M., Barton G. J. (2009) Jalview Version 2–a multiple sequence alignment editor and analysis workbench. Bioinformatics 25, 1189–1191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Martí-Renom M. A., Stuart A. C., Fiser A., Sánchez R., Melo F., Sali A. (2000) Comparative protein structure modeling of genes and genomes. Annu. Rev. Biophys. Biomol. Struct. 29, 291–325 [DOI] [PubMed] [Google Scholar]
- 14. Gkountelias K., Papadokostaki M., Javitch J. A., Liapakis G. (2010) Exploring the binding site crevice of a family B G protein-coupled receptor, the type 1 corticotropin releasing factor receptor. Mol. Pharmacol. 78, 785–793 [DOI] [PubMed] [Google Scholar]
- 15. Gkountelias K., Tselios T., Venihaki M., Deraos G., Lazaridis I., Rassouli O., Gravanis A., Liapakis G. (2009) Alanine scanning mutagenesis of the second extracellular loop of type 1 corticotropin-releasing factor receptor revealed residues critical for peptide binding. Mol. Pharmacol. 75, 793–800 [DOI] [PubMed] [Google Scholar]
- 16. Park J. H., Scheerer P., Hofmann K. P., Choe H. W., Ernst O. P. (2008) Crystal structure of the ligand-free G protein-coupled receptor opsin. Nature 454, 183–187 [DOI] [PubMed] [Google Scholar]
- 17. Rasmussen S. G., Choi H. J., Fung J. J., Pardon E., Casarosa P., Chae P. S., Devree B. T., Rosenbaum D. M., Thian F. S., Kobilka T. S., Schnapp A., Konetzki I., Sunahara R. K., Gellman S. H., Pautsch A., Steyaert J., Weis W. I., Kobilka B. K. (2011) Structure of a nanobody-stabilized active state of the β2 adrenoceptor. Nature 469, 175–180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Rasmussen S. G., DeVree B. T., Zou Y., Kruse A. C., Chung K. Y., Kobilka T. S., Thian F. S., Chae P. S., Pardon E., Calinski D., Mathiesen J. M., Shah S. T., Lyons J. A., Caffrey M., Gellman S. H., Steyaert J., Skiniotis G., Weis W. I., Sunahara R. K., Kobilka B. K. (2011) Crystal structure of the β2 adrenergic receptor-Gs protein complex. Nature 477, 549–555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ballesteros J., Weinstein H. (1995) Integrated methods for the construction of three-dimensional models of structure-function relations in G protein-coupled receptors. Methods Neurosci. 25, 366–428 [Google Scholar]
- 20. Javitch J. A., Shi L., Liapakis G. (2002) Use of the substituted cysteine accessibility method to study the structure and function of G protein-coupled receptors. Methods Enzymol. 343, 137–156 [DOI] [PubMed] [Google Scholar]
- 21. Sansuk K., Deupi X., Torrecillas I. R., Jongejan A., Nijmeijer S., Bakker R. A., Pardo L., Leurs R. (2011) A structural insight into the reorientation of transmembrane domains 3 and 5 during family A G protein-coupled receptor activation. Mol. Pharmacol. 79, 262–269 [DOI] [PubMed] [Google Scholar]
- 22. Rasmussen S. G., Choi H. J., Rosenbaum D. M., Kobilka T. S., Thian F. S., Edwards P. C., Burghammer M., Ratnala V. R., Sanishvili R., Fischetti R. F., Schertler G. F., Weis W. I., Kobilka B. K. (2007) Crystal structure of the human β2 adrenergic G protein-coupled receptor. Nature 450, 383–387 [DOI] [PubMed] [Google Scholar]
- 23. Choe H. W., Kim Y. J., Park J. H., Morizumi T., Pai E. F., Krauss N., Hofmann K. P., Scheerer P., Ernst O. P. (2011) Crystal structure of metarhodopsin II. Nature 471, 651–655 [DOI] [PubMed] [Google Scholar]
- 24. Yaqub T., Tikhonova I. G., Lättig J., Magnan R., Laval M., Escrieut C., Boulègue C., Hewage C., Fourmy D. (2010) Identification of determinants of glucose-dependent insulinotropic polypeptide receptor that interact with N-terminal biologically active region of the natural ligand. Mol. Pharmacol. 77, 547–558 [DOI] [PubMed] [Google Scholar]
- 25. Lu Z. L., Hulme E. C. (1999) The functional topography of transmembrane domain 3 of the M1 muscarinic acetylcholine receptor, revealed by scanning mutagenesis. J. Biol. Chem. 274, 7309–7315 [DOI] [PubMed] [Google Scholar]
- 26. Perlman J. H., Laakkonen L. J., Guarnieri F., Osman R., Gershengorn M. C. (1996) A refined model of the thyrotropin-releasing hormone (TRH) receptor binding pocket. Experimental analysis and energy minimization of the complex between TRH and TRH receptor. Biochemistry 35, 7643–7650 [DOI] [PubMed] [Google Scholar]
- 27. Ersoy B. A., Pardo L., Zhang S., Thompson D. A., Millhauser G., Govaerts C., Vaisse C. (2012) Mechanism of N-terminal modulation of activity at the melanocortin-4 receptor GPCR. Nat. Chem. Biol. 8, 725–730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Grigoriadis D. E., Haddach M., Ling N., Saunders J. (2001) The CRF receptor: structure, function and potential for therapeutic intervention. Curr. Med. Chem. Syst. Agents 1, 63–97 [Google Scholar]
- 29. Winslow J. T., Newman J. D., Insel T. R. (1989) CRH and α-helical-CRH modulate behavioral measures of arousal in monkeys. Pharmacol. Biochem. Behav. 32, 919–926 [DOI] [PubMed] [Google Scholar]
- 30. Perrin M. H., Sutton S. W., Cervini L. A., Rivier J. E., Vale W. W. (1999) Comparison of an agonist, urocortin, and an antagonist, astressin, as radioligands for characterization of corticotropin-releasing factor receptors. J. Pharmacol. Exp. Ther. 288, 729–734 [PubMed] [Google Scholar]
- 31. Madabushi S., Gross A. K., Philippi A., Meng E. C., Wensel T. G., Lichtarge O. (2004) Evolutionary trace of G protein-coupled receptors reveals clusters of residues that determine global and class-specific functions. J. Biol. Chem. 279, 8126–8132 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.