

Background: Intracellular calcium and metabolic signals mediate glucose-induced insulin secretion.
Results: In metabolically compromised islets, calcium signaling was robust, whereas reduction and translocation of cytochrome c were diminished.
Conclusion: Data are consistent with a rate-determining role at the level of cytochrome c for insulin secretion.
Significance: Impairment of metabolic control preferentially affects cytochrome c versus calcium signaling.
Keywords: Calcium, Calcium Signaling, Cytochrome c, Insulin Secretion, Mitochondrial Metabolism, Pancreatic Islet, Cytochrome c Translocation, Oxygen Consumption
Abstract
The aim of the study was to assess the relative control of insulin secretion rate (ISR) by calcium influx and signaling from cytochrome c in islets where, as in diabetes, the metabolic pathways are impaired. This was achieved either by culturing isolated islets at low (3 mm) glucose or by fasting rats prior to the isolation of the islets. Culture in low glucose greatly reduced the glucose response of cytochrome c reduction and translocation and ISR, but did not affect the response to the mitochondrial fuel α-ketoisocaproate. Unexpectedly, glucose-stimulated calcium influx was only slightly reduced in low glucose-cultured islets and was not responsible for the impairment in glucose-stimulated ISR. A glucokinase activator acutely restored cytochrome c reduction and translocation and ISR, independent of effects on calcium influx. Islets from fasted rats had reduced ISR and cytochrome c reduction in response to both glucose and α-ketoisocaproate despite normal responses of calcium. Our data are consistent with the scenario where cytochrome c reduction and translocation are essential signals in the stimulation of ISR, the loss of which can result in impaired ISR even when calcium response is normal.
Introduction
Insulin secretion rate (ISR)3 is determined both by KATP-dependent pathways involving increased calcium (Ca2+) influx, as well as by KATP-independent pathways involving the generation of metabolic factors (1–5). Increased Ca2+ in the absence of elevated metabolic rate does not elicit second-phase insulin secretion (6, 7). Outstanding questions remain as to the relative control of ISR by Ca2+ influx versus contribution of metabolic factors and as to which metabolic factors are most responsible for determining ISR (8–10). We have recently presented evidence that signaling by cytochrome c is an essential component of the KATP-independent pathway (11) in addition to its role in the electron transport chain.
Multiple quantitative criteria for establishing cytochrome c signaling as a control point in the regulation of ISR were met. Importantly, under a wide range of experimental conditions resulting in elevated Ca2+ influx, ISR is temporally and proportionally related to the response in reduced cytochrome c. Moreover, the reduced state of cytochrome c has a remarkable dynamic sensitivity to nutrient secretagogues, whereas this metabolic factor is tightly maintained in the absence of changes in levels of nutrient secretagogues (12). This intrinsic attribute of islets allows cytochrome c reduction to function as a signal that relegates control of ISR to extracellular nutrient levels unaffected by the energetic needs of the cells. In addition, cytochrome c is unique among the electron transport chain components that manifest this behavior. Finally, the metabolic factor must directly or indirectly interact with exocytotic machinery in the cytosol. Cytochrome c is the only mobile protein in the electron transport chain, and it translocates to the cytosol in response to nutrient secretagogues (13), thus providing the opportunity for direct action on downstream processes of exocytosis.
The pathway of KATP-independent insulin secretion has been conceptualized as a system that increases the efficacy of Ca2+ to stimulate ISR (14). The fact that both Ca2+ and a metabolic factor need to be increased suggested to us a model where these two signals interact downstream, presumably via a regulatory protein complex. We have previously delineated this scenario by defining a process (the Ca2+/metabolic coupling process (CMCP)) that is activated only when both Ca2+ influx is stimulated and cytochrome c is reduced and perhaps translocated (11, 15). The proteins comprising the CMCP have not been identified, but its activity can be measured as Ca2+-sensitive oxygen consumption rate (OCR), a parameter representing the work carried out by the regulatory process. Functionally, cytochrome c reduction and Ca2+ influx both have to increase to activate the CMCP and ISR.
Although it is known that islets initially compensate for insulin resistance, impairment of glucose metabolism in islets is associated with long standing diabetes (16–18). We therefore strove to determine the relative control of ISR by Ca2+ and cytochrome c signaling in islets that have lower metabolic activity. We reasoned that the loss of metabolic response would differentially affect Ca2+ influx versus cytochrome c reduction allowing evaluation of the relative importance of these two signals in governing ISR. To do this, two islet models with impaired metabolic response were used. The first model was induced by culturing islets in the presence of low glucose, which causes the loss of glucokinase (GK) activity, thereby decreasing the metabolic response to glucose (19, 20). The second model of impaired metabolism was generated by fasting rats prior to islet isolation (21). The major findings of the study from both islet models were that although glucose-stimulated Ca2+ influx remained robust in the islets, OCR, cytochrome c reduction, and ISR were all decreased. The implication is that when islet metabolism is moderately compromised, there is still enough production of energy to promote stimulatory levels of Ca2+ influx, but the impairment of cytochrome c signaling results in decreased ISR.
EXPERIMENTAL PROCEDURES
Chemicals
Krebs-Ringer bicarbonate was used for the perifusion analyses as described (12). Antimycin A, potassium cyanide (KCN), glucose, KIC, (S)-(−)-BayK8644, and nimodipine were purchased from Sigma-Aldrich. A high affinity glucokinase activator (GKa) (PF-05029514), [(S)-6-(3-cyclopentyl-2-(2-oxo-5(trifluoromethyl)pyrazin-1(2H)-yl)propanamido)nicotinic acid] (22), was kindly provided by Pfizer.
Islet Isolation and Culture
Islets were harvested from Sprague-Dawley rats (∼250 g, Charles River) anesthetized by intraperitoneal injection of Beuthanasia-D (35 mg of pentobarbital sodium and 5.5 mg of phenytoin sodium/230 g of rat; Schering-Plough, Union, NJ). All procedures were approved by the University of Washington Institutional Animal Care and Use Committee. Islets were prepared and purified as described (23, 24) and then used either fresh or cultured at 37 °C in RPMI medium 1640 (containing no glucose) supplemented with 10% heat-inactivated fetal bovine serum (Invitrogen) and either 3 mm or 11 mm glucose. Islets were also harvested from a novel mouse model in which we induced the conditional double knock-out (DKO) of islet Bax and Bak, as recently described (25), as well as from age- and background-matched Cre-negative Bax-floxed (i.e. wild type) mice that were used as controls.
Measurement of OCR, Cytochrome c Reduction, and ISR
A flow culture system was used that concomitantly measured OCR and cytochrome c reduction while collecting outflow fractions for subsequent measurement of insulin (described previously (23, 26, 27)). OCR was calculated as the flow rate (∼80 μl/min) times the difference between inflow and outflow oxygen tension and measured by detecting signal from an oxygen-sensitive dye painted on the inside of the perifusion chamber (23). Reduced cytochrome c was measured as absorbance at 550 nm by the layer of intact islets in the perifusion chamber (23). Insulin in the outflow fractions was measured using an RIA kit (Linco Research Inc., Billerica, MA).
Imaging and Quantification of Cytosolic Ca2+
Cytosolic Ca2+ was measured by fluorescent imaging of islets after loading them with Fura-2 AM (Invitrogen) as described previously (28). Dyed islets were pipetted into a temperature-controlled perifusion dish as a monolayer (Bioptechs, Butler, PA) that was mounted onto the stage of a Nikon Eclipse TE-200 inverted microscope, and fluorescent emission was detected at 510 nm during alternating excitation at either 340 nm or 380 nm. Flow rate was 120 μl/min, and the perifusion chamber volume was 40 μl.
Measurement of Ca2+ Influx
Islets were preincubated in Krebs-Ringer bicarbonate containing 3 mm glucose for 60 min at 37 °C/5% CO2. Subsequently, islets were picked into 12 × 75 test tubes containing 90 μl of Krebs-Ringer bicarbonate (with 0.5 mm Ca2+) and incubated for 30 min. Ten μl of 45Ca2+ (1 μCi) were then added followed by the addition of the indicated reagents. The free and cell-associated radioactivity were separated at exactly 15 min by centrifuging the islets through an oil layer and measuring the radioactivity (29).
Measurement of ATP and Insulin Content
ATP was measured using an ATP Lite kit (PerkinElmer), where luminescence was measured using a Synergy4 microplate reader (BioTek, Winooski, VT). Insulin content per islet was determined after adding islets (5/sample) to 1 ml of a hydrochloric acid, 95% ethanol mixture (2% v/v) and incubating for 60 min. The supernatant was collected and assayed for insulin.
Western Blot Analysis of Cytosolic Cytochrome c
To avoid mitochondrial contamination, extraction of cytosol was accomplished by permeabilization of the islet plasma membrane (30, 31). Islets were incubated in 24-well plates (500/well) containing 500 μl of Krebs-Ringer bicarbonate under various conditions. To extract cytosol, 480 μl of supernatant were removed (leaving 20 μl), and 1 ml of ice-cold PBS was added. After the islets settled, the wash buffer was replaced with 70 μl of ice-cold permeabilization buffer (20 mm HEPES, 2.5 mm KH2PO4, 10 mm KCl, 1.5 mm MgCl2, 250 mm sucrose, pH 7.5, with 50 μg/ml digitonin, and protease inhibitors (Clontech)). After 5 min on ice, islets were centrifuged (13,000 × g for 10 min at 4 °C), and the supernatant was analyzed for presence of cytochrome c and cytochrome c oxidase subunit IV (antibodies from Clontech) and actin (Santa Cruz Biotechnology, Santa Cruz, CA) by Western blot. Bands were quantified using ImageJ (National Institutes of Health). Lack of appearance of cytochrome c oxidase in the cytosolic fraction demonstrated a lack of contamination by mitochondria (data not shown).
Data Analysis
Statistical significance was determined using either Student's t tests or analysis of variance with a Bonferroni's post hoc test (as indicated) using KaleidaGraph (Synergy Software, Reading, PA).
RESULTS
Retention of KIC-stimulated ISR despite Loss of Glucose-stimulated ISR by Islets Cultured at 3 mm Glucose for 40 h
To establish the premise of the model system that islets cultured in 3 mm glucose have decreased glycolytic response relative to islets cultured optimally (i.e. at 11 mm glucose), we compared the effect of glucose and a mitochondrial fuel (KIC) that would bypass GK on ISR (Fig. 1). ISR by islets cultured at 11 mm glucose responded similarly to 20 mm glucose or 10 mm KIC. In contrast, ISR by islets cultured at 3 mm glucose increased only slightly (about 10% of the control response to 20 mm glucose). Secretory response of these islets to 10 mm KIC was still intact, although somewhat reduced (about 50% of the 11 mm glucose control), indicating that intrinsic mechanisms of secretion were still operational. This appeared to be the case despite diminished insulin content (0.44 ± 0.19 versus 0.64 ± 0.27 ng/10 islets, n = 8, although not statistically significant) and ATP (21 ± 1.5 versus 33 ± 1.7 pmol/10 islets, n = 12, p < 0.05) in the islets cultured in 3 mm glucose.
FIGURE 1.
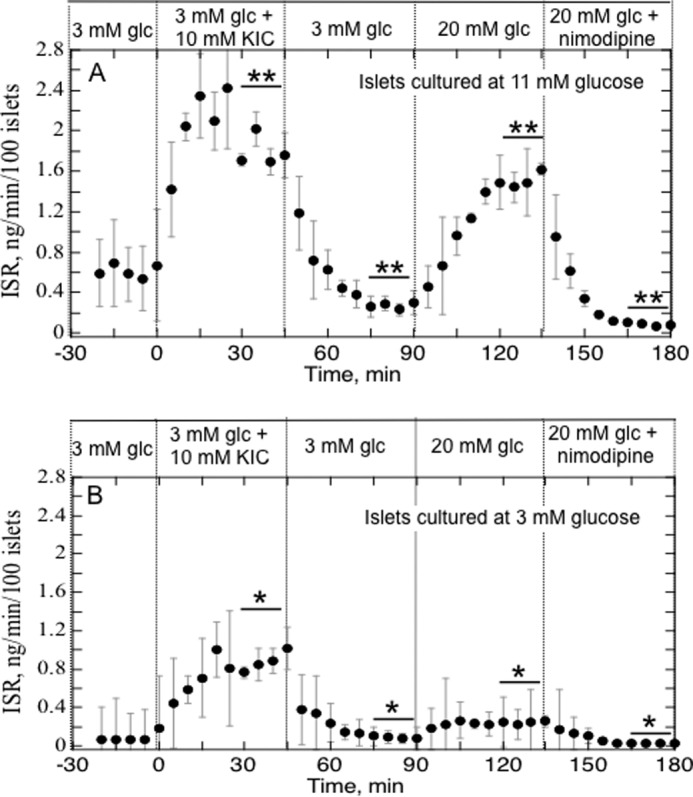
KIC- and glucose-stimulated ISR by islets cultured in RPMI 1640 medium containing either 11 mm (A) or 3 mm (B) glucose for 40 h. Islets were perifused in the presence of 3 mm glucose for 90 min. Subsequently, 10 mm KIC was added and removed, and 20 mm glucose and nimodipine were added to and removed from the inflow at the times shown on the graph. Fractions were collected and insulin was assayed to determine the ISR (average ± S.E., n = 4). Statistical analysis was carried out by comparing steady-state values (determined by averaging data obtained in the final 15 min of each experimental condition) before and after each change in medium composition using a paired t test (*, p < 0.05; **, p < 0.01).
Note that no first phase response of ISR was seen when glucose or KIC was increased due to the low flow rates needed to resolve OCR relative to the dead space volume of the perifusion chamber and tubing (80 μl/min versus 800-μl chamber). As a result, changes in fuels resemble a ramp, a waveform that does not induce first phase insulin secretion (32).
Metabolic Responses to Glycolytic and Mitochondrial Fuel
The increase in OCR observed in response to KIC was similar in islets cultured in either 3 mm or 11 mm glucose (Fig. 2), although the baseline OCR was lower in the islets culture in 3 mm glucose. Glucose response, however, in islets cultured in 3 mm glucose was about 50% of control, consistent with decreased glycolytic capacity. We have previously observed that Ca2+ entering the cell via L-type Ca2+ channels activates a highly energetic process that couples Ca2+ and metabolic factors to the secretion of insulin (termed the CMCP (15)). If the process is blocked, then the islet does not generate the ATP needed to drive the process, and OCR decreases. This can be quantified as the decrement in OCR in response to a blocker of L-type Ca2+ channels (nimodipine) (Fig. 2), which in control islets is typically about 30% of glucose-stimulated OCR (12). Remarkably, in islets cultured at 3 mm glucose, the decrement was almost completely absent. We also tested whether the loss of glucose-stimulated OCR was due to decreased glucose transport or a switch to utilization of fatty acids as a fuel. However, glucose transport (measured as uptake of 3-O-methylglucose uptake) was not decreased, and palmitate had little effect on OCR by islets cultured in either 3 mm or 11 mm glucose (data not shown). Thus, it appears that culturing islets in 3 mm glucose reduces their capacity to generate factors needed to stimulate the CMCP and ISR.
FIGURE 2.
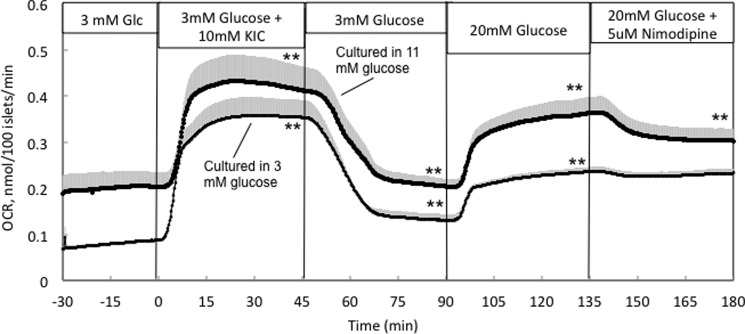
KIC- and glucose-stimulated OCR by islets cultured in RPMI 1640 medium containing either 11 mm or 3 mm glucose for 40 h. Islets were perifused in the presence of 3 mm glucose for 90 min. Subsequently, 10 mm KIC was added and removed, and 20 mm glucose and nimodipine were added to and removed from the inflow at the times shown on the graph. *, p < 0.05; **, p <0.01. Statistics were also calculated comparing islets cultured at 3 and 11 mm glucose. Difference in response to KIC is nonsignificant; responses to 20 mm glucose and nimodipine were different for the two culture conditions (p < 0.05).
Intact Ca2+ Responses to Glucose and KIC in Islets Cultured at 3 mm Glucose
Our initial hypothesis was that a loss of glycolytic capacity due to culture at low glucose would impair Ca2+ influx in response to glucose, which would explain the loss of glucose-stimulated ISR. To test this, cytosolic Ca2+ was imaged after loading islets with a Ca2+-sensitive dye. Responses to KIC were nearly identical in islets cultured at 3 or 11 mm glucose (Fig. 3, A and B); consistent with the decrease in oxidative response to glucose, Ca2+ increment was less in the 3 mm glucose culture islets, but only by about 20% (similar to results obtained by Jonas and co-workers (20), although our data did not achieve statistical significance). Results were also calculated after converting the fluorescence ratios to Ca2+ concentrations using equations provided by (33), but the results differed by only a few percentage points. The contribution of L-type Ca2+ channels to glucose-induced changes in cytosolic Ca2+, about 50%, was similar in islets cultured in either glucose concentration (and consistent with previous studies (34)).
FIGURE 3.

KIC- and glucose-stimulated cytosolic Ca2+ or 45Ca2+ influx by islets cultured in RPMI 1640 medium containing either 11 mm or 3 mm glucose for 40 h. A and B, islets were perifused in the presence of 3 mm glucose for 90 min. Subsequently, 10 mm KIC was added and removed, and 20 mm glucose and nimodipine were added to and removed from the inflow at the times shown on the graph. Cytosolic Ca2+ was imaged fluorescently in real time and displayed as the change in relative fluorescence relative to the steady-state value obtained at 3 mm glucose (average ± S.E., n = 3). *, p < 0.05; **, p < 0.01. C, Ca2+ influx was measured on islets in 24-well plates in the presence of the indicated compounds. Analysis of variance with a Bonferroni's post hoc test was carried out on all conditions; no differences were seen between the islets cultured at 3 mm glucose versus those cultured at 11 mm glucose.
Measured increases of cytosolic Ca2+ response to glucose reflect activation of L-type Ca2+ channels, but also reflect activation by influx through other plasma membrane channels and exchange between intracellular compartments such as efflux from the endoplasmic reticulum. We therefore used a second approach, one that is based on the uptake of 45Ca2+ from extracellular buffer (Fig. 3C). In contrast to the results obtained by cytosolic imaging, however, Ca2+ influx in response to glucose was actually bigger in islets cultured in low versus high glucose, and Ca2+ influx in response to glucose was totally suppressed by nimodipine.
Thus, it appeared that Ca2+ entry into the islets through L-type Ca2+ channels remains intact in low glucose-cultured islets, despite the diminished metabolic response to glucose, suggesting that the impaired ISR in low glucose-cultured islets was not caused by a decrease in Ca2+ influx. To further test this interpretation, Ca2+ and ISR were measured in response to a direct activator of L-type Ca2+ channels. Past studies have shown that in islets cultured at 11 mm glucose, BayK8644 increases both Ca2+ influx and glucose-stimulated ISR (11). The glucose-induced Ca2+ response to the BayK compound in islets cultured in 3 mm glucose (Fig. 4A) was much larger than that stimulated by glucose in islets cultured at 11 mm glucose (Fig. 3A). However, ISR increased only marginally (Fig. 4B), indicating that Ca2+ influx stimulated by 20 mm glucose was nearly at maximally effective levels with respect to ISR. Moreover, the impairment of ISR in the low glucose-cultured islets was not mediated by a loss of Ca2+ response.
FIGURE 4.

Effect of L-type Ca2+ channel flux on glucose-stimulated cytosolic Ca2+ (A) and ISR (B) by islets cultured in RPMI 1640 medium containing 3 mm glucose for 40 h. Islets were perifused in the presence of 3 mm glucose for 90 min. Subsequently, glucose was raised to 20 mm glucose followed by 10 μm BayK8644 and by 5 μm nimodipine for the Ca2+ measurements. Cytosolic Ca2+ was imaged fluorescently in real time and displayed as the change in relative fluorescence relative to the steady-state value obtained at 3 mm glucose (average ± S.E., n = 3). *, p < 0.05; **, p < 0.01.
Culture of Islets in Low Glucose Leads to Loss of Cytochrome c Response to Glucose
The retention of Ca2+ response to glucose suggested that the loss of ISR in low glucose culture islets was due to an inability of islets to generate essential metabolic coupling factors. The reduced state of cytochrome c was measured in response to glucose and KIC (Fig. 5). Reduction of cytochrome c in response to 20 mm glucose in the low glucose-cultured islets was virtually absent, despite a normal response to KIC. This is consistent with the notion that conversion of cytochrome c to the reduced state, independent of calcium signaling, is essential for glucose-stimulated ISR.
FIGURE 5.
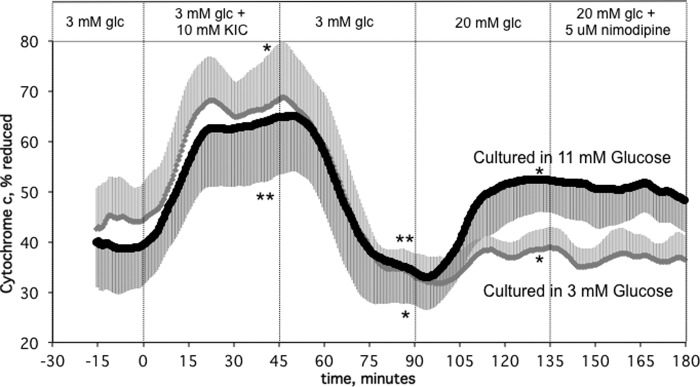
KIC- and glucose-stimulated reduced cytochrome c by islets cultured in RPMI 1640 medium containing either 11 mm or 3 mm glucose for 40 h. Islets were perifused in the presence of 3 mm glucose for 90 min. Subsequently, 10 mm KIC was added and removed, and 20 mm glucose and nimodipine were added to and removed from the inflow at the times shown on the graph. Data are displayed as the change in cytochrome c reduction relative to a blank and maximal signal (in the presence of antimycin A and KCN) (average ± S.E., n = 4). Asterisks denote significance of the change relative to the preceding condition as described in the legend for Fig. 1. *, p < 0.05; **, p <0.01. Statistics were also calculated comparing islets cultured at 3 and 11 mm glucose. Difference in response to KIC is nonsignificant; responses to 20 mm glucose and nimodipine were different for the two culture conditions (p < 0.05).
To see whether ISR in low glucose-cultured islets could be rescued by conversion of cytochrome c to the reduced state, the effects of increasing GK activity with a GKa were assessed. As predicted, the addition of the GKa increased OCR, cytochrome c reduction, and ISR to levels nearly as high as those seen in response to KIC (Fig. 6, A–C). In contrast to cytochrome c, the reductive state of cytochrome c oxidase, a step downstream from cytochrome c in the electron transport chain, did not change in response to the GKa (data not shown). Remarkably, the GKa also activated the CMCP as reflected by the reappearance of Ca2+-sensitive OCR similar to that seen for control islets (Fig. 6, D and E). The GKa increased glucose-stimulated Ca2+ by about 35% (Fig. 6F), but much less than the increase observed in response to BayK8644, ruling out the possibility that the increase in ISR induced by GKa was in part mediated by stimulation of cytosolic Ca2+ levels.
FIGURE 6.

Effect of a GKa on cytochrome c reduction, OCR, ISR, and cytosolic Ca2+, in islets with impaired glucose-stimulated ISR. A–C, islets cultured for 40 h in the presence of 3 mm glucose were perifused in the presence of 3 mm glucose for 90 min. Subsequently, 10 mm KIC was added and removed, and 20 mm glucose and the GKa were added to the inflow at the times shown on the graph. Cytochrome c reduction, OCR, and ISR were measured concomitantly using the flow culture system and expressed as average ± S.E., n = 4. OCR data are displayed as the change in OCR relative to the steady-state value obtained at 3 mm glucose. Steady-state values of OCR at 3 mm glucose were 0.11 ± 0.03 nmol/min/100 islets (n = 4). D, effect of GKa on Ca2+-sensitive OCR by islets cultured at 3 mm glucose for 40 h. Islets were perifused in the presence of 3 mm glucose for 90 min. Subsequently, 20 mm glucose, a GKa, and a Ca2+ channel blocker (nimodipine) were added sequentially to the inflow at the times shown on the graph. OCR and Ca2+ were expressed as average ± S.E., n = 4. OCR data are displayed as the change in OCR relative to the steady-state value obtained at 3 mm glucose (0.19 ± 0.04 nmol/min/100 islets (n = 4)). E, comparison of Ca2+-sensitive OCR by islets cultured for 40 h in 11 mm glucose or 3 mm glucose. Large black symbols from Fig. 3, OCR response to nimodipine from in islets cultured at 3 mm glucose; small black symbols from panel D), islets cultured at 3 mm glucose and exposed to the GKa; gray symbols from Fig. 2), islets cultured at 11 mm glucose. Data were normalized by subtracting the steady-state value for the final 5 min prior to the exposure to nimodipine. F, effect of GKa on Ca2+ by islets cultured at 3 mm glucose for 40 h. Islets were perifused in the presence of 3 mm glucose for 90 min. Subsequently, 20 mm glucose, the GKa, and an L-type Ca2+ channel blocker (nimodipine) were added sequentially to the inflow at the times shown on the graph. Ca2+ was expressed as average ± S.E., n = 4 (*, p < 0.05; **, p < 0.01).
Culture of Islets in Low Glucose Leads to Loss of Cytochrome c Translocation in Response to Glucose
Based on the previously published conclusions that conversion of cytochrome c to the reduced state by glucose and other nutrient secretagogues results in the appearance of cytochrome c in the cytosol (11), we measured the translocation of cytochrome c in low and high glucose-cultured islets (Fig. 7A). Although 20 mm glucose increased cytosolic cytochrome c in islets cultured in standard glucose levels (11 mm), this effect was not seen in the low glucose-cultured islets. As with the rescue of ISR and reduced cytochrome c response, the GKa rescued glucose-stimulated translocation of cytochrome c in islets cultured in 3 mm glucose in response to 20 mm glucose (Fig. 7A). These results were not caused by variability in gel loading of protein as confirmed by actin blots, nor were they caused by differences in apoptotic rates due to the extended culture at 3 mm glucose; levels of caspase-3 activity were similar in islets cultured at either 3 mm or 11 mm glucose, and these levels were low as compared with staurosporine-treated islets (data not shown). To confirm that glucose-stimulated translocation of cytochrome c was mediated by a mechanism independent of the apoptotic translocation of cytochrome c, we harvested islets from apoptosis-deficient Bax/Bak DKO mice. As expected, staurosporine-induced translocation of cytochrome c was decreased by 82% in islets from the Bax/Bak DKO relative to control islets (Fig. 7B). In contrast, glucose-induced translocation of cytochrome c in DKO islets was only decreased by 29%, which was not statistically significant. Thus, the majority of glucose-stimulated translocation of cytochrome c is independent of mechanisms mediating apoptosis.
FIGURE 7.

Effect of glucose, a GKa, and staurosporine on appearance of cytochrome c in cytosol. Extracted cytosolic protein from islets following permeabilization was analyzed by Western blot for the presence of cytochrome c, and the density of the cytochrome c bands was quantified. The blot was subsequently reprobed for actin. A, rat islets (500 per condition) were incubated in the presence of the indicated agents (n = 4). Extracts equivalent to 200 islets were loaded on to the gel. Due to the variability in quantifying Western bands, data were normalized to the band densities generated in response to 3 mm glucose; the ratio of the bands was 1.04 ± 0.28 for islets cultured at 3 mm versus 11 mm glucose, which was not statistically different from 1. *, p < 0.05; **, p < 0.01. B, mouse islets harvested from either Bax/Bak DKO or control mice were incubated in the presence of 20 mm glucose (500 islets) as in A or cultured overnight in the presence of 3 μm staurosporine (60 islets) (n = 3). Extracts equivalent to 30 islets were loaded on to the gel. Note that the actin bands for staurosporine-treated islet extracts were not visible. Statistics were carried out comparing the effects of agents as compared with their normalized controls using paired t tests where * denotes p < 0.05. NS indicates that difference in response to 20 mm glucose is nonsignificant.
Effect of Fasting on ISR, OCR, Ca2+, and Cytochrome c Reduction
To assess whether the contribution of cytochrome c signaling to regulation of ISR seen in vitro could occur under in vivo conditions, we analyzed islets from rats fasted for 40 h, a state known to result in decreased glucose-induced ISR (35). Blood glucose levels at the time of the islet isolation were 103 ± 3 versus 161 ± 7 mg/dl (n = 16, p < 0.01) for fasted and fed rats, respectively. Islets from fasted rats had diminished responses of OCR, ISR, and cytochrome c reduction to both KIC and glucose (Fig. 8). However, Ca2+ responses to the two fuels were identical to the control. Therefore, fasting caused a general decrease in mitochondrial metabolism that was enough to reduce the response of cytochrome c but not Ca2+ influx. Loss of the nimodipine-sensitive OCR was observed in the fasted islets, but this seemed to be partially mediated by compensation by the islet that reversed the inhibitory effect of nimodipine on Ca2+.
FIGURE 8.
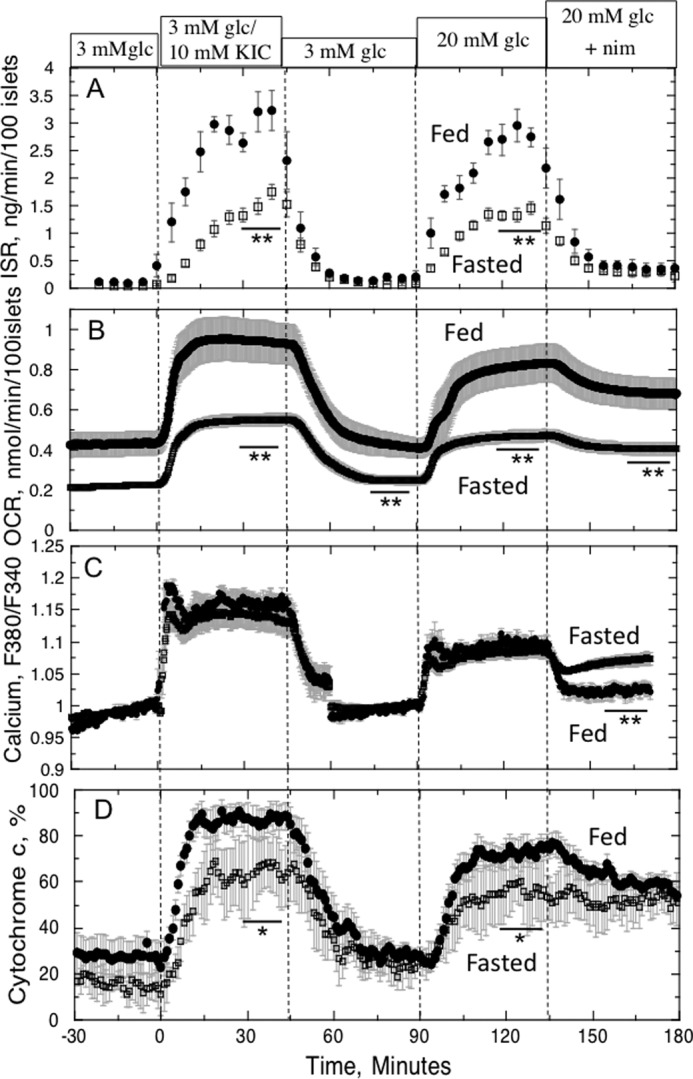
Effect of fasting on ISR (A), OCR (B), calcium (C), and cytochrome c signaling (D). Islets were harvested from rats that were either fed or fasted for 40 h prior to isolation. Perifusions of the islets were initiated within 1 h of isolation using the same protocol used for islets cultured for 40 h. Data are displayed as the average ± S.E., where OCR, cytochrome c, and ISR have n = 4–7, and Ca2+ data are n = 3–4 (*, p < 0.05; **, p < 0.01).
DISCUSSION
Relative Control of ISR by Ca2+ Influx and Cytochrome c Reduction
To investigate the role of factors generated in the metabolism of glucose in the stimulation of ISR, we used islets cultured in low glucose, previously shown to result in low GK activities and near absent glucose stimulation of ISR (19, 20). As expected with islets with low GK activity, glucose-stimulated OCR was only 50% as compared with that in islets cultured in 11 mm glucose, but the response to a mitochondrial fuel that bypasses GK (KIC) was nearly identical in the low and standard glucose-cultured islets, indicating that mitochondrial function was not diminished in the low glucose-cultured islets.
We had originally predicted that loss of glucose-stimulated Ca2+ influx due to the inadequate closure of KATP channels was mediating the lack of glucose-stimulated ISR in the low glucose-cultured islets as this is consistent with previous studies (19, 20) and the well accepted view that Ca2+ is the primary trigger for ISR. Indeed, the Ca2+ response to glucose in the low glucose-treated islets was slightly reduced as expected based on lowered oxidative rate. However, a major finding of our study was the finding that loss of Ca2+ signaling did not correlate with the loss of glucose-stimulated ISR; increasing Ca2+ influx by the L-type Ca2+ channel agonist BayK8644 did not restore secretory function. This conclusion is seemingly contrary to the observations that K+-induced increases in Ca2+ influx did restore ISR (20). However, the two studies are reconciled after considering the recent findings that in addition to increasing Ca2+ influx, K+ also increases mitochondrial respiration (12, 36, 37) and cytochrome c reduction in islets with no effect on components upstream of the electron transport chain such as NADH (11). Thus, K+ cannot be used as a tool to distinguish the contribution of Ca2+- versus cytochrome c-mediated processes. In contrast, the agent BayK8644 strongly stimulated Ca2+, but had no effect on cytochrome c reduction. To test the regulatory control strength of cytochrome c reduction on ISR in islets with impaired metabolic activity, we used a GKa, which did increase Ca2+ influx, but to a lesser extent than the BayK8644 compound. Significantly, it greatly increased the response of cytochrome c reduction and acutely restored glucose-stimulated ISR.
Taken together, the ability of glucose to increase the reductive state and the translocation of cytochrome c from the mitochondria to the cytosol was greatly diminished in the low glucose-cultured islets, both of which recovered in the presence of a GKa, but not in response to a specific agonist of Ca2+ influx. The GKa had little effect on a metabolic step downstream of cytochrome c, the reductive state of cytochrome c oxidase (data not shown). In this study, it was not possible to study the direct effect of changes in cytochrome c reduction alone on ISR because the approach previously used based on inhibition of cytochrome c oxidase with cyanide was variably toxic to the islets that were cultured at low glucose. However, combined with previous data showing a causal relationship between reduced cytochrome c and ISR (11), the agreement between cytochrome c data and ISR independent of Ca2+ and changes in other components of the electron transport chain supports a critical contribution of cytochrome c to regulation of ISR. This conclusion was further supported by parallel decreases in ISR and cytochrome c reduction observed in islets from fasted rats, whereas Ca2+ responses remained intact, demonstrating that cytochrome c reduction can displace Ca2+ as the primary regulator of ISR. We do not rule out contributions from other metabolic coupling factors such as products of pyruvate cycling (NADPH and α-ketoglutarate) (5), anaplerosis (38), as well as KATP channel-independent effects of ATP/ADP (39) and long chain acyl CoA (40), which were not measured in this study.
Interrelationship between Cytochrome c, CMCP, and ISR
The mechanisms mediating the stimulation of ISR by Ca2+ are not well understood, although several regulatory proteins in the beta cell have been proposed to act as Ca2+ sensors including synaptotagmins (41) and L-type Ca2+ channels (42). We have previously established that Ca2+, along with cytochrome c signaling, co-stimulate a highly energetic process, the CMCP, whose activation is essential for stimulating ISR. This is based on observations that the glucose dose response of the CMCP is superimposable to that of ISR (12), the CMCP is blocked by an inhibitor of Ca2+-dependent kinases (15), and like ISR, the CMCP is only activated when both the reductive state of cytochrome c, and the activity of L-type Ca2+ channels, are increased (11). In the present study, Ca2+-sensitive OCR was completely absent in the islets precultured in low glucose and quantitatively restored with the GKa. That stimulation of the OCR response by the GKa was nearly identical in magnitude to the decrease in OCR caused by blocking the Ca2+ response is consistent with activation and deactivation of a common step. We have recently published an in vivo study showing that the loss of CMCP regulation in a rat model of diabetes, where islet Ca2+ response was normal, was linked to decreased ISR during the transition to hyperglycemia (43). In the present study, the loss of CMCP regulation also accompanied the loss of ISR in islets from fasted rats, similar to the diabetic state. Taken together, both the in vitro and the in vivo results of this study add to the body of evidence supporting the existence and role of the CMCP and its dual control by Ca2+ and cytochrome c signaling. Previous studies revealed that the operation of the CMCP is not linked to uncoupling or changes in maximal respiration (43).
Dual Roles for Cytochrome c Signaling
As the majority of cytochrome c exists in the mitochondria, we propose that fuel-induced translocation of this protein to the cytosol must occur for it to have action on exocytosis (11). However, cytochrome c translocation is also a signaling system that can lead to apoptosis (44). We have postulated that the physiological role of cytochrome c translocation can operate without inducing cell death because the physiological and apoptotic processes operate with different cytochrome c thresholds for activation (the amount of cytochrome c in the cytosol after inducing apoptosis was 12 times higher than that caused by high glucose). In addition, the cytochrome c translocation induced by glucose appeared to operate with a mechanism independent of Bax/Bak-mediated (apoptotic) translocation of cytochrome c. Our data argue against the possibility that glucose and the GKa lead to the observed increase in cytosolic cytochrome c due to apoptotic death occurring in a small number of cells. The recovery of the CMCP (i.e. Ca2+-sensitive OCR) in response to GKa would not arise from a small number of apoptotic cells, which would decrease and not increase OCR.
Evidence for an Interactive Signaling Role between GK and Cytochrome c
Activation of GK, the rate-limiting step in glycolysis in the islet (45, 46), by GKas is thought to increase ISR due to increased flux through metabolic pathways (47). We also found that GK activation increased OCR 30% over the response induced by 20 mm glucose. Notably, reduced cytochrome c and ISR both increased 4-fold over their glucose responses consistent with the established nonlinearity of the relation between reduced cytochrome c and OCR (48), increasing the sensitivity of ISR to GK. Interrelations between mitochondrial proteins in the Bcl-2 family that regulate cytochrome c translocation and affect ISR have been reported (25, 49, 50), and mitochondrial protein Bcl-2-associated death promoter (BAD) bound and activated GK, which leads to stimulation of glucose metabolism and ISR (51). Although our results show that glucose-induced cytochrome c translocation was not mediated by Bax or Bak, it does appear that mitochondrial signaling involving cytochrome c may feed back on GK and metabolic rate. Future studies will investigate the involvement of other channels, such as voltage-dependent anion channels, in transporting cytochrome c.
Acknowledgment
We thank Dr. Nika Danial for important intellectual contributions to the manuscript involving Bax/Bak and cytochrome c translocation.
This work was supported in part by National Institutes of Health Grant DK17047 (the Diabetes Research Center (DRC) Islet Core and the Immunology and Inflammation Core, and an R24 seed grant supplement), funding from the University of Washington Diabetes and Obesity Center and the Washington State Life Sciences Discovery Fund, National Science Foundation Grant IIP-0750508, and a grant from the American Diabetes Association/Gail Patrick Estate (Grant 1-13-IN-53).
- ISR
- insulin secretion rate
- CMCP
- Ca2+/metabolic coupling process
- DKO
- double knockout
- GK
- glucokinase
- GKa
- glucokinase activator
- KATP
- ATP-sensitive potassium channel
- KIC
- α-ketoisocaproate
- OCR
- oxygen consumption rate.
REFERENCES
- 1. Prentki M., Tornheim K., Corkey B. E. (1997) Signal transduction mechanisms in nutrient-induced insulin secretion. Diabetologia 40, Suppl. 2, S32–S41 [DOI] [PubMed] [Google Scholar]
- 2. Henquin J. C. (2000) Triggering and amplifying pathways of regulation of insulin secretion by glucose. Diabetes 49, 1751–1760 [DOI] [PubMed] [Google Scholar]
- 3. Straub S. G., Sharp G. W. (2002) Glucose-stimulated signaling pathways in biphasic insulin secretion. Diabetes Metab. Res. Rev. 18, 451–463 [DOI] [PubMed] [Google Scholar]
- 4. Henquin J. C. (2011) The dual control of insulin secretion by glucose involves triggering and amplifying pathways in β-cells. Diabetes Res. Clin Pract. 93, Suppl. 1, S27–S31 [DOI] [PubMed] [Google Scholar]
- 5. Jensen M. V., Joseph J. W., Ronnebaum S. M., Burgess S. C., Sherry A. D., Newgard C. B. (2008) Metabolic cycling in control of glucose-stimulated insulin secretion. Am. J. Physiol. Endocrinol. Metab. 295, E1287–E1297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Henquin J. C. (2009) Regulation of insulin secretion: a matter of phase control and amplitude modulation. Diabetologia 52, 739–751 [DOI] [PubMed] [Google Scholar]
- 7. Prentki M. (1996) New insights into pancreatic beta-cell metabolic signaling in insulin secretion. Eur. J. Endocrinol. 134, 272–286 [DOI] [PubMed] [Google Scholar]
- 8. Newgard C. B., McGarry J. D. (1995) Metabolic coupling factors in pancreatic β-cell signal transduction. Annu. Rev. Biochem. 64, 689–719 [DOI] [PubMed] [Google Scholar]
- 9. Prentki M., Matschinsky F. M., Madiraju S. R. (2013) Metabolic signaling in fuel-induced insulin secretion. Cell Metab. 18, 162–185 [DOI] [PubMed] [Google Scholar]
- 10. Wiederkehr A., Wollheim C. B. (2012) Mitochondrial signals drive insulin secretion in the pancreatic β-cell. Mol. Cell. Endocrinol. 353, 128–137 [DOI] [PubMed] [Google Scholar]
- 11. Jung S. R., Kuok I. T., Couron D., Rizzo N., Margineantu D. H., Hockenbery D. M., Kim F., Sweet I. R. (2011) Reduced cytochrome c is an essential regulator of sustained insulin secretion by pancreatic islets. J. Biol. Chem. 286, 17422–17434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sweet I. R., Gilbert M. (2006) Contribution of calcium influx in mediating glucose-stimulated oxygen consumption in pancreatic islets. Diabetes 55, 3509–3519 [DOI] [PubMed] [Google Scholar]
- 13. Hüttemann M., Pecina P., Rainbolt M., Sanderson T. H., Kagan V. E., Samavati L., Doan J. W., Lee I. (2011) The multiple functions of cytochrome c and their regulation in life and death decisions of the mammalian cell: from respiration to apoptosis. Mitochondrion 11, 369–381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Henquin J. C., Ravier M. A., Nenquin M., Jonas J. C., Gilon P. (2003) Hierarchy of the β-cell signals controlling insulin secretion. Eur. J. Clin. Invest. 33, 742–750 [DOI] [PubMed] [Google Scholar]
- 15. Jung S. R., Reed B. J., Sweet I. R. (2009) A highly energetic process couples calcium influx through L-type calcium channels to insulin secretion in pancreatic β-cells. Am. J. Physiol. Endocrinol. Metab. 297, E717–E727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Doliba N. M., Fenner D., Zelent B., Bass J., Sarabu R., Matschinsky F. M. (2012) Repair of diverse diabetic defects of β-cells in man and mouse by pharmacological glucokinase activation. Diabetes Obes. Metab. 14, Suppl. 3, 109–119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Remedi M. S., Koster J. C., Patton B. L., Nichols C. G. (2005) ATP-sensitive K+ channel signaling in glucokinase-deficient diabetes. Diabetes 54, 2925–2931 [DOI] [PubMed] [Google Scholar]
- 18. Del Guerra S., Lupi R., Marselli L., Masini M., Bugliani M., Sbrana S., Torri S., Pollera M., Boggi U., Mosca F., Del Prato S., Marchetti P. (2005) Functional and molecular defects of pancreatic islets in human type 2 diabetes. Diabetes 54, 727–735 [DOI] [PubMed] [Google Scholar]
- 19. Liang Y., Najafi H., Smith R. M., Zimmerman E. C., Magnuson M. A., Tal M., Matschinsky F. M. (1992) Concordant glucose induction of glucokinase, glucose usage, and glucose-stimulated insulin release in pancreatic islets maintained in organ culture. Diabetes 41, 792–806 [DOI] [PubMed] [Google Scholar]
- 20. Bensellam M., Van Lommel L., Overbergh L., Schuit F. C., Jonas J. C. (2009) Cluster analysis of rat pancreatic islet gene mRNA levels after culture in low-, intermediate- and high-glucose concentrations. Diabetologia 52, 463–476 [DOI] [PubMed] [Google Scholar]
- 21. Burch P. T., Trus M. D., Berner D. K., Leontire A., Zawalich K. C., Matschinsky F. M. (1981) Adaptation of glycolytic enzymes: glucose use and insulin release in rat pancreatic islets during fasting and refeeding. Diabetes 30, 923–928 [DOI] [PubMed] [Google Scholar]
- 22. Guzman-Perez A., Pfefferkorn J. A., Litchfield J., Aiello R., Treadway J. L., Minich M. L., Filipski K. J., Jones C. S., Tu M., Aspnes G. E., Risley H., Wright S. W., Li J.-C., Bian J., Benbow J., Dow R. L., Didiuk M. T., Perreault C., Bourbonais F., Derksen D. R., MacDougall M., Cabrera O., Landro J., Atkinson K., Yao L., Kosa R. E., Haddish-Berhane N., Feng B., Diugnan D. B., Liu S., Ammirati M. J., Knafels J. D. (2011) Designing for liver selective drug distribution: hepatoselective glucokinase activators as a case study. 242nd ACS National Meeting, Denver, CO, Aug 28–Sept 1, 2011, American Chemical Society, Washington, D.C [Google Scholar]
- 23. Sweet I. R., Cook D. L., DeJulio E., Wallen A. R., Khalil G., Callis J., Reems J. (2004) Regulation of ATP/ADP in pancreatic islets. Diabetes 53, 401–409 [DOI] [PubMed] [Google Scholar]
- 24. Matsumoto S., Shibata S., Kirchhof N. (1999) Immediate reversal of diabetes in primates following intraportal transplantation of porcine islets purified on a new histidine-lactoioniate-iodixanol gradient. Transplantation 67, S220 [Google Scholar]
- 25. Luciani D. S., White S. A., Widenmaier S. B., Saran V. V., Taghizadeh F., Hu X., Allard M. F., Johnson J. D. (2013) Bcl-2 and Bcl-xL suppress glucose signaling in pancreatic β-cells. Diabetes 62, 170–182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sweet I. R., Cook D. L., Wiseman R. W., Greenbaum C. J., Lernmark A., Matsumoto S., Teague J. C., Krohn K. A. (2002) Dynamic perifusion to maintain and assess isolated pancreatic islets. Diabetes Technol. Ther. 4, 67–76 [DOI] [PubMed] [Google Scholar]
- 27. Sweet I. R., Khalil G., Wallen A. R., Steedman M., Schenkman K. A., Reems J. A., Kahn S. E., Callis J. B. (2002) Continuous measurement of oxygen consumption by pancreatic islets. Diabetes Technol. Ther. 4, 661–672 [DOI] [PubMed] [Google Scholar]
- 28. Jung S. R., Reed B. J., Sweet I. R. (2009) PMCID: PMC2739700) A highly energetic process couples calcium influx through L-type calcium channels to insulin secretion in pancreatic β-cells. Am. J. Physiol. Endocrinol. Metab. 297, E717–E727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Sweet I. R., Cook D. L., Lernmark A., Greenbaum C. J., Wallen A. R., Marcum E. S., Stekhova S. A., Krohn K. A. (2004) Systematic screening of potential β-cell imaging agents. Biochem. Biophys. Res. Commun. 314, 976–983 [DOI] [PubMed] [Google Scholar]
- 30. Wolf B. A., Comens P. G., Ackermann K. E., Sherman W. R., McDaniel M. L. (1985) The digitonin-permeabilized pancreatic islet model: effect of myo-inositol 1,4,5-trisphosphate on Ca2+ mobilization. Biochem. J. 227, 965–969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Gottlieb R. A., Granville D. J. (2002) Analyzing mitochondrial changes during apoptosis. Methods 26, 341–347 [DOI] [PubMed] [Google Scholar]
- 32. Sweet I. R., Li G., Najafi H., Berner D., Matschinsky F. M. (1996) Effect of a glucokinase inhibitor on energy production and insulin release in pancreatic islets. Am. J. Physiol. 271, E606–E625 [DOI] [PubMed] [Google Scholar]
- 33. Grynkiewicz G., Poenie M., Tsien R. Y. (1985) A new generation of Ca2+ indicators with greatly improved fluorescence properties. J. Biol. Chem. 260, 3440–3450 [PubMed] [Google Scholar]
- 34. Gilbert M., Jung S. R., Reed B. J., Sweet I. R. (2008) Islet oxygen consumption and insulin secretion tightly coupled to calcium derived from L-type calcium channels but not from the endoplasmic reticulum. J. Biol. Chem. 283, 24334–24342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Zawalich W. S., Zawalich K. C. (2000) Glucose-induced insulin secretion from islets of fasted rats: modulation by alternate fuel and neurohumoral agonists. J. Endocrinol. 166, 111–120 [DOI] [PubMed] [Google Scholar]
- 36. Erecińska M., Nelson D., Chance B. (1991) Depolarization-induced changes in cellular energy production. Proc. Natl. Acad. Sci. U.S.A. 88, 7600–7604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Barnes W. S. (1993) Effects of Ca2+-channel drugs on K+-induced respiration in skeletal muscles. Med. Sci. Sports Exerc. 25, 473–478 [PubMed] [Google Scholar]
- 38. MacDonald M. J., Fahien L. A., Brown L. J., Hasan N. M., Buss J. D., Kendrick M. A. (2005) Perspective: emerging evidence for signaling roles of mitochondrial anaplerotic products in insulin secretion. Am. J. Physiol. Endocrinol. Metab. 288, E1–E15 [DOI] [PubMed] [Google Scholar]
- 39. Henquin J. C., Gembal M., Detimary P., Gao Z. Y., Warnotte C., Gilon P. (1994) Multisite control of insulin release by glucose. Diabete Metab. 20, 132–137 [PubMed] [Google Scholar]
- 40. Deeney J. T., Prentki M., Corkey B. E. (2000) Metabolic control of β-cell function. Semin. Cell Dev. Biol. 11, 267–275 [DOI] [PubMed] [Google Scholar]
- 41. Barg S., Rorsman P. (2004) Insulin secretion: a high-affinity Ca2+ sensor after all? J. Gen. Physiol. 124, 623–625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Trus M., Corkey R. F., Nesher R., Richard A. M., Deeney J. T., Corkey B. E., Atlas D. (2007) The L-type voltage-gated Ca2+ channel is the Ca2+ sensor protein of stimulus-secretion coupling in pancreatic beta cells. Biochemistry 46, 14461–14467 [DOI] [PubMed] [Google Scholar]
- 43. Rountree A. M., Reed B. J., Cummings B. P., Jung S. R., Stanhope K. L., Graham J. L., Griffen S. C., Hull R. L., Havel P. J., Sweet I. R. (2013) Loss of coupling between calcium influx, energy consumption and insulin secretion associated with development of hyperglycaemia in the UCD-T2DM rat model of type 2 diabetes. Diabetologia 56, 803–813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ow Y. P., Green D. R., Hao Z., Mak T. W. (2008) Cytochrome c: functions beyond respiration. Nat. Rev. Mol. Cell Biol. 9, 532–542 [DOI] [PubMed] [Google Scholar]
- 45. Matschinsky F. M. (1996) Banting Lecture 1995. A lesson in metabolic regulation inspired by the glucokinase glucose sensor paradigm. Diabetes 45, 223–241 [DOI] [PubMed] [Google Scholar]
- 46. Sweet I. R., Matschinsky F. M. (1995) Mathematical model of beta-cell glucose metabolism and insulin release. I. Glucokinase as glucosensor hypothesis. Am. J. Physiol. 268, E775–E788 [DOI] [PubMed] [Google Scholar]
- 47. Matschinsky F. M., Porte D. (2010) Glucokinase activators (GKAs) promise a new pharmacotherapy for diabetics. F1000 Med. Rep. 2, 43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Wilson D. F., Owen C. S., Erecińska M. (1977) Regulation of mitochondrial respiration in intact tissues: a mathematical model. Adv. Exp. Med. Biol. 94, 279–287 [DOI] [PubMed] [Google Scholar]
- 49. Philipson L. H., Roe M. W. (2008) When BAD is good for β cells. Cell Metab. 7, 280–281 [DOI] [PubMed] [Google Scholar]
- 50. Johnson J. D., Ahmed N. T., Luciani D. S., Han Z., Tran H., Fujita J., Misler S., Edlund H., Polonsky K. S. (2003) Increased islet apoptosis in Pdx1+/− mice. J. Clin. Invest. 111, 1147–1160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Danial N. N., Walensky L. D., Zhang C. Y., Choi C. S., Fisher J. K., Molina A. J., Datta S. R., Pitter K. L., Bird G. H., Wikstrom J. D., Deeney J. T., Robertson K., Morash J., Kulkarni A., Neschen S., Kim S., Greenberg M. E., Corkey B. E., Shirihai O. S., Shulman G. I., Lowell B. B., Korsmeyer S. J. (2008) Dual role of proapoptotic BAD in insulin secretion and beta cell survival. Nat. Med. 14, 144–153 [DOI] [PMC free article] [PubMed] [Google Scholar]