

Background: Myb1 mediates transcription suppression of an ap65-1 gene.
Results: TvCyP1 may accelerate conformational changes in Myb1 to enable its release from certain vesicles.
Conclusion: TvCyP1 is essential for moving Myb1 toward the nucleus.
Significance: This study elucidates a key step in nuclear translocation of Myb1 and provides tools to study the physiological role of TvCyP1.
Keywords: Cyclophilin, Membrane Trafficking, Nuclear Translocation, Parasitology, Protein-Protein Interaction, Transcription Regulation, Myb1, Peptidyl-prolyl Isomerase, Trichomonas vaginalis, TvCyP1
Abstract
In Trichomonas vaginalis, a Myb1 protein was previously demonstrated to repress transcription of an iron-inducible ap65-1 gene. In this study, a human cyclophilin A homologue, TvCyclophilin 1 (TvCyP1), was identified as a Myb1-binding protein using a bacterial two-hybrid library screening system. The recombinant TvCyP1 exhibited typical peptidyl-prolyl isomerase activity with kcat/Km of ∼7.1 μm−1 s−1. In a pulldown assay, the His-tagged Myb1 interacted with a GST-TvCyP1 fusion protein, which had an enzymatic proficiency half that of recombinant TvCyP1. Both the enzymatic proficiency of GST-TvCyP1 and its binding to His-Myb1 were eliminated by mutation of Arg63 in the catalytic motif or inhibited by cyclosporin A. TvCyP1 was primarily localized to the hydrogenosomes by immunofluorescence assay, but it was also co-purified with Myb1 in certain vesicle fractions from differential and gradient centrifugations. Transgenic cells overexpressing HA-TvCyP1 had a higher level of nuclear Myb1 but a much lower level of Myb1 associated with the vesicles than control and those overexpressing HA-TvCyP1(R63A). Myb1 was detected at a much higher level in the HA-TvCyP1 protein complex than in the HA-TvCyP1(R63A) protein complex immunoprecipitated from P15 and P100, but not S100, fractions of postnuclear lysates. A TvCyP1-binding motif, 105YGPKWNK111, was identified in Myb1 in which Gly106 and Pro107 were essential for its binding to TvCyP1. Mutation of Gly106 and Pro107, respectively, in HA-Myb1 resulted in cytoplasmic retention and elevated nuclear translocation of the overexpressed protein. These results suggest that TvCyP1 may induce the release of Myb1 that is restrained to certain cytoplasmic vesicles prior to its nuclear translocation.
Introduction
The parasitic protozoan Trichomonas vaginalis is the most common cause of nonviral sexually transmitted diseases of humans (1). The infection usually manifests mild symptoms or is asymptomatic, but it can cause adverse events during pregnancy that range from low birth weight at birth to abortion and stillbirth (2). Curative chemotherapy is available, but transmission is difficult to control, and reports on clinical drug-resistant isolates have been increasing in recent years (2, 3). Upon infection, the parasite trophozoites can breach the mucosal and epithelial barriers, lyse red blood cells, degrade complements and other humoral factors in host defense, and elicit chronic inflammation (1, 4). The infection has also been linked to the elevated transmission of human immunodeficiency virus (HIV) and human papillomaviruses (1, 3, 5, 6), making this overlooked pathogen a threat to public health.
The parasite persistently colonizes human urogenital tracts where oxygen tension is low and iron supply varies tremendously in reproductive stage females. The organism utilizes fermentative metabolism for energy production, which primarily takes place in the hydrogenosomes (2). Intriguingly, iron overloading, toxic to most organisms, is beneficial to the parasite by increasing the expression of many hydrogenosomal enzymes and promoting its cytoadherence to human vaginal epithelial cells (7–9). Several hydrogenosomal enzymes have previously been shown to be expressed on cell surfaces and mediate the cytoadherence of T. vaginalis when its growth medium was supplied with an excessive amount of iron (7, 8, 10). Although the secondary roles of these hydrogenosomal enzymes remain arguable, biochemical and genetic evidence suggests that a 65-kDa malic enzyme, referred to as AP65 and encoded by the ap65-1 gene, is likely involved in parasitic cytoadherence to human vaginal epithelial cells (11–13).
We have previously demonstrated that iron up-regulates the transcription of the ap65-1 gene via the coordinated actions of Myb1, Myb2, and Myb3 transcription factors (14–18). These Myb proteins share the conserved R2R3 DNA-binding domains (DBDs)3 of the eukaryotic Myb protein family (19), but each has a variable N or C terminus that is much shorter than the variable C-terminal regulatory regions of vertebrate c-Myb, A-Myb, and B-Myb (20). Similar to c-Myb, the DBDs in Myb1, Myb2, and Myb3 are composed of six helices arranged in two bundles with two antiparallel β-sheets unique to Myb3 forming a small hairpin following the helices (21–23). Intriguingly, the DBDs of Myb2 and Myb3 were also shown to mediate their own nuclear translocation (24, 25). A similar structural entity in Myb1 may also mediate its own nuclear translocation.4 Much remains to be learned about the cellular machinery mediating the nuclear importation of Myb proteins in T. vaginalis.
Cyclophilins (CyPs) are a group of ubiquitously expressed peptidyl-prolyl cis/trans isomerases (PPIs) catalyzing the interconversion of the peptidyl-prolyl imide bond responsible for conformation changes or correct folding of substrate proteins (26). A cyclophilin-like domain (CLD) for both the enzyme activity and substrate binding is conserved among eight different types of CyPs in humans (hCyPs) and yeasts (yCPRs). CLD is also the site that binds the immunosuppressive drug cyclosporin A (CsA) (27). The ∼18-kDa hCyPA comprises a single CLD and is the major receptor for CsA in vivo. It normally resides in the cytoplasm and is known to regulate a wide range of cellular activities dependent on or independent of the enzyme activity (28, 29). hCyPA can also be assembled into the viral particles of HIV and a few other viruses to facilitate viral replications (30, 31).
In the present study, an hCyPA-like protein, referred to as TvCyP1, was identified as a Myb1-binding protein via the bacterial two-hybrid screening of a T. vaginalis cDNA expression library and glutathione S-transferase (GST) pulldown assay. We examined the biochemical properties of TvCyP1 such as its enzymatic proficiency, expression profile, interaction with Myb1, and role in regulating the function of Myb1. Interestingly, TvCyP1 was localized to multiple membranous compartments, including the hydrogenosomes and some Myb1-associated vesicles. A TvCyP1-binding motif in Myb1 with a 105YGP107 tripeptide sequence essential for its binding to TvCyP1 was identified. Our observations suggest that TvCyP1 may facilitate the cis-trans conversion of a glycyl-prolyl imide bond in Myb1 to facilitate its nuclear translocation and achieve its role as a transcription repressor.
EXPERIMENTAL PROCEDURES
Cultures
T. vaginalis T1 isolate and its derived transfectants were maintained in TYI medium supplemented with 10% calf serum as described (18), and cells with an initial density of 1.0 × 105 ml−1 were grown to 1.5 × 106 ml−1 for experiments. Iron depletion and repletion were achieved by adding 50 μm 2,2′-dipyridyl and 250 μm ferrous sulfate, respectively, to normal growth medium as described (18).
DNA Transfection and Selection of Stable Transfectants
Plasmid DNA was electroporated into T. vaginalis, and a stable cell line was selected by paromomycin as described previously (18).
Oligonucleotides
The sequences of oligonucleotides used in the present study are listed in Table 1 unless otherwise specified in the text.
TABLE 1.
Sequences of oligonucleotides used in this study
| Name | Sequence (5′–3′) | |
|---|---|---|
| For two-hybrid plasmid construction | ||
| pBait-Myb1 | ||
| EcoRI-1-pBT-5′ | AGAATTCGATGATGTTTGACGGCCTTTCC | |
| BglII/SpeI-206-pBT-3′ | AAGATCTACTAGTTTAGAAGAATGAAAAATCGTTCCAAAG | |
| pBait-Myb1/N | ||
| EcoRI-1-pBT-5′ | AGAATTCGATGATGTTTGACGGCCTTTCC | |
| BglII/SpeI-52-pBT-3′ | AAGATCTACTAGTTCTCATTACTAATTGTTGAAGCTTAAGATC | |
| pBait-Myb1/R2R3(1) | ||
| EcoRI-53-pBT-5′ | AGAATTCGTATGGAGCTAAAGACTGGATCCGCATC | |
| BglII/SpeI-103-pBT-3′ | AAGATCTACTAGTTGCATACTTCTGATCAAGAAGCATATC | |
| pBait-Myb1/R3(2)C | ||
| EcoRI-104-pBT-5′ | AAGAATTCGGAGTACGGTCCTAAGTGGAACAAG | |
| BglII/SpeI-206-pBT-3′ | AAGATCTACTAGTTTAGAAGAATGAAAAATCGTTCCAAAG | |
| pBait-Myb1/R3(2)C-C112 | ||
| EcoRI-112-pBT-5′ | AGAATTCGATTTCAAAGTTCTTAAAGAATCGTTCT | |
| BglII/SpeI-206-pBT-3′ | AAGATCTACTAGTTTAGAAGAATGAAAAATCGTTCCAAAG | |
| pBait-Myb1/R3(2)C-C121 | ||
| EcoRI-121-pBT-5′ | AGAATTCGGACAATAATATTAGAAATCGCTGGATG | |
| BglII/SpeI-206-pBT-3′ | AAGATCTACTAGTTTAGAAGAATGAAAAATCGTTCCAAAG | |
| pBait-Myb1/R3(2)C-C127 | ||
| EcoRI-127-pBT-5′ | AGAATTCGCGCTGGATGATGATTGCTCG | |
| BglII/SpeI-206-pBT-3′ | AAGATCTACTAGTTTAGAAGAATGAAAAATCGTTCCAAAG | |
| For site-directed mutagenesis in two-hybrid screening | ||
| pBT-5′ | TCCGTTGTGGGGAAAGTTATC | |
| pBT-3′ | GGGTAGCCAGCAGCATCC | |
| Myb1-E104A-5′ | GCATCGAATTCGGCTTACGGTCCTAAG | |
| Myb1-E104A-3′ | CTTAGGACCGTAAGCCGAATTCGATGC | |
| Myb1-Y105A-5′ | TCGAATTCGGAGGCTGGTCCTAAGTGG | |
| Myb1-Y105A-3′ | CCACTTAGGACCAGCCTCCGAATTCGA | |
| Myb1-G106A-5′ | GAATTCGGAGTACGCTCCTAAGTGGAAC | |
| Myb1-G106A-3′ | GTTCCACTTAGGAGCGTACTCCGAATTC | |
| Myb1-P107A-5′ | CGGAGTACGGTGCTAAGTGGAACAAG | |
| Myb1-P107A-3′ | CTTGTTCCACTTAGCACCGTACTCCG | |
| Myb1-K108A-5′ | GGAGTACGGTCCTGCTTGGAACAAGATTTC | |
| Myb1-K108A-3′ | GAAATCTTGTTCCAAGCAGGACCGTACTCC | |
| Myb1-W109A-5′ | GGAGTACGGTCCTAAGGCTAACAAGATTTC | |
| Myb1-W109A-3′ | GAAATCTTGTTAGCCTTAGGACCGTACTCC | |
| Myb1-N110A-5′ | GGTCCTAAGTGGGCTAAGATTTCAAAGTTC | |
| Myb1-N110A-3′ | GAACTTTGAAATCTTAGCCCACTTAGGACC | |
| For overexpression | ||
| FLP-PPI-f | AGGATCCATGCTGAAGCGTCCAAAAACATTC | |
| FLP-PPI-r | AGAATTCTTATTTCATTTCGCCGCAATCTGT | |
| FLP-seq | TTGGGTTGAAAAAATCGGTTACCG | |
| sp6 | CATACGATTTAGGTGACACTATAG | |
| Myb1-G106A-5′ | GTATGCAGAGTACGCTCCTAAGTGGAACAAGATTTC | |
| Myb1-G106A-3′ | GAAATCTTGTTCCACTTAGGAGCGTACTCTGCATAC | |
| Myb1-P107A-5′ | GTATGCAGAGTACGGTGCTAAGTGGAACAAGATTTC | |
| Myb1-P107A-3′ | GAAATCTTGTTCCACTTAGCACCGTACTCTGCATAC | |
| CyPA1-R63A-5′ | AATGTTCCACGCTATTATTCCTCAGTTCATGATCCAA | |
| CyPA1-R63A-3′ | GACGAATAATAGCGTGGAACATTGTTCCCTTGTAGCT | |
| tub90f | GAATGCCGTCCGTTAAGAC | |
| T7p | TAATACGACTCACTATAGGG | |
| T7t | GCTAGTTATTGCTCAGCGG | |
Purification of DNA and RNA from T. vaginalis
Genomic DNA from T. vaginalis T1 cells was purified from the parasite as described previously (32), and total RNA was extracted by TRIzol and purified as described by the supplier (Invitrogen).
Construction of cDNA Expression Library and Bacterial Two-hybrid Screening for Myb1-interacting Proteins
The messenger RNA was purified from total RNA using oligo(dT) cellulose column chromatography as described previously (18). The purified messenger RNA was then reverse transcribed for subsequent cDNA synthesis using a SuperScript III reverse transcription kit (Invitrogen). The cDNA was inserted into a pTRG vector using a BacterioMatch II two-hybrid system vector kit to generate a cDNA expression library (Stratagene). The cDNA expression library was screened with the baits from various fragments of Myb1 (see Fig. 1A) subcloned into the pBait vector as described below, and the screening was performed as described by the supplier (Stratagene). In brief, pBait-R3(2)/C as described below was transformed into Escherichia coli competent cells along with pTRG-cDNA library plasmids and selected on the selective screening medium containing 5 mm 3-amino-1,2,4-triazole. The positive clones were further screened on a dual selective medium containing 5 mm 3-amino-1,2,4-triazole and streptomycin. A pairwise interaction assay using pBait-R3(2)/C and each positive clone in the pTRG vector for two-hybrid selection was performed to confirm the interacting pair.
FIGURE 1.

Interaction of TvCyP1 with various regions of Myb1 and sequence analysis of TvCyP1. In A, DNA fragments from various regions of the myb1 gene were cloned into pBait to react with TvCyP1-expressing pTRG-c21 in a bacterial two-hybrid assay. The relative strength of the interaction as revealed by the formation of colonies in each assay (−, no colony formation; +++, >300 colonies) is summarized in the right panel. In B, the sequence of TvCyP1 was aligned to that of CeCyP3 (P52011), yCPR1 (P14832), and hCyPA (P62937). The amino acids involved in the enzyme activity and CsA binding are indicated by asterisks. The amino acids involved in hydrogen bond formation are indicated by arrows, and those in disulfide bond formation by arrowheads. In C, the predicted three-dimensional structure of TvCyP1 using CeCyP3 as a template is depicted in the left panel. Local aa sequences in TvCyP1 are respectively depicted in ball-and-stick modes. The dotted lines represent hydrogen bond formation between Glu84 and Lys49/Ser50 or disulfide bond formation between Cys41 and Cys169.
Construction of Plasmids
To construct baits for screening Myb1-interacting proteins using a bacterial two-hybrid system (Stratagene), DNA fragments spanning various regions of the Myb1 coding sequence (see Fig. 1A) were each amplified from pET28-Myb1 (16) by a polymerase chain reaction (PCR) using a primer pair, EcoRI-(x)-pBT-5′ and BglII/SpeI-(y)-pBT-3′ (x and y indicate the location of the N- and C-terminal amino acid (aa), respectively). The PCR product was gel-purified and cloned into pGEM-T Easy (Promega). The EcoRI and BglII insert was cloned into a EcoRI/BglII-restricted pBT backbone to produce pBait-Myb1, pBait-Myb1/N, pBait-Myb1/R2R3(1), or pBait-Myb1/R3(2)C (Fig. 1A).
To map the TvCyP1-binding region in Myb1, a series of deletion mutants in pBait-Myb1/R3(2)C were constructed by an approach similar to that described above using a specific 5′-primer and a common 3′-primer for individual constructs (see Fig. 7A). Site-directed mutagenesis was then performed in pBait-Myb1/R3(2)C by a two-step PCR approach (see Fig. 7B). Briefly, from pBait-Myb1/R3(2)C, a 5′-DNA fragment was amplified using the primer pair pBT-5′ and Myb1-(XnA)-3′, and a 3′-DNA fragment was amplified using primer pair Myb1-(XnA)-5′ and pBT-3′ (X indicates the aa to be mutated, n indicates the numeric location of the residue, and A is alanine). The gel-purified PCR products were mixed, denatured, and annealed for a second PCR using the primer pair pBT-5′ and pBT-3′. The PCR product digested by EcoRI and SpeI was then cloned into an EcoRI/SpeI-restricted pBait-Myb1/R3(2)C backbone to generate the mutant plasmids as shown in Fig. 7.
FIGURE 7.

Identification of a TvCyP1-binding motif in Myb1. N-terminal deletion (A) and site-directed mutagenesis (B) were introduced into pBait-Myb1/R3(2)C to identify the TvCyP1-binding motif in Myb1 by a bacterial two-hybrid assay in which the bait plasmids were each incubated with pTRG-c21. The relative strength of the interactions as revealed by the formation of colonies in each assay is summarized in the right panel (− indicates no colony formation, + indicates <50 colonies, ++ indicates 50–100 colonies, and +++ indicates >300 colonies). In C, recombinant His-Myb1 and its derived mutant proteins His-Myb1(G106A) and His-Myb(P107A) were respectively incubated with GST and GST-TvCyP1 and immobilized on glutathione beads. The reaction products and 10% input of His-Myb1 and derived mutants were examined by Western blotting using the anti-His6 antibody.
To overexpress the hemagglutinin (HA)-tagged TvCyP1 in the parasite, the coding sequence of Tvcyp1 was amplified from the genomic DNA by a PCR using the primer pair FLP-PPI-f and FLP-PPI-r. The gel-purified PCR product was cloned into pGEM-T Easy (Promega), and the insert restricted by BamHI/EcoRI was ligated with SacII/EcoRI-restricted pAP65–2.2-ha-myb1 (14) and SacII/BamHI-restricted pTA-FLP-ha (16) to generate pFLP-ha-TvCyP1. A two-step PCR as described above was used to mutate the Arg63 residue in TvCyP1 to alanine. Briefly, from pFLP-ha-TvCyP1, a 5′-DNA fragment was amplified using the primer pair FLP-seq and CyPA1-R63A-3′ and a 3′-DNA fragment using the primer pair CyPA1-R63A-5′ and sp6. The gel-purified PCR products were mixed, denatured, and annealed for a second PCR using the primer pair FLP-seq and sp6. The PCR product digested by BamHI/EcoRI was then cloned into a BamHI/EcoRI-restricted pFLP-ha-TvCyP1 backbone to generate pFLP-ha-TvCyP1(R63A).
To produce point mutation in HA-Myb1, a 5′-DNA fragment was amplified from pFLP-ha-myb1 (16) using a primer pair, tub90f and Myb1-(XnA)-3′ (X indicates the aa to be mutated, n indicates the location of the residue, and A is alanine), and a 3′-DNA fragment was amplified by a primer pair, Myb1-(XnA)-5′ and SP6. The gel-purified PCR products were mixed, denatured, and annealed for a second PCR using the primer pair tub90f and SP6. The PCR product digested by BglII and NsiI was cloned into a BglII/NsiI-restricted pAP65–2.2.ha-Myb1 backbone (14) to generate pAP65–2.2.ha-Myb1(G106A) and pAP65–2.2.ha-Myb1(P107A).
In our preliminary study, His-tagged TvCyP1 that was produced and purified from an E. coli protein expression system became insoluble in a PPI enzymatic proficiency assay performed at 10 °C (data not shown). To produce a tagless recombinant TvCyP1 (rTvCyP1), a DNA fragment spanning the coding region of the Tvcyp1 gene was amplified from genomic DNA using the primer pair FLP-PPI-f and FLP-PPI-r. The PCR product was gel-purified and cloned into pGEM-T Easy (Promega), and the BamHI/EcoRI insert was cloned into a BamHI/EcoRI-restricted pET32a backbone (Novagen) to generate pET-rTvCyP1.
To produce GST-fused TvCyP1 (GST-TvCyP1), pFLP-ha-TvCyP1 or pFLP-ha-TvCyP1(R63A) was digested by BamHI and EcoRI. The insert was cloned into a BamHI/EcoRI-restricted pGEX-2T (GE Healthcare) to generate pGST-TvCyP1 or pGST-TvCyP1(R63A), respectively.
To express Myb1 mutant proteins, a 5′-DNA fragment was amplified from pET28-Myb1 (16) by PCR using a primer pair, T7p and Myb1(XnA)-3′, and a 3′ DNA fragment was amplified using a primer pair, Myb1(XnA)-5′ and T7t. PCR products were gel-purified, mixed, denatured, and annealed for a second PCR using the primer pair T7p and T7t. The PCR product thus produced was digested by NdeI and SalI and subcloned into an NdeI/SalI-restricted pET28-Myb1 backbone to generate pET28-Myb1(G106A) or pET28-Myb1(P107A).
Expression and Purification of Recombinant Proteins
pET-rTvCyP1 was transformed into E. coli BL21(DE3). pET28-Myb1, pET28-Myb1(G106A), and pET28-Myb1(P107A) were transformed into E. coli BL21-CodonPlus(DE3)-RIL. pGST-TvCyP1 and pGST-TvCyP1(R63A) were transformed into E. coli DH5α. A colony from each transformation was inoculated into LB broth containing 50 μg/ml ampicillin and incubated at 37 °C with constant shaking.
The protein expression of rTvCyP1 was induced at an A600 of 0.8 in the presence of 0.5 mm isopropyl thio-β-d-galactoside for 5 h at 30 °C. The cell mass was harvested and resuspended in buffer A (30 mm Tris, 0.5 mm tris(2-carboxyethyl)phosphine hydrochloride, pH 9.4) for subsequent disruption using a Microfluidizer (Microfluidics). The insoluble fraction of the lysates was removed by centrifugation at 13,500 × g for 30 min. The protein was purified by ion-exchange chromatography using a Q-Sepharose (GE Healthcare) column and eluted in buffer B (20 mm Tris, 0.5 mm tris(2-carboxyethyl)phosphine hydrochloride, pH 8). rTvCyP1 in the eluate was further purified by size exclusion chromatography using a Superdex-75 (GE Healthcare) column and concentrated in a buffer containing 20 mm NaH2PO4, 50 mm NaCl, 0.5 mm NaN3, and 1 mm dithiothreitol at pH 6. The purity of rTvCyP1 was verified by sodium dodecylsulfate-polyacrylamide gel electrophoresis (SDS-PAGE) (see Fig. 3A).
FIGURE 3.

The PPI enzymatic proficiency of recombinant TvCyP1. In A, purified rTvCyP1 was separated by SDS-PAGE in a 12% gel and stained by Coomassie Blue. In the enzyme reaction, 0–4 nm rTvCyP1 (B), GST-TvCyP1 (C), and GST-TvCyP1(R63A) (D) were incubated with a chromogenic substrate. The enzyme reaction was measured at 1-s intervals over a 10-min period by monitoring A390 using a spectrophotometer. The logarithmic phase of the enzyme reaction is plotted against various concentrations of rTvCyP1, and the calculated kcat/Km value is listed below each panel The inhibitory effect of CsA (0–40 nm) on the enzyme reaction using 4 nm rTvCyP1 is depicted in E. Error bars represent S.D. from three separate experiments.
For His-tagged proteins, expression was induced at an A600 of 0.6 in the presence of 0.4 mm isopropyl thio-β-d-galactoside for 2 h at 30 °C and purified using a His-Bind nickel column as described by the supplier (Novagen). The production and purification of GST fusion proteins was performed as described by the supplier (GE Healthcare).
The GST Pulldown Assay
Eighty picomoles of GST and GST fusion proteins, respectively, were incubated with 20 μl of glutathione-Sepharose 4B beads (GE Healthcare) in 1 ml of TEN200 buffer (1 mm EDTA, 200 mm NaCl, 20 mm Tris-HCl, pH 7.4) at 4 °C for 30 min. The beads were washed three times in washing buffer (0.5% Triton X-100 in TEN200 buffer) and then incubated with 35 nm His-tagged protein in 1 ml of TEN200 buffer at 4 °C for 30 min. The beads were washed three times in washing buffer and boiled in SDS sample buffer. Proteins were separated by SDS-PAGE for Coomassie Blue staining and Western blotting.
PPI Enzymatic Proficiency Assay
The PPI enzymatic proficiency of TvCyP1 was analyzed by measuring cis-trans isomerization of a chromogenic peptide, N-succinyl-Ala-Ala-Pro-Phe-p-nitroanilide (Sigma), using a commonly used spectrophotometric method as described (33). Briefly, 10 μl of the substrate (2.2 mm peptide in trifluoroethanol and 0.45 m LiCl) and 30 μl of chymotrypsin (0.5 m) were mixed with 250 μl of 40 mm HEPES, pH 7.8 in a prechilled cuvette. The reaction was initiated at 10 °C and lasted for 10 min in a spectrophotometer (DU800, Beckman Coulter) with the addition of 10 μl of TvCyP1 diluted to the desired concentration. The A390 value was recorded at 1-s intervals. Variation of the concentration of added enzyme made a series of reaction curves as shown in Fig. 3 B, C, and D. The natural logarithms of differences between the A390 and final A390 were plotted versus time over a span of 60 s to derive the first-order reaction constant (kobs). kobs was then graphed against the concentrations of enzyme based on kobs = kcat/Km × [rTvCyP1] to give the enzymatic proficiency, kcat/Km (33). To determine the 50% inhibition concentration (IC50) of the inhibitor, 0–80 nm CsA serially diluted from a stock solution of 8.3 mm CsA in ethanol was added in the enzyme reaction containing 4 nm rTvCyP1.
Antibody Production
Purified recombinant proteins TvCyP1, TvCyP2 (TVAG_062520),4 glycosyltransferase (TvGT) (TVAG_258220),4 and heat shock protein (Hsp) 70 (TvHsp70-1) (TVAG_044510)4 (34) and a protein with seven ankyrin repeats (TvAnk-1) (TVAG_016370)4 (34) were used for immunization in mice and rats following a standard protocol (35). Antisera were collected and purified by protein A affinity chromatography as described by the supplier (Sigma).
The Immunofluorescence Assay (IFA)
Cells on a slide were fixed in 4% paraformaldehyde in PBS for 30 min and permeabilized with 0.2% Triton X-100 in PBS for 15 min. The primary immunoreaction was performed using mouse (400×) (HA-7, Sigma) or rat (200×) (3F10, Roche Applied Science) monoclonal anti-HA antibodies, mouse (1200×) or rat (1200×) anti-TvCyP1 polyclonal antibodies, or 12G4 (500×) (a gift from Dr. John Alderete, Washington State University), the mouse monoclonal antibody against malic enzyme, also referred to as AP65 (7), as specified in text. Secondary immunoreactions were performed using FITC- or Cy3-conjugated secondary antibodies (Jackson ImmunoResearch Laboratories). The nuclei were stained using DAPI. Fluorescence signals were documented with a confocal microscope (LSM700, Zeiss), and cell morphology was documented by phase-contrast microscopy.
Subcellular Fractionation Using Detergents
Cell lysates were fractionated using a subcellular fractionation kit as described by the supplier (Merck) but with modifications. Briefly, cells from 15-ml cultures were harvested by centrifugation at 6000 × g at 4 °C for 15 min (Beckman, Allegra 6R). Cell pellets were washed with 1 ml of cold PBS and extracted with 100 μl of buffer I (buffers I–IV are all from the ProteoExtract subcellular proteome extraction kit, Calbiochem). The pellets were extracted once more using 100 μl of fresh buffer I, and lysates from both extractions were combined to obtain cytosolic fractions. The resulting pellets were then extracted with 100 μl of buffer II twice as described above to obtain membranous fractions. The pellets were again extracted with 100 μl of buffer III twice to obtain nuclear fractions. The final pellets were dissolved in 50 μl of buffer IV to obtain cytoskeletal fractions.
Subcellular Fractionation by Differential and Gradient Centrifugations
Membrane vesicles were enriched from 250 ml of cells by differential and gradient centrifugations as described previously (36) with some modifications. Briefly, cells were harvested by centrifugation at 6000 × g at 4 °C for 15 min (Beckman, JA10), and pellets were washed with 10 ml of cold PBS. Cells resuspended in 2 ml of a hypotonic lysis buffer (0.32 m sucrose, 10 mm Tris-HCl, pH 7.8, 2 mm MgCl2, 10 mm β-mercaptoethanol, 10 mm KCl, 1 mm EDTA, 200 μg ml−1 TLCK) were kept on ice for 15 min and homogenized by a Dounce type homogenizer until >90% of cells were broken as observed under a light microscope. The nuclei and unbroken cells were removed by centrifugation at 1000 × g at 4 °C for 15 min (Beckman, JA20). The postnuclear lysate was centrifuged again at 15,000 × g at 4 °C for 30 min (Beckman, JA20). The pellet, P15, which comprises hydrogenosomes and other smaller vesicles (see Fig. 9), was resuspended in 0.3 ml of buffer (0.25 m sucrose, 1 mm EDTA, 10 mm Tris-HCl, pH 7.4, 200 μg ml−1 TLCK) and layered on top of a stepwise (12–30%) OptiPrep gradient gel as described elsewhere (36). The samples were centrifuged at 200,000 × g at 4 °C for 12 h (Beckman, SW60). The gradient was fractionated into 200-μl fractions from top. The post-P15 supernatant was further separated into S100 and P100 fractions by centrifugation at 100,000 × g at 4 °C for 1 h (Beckman, TLA100.3). For immunoprecipitation, proteins in P15 and P100 were extracted using extraction buffer II of a cellular fractionation kit (Merck). Extracts were diluted in PBS by a factor of 5.
FIGURE 9.

Subcellular distribution of TvCyP1 and the trafficking of Myb1. In A, the P15 fraction from postnuclear lysates of T. vaginalis was separated by gradient centrifugation for Western blotting using the anti-Myb1, anti-Myb3, anti-TvCyP1, and 12G4 antibodies as indicated. In B, samples from fractions 4 and 10 were adsorbed onto Formvar-coated grids, fixed, negatively stained, and observed under a transmission electron microscope. The bar in the micrographs represents 200 nm. In C, samples from fractions 4 and 10 were examined by a protease protection assay in which samples were digested by the protease K with or without permeation by 0.5% Triton X-100 and examined by Western blotting using anti-TvCyP1, anti-Myb1, anti-Myb3, anti-Myb2, anti-TvAnk-1, and 12G4 antibodies as indicated. In D, vesicle fractions 3 and 4 from controls and cells overexpressing TvCyP1 and TvCyP1(R63A) were assayed by Western blotting using the antibodies to detect various proteins as indicated. The relative intensities of Myb1, Myb2, Myb3, and TvCyP1 versus TvHsp70-1 from three separate experiments are quantified in histograms. In E, the role of TvCyP1 in regulating the nuclear translocation of Myb1 and Myb3 is proposed. In this scheme, TvCyP1 may form a protein complex with TvAnk-1 and Myb1 or Myb3 on the outer surface of a novel vesicle, referred to as VMyb, to accelerate cis-trans interconversion of a glycinyl-prolyl bond in Myb1 and possibly in Myb3, resulting in the release of restrained Myb1 and Myb3 into the cytoplasm. In the cytoplasm, TvCyP1 forms a protein complex with TvHSP70-1 and Myb3 but not Myb1. Myb1 and Myb3 are then translocated into the nucleus where they regulate the transcription of the ap65-1 gene. Error bars represent S.D. from three separate experiments.
Western Blotting
Protein samples were separated by SDS-PAGE in a 12% gel. Proteins were stained by Coomassie Blue or transferred to polyvinylidene difluoride (PVDF) membranes (Millipore) by a semidry electroblotter for Western blotting. Antibodies from commercial sources, including rabbit anti-acetylhistone H3K9 (3000×) (Upstate), rat monoclonal anti-HA antibody (2000×) (3F10, Roche Applied Science), and mouse monoclonal anti-α-tubulin antibody (10,000×) (DM1A, Sigma), were used as described by the suppliers. AP65, Myb1, Myb2, Myb3, TvCyP1, TvCyP2, TvGT, TvHsp70-1, TvAnk-1, and hexokinase (TvHK), respectively, were detected using mouse monoclonal 12G4 (1000×) (7), mouse anti-Myb1 (1000×) (16), rabbit anti-Myb2 (4000×) (17), rabbit anti-Myb3 (3000×) (14), mouse anti-TvCyP1 (5000×), rat anti-TvCyP2 (1000×), rat anti-TvGT (3000×), rat anti-TvHsp70-1 (10,000×), rat anti-TvAnk-1 (5000×), and rabbit anti-TvHK (1000×) (a gift from Dr. Rossana Arroyo, Centro de Investigación y de Estudios Avanzados del Instituto Politécnico Nacional, Mexico City, Mexico) (10). Signals on blots were detected by an enhanced chemiluminescence (ECL) system as described by the supplier (Thermo Scientific). The relative intensities of signals were quantified and analyzed by MetaMorph software (Molecular Devices).
Immunoprecipitation
For each sample, 20 μl of the agarose-conjugated anti-HA antibody (Sigma) were added, and the reactions were incubated at 4 °C for 2 h with constant agitation. Agarose beads recovered by low speed centrifugation were washed in PBS containing 0.1% Triton X-100 for 10 min three times, and the proteins were eluted with 50 μl of 50 mm acidic glycine, pH 2.8 and immediately neutralized with 3 μl of 1 m Tris base, pH 9.0. The elution was repeated three times, and the eluates were combined.
Structure Modeling
The structural model of TvCyP1 was predicted using SWISS-MODEL Workspace in automated mode and visualized using the PyMOL Molecular Graphics System version 1.3 (Schrödinger, LLC) (37).
Negative Staining and Transmission Electron Microscopy
Samples adsorbed onto Formvar-coated copper grids (300 mesh, Electron Microscopy Sciences) were fixed in 1% glutaraldehyde for 10 min and negatively stained with 2% uranyl acetate for 1 min (38). Images were captured by transmission electron microscopy (JEM 1200 EX, EM Lab Services).
RESULTS
Screening of the Myb1-interacting Proteins
A T. vaginalis cDNA expression library was constructed in pTRG. Screening of this library with a pBait-Myb1/R3(2)C plasmid that can express the R3(2)C fragment (aa 103–206) of Myb1 in the bacterial host resulted in the identification of two identical partial cDNA clones, pTRG-c21 and pTRG-c22. No positive cDNA clone was obtained when screening the library with pBait-Myb1, which encodes the full-length coding sequence of the myb1 gene. In a pairwise two-hybrid interaction assay, pTRG-c21 revealed no interaction with pBait, pBait-Myb1/N, or pBait-Myb1/R2R3(1) plasmid, which harbor empty, N-terminal (aa 1–51), or R2R3(1) (aa 52–102) moieties, respectively, of the Myb1 coding sequence (Fig. 1A). The sequence analysis of pTRG-c21 revealed a human cyclophilin A-like gene (TVAG_004440) (34), which encodes an open reading frame of 173 aa (Fig. 1B) with an estimated mass of 19 kDa and a pI value of 7.8. This gene is referred to as Tvcyp1 because it is the first CyP-like gene to be functionally characterized in T. vaginalis. TvCyP1 comprises a CsA-binding motif within a conserved CLD (aa 18–160) and shares sequence identities with Caenorhabditis elegans CeCyP3 (69%), hCyPA (61%), and yCPR1 (62%). All aa residues (Arg63, Phe68, Met69, Gln71, Gly80, Ala109, Asn110, Ala111, Gln119, Phe121, Trp129, Leu130, and His134) crucial for PPI enzyme activity and CsA binding are conserved in TvCyP1. Like CeCyP3 and a few other hCyPA homologues of worms and plants (39), TvCyP1 possesses an extra sequence, 49KSGMPLS55, that is not present in hCyPA or yCPR1 (Fig. 1B). As predicted by structural modeling using CeCyP3 as a template (SWISS-MODEL server, Protein Data Bank code 1DYW) (39), TvCyP1 may comprise eight antiparallel β-strands (β1–β8) capped by two α-helices (α1 and α3) and the extra sequence in the loop region connecting α1 and β3 (Fig. 1 C, left panel). This extra loop sequence may provide intramolecular hydrogen bond formation between Glu84 and Lys49 or Ser50 or disulfide bond formation between Cys41 and Cys169 (Fig. 1C, right panel).
The GST Pulldown Assay
The interaction between Myb1 and TvCyP1 was examined in vitro by a GST pulldown assay in which His-tagged Myb1 and His-Myb2 were separately incubated with GST-TvCyP1 and GST, respectively. Pulldown products were examined by Western blotting using the anti-His6 antibody, and a duplicate gel was stained by Coomassie Blue to show input levels of GST-TvCyP1 and GST. A major band was detected in samples from the pulldown of His-Myb1 by GST-TvCyP1 but not GST (Fig. 2A, left panel). In contrast, no pulldown product was detected in the reaction of His-Myb2 with either GST-TvCyP1 or GST (Fig. 2A, right panel), demonstrating a stringent interaction between Myb1 and TvCyP1. The interaction was inhibited by 2 μm CsA (Fig. 2B). In the pulldown assay, His-Myb1 did not interact with GST-TvCyP1(R63A) in which Arg63 in the catalytic domain of TvCyP1 was mutated to alanine. These results suggest that Myb1 and CsA likely share a common binding site in the catalytic domain of TvCyP1.
FIGURE 2.

In vitro interaction of His-Myb1 and GST-TvCyP1. In A, GST (lane 1) and GST-TvCyP1 (lane 2) were incubated with His-Myb1 (left panel) and His-Myb2 (right panel), respectively. In B, GST, GST-TvCyP1, and GST-TvCyP1(R63A) were incubated with His-Myb1 with (lanes 4–6) and without (lanes 1–3) 2 μm CsA, respectively. The reaction products and 10% input of His-Myb1 and His-Myb2 were separated by SDS-PAGE in a 12% gel in duplicate for Coomassie Blue staining and Western blotting, respectively, using the anti-His6 antibody (α-6xHis).
The PPI Enzymatic Proficiency of rTvCyP1
rTvCyP1 was produced and purified to near homogeneity (Fig. 3A). In a reaction with a standard peptide substrate, the enzymatic proficiency (kcat/Km) of rTvCyP1 was 7.1 μm−1 s−1 (Fig. 3B). Meanwhile, the enzymatic proficiency of GST-TvCyP1 was ∼4.0 μm−1 s−1 (Fig. 3C), whereas that of GST-TvCyP1(R63A) was barely detectable (Fig. 3D). The catalytic efficiency of rTvCyP1 was inhibited by CsA in a dose-dependent manner with an IC50 of ∼7.5 nm (Fig. 3E). The recombinant His-tagged TvCyP1 was unsuitable for the enzyme assay because it was mostly precipitated at 10 °C (data not shown), the temperature for the enzyme assay.
Expression Profile and Subcellular Localization of TvCyP1
When examined by Western blotting (Fig. 4A), a single 19-kDa band was detected in total lysates from T. vaginalis using the mouse anti-TvCyP1 antibody. The expression level of TvCyP1 was similar in cells exposed to variable iron concentrations for 8 h (Fig. 4B); however, a slightly lower expression was detected in total lysates from cells depleted of iron compared with those grown in normal or high iron conditions for 12 h. The expression levels of Myb1, Myb3, and α-tubulin in these samples as detected by respective antibodies were little changed. By contrast, the expression of AP65 as detected by the 12G4 monoclonal antibody was induced by iron. Total lysates were then fractionated using a cellular fractionation kit for Western blotting (Fig. 4C). TvCyP1 was enriched in the membranous fraction with a slight amount in the soluble cytosolic fraction but not in the nuclear fraction. AP65 was detected primarily in membranous fractions and to a much lesser extent in cytosolic fractions. The purity of each cellular fraction was validated by respective detection of TvHK, TvCyP2, H3K9-Ac, and α-tubulin only in cytosolic, membranous, nuclear, and cytoskeletal fractions.
FIGURE 4.

Expression profile and subcellular localization of TvCyP1 in T. vaginalis. In A, the cell lysates from T. vaginalis in normal growth medium were examined by Western blotting using the anti-TvCyP1 antibody. In B, cell lysates from T. vaginalis grown in iron-depleted (d; lanes 1 and 4), normal (n; lanes 2 and 5), and high iron (h; lanes 3 and 6) media for 8 (lanes 1–3) and 12 h (lanes 4–6) were examined by Western blotting using the antibodies to detect various proteins as indicated. In C, total cell lysates (TL) from T. vaginalis under normal growth conditions were fractionated into cytosolic (C), membranous (M), nuclear (N), and insoluble cytoskeletal (Cs) fractions for Western blotting using the antibodies to detect various proteins as indicated. In D, IFAs of T. vaginalis cells were performed using rat anti-TvCyP1, normal rat serum, normal mouse serum, and 12G4. Cells were then incubated with FITC- or Cy3-conjugated rat and mouse IgG, respectively. The nuclei were stained with DAPI. Fluorescence signals were each recorded under confocal microscopy and merged. Cell morphology was recorded under phase-contrast microscopy. The bar in the micrographs represents 5 μm. Tub, tubulin.
When examined by an IFA, strong punctate signals and some weaker signals were detected in the cytoplasm using the rat anti-TvCyP1 antibody (Fig. 4D, top panel). Only strong punctate signals were detected using 12G4 (Fig. 4D, middle panel), which had been used to detect the hydrogenosomal malic enzyme that is also reputed to be AP65 (7). When double stained with rat anti-TvCyP1 and 12G4, not all signals derived from TvCyP1 were co-localized with AP65 (Fig. 4D, bottom panel), implying that TvCyP1 is localized to multiple membranous compartments in the cytoplasm. The unusual subcellular distribution of TvCyP1 was further examined below (see Fig. 6).
FIGURE 6.

Purification of the TvCyP1 protein complexes by immunoprecipitation. Postnuclear lysates from cells overexpressing TvCyP1 or TvCyP1(R63A) and controls were separated into P15, P100, and S100 fractions by differential centrifugations (A) and immunoprecipitated by the anti-HA antibody (B). Protein samples were examined by Western blotting using the antibodies to detect various proteins as indicated. TL, total lysates.
TvCyP1 and Nuclear Translocation of Myb1
To study the potential roles of TvCyP1, stable cell lines overexpressing HA-TvCyP1 and HA-TvCyP1(R63A) were established from transfection of pFLPha-TvCyP1 and pFLPha-TvCyP1(R63A), respectively (Fig. 5A). When cells were examined by the IFA (Fig. 5B), subcellular localizations of HA-TvCyP1 and TvCyP1(R63A) as double stained with the rat anti-HA and 12G4 antibodies were similar to that of endogenous TvCyP1 (see Fig. 4D). Signal intensities of AP65 as detected by 12G4 were similar in transgenic cell lines and controls, indicating that the overall expression of AP65 was not affected by the overexpression of TvCyP1.
FIGURE 5.
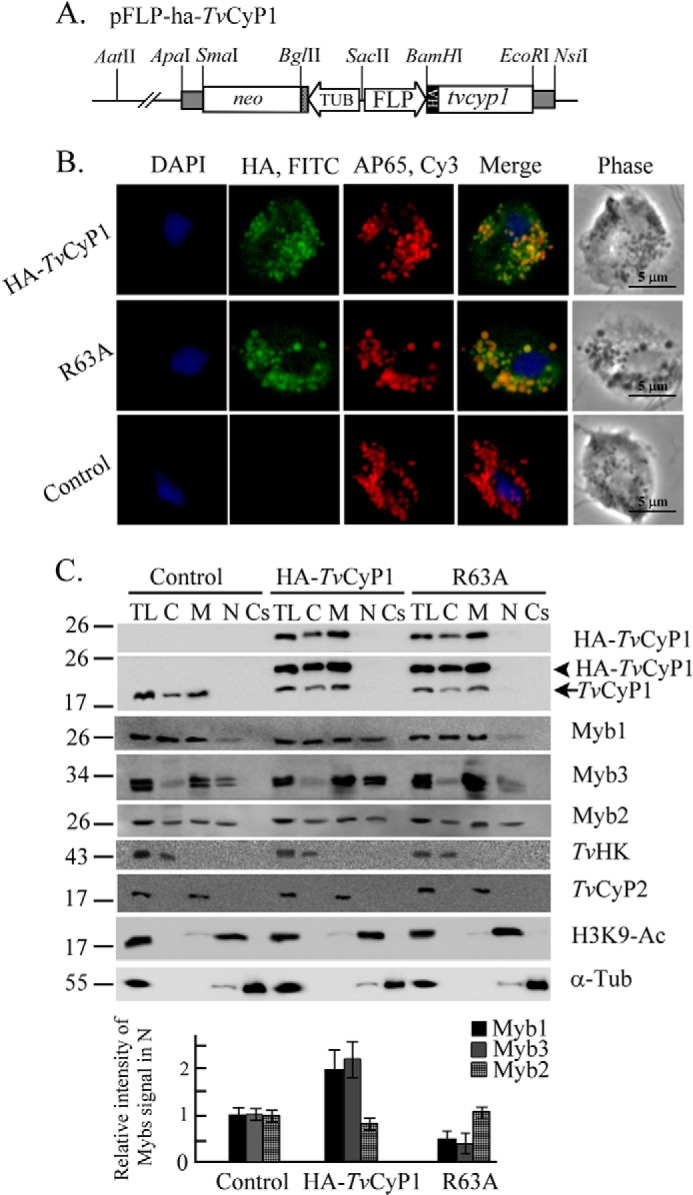
Regulation of nuclear translocation of Myb1 by TvCyP1. A stably expressed plasmid, pFLP-ha-TvCyP1, that overexpresses HA-TvCyP1 in T. vaginalis is depicted in A. A mutation was introduced to produce pFLP-ha-TvCyP1(R63A). In B, transfected cells overexpressing HA-TvCyP1 or HA-TvCyP1(R63A) and nontransfected controls were incubated sequentially with a rat anti-HA antibody paired with FITC-conjugated rat IgG and 12G4 paired with Cy3-conjugated mouse IgG. Nuclei were stained with DAPI. Fluorescence signals (FITC, Cy3, and DAPI) were recorded under confocal microscopy and merged. Cell morphology was recorded under phase-contrast microscopy. The bar in the micrographs represents 5 μm. In C, total cell lysates (TL) from cells overexpressing HA-TvCyP1 and HA-TvCyP1(R63A) and controls were fractionated into cytosolic (C), membranous (M), nuclear (N), and cytoskeletal (Cs) fractions for Western blotting using the antibodies to detect various proteins as indicated. HA-TvCyP1 and endogenous TvCyP1 that were simultaneously detected by anti-TvCyP1 are indicated by left arrowhead and left arrow, respectively. The relative intensities of Myb1, Myb2, and Myb3 versus H3K9-Ac in nuclear samples from three separate experiments are quantified and shown as histograms at the bottom. Error bars represent S.D. from three separate experiments. TUB, tubulin.
When assayed by Western blotting, HA-TvCyP1 and HA-TvCyP1(R63A) were detected to similar extents in cell lysates from their respective transgenic cell lines using the anti-HA antibody with levels severalfold higher than endogenous TvCyP1 that was detected by the anti-TvCyP1 antibody (Fig. 5C). The expression levels of Myb1, Myb2, and Myb3 relative to α-tubulin changed little in cells overexpressing HA-TvCyP1 and HA-TvCyP1(R63A). Similar to TvCyP1, HA-TvCyP1 and HA-TvCyP1(R63A) were enriched more in membranous than cytosolic fractions. In nontransfected control, Myb1 was detected mostly in membranous and cytosolic fractions and to a much lesser degree in nuclear fractions. Myb3 was also enriched in the membranous fraction, but it was detected at similar low levels in cytosolic and nuclear fractions. Myb2 was detected at similar levels in the nuclear and membranous fractions but to a much lesser extent in the cytosolic fraction. The levels of Myb1 and Myb3 in nuclear fractions increased substantially in samples from cells overexpressing HA-TvCyP1, but nuclear translocation was repressed by the overexpression of HA-TvCyP1(R63A). The amounts of Myb2 remained unchanged, suggesting that TvCyP1 might facilitate nuclear translocation of Myb1 and Myb3. The purity of each cellular fraction was validated by respective detection of TvHK, TvCyP2, H3K9-Ac, and α-tubulin only in cytosolic, membranous, nuclear, and cytoskeletal fractions.
The TvCyP1 Protein Complex
Postnuclear lysates were separated into P15, P100, and S100 fractions by differential centrifugations to study the formation of the TvCyP1 protein complex. Western blotting revealed that HA-TvCyP1 and HA-TvCyP1(R63A) were expressed to similar levels in total lysates, and overall expression of various endogenous proteins varied slightly in control and transgenic cells (Fig. 6A). Like TvCyP1, they were detected at higher levels in S100 than in P15 with the lowest levels in P100. Myb1 was enriched more in P100 than in P15 but not in S100, whereas Myb2 and Myb3 were each detected to similar levels in P15 and P100 but to a lower level in S100. TvHsp70-1 was detected at a higher level in P100 than in S100 with the lowest level in P15. TvAnk-1 was detected to similar levels in S100 and P15 with only a slight amount in P100. The purity of each cellular fraction was shown by the presence of TvCyP2 only in P15, TvHK only in S100, and TvGT only in P100, indicating that the presence of Myb1 and Myb3 in P100 is not due to cross-contamination from P15. The levels of Myb1 and Myb3 in P15 and P100 were lower in samples from cells overexpressing HA-TvCyP1 than in control and in those from cells overexpressing HA-TvCyP1(R63A), implying that TvCyP1 may facilitate trafficking of Myb1 and Myb3 from multiple membranous compartments to cytoplasm.
Protein samples were immunoprecipitated by anti-HA antibody for Western blotting (Fig. 6B). Consistent with their subcellular distributions (Fig. 6A), the amounts of HA-TvCyP1 and HA-TvCyP1(R63A) in the pulldowns from S100 were substantially higher than those from P15 and P100. A degraded form was observed only in samples from S100. Myb1 was detected to similar extents in the protein complex associated with TvCyP1, but not TvCyP1(R63A), from P15 and P100 but not S100. By contrast, Myb3 was detected largely in S100 but to a lesser extent in P15 and P100 and at a level much higher in the protein complex associated with TvCyP1 than that with TvCyP1(R63A). TvAnk-1 was detected at a level much higher in P100 than in P15 but was barely detected in S100, whereas TvHSP70-1 was detected mostly in S100. Unlike Myb1 and Myb3, TvHsp70-1 and TvAnk-1 were associated with equivalent amounts of HA-TvCyP1 and HA-TvCyP1(R63A). Myb2 was not detected in these protein complexes. None of these protein signals were detected in samples from controls. These observations suggest that TvCyP1 associates with different accessory proteins in distinct cellular compartments.
The TvCyP1-interacting Domain in Myb1
The motif in Myb1 that interacts with TvCyP1 was mapped by a pairwise bacterial two-hybrid assay using pTRG-c21 in a reaction with each of a series of pBait-Myb1/R3(2)C deletion mutants (Fig. 7A). The interaction was completely lost with the deletion of a small region, 104EYGPKWNKI112, from R3(2)C. Scanning mutagenesis was then performed to define the aa residues in this region that are essential for Myb1-TvCyP1 interaction. The mutation of any one of the aa residues in 105YGP107 resulted in a complete loss of protein-protein interaction, whereas the mutation of either Lys108 or Lys111 resulted in a substantial loss of interaction (Fig. 7B). Interactions were slightly reduced with the mutation of either Glu104 or Asn110, whereas the mutation of either Trp109 or Ile114 had no effect, indicating that 104EYGPKWNK111 is the TvCyP1-binding motif. Point mutation was introduced into His-Myb1 to produce His-Myb1(G106A) and His-Myb1(P107A) for the GST pulldown assay. Consistent with previous findings, His-Myb1 interacted with GST-TvCyP1 but not GST; however, no interaction was detected between GST-TvCyP1 and His-Myb1(G106A) or Myb1(P107A) (Fig. 7C).
An HA-Myb1-overexpressing plasmid, pFLPha-myb1/TUBneo (16), was explored to study the potential function of the TvCyP1-binding motif in Myb1. Point mutation was introduced into the plasmid to overexpress HA-Myb1(G106A) and HA-Myb1(P107A). The expression of these proteins in T. vaginalis was examined overnight after transfection because many attempts to establish stable cell lines overexpressing HA-Myb1 and HA-Myb1(G106A) had failed. When examined by double staining in the IFA using rat anti-TvCyP1 and mouse anti-HA antibodies, HA-Myb1 was visualized as punctate signals mostly in the nucleus (Fig. 8A), whereas HA-Myb1(P107A) was primarily localized to the nucleus with much greater intensity than HA-Myb1. In contrast, HA-Myb1(G106A) was exclusively localized to the cytoplasm as punctate signals. A bright fluorescent spot was always observed in the cytoplasm of cells overexpressing HA-Myb1(G106A) and sometimes in HA-Myb1-overexpressing cells but not in cells overexpressing HA-Myb1(P107A). In all three transgenic cell lines, TvCyP1 was localized to the cytoplasm in similar patterns by the rat anti-TvCyP1 antibody, and TvCyP1 was localized close to HA-Myb1(G106A). When cells were double stained with rat anti-HA and 12G4 antibodies, the expression of AP65 was repressed in cells overexpressing HA-Myb1 or HA-Myb1(P107A) versus nontransfected neighboring cells, but the repression was not seen in cells overexpressing HA-Myb1(G106A), which was largely localized close to the hydrogenosomes (Fig. 8B). These results are consistent with the role of Myb1 as a transcription repressor of the ap65-1 gene (16).
FIGURE 8.

The role of TvCyP1-binding motif in the trafficking of Myb1 to the nucleus. Transfected cells overexpressing HA-Myb1, HA-Myb1(G106A), and HA-Myb1(P107A) were examined by IFA (A and B). Slides were double stained, first with either rat anti-TvCyP1 (A) or rat anti-HA (B) followed by FITC-conjugated rat IgG and then with either mouse anti-HA (A) or 12G4 antibody (B) followed by Cy3-conjugated mouse IgG as indicated at the top. Nuclei were stained with DAPI. Fluorescence signals were each recorded under confocal microscopy and merged. Transfected and nontransfected cells in B are indicated by open arrows and open arrowheads, respectively. Cell morphology was recorded under phase-contrast microscopy. The bar in the micrographs represents 3 μm. In C, total cell lysates (TL) from transfected cells and controls were fractionated into cytosolic (C), membranous (M), nuclear (N), and cytoskeletal (Cs) fractions for Western blotting using rat anti-HA and mouse anti-Myb1 antibodies to detect overexpressed (exo-Myb1) and endogenous Myb1 (endo-Myb1), respectively. Blots were also probed using the antibodies to detect various proteins as indicated. The relative intensities of overexpressed Myb1 versus endogenous Myb1 in nuclear samples from three separate experiments are quantified in histograms. Error bars represent S.D. from three separate experiments. Tub, tubulin.
When cell lysates were examined by Western blotting using the anti-HA antibody (Fig. 8C), HA-Myb1 was detected at similar levels in cytosolic and membranous fractions, but it was detected at a much lower level in the nuclear fraction. HA-Myb1(G106A) was not detected in the nuclear fraction, and its level in the membranous fraction was substantially higher than that of HA-Myb1. By contrast, levels of HA-Myb1(P107A) in the membranous and nuclear fractions were slightly lower and substantially higher than those of HA-Myb1, respectively (Fig. 8C). When a duplicate blot was examined using anti-Myb1 antibody, endogenous Myb1 was distributed in a profile similar to that of HA-Myb1. The purity of each cellular fraction was validated by respective detection of TvHK, TvCyP2, H3K9-Ac, and α-tubulin only in cytosolic, membranous, nuclear, and cytoskeletal fractions. These results suggest that nuclear translocation of Myb1 may be regulated by cis-trans conversion of a glycinyl-prolyl imide bond.
TvCyP1 and Trafficking of Myb1
To further study the role of TvCyP1 in translocating Myb1, the cellular distribution of TvCyP1 was re-examined by multiple rounds of centrifugation in which the organelle fraction, P15, enriched from the postnuclear lysates was refractionated by gradient centrifugation. As shown by Western blotting (Fig. 9A), AP65 was detected at various levels across all fractions in the gradient and enriched in fractions 8–10, the hydrogenosomal fractions, as reported previously (36). TvCyP1 was detected in hydrogenosomal fractions as well as in fractions 1–4 among which Myb1 and Myb3 were consistently detected in fractions 3 and 4, and a slight amount of Myb3 was also detected in fraction 5. Similar results were observed in samples from cells overexpressing HA-TvCyP1 and HA-TvCyP1(R63A) (data not shown). A sample from fraction 4 was negatively stained, and two different sizes of vesicles in the range of 100–200 nm were observed under a transmission microscope (Fig. 9B). The Myb-associated vesicles are referred to as VMyb below for the convenience of discussion. In contrast, vesicles of ∼500 nm were observed in samples from fraction 10, suggesting that TvCyP1 is most likely localized to multiple membrane vesicles with different buoyant densities.
A protease protection assay was explored to examine the localization of TvCyP1 in these vesicles (Fig. 9C). In this assay, samples were degraded by the protease K with or without Triton X-100 and analyzed by Western blotting. In samples from Myb1-containing fraction 4, AP65 was protected from protease degradation, but it was largely degraded in the presence of Triton X-100. By contrast, TvCyP1 was largely but not completely degraded by the protease, and the degradation was complete in the presence of Triton X-100, whereas Myb1 and Myb3 were completely degraded by the protease with or without detergent. In samples from hydrogenosomal fraction 10, AP65 was protected from protease degradation but mostly degraded in the presence of Triton X-100. TvCyP1 was also protected from protease digestion but completely degraded in the presence of Triton X-100. Together, these observations suggest that TvCyP1 may reside inside hydrogenosomes but is mostly on the surface of the VMyb, whereas AP65 resides inside both of these vesicles.
Two consecutive Myb1-containing gradient fractions enriched from cell lines overexpressing HA-TvCyP1 and its mutant were then examined by Western blotting in the same blot (Fig. 9D). The amounts of Myb1 versus TvHsp70-1 were much lower in HA-TvCyP1 samples than in controls, which were in turn lower than HA-TvCyP1(R63A) samples. The levels of overall TvCyP1 (endogenous plus overexpressed) in samples from HA-TvCyP1- or HA-TvCyP1(R63A)-overexpressing cells were 2-fold higher than in controls. These observations suggest that TvCyP1 probably facilitates the release of Myb1 from specific membrane vesicles into the cytoplasm prior to nuclear importation.
DISCUSSION
The function of a transcription factor is often regulated at multiple cellular steps via intricate interactions with regulatory partners in a spatial and temporal manner (40). To understand the regulation of Myb1-mediated transcription repression, a Myb1-interacting protein, TvCyP1, was identified using a bacterial two-hybrid system to screen a T. vaginalis cDNA expression library in the present study. The initial screening using a bait derived from the sequence encoding the R3(2)/C moiety (aa 120–205) but not that of the full coding sequence of the myb1 gene resulted in the identification of a TvCyP1 protein (Fig. 1). Consistent with these findings, the binding of Myb1 to TvCyP1 in the GST pulldown assay was greatly enhanced with the deletion of either the N or C terminus from Myb1.5 The binding of a CyPA homologue to a full-length substrate protein that is weaker than its deleted mutant proteins is not unusual as it is also reported in the binding of hCyPA to the N-terminal SH3 domain of a mammalian Vav protein and other cellular targets (41). Because Myb1 exhibits a central helical structure flanked by nonstructural regions (22), it is likely folded into conformation with its N and C termini juxtaposed to mask the TvCyP1-binding motif (Fig. 7B). Spontaneous or inducible changes of conformation may be required for Myb1 to expose this motif, providing a way to regulate the function of the protein.
TvCyP1 exhibits a PPI enzymatic proficiency comparable with hCyPA and its homologues in various biological systems (33, 42–45) and specific binding to Myb1 that can be inhibited by CsA and eliminated by mutation of an active residue, Arg63, in the catalytic motif (Fig. 2B), indicating that the catalytic motif and substrate-binding motif of TvCyP1 may overlap as in hCyPA (27). In contrast to hCyPA, TvCyP1 has an extra sequence in the loop region connecting α1 and β3 (Fig. 1B). Like those of CeCyP3, the nitrogen atoms in the amide of Lys49 and Ser50 in TvCyP1 may form hydrogen bonds with the side-chain carboxylic oxygen of Glu84 to maintain the extra loop sequence in a particular conformation that may provide a platform for intermolecular protein-protein interactions with substrate proteins (Fig. 1C, upper right panel) (39). In this structure, Cys41 and Cys169 (Fig. 1C, lower right panel) may form a disulfide bond, presumably for sensing redox status in cellular environments (39). In CeCyP3, the postulated disulfide bond in coordination with His54 at the C terminus of the extra loop sequence may form a zinc-binding site (39). The proposed metal-binding site is probably absent in TvCyP1 because His54 in CeCyP3 is replaced by a serine in TvCyP1. Although these residues are highly conserved in TvCyP1, their roles remain elusive. In our enzyme assay, His-TvCyP1, but neither rTvCyP1 nor GST-TvCyP1, was precipitated under assay conditions likely due to the perturbation of proper TvCyP1 folding by fusion with the N-terminal His tag (46).
With regard to substrate specificity, TvCyP1 binds to Myb1 at a sequence motif, 104EYGPKWNK111 (Fig. 7B), in which Tyr105, Gly106, and Pro107 reside in the loop region connecting helix 4 and helix 5 of the bundled helical structure of the R2R3 DBD and are each essential for Myb1 binding to TvCyP1 (Fig. 7C). The tripeptide sequence is similar to that of hCyPA in which the substrate-binding site directly contacts the Ala88, Gly89, and Pro90 residues in the N-terminal domain of HIV capsid protein to induce cis-trans interconversion of the glycinyl-prolyl bond (47). TvCyP1 may slightly deviate from hCyPA in the substrate-binding site because Tyr105 in the TvCyP1-binding motif of Myb1 is bulkier than Ala88 in the hCyPA-binding motif of the HIV capsid protein, and the binding of Myb1 to TvCyP1 is lost in Y105A (Fig. 7C). Because of the potential use of the CsA-related compounds as antiparasitic drugs (48), the slight difference in substrate binding may provide an opportunity to design an inhibitor specific for TvCyP1 and test its cytotoxic effects against the parasite. Nevertheless, these biochemical features suggest that TvCyP1 may facilitate a change of conformation in Myb1 as a way to regulate the function of Myb1.
TvCyP1 and Myb proteins were all detected in membranous and cytosolic fractions to various extents using different extraction methods (Figs. 5C and 6A). For example, Myb1 was detected in the cytosolic fraction extracted by detergent but not seen in S100, whereas Myb3 was detected in S100 but barely detectable in cytosolic fraction extracted by detergent. The discrepancy is likely due to basic principles of detergent-based extraction and centrifugation-based separation. Some proteins on the outer surface of vesicles or organelles are likely to dissociate from the membranous compartments and be released into cytosolic fractions during extraction by detergents. It the meanwhile, proteasomal degradation might be activated by detergents (49). In this regard, stability of Myb3, but not Myb1, may be regulated by proteasomal degradation.5 These possibilities may account for the inconsistent cellular distribution of Myb1 and Myb3 derived from cellular fractionation by detergents versus centrifugation.
The most striking feature of TvCyP1 is its localization to multiple membranous compartments (Figs. 6 and 9A). In general, each type of hCyP is usually sorted to a particular cellular compartment depending on the signal peptide in that protein (26). Without a signal peptide, hCyPA is normally localized to the cytoplasm but can be transported to the nucleus along with interacting proteins or secreted in response to stimuli (26, 50). In contrast, TvCyP1 was localized to hydrogenosomes and other smaller vesicles (Figs. 4D and 9, A and B) with the latter also containing Myb1, Myb2, and Myb3 (Fig. 9A). As shown by a protease protection assay (Fig. 9C), TvCyP1 may reside on the surface of VMyb along with Myb1, Myb2, and Myb3. In cells overexpressing TvCyP1 versus controls (Fig. 9D), the levels of Myb1 and Myb3 in such vesicle fractions were inversely related to the total amount of TvCyP1 in the same fractions, suggesting that TvCyP1 may assist in the release of Myb1 and Myb3 from VMyb. This activity is dependent on PPI activity because higher levels of Myb1 and Myb3 were associated with VMyb from cells overexpressing TvCyP1(R63A). Intriguingly, TvCyP1 might also complex with Myb1 and Myb3 in P100, which is enriched in plasma membrane and small vesicles like endosomes4 but devoid of the hydrogenosomes and VMyb in P15 (Fig. 6). Exact localization of these proteins and their physiological significance remain to be studied; however, TvCyP1 might also assist in the release of Myb1 and Myb3 from these membranous compartments (Fig. 6A).
The TvCyP1-mediated release of Myb1 from VMyb was also supported by subcellular localization of Myb1 mutants G106A and P107A (Fig. 7A) in which the original peptidyl-prolyl imide bond between Gly106 and Pro107 in Myb1 was respectively changed into a bulkier version and eliminated. The binding to TvCyP1 was eliminated in both mutants, but Gly106 was constrained to vesicles, and Pro107 was imported into the nucleus at a much higher level than Myb1 without losing its repressive activity on the transcription of the ap65-1 gene. It is notable that the expression of AP65 changed little in transgenic cell lines overexpressing HA-TvCyP1 and HA-TvCyP1(R63A) and control (Fig. 5B). This observation is contradictory to the effect of TvCyP1 on the nuclear importation of Myb1 (Fig. 8B) unless TvCyP1 also facilitates the nuclear translocation of a transcription activator to counteract Myb1 in the transcription of the ap65-1 gene as implicated in an earlier report (14). In this regard, a higher level of Myb3 was detected in the nuclear fractions from cells overexpressing TvCyP1 (Fig. 5C). Intriguingly, TvCyP1 can form distinct protein complexes with Myb3 along with different accessory proteins in the membranous and cytosolic compartments (Fig. 6B). With only two glycinyl-prolyl bonds in Myb3, one preceding helix 1 and the other in the loop region connecting helices 1 and 2 (23), it is tempting to speculate that TvCyP1 may regulate cis-trans conversion of respective glycinyl-prolyl bonds in different cellular compartments for a two-step trafficking of Myb3 from certain membranous compartments and VMyb toward the nucleus. It remains to be studied whether direct interaction between TvCyP1 and Myb3 exists. Nonetheless, our observations underscore the importance of a glycinyl-prolyl bond in the subcellular localization of Myb1 and possibly Myb3.
Myb1 was associated with a TvCyP1 protein complex in a spatial manner, and this association was also Arg63-dependent (Fig. 6B), suggesting that TvCyP1 probably binds to Myb1 on the surface of vesicles to induce conformational changes and trafficking of Myb1 toward the nucleus (Fig. 9E). It is possible that a third party is needed for the spatial interaction because the TvCyP1 complex that was purified from either P15 or P100 had an accessory protein different from the one that was purified from S100 (Fig. 6B). In this regard, TvCyP1 might display an Arg63-independent complex formation with TvHsp70-1 and TvAnk-1 in the cytoplasm and on different membranous compartments, respectively (Fig. 6B). Such an enzyme-independent binding activity is not unusual as it is also seen in hCyPA in which the conserved Arg55 that is essential for enzyme activity is not involved in binding to CD147 (29). Hsp70 in human cells can bind to the tetratricopeptide repeats in the C terminus of hCyP40 to assist in the formation of an inactivated multicomponent steroid receptor (51), whereas the enzyme activity of hCyPA catalyzes the proper folding of ankyrin repeats in a protein for the assembly of protein complexes (52, 53). Although TvCyP1 has no functional domain other than the CLD, it may exploit TvAnk-1 as a scaffold for the assembly of the TvCyP1-Myb1 protein complex on vesicle surfaces, and TvHsp70-1 likely serves as a co-chaperone for TvCyP1 for conformational changes in unknown targets. These possibilities remain to be examined. Interestingly, Myb2 was also on the surface of the same vesicles as Myb1 (Fig. 9D), implying that such vesicles are the sites that restrain Myb1 and possibly other Myb transcription factors before their release into the cytoplasm and importation into the nucleus.4 TvCyP1 was most abundant inside hydrogenosomes, implying that TvCyP1 might assist in the proper folding of some hydrogenosomal proteins for regulating energy metabolism. These speculations can be tested in future studies.
Consistent with our previous findings (16), attempts to establish stable cell lines overexpressing HA-Myb1 and HA-Myb1(G106A) were unsuccessful.5 Myb1 mutants that can be stably expressed in T. vaginalis mostly accumulated within inclusion bodies5 except P107A as reported here, implying that a large portion of overexpressed Myb1 was not correctly folded possibly due to the limited amount of TvCyP1 in cells. Like HA-Myb1, HA-Myb1(P107A) could repress AP65 expression (Fig. 8B), suggesting that its role in transcription regulation remains unchanged. In this regard, the transfected cell line overexpressing HA-Myb1(P107A) may provide a tool to study the downstream target genes of Myb1. The genome of T. vaginalis encodes >400 Myb-like proteins among which Myb2 and Myb3 exploit highly ordered helical structures spanning the DBD for nuclear translocation (24, 25). In addition, Myb1 may use a similar entity for nuclear translocation.5 The effect of TvCyP1 on Myb1 nuclear translocation implies that specific factors other than structural components as observed previously in Myb2 and Myb3 are also involved in regulating the nuclear translocation of individual small Myb proteins. In summary, TvCyP1 may form separate protein complexes with Myb1 and Myb3 on the outer surfaces of VMyb to regulate conformational changes in Myb1 and Myb3 and release them to the cytoplasm prior to nuclear translocation and transcription regulation of the ap65-1 gene (Fig. 9E).
Acknowledgments
We thank Buford Pruitt Jr. for correcting the manuscript. We are grateful to Drs. Sven Gould (Heinrich-Heine-Universität Düsseldorf, Düsseldorf, Germany) for α-succinyl CoA synthetase, Rossana Arroyo (Centro de Investigación y de Estudios Avanzados del Instituto Politécnico Nacional, Mexico City, Mexico) for α-HK, Soon-Jung Park and Hyeyeon Lee (Yonsei University, Seoul, Korea) for α-actinin and α-AP33, Petrus Tang (Chun Gan University, Tao Yuan, Taiwan) for α-rubrerythrin antibodies in testing suitable markers in cellular fractionation experiments, and Drs. Hung Yin Deng and Ru-Chi Shieh for advice on the PPI enzymatic assay.
This work was supported by National Science Council Grant NSC101-2320-B-001-026 and the Institute of Biomedical Sciences, Academia Sinica.
H. M. Hsu and J. H. Tai, unpublished data.
C. H. Chu and J. H. Tai, unpublished data.
- DBD
- DNA-binding domain
- aa
- amino acid(s)
- Ank
- ankyrin repeat-containing protein
- CyP
- cyclophilin
- CLD
- cyclophilin-like domain
- CsA
- cyclosporin A
- Hsp70
- heat shock protein 70
- IFA
- immunofluorescence assay
- PPI
- peptidyl-prolyl cis/trans isomerase
- VMyb
- Myb-associated vesicles
- Tv
- T. vaginalis
- Ce
- C. elegans
- TvCyP1
- TvCyclophilin 1
- rTvCyP1
- recombinant TvCyP1
- GT
- glycosyltransferase
- TLCK
- Nα-p-tosyl-l-lysine chloromethyl ketone
- HK
- hexokinase
- h
- human
- y
- yeast.
REFERENCES
- 1. Fichorova R. N. (2009) Impact of T. vaginalis infection on innate immune responses and reproductive outcome. J. Reprod. Immunol. 83, 185–189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Petrin D., Delgaty K., Bhatt R., Garber G. (1998) Clinical and microbiological aspects of Trichomonas vaginalis. Clin. Microbiol. Rev. 11, 300–317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Shafir S. C., Sorvillo F. J., Smith L. (2009) Current issues and considerations regarding trichomoniasis and human immunodeficiency virus in African-Americans, Clin. Microbiol. Rev. 22, 37–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Alderete J. F., Provenzano D., Lehker M. W. (1995) Iron mediates Trichomonas vaginalis resistance to complement lysis. Microb. Pathog. 19, 93–103 [DOI] [PubMed] [Google Scholar]
- 5. Thurman A. R., Doncel G. F. (2011) Innate immunity and inflammatory response to Trichomonas vaginalis and bacterial vaginosis: relationship to HIV acquisition. Am. J. Reprod. Immunol. 65, 89–98 [DOI] [PubMed] [Google Scholar]
- 6. Weinstock H., Berman S., Cates W., Jr. (2004) Sexually transmitted diseases among American youth: incidence and prevalence estimates, 2000. Perspect. Sex Reprod. Health 36, 6–10 [DOI] [PubMed] [Google Scholar]
- 7. Garcia A. F., Chang T. H., Benchimol M., Klumpp D. J., Lehker M. W., Alderete J. F. (2003) Iron and contact with host cells induce expression of adhesions on surface of Trichomonas vaginalis. Mol. Microbiol. 47, 1207–1224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Moreno-Brito V., Yáñez-Gómez C., Meza-Cervantez P., Avila-González L., Rodríguez M. A., Ortega-López J., González-Robles A., Arroyo R. (2005) A Trichomonas vaginalis 120 kDa protein with identity to hydrogenosome pyruvate:ferredoxin oxidoreductase is a surface adhesin induced by iron. Cell. Microbiol. 7, 245–258 [DOI] [PubMed] [Google Scholar]
- 9. Torres-Romero J. C., Arroyo R. (2009) Responsiveness of Trichomonas vaginalis to iron concentrations: evidence for a post-transcriptional iron regulation by an IRE/IRP-like system. Infect. Genet. Evol. 9, 1065–1074 [DOI] [PubMed] [Google Scholar]
- 10. Meza-Cervantez P., González-Robles A., Cárdenas-Guerra R. E., Ortega-López J., Saavedra E., Pineda E., Arroyo R. (2011) Pyruvate:ferredoxin oxidoreductase (PFO) is a surface-associated cell-binding protein in Trichomonas vaginalis and is involved in trichomonal adherence to host cells. Microbiology 157, 3469–3482 [DOI] [PubMed] [Google Scholar]
- 11. Garcia A. F., Alderete J. (2007) Characterization of the Trichomonas vaginalis surface-associated AP65 and binding domain interacting with trichomonads and host cells. BMC Microbiol. 7, 116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kucknoor A. S., Mundodi V., Alderete J. F. (2005) Heterologous expression in Tritrichomonas foetus of functional Trichomonas vaginalis AP65 adhesin. BMC Mol. Biol. 6, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Mundodi V., Kucknoor A. S., Klumpp D. J., Chang T. H., Alderete J. F. (2004) Silencing the ap65 gene reduces adherence to vaginal epithelial cells by Trichomonas vaginalis. Mol. Microbiol. 53, 1099–1108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hsu H. M., Ong S. J., Lee M. C., Tai J. H. (2009) Transcriptional regulation of an iron-inducible gene by differential and alternate promoter entries of multiple Myb proteins in the protozoan parasite Trichomonas vaginalis. Eukaryot. Cell 8, 362–372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ong S. J., Huang S. C., Liu H. W., Tai J. H. (2004) Involvement of multiple DNA element in iron-inducible transcription of the ap65-1 gene in the protozoan parasite Trichomonas vaginalis. Mol. Microbiol. 52, 1721–1730 [DOI] [PubMed] [Google Scholar]
- 16. Ong S. J., Hsu H. M., Liu H. W., Chu C. H., Tai J. H. (2006) Multifarious transcriptional regulation of adhesion protein gene ap65-1 by a novel Myb1 protein in the protozoan parasite Trichomonas vaginalis. Eukaryot. Cell 5, 391–399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ong S. J., Hsu H. M., Liu H. W., Chu C. H., Tai J. H. (2007) Activation of multifarious transcription of an adhesion protein ap65-1 gene by a novel Myb2 protein in the protozoan parasite Trichomonas vaginalis. J. Biol. Chem. 282, 6716–6725 [DOI] [PubMed] [Google Scholar]
- 18. Tsai C. D., Liu H. W., Tai J. H. (2002) Characterization of an iron-responsive promoter in the protozoan pathogen Trichomonas vaginalis. J. Biol. Chem. 277, 5153–5162 [DOI] [PubMed] [Google Scholar]
- 19. Bergholtz S., Andersen T. O., Andersson K. B., Borrebaek J., Lüscher B., Gabrielsen O. S. (2001) The highly conserved DNA-binding domains of A-, B- and c-Myb differ with respect to DNA-binding, phosphorylation and redox properties. Nucleic Acids Res. 29, 3546–3556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Oh I. H., Reddy E. P. (1999) The myb gene family in cell growth, differentiation and apoptosis. Oncogene 18, 3017–3033 [DOI] [PubMed] [Google Scholar]
- 21. Jiang I., Tsai C. K., Chen S. C., Wang S. H., Amiraslanov I., Chang C. F., Wu W. J., Tai J. H., Liaw Y. C., Huang T. H. (2011) Molecular basis of the recognition of the ap65-1 gene transcription promoter elements by a Myb protein from the protozoan parasite Trichomonas vaginalis. Nucleic Acids Res. 39, 8992–9008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lou Y. C., Wei S. Y., Rajasekaran M., Chou C. C., Hsu H. M., Tai J. H., Chen C. (2009) NMR structural analysis of DNA recognition by a novel Myb1 DNA-binding domain in the protozoan parasite Trichomonas vaginalis. Nucleic Acids Res. 37, 2381–2394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wei S. Y., Lou Y. C., Tsai J. Y., Ho M. R., Chou C. C., Rajasekaran M., Hsu H. M., Tai J. H., Hsiao C. D., Chen C. (2012) Structure of the Trichomonas vaginalis Myb3 DNA-binding domain bound to a promoter sequence reveals a unique C-terminal β-hairpin conformation. Nucleic Acids Res. 40, 449–460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chu C. H., Chang L. C., Hsu H. M., Wei S. Y., Liu H. W., Lee Y., Kuo C. C., Indra D., Chen C., Ong S. J., Tai J. H. (2011) A highly organized structure mediating nuclear localization of a Myb2 transcription factor in the protozoan parasite Trichomonas vaginalis. Eukaryotic Cell 10, 1607–1617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hsu H. M., Lee Y., Indra D., Wei S. Y., Liu H. W., Chang L. C., Chen C., Ong S. J., Tai J. H. (2012) Iron-inducible nuclear translocation of a Myb3 transcription factor in the protozoan parasite Trichomonas vaginalis. Eukaryot. Cell 11, 1441–1450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wang P., Heitman J. (2005) The cyclophilins. Genome Biol. 6, 226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kallen J., Spitzfaden C., Zurini M. G., Wider G., Widmer H., Wüthrich K., Walkinshaw M. D. (1991) Structure of human cyclophilin and its binding site for cyclosporin A determined by x-ray crystallography and NMR spectroscopy. Nature 353, 276–279 [DOI] [PubMed] [Google Scholar]
- 28. Brazin K. N., Mallis R. J., Fulton D. B., Andreotti A. H. (2002) Regulation of the tyrosine kinase Itk by the peptidyl-prolyl isomerase cyclophilin A. Proc. Natl. Acad. Sci. U.S.A. 99, 1899–1904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Song F., Zhang X., Ren X. B., Zhu P., Xu J., Wang L., Li Y. F., Zhong N., Ru Q., Zhang D. W., Jiang J. L., Xia B., Chen Z. N. (2011) Cyclophilin A (CyPA) induces chemotaxis independent of its peptidylprolyl cis-trans isomerase activity: direct binding between CyPA and the ectodomain of CD147. J. Biol. Chem. 286, 8197–8203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bosco D. A., Eisenmesser E. Z., Pochapsky S., Sundquist W. I., Kern D. (2002) Catalysis of cis/trans isomerization in native HIV-1 capsid by human cyclophilin A. Proc. Natl. Acad. Sci. U.S.A. 99, 5247–5252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Dietrich L., Ehrlich L. S., LaGrassa T. J., Ebbets-Reed D., Carter C. (2001) Structural consequences of cyclophilin A binding on maturational refolding in human immunodeficiency virus type 1 capsid protein. J. Virol. 75, 4721–4733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Chou C. F., Tai J. H. (1996) Simultaneous extraction of DNA and RNA from nuclease rich pathogenic protozoan Trichomonas vaginalis. BioTechniques. 20, 790–791 [DOI] [PubMed] [Google Scholar]
- 33. Harrison R. K., Stein R. L. (1990) Substrate specificities of the peptidyl prolyl cis-trans isomerase activities of cyclophilin and FK-506 binding protein: evidence for the existence of a family of distinct enzymes. Biochemistry. 29, 3813–3816 [DOI] [PubMed] [Google Scholar]
- 34. Carlton J. M., Hirt R. P., Silva J. C., Delcher A. L., Schatz M., Zhao Q., Wortman J. R., Bidwell S. L., Alsmark U. C., Besteiro S., Sicheritz-Ponten T., Noel C. J., Dacks J. B., Foster P. G., Simillion C., Van de Peer Y., Miranda-Saavedra D., Barton G. J., Westrop G. D., Müller S., Dessi D., Fiori P. L., Ren Q., Paulsen I., Zhang H., Bastida-Corcuera F. D., Simoes-Barbosa A., Brown M. T., Hayes R. D., Mukherjee M., Okumura C. Y., Schneider R., Smith A. J., Vanacova S., Villalvazo M., Haas B. J., Pertea M., Feldblyum T. V., Utterback T. R., Shu C. L., Osoegawa K., de Jong P. J., Hrdy I., Horvathova L., Zubacova Z., Dolezal P., Malik S. B., Logsdon J. M., Jr., Henze K., Gupta A., Wang C. C., Dunne R. L., Upcroft J. A., Upcroft P., White O., Salzberg S. L., Tang P., Chiu C. H., Lee Y. S., Embley T. M., Coombs G. H., Mottram J. C., Tachezy J., Fraser-Liggett C. M., Johnson P. J. (2007) Draft genome sequence of the sexually transmitted pathogen Trichomonas vaginalis. Science 315, 207–212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Harlow E., Land D. (1988) Antibodies: a Laboratory Manual, pp. 55–137, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 36. Beltrán N. C., Horváthová L., Jedelský P. L., Sedinová M., Rada P., Marcinčiková M., Hrdý I., Tachezy J. (2013) Iron-induced changes in the proteome of Trichomonas vaginalis hydrogenosomes. PLoS One 8, e65148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Schwede T., Kopp J., Guex N., Peitsch M. C. (2003) SWISS-MODEL: an automated protein homology-modeling server. Nucleic Acids Res. 31, 3381–3385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Théry C., Amigorena S., Raposo G., Clayton A. (2006) Isolation and characterization of exosomes from cell culture supernatants and biological fluids. Curr. Protoc. Cell Biol. Chapter 3, Unit 3.22 [DOI] [PubMed] [Google Scholar]
- 39. Dornan J., Page A. P., Taylor P., Wu S.-y., Winter A. D., Husi H., Walkinshaw M. D. (1999) Biochemical and structural characterization of a divergent loop cyclophilin from Caenorhabditis elegans. J. Biol. Chem. 274, 34877–34883 [DOI] [PubMed] [Google Scholar]
- 40. Narlikar G. J., Fan H. Y., Kingston R. E. (2002) Cooperation between complexes that regulate chromatin structure and transcription. Cell 108, 475–487 [DOI] [PubMed] [Google Scholar]
- 41. Piotukh K., Gu W., Kofler M., Labudde D., Helms V., Freund C. (2005) Cyclophilin A binds to linear peptide motifs containing a consensus that is present in many human proteins. J. Biol. Chem. 280, 23668–23674 [DOI] [PubMed] [Google Scholar]
- 42. Schönbrunner E. R., Mayer S., Tropschug M., Fischer G., Takahashi N., Schmid F. X. (1991) Catalysis of protein folding by cyclophilins from different species. J. Biol. Chem. 266, 3630–3635 [PubMed] [Google Scholar]
- 43. Kofron J. L., Kuzmic P., Kishore V., Colón-Bonilla E., Rich D. H. (1991) Determination of kinetic constants for peptidyl prolyl cis-trans isomerases by an improved spectrophotometric assay. Biochemistry 30, 6127–6134 [DOI] [PubMed] [Google Scholar]
- 44. Berriman M., Fairlamb A. H. (1998) Detailed characterization of a cyclophilin from the human malaria parasite Plasmodium falciparum. Biochem. J. 334, 437–445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Gourlay L. J., Angelucci F., Baiocco P., Boumis G., Brunori M., Bellelli A., Miele A. E. (2007) The three-dimensional structure of two redox states of cyclophilin A from Schistosoma mansoni. Evidence for redox regulation of peptidyl-prolyl cis-trans isomerase activity. J. Biol. Chem. 282, 24851–24857 [DOI] [PubMed] [Google Scholar]
- 46. Terpe K. (2003) Overview of tag protein fusions: from molecular and biochemical fundamentals to commercial systems. Appl. Microbiol. Biotechnol. 60, 523–533 [DOI] [PubMed] [Google Scholar]
- 47. Vajdos F. F., Yoo S., Houseweart M., Sundquist W. I., Hill C. P. (1997) Crystal structure of cyclophilin A complexed with a binding site peptide from the HIV-1 capsid protein. Protein Sci. 6, 2297–2307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Bell A., Monaghan P., Page A. P. (2006) Peptidyl-prolyl cis-trans isomerases (immunophilins) and their roles in parasite biochemistry, host-parasite interaction and antiparasitic drug action. Int. J. Parasitol. 36, 261–276 [DOI] [PubMed] [Google Scholar]
- 49. Arribas J., Castaño J. G. (1990) Kinetic studies of the differential effect of detergents on the peptidase activities of the multicatalytic proteinase from rat liver. J. Biol. Chem. 265, 13969–13973 [PubMed] [Google Scholar]
- 50. Galat A. (1999) Variations of sequences and amino acid compositions of proteins that sustain their biological functions: an analysis of the cyclophilin family of proteins. Arch. Biochem. Biophys. 371, 149–162 [DOI] [PubMed] [Google Scholar]
- 51. Carrello A., Allan R. K., Morgan S. L., Owen B. A., Mok D., Ward B. K., Minchin R. F., Toft D. O., Ratajczak T. (2004) Interaction of the Hsp90 cochaperone cyclophilin 40 with Hsc70. Cell Stress Chaperones 9, 167–181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Mosavi L. K., Cammett T. J., Desrosiers D. C., Peng Z. Y. (2004) The ankyrin repeat as molecular architecture for protein recognition. Protein Sci. 13, 1435–1448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Li J., Mahajan A., Tsai M. D. (2006) Ankyrin repeat: a unique motif mediating protein-protein interactions. Biochemistry 45, 15168–15178 [DOI] [PubMed] [Google Scholar]