

Abstract
The antiallergy and potential anticancer drug tranilast has been patented for treating Alzheimer’s disease (AD), in which amyloid β-protein (Aβ) plays a key pathogenic role. We used solution NMR to determine that tranilast binds to Aβ40 monomers with ~300 μM affinity. Remarkably, tranilast increases Aβ40 fibrillation more than 20-fold in the thioflavin T assay at a 1:1 molar ratio, as well as significantly reducing the lag time. Tranilast likely promotes fibrillation by shifting Aβ monomer conformations to those capable of seed formation and fibril elongation. Molecular docking results qualitatively agree with NMR chemical shift perturbation, which together indicate that hydrophobic interactions are the major driving force of the Aβ–tranilast interaction. These data suggest that AD may be a potential complication for tranilast usage in elderly patients.
Alzheimer’s disease (AD) accounts for 60–80% of all dementia cases, affecting ~5.3 million people in the United States alone.1 AD is pathologically characterized by neurofibrillary tangles and senile plaques. The major component of senile plaques is aggregated, fibrillar, insoluble Aβ. Aβ is a cleavage product of the amyloid precursor protein (APP). First, β-secretase cleaves APP at what will be the N-terminus of Aβ, followed by γ-secretase cleavage to generate the C-terminus of Aβ. The most common forms of Aβ are 40 and 42 amino acid residues in length and are called Aβ40 and Aβ42, respectively. Aβ42 aggregates much faster than Aβ40 and is more toxic. Though Aβ is considered benign in its monomeric form,2–4 Aβ fibrils,5–7 protofibrils,8 and various oligomers including dimers,9 trimers,10 and 12-mers11,12 are toxic, and their presence in the AD brain correlates with the clinical manifestation of dementia.13–15
Tranilast is a small molecule currently marketed in Korea and Japan as an antiallergy drug. It is being investigated for a wide variety of other uses,16 from reducing the pathological fibrosis associated with myocardial infarctions,17 to reducing human breast cancer cell migration and colony formation.18 The drug is particularly promising as a potential anticancer agent19 due to its antiproliferative effects in prostate,20 breast,21 and pancreatic cancers.22 Usage of tranilast and its derivatives in the treatment of neurodegenerative conditions, including AD, has been patented.23 Oral administration of tranilast inhibited the growth of rat gliomas in vivo, suggesting tranilast can cross the blood–brain barrier.24 Tranilast also had an antiapoptotic effect on neurons and increased neurogenesis, both in a dose-dependent manner.23 Given tranilast’s potential use in the treatment for AD, we decided to explore its possible interaction with Aβ.
We combined NMR, aggregation assays, and docking simulations to study the tranilast–Aβ interaction in a multidisciplinary approach. First, NMR spectroscopy was used to study tranilast’s interaction with monomeric Aβ and to map tranilast’s binding site on the Aβ monomer with residue-specific resolution. Second, ThT fluorescence and dot blots with the antioligomer antibody A11 determined tranilast’s effect on Aβ self-assembly. Fibril morphology was confirmed using atomic force microscopy (AFM). Finally, molecular docking of tranilast to REMD-simulated structures of Aβ was applied to gain further insight into the interaction between tranilast and Aβ.
MATERIALS AND METHODS
NMR Experiments
NMR samples were made from 15N-labeled, HFIP-treated Aβ40 or Aβ42 (rPeptide, Bogart, GA, USA). Peptide films were resuspended in 1 mL of 10 mM NaOH. Aliquots of 125 μL were then added to 20 mM potassium phosphate, pH 7.3, and 10% D2O to prepare initial NMR samples for titration. Tranilast was purchased from Cayman Chemical and solubilized in either DMSO or DMSO-d6 to create a stock solution of 20 mg/mL. Chemical shift perturbation (CSP) was monitored using 15N–1H-HSQC spectra at increasing molar ratios of tranilast to Aβ. A control sample of DMSO without tranilast was also titrated into Aβ samples and CSP due to the addition of DMSO was subtracted from CSP observed during tranilast titrations. CSP was calculated as Δδ = ((10ΔδH)2 + (ΔδN)2)1/2. All NMR experiments were performed on an 800 or 600 MHz spectrometer equipped with a cryogenic probe at 277 K, to minimize the aggregation of Aβ. This protocol of Aβ sample preparation and NMR setup ensures that NMR signals are from Aβ monomers.25,26 CSP values were fitted to the following equation to determine Kd for the Aβ/tranilast interaction:
where Δδobs is observed CSP of Aβ, Δδmax is the CSP of Aβ in the bound state, [P]t is total Aβ peptide concentration, [L]t is total ligand concentration, and Kd is the disassociation constant.
Thioflavin T (ThT) Assay and Dot Blot
For aggregation experiments, samples of HFIP-treated Aβ40 or Aβ42 first were resuspended to 1 mg/mL in DMSO, then diluted in PBS to a final concentration of 20 μM and incubated at 37 °C. ThT assays were performed in triplicate to measure fibril formation using 50 μL of incubated sample and 350 μL of 10 μM ThT in 50 mM glycine, pH 8.0. Samples were excited at 440 nm and emission at 485 nm was measured using a Hitachi F-4500 fluorescence spectrophotometer (Hitachi High-Technologies Co, Tokyo, Japan). Dot blots were performed using the oligomer-specific antibody A11 (Invitrogen, Carlsbad, CA, USA) and the WesternBreeze Chemiluminescent kit (Invitrogen, Carlsbad, CA, USA). Blots were visualized on a ChemiDoc MP system (Bio-Rad, Hercules, CA, USA).
Atomic Force Microscopy (AFM)
Aggregated samples of Aβ were prepared in the same manner as for the ThT assay. For each sample, 20 μL was placed on a mica surface for adsorption for 5 min. Nonadsorbed protein was washed away with abundant water. Three-dimensional measurements at the nanometer scale were collected in air using the tapping mode technique with an MFP-3D atomic force microscope (Asylum Research, Santa Barbara, CA, USA) and standard Si cantilevers (AC240TS, Olympus America Inc., Center Valley, PA, USA). Images were then analyzed with IGOR Pro 6 (WaveMetrics, Inc., Lake Oswego, OR, USA).
Molecular Docking
Tranilast was docked onto previously published centroid structures from all-atom REMD simulations of Aβ40.27 The structure of tranilast was obtained from the Pub-Chem database (CID: 5282230)28 and subsequently minimized in vacuo with MOE29 by using the MMFF94x force field (a modified version of MMFF94s30), stopping at a gradient of 10−3. This ligand structure was blindly docked onto each Aβ centroid MOE’s docking feature29 with the entire centroid as the receptor structure. Placement was done using the triangle matcher algorithm. The top 30 best placements (according to London dG scores) were refined with minimization (stopping at a gradient 10−3) using the OPLS-AA force field with Born solvation. The top scoring pose (after rescoring with the London dG algorithm) was then retained. Residue-indexed surface area of contact between Aβ and tranilast for each of the MOE docks were calculated using LPC software.31
RESULTS
Chemical Shift Perturbation
NMR titrations of tranilast into 15N-labeled Aβ40 or Aβ42 yielded residue-specific chemical shift perturbation (CSP) (Figure 1). The greatest CSP was observed at the C-terminus of both Aβ40 and Aβ42. Relatively large CSP was also observed in regions comprising residues 11–21 and 30–37 in Aβ40, and to a lesser extent, in similar regions of Aβ42. Largest CSP are observed for residues E11, V18, F19, F20, I31, I32, G33 M35, V36, and V40. These residues are largely nonpolar, indicating that hydrophobic interaction may play a key role in tranilast binding to Aβ. CSP in Aβ40 was found to be substantially greater than in Aβ42. 1H and 15N perturbations for tranilast at 0–8 molar ratio equivalents were then fit to determine the Kd values for the tranilast–Aβ interaction. Because of the low affinity of binding between Aβ and tranilast and limitation of tranilast solubility in aqueous solution, titrating Aβ to near complete saturation was not possible. For the Aβ40–tranilast interaction, CSP fittings for residues I31, I32, and V36, where the titration is closer to saturation and there was apparent curvature in the fitted titration curve, were used to calculate an average Kd 0.31 ± 0.13 mM. For Aβ42–tranilast interaction, due to the lower affinity, none of the residues were titrated close to saturation. But for a comparison with Aβ40, the average Kd of the three residues in Aβ42 with lowest apparent Kd (F4, Y10, and G33) was 2.4 ± 0.2 mM, still significantly higher than the measured Kd for Aβ40.
Figure 1.

NMR chemical shift perturbations due to tranilast binding to Aβ40 or Aβ42. (A) A 2D representation of tranilast, which was titrated from 1 to 8 mol equiv of (B) Aβ40 and (C) Aβ42. CSP was calculated as Δδ = ((10ΔδH)2 + (ΔδN)2)1/2, where ΔδH and ΔδN are CSPs of amide proton and nitrogen, respectively. The 10 residues with the largest CSP in each peptide are labeled. Aβ40 CSP due to tranilast was much greater than that of Aβ42, suggesting greater affinity for Aβ40. (D) Examples of fitted Kd curves calculated using CSP. Samples of Aβ monomer were prepared at 60 μM in 20 mM potassium phosphate (pH 7.3) and 10% D2O. All NMR experiments were carried out at a temperature of 277 K.
Aggregation Assays and AFM
To further confirm the Aβ–tranilast interaction and to probe how tranilast affects Aβ self-assembly, we carried out ThT assays to study Aβ β-sheet formation, and dot blots with antibody A11 to study Aβ oligomer formation. AFM was used to confirm aggregate morphology.
Despite the measured millimolar affinity of tranilast to Aβ40 and Aβ42, there was a significant effect of tranilast binding on Aβ fibrillation. ThT assays of Aβ40 and Aβ42 both showed significant increases in fibril formation with the addition of tranilast (Figure 2). There was nearly no increase in ThT fluorescence for Aβ40 in the absence of tranilast, whereas the addition of 1 equiv of tranilast caused more than a 20-fold increase in maximum ThT signal (Figure 2A). This increase was dose-dependent up to a 5:1 molar ratio, the highest ratio tested. There was also a significant decrease in the lag phase of fibril formation between 1:1 and 3:1, as well as between 3:1 and 5:1 molar ratios of tranilast to Aβ40 (Figure 2A). This decrease in the lag phase suggests tranilast not only increased the overall amount of fibrils formed but also promoted formation of fibril seeds. The effect on Aβ42 fibril formation was less pronounced (Figure 2B), with only a ~1.7-fold increase in maximum ThT signal at a molar ratio of 5:1, possibly due to the greater affinity of tranilast for Aβ40 and the faster aggregation propensity of Aβ42 on its own.
Figure 2.

Tranilast promotes Aβ fibrillation. ThT assay measuring the effect of varying molar ratios (0:1, 1:1, 3:1, 5:1) of tranilast to Aβ on fibril formation. (A) Tranilast significantly increased Aβ40 maximum fibrillation in a dose-dependent manner. There was also a significant decrease in the lag phase between 1:1 and 3:1, and 3:1 and 5:1 molar ratios of tranilast to Aβ40. (B) Tranilast significantly increased Aβ42 fibrillation, though the effect was less pronounced. The assay was carried out in triplicate with 20 μM Aβ in PBS + 5% DMSO, incubated at 37 °C without agitation. Error bars represent ±1 standard deviation.
To confirm that the increase in ThT signal was due to increased fibril formation, we observed Aβ samples with AFM. These samples were prepared under the same conditions as the samples for the ThT assay. No fibrilar aggregates where observed in the initial Aβ samples, both with and without 100 μM tranilast. Additionally, fibrils were not observed in the Aβ40 without tranilast at any time point, consistent with the very small change in ThT signal. After incubation at 37 °C for 12 h, fibrils formed in both Aβ42 samples. To a lesser extent, fibrils formed in the Aβ40 sample with tranilast. After 72 h, the time where ThT signal plateaued fibrils were seen to form in both Aβ42 samples and Aβ40 with tranilast, a result consistent with the significant increases in ThT signal observed for these samples.
To determine if tranilast was incorporated into the Aβ fibrils, we used 1H NMR on disaggregated fibril samples formed in the presence of tranilast. Fibril samples were first centrifuged. The supernatant was then decanted, leaving only precipitated fibrils, which were washed with PBS two times and then resuspended in disaggregation buffer (8 M urea, 20 mM potassium phosphate, pH 7.3). 1H NMR spectra were then recorded of the disaggregated samples to determine the presence of tranilast signal. 1H NMR showed no tranilast signal in the disaggregated fibril samples, while there was clear signal for disaggregated Aβ monomers (data not shown). Therefore, tranilast was not incorporated into Aβ fibrils formed in its presence.
A11 dot blots were used to study Aβ oligomer formation in the absence or presence of tranilast (Figure 4). There was no noticeable difference in oligomer formation between the samples containing tranilast and control samples, suggesting that tranilast binding did not affect Aβ40 or Aβ42 A11-active oligomerization.
Figure 4.

Aβ dot blots with oligomer-specific antibody A11 in the absence or presence of tranilast. At a 5:1 molar ratio of tranilast to Aβ, after 24 h of incubation at room temperature without agitation, there is no significant difference in oligomer formation between samples containing tranilast and control samples. Samples consisted of 20 μM Aβ in PBS + 5% DMSO.
Molecular Docking Studies
To further probe the binding modes of tranilast’s interaction with Aβ, we employed molecular docking with centroid structures of Aβ40 derived from replica exchange molecular dynamics (REMD) in explicit water that have been validated by NMR-observed constraints.27 The many structures from these simulations were grouped into clusters comprising structures within a 3 Å RMSD cutoff for Aβ40.27 These clusters were then represented by a centroid structure, which had the lowest RMSD to all members within the cluster. Using MOE, we docked tranilast to two centroid structures representing ~30% of all simulated Aβ40 structures (Figure 5).
Figure 5.

Binding pocket from the top scoring docking simulation produced by MOE between tranilast and the two most representative centroids of Aβ40. Only Aβ atoms (carbon in green, oxygen in red, nitrogen in blue, and hydrogen in white) within 5 Å of tranilast (carbon in cyan, oxygen in red, nitrogen in blue, and hydrogen in white) are depicted. Black dashed lines indicate π-H interactions. Magenta dashed lines indicate hydrogen bonds. This docking pose was the highest scoring dock to tranilast among all of the docks determined for both of the Aβ centroids used.
We found qualitative agreement between the binding sites determined by NMR and in silico docking. Residues Y10, E11, L17, V18, F19, G29, A30, I31, I32, G33, L34, M35, V36, and G37 are in direct contact with tranilast, with atoms within 5 Å of tranilast (Figure 5), as calculated by LPC software.31 These residues all exhibited amide CSP greater than 0.3 ppm in the presence of 6 mol equiv of tranilast (Figure 1). Among the 12 residues with largest CSP, indicated in Figure 1B, 10 could be explained by their proximity to tranilast in Figure 5. Significant CSP of other two residues, F20 and V40, could not be explained by docking results.
Docking revealed key interactions that drive Aβ–tranilast binding (Figure 5). The side chain of I32 interacted with the benzoic acid ring of tranilast, while the side chain of I31 showed a similar interaction with the other aromatic ring of tranilast. There was a π-H interaction between the aromatic ring of F19 and the amide group of tranilast. In addition, there was bifurcated hydrogen bonding between the carboxylic acid group of tranilast to its own amide group and to the amide of A30.
DISCUSSION
Implication for Clinical Use of Tranilast in Patients at Risk for AD
The patent for tranilast’s use in neuro-degenerative diseases is based on its antiapoptotic and pro-neurogenesis effects regardless of the presence of Aβ.23 Given its potential use in the treatment of neurodegenerative diseases and in other diseases commonly affecting elderly patients (e.g., cancer18–21,32–44), the characterization of tranilast’s interaction with Aβ is important. The toxic effect of Aβ fibrils in AD patients is well documented.45 An increase in fibrilization of Aβ40 in particular due to tranilast could lead to elevated cerebral amyloid angiopathy and become a devastating side-effect. We showed that there is a ~20-fold increase in Aβ fibril formation in the presence of an equimolar amount of tranilast, as well as a significant increase in fibril seeding. Thus, in treating elderly patients with tranilast, new signs or the aggravation of dementia need to be carefully monitored. Ideally, a statistical analysis in elderly patient population should be carried out to ascertain whether tranilast indeed affects the onset and progression of AD.
Mechanism of Tranilast Promotion of Aβ Fibrillation
Although tranilast binds Aβ monomers with low affinity, it significantly increases Aβ40 fibrillation. These ~millimolar binding affinities of tranilast to both Aβ40 and 42 monomers are in a range that is traditionally considered too weak for effective drug interaction. However, taking into account that IDPs, such as Aβ, sample a large number of conformations, drugs like tranilast may only bind certain populations of conformations with relatively high affinity. The observed binding affinities reffect binding to all monomer conformations, including those that do not bind, and thus may be deceptively low. These observed low binding affinities also do not necessarily correlate with the extent of the effect of binding on aggregation, as is the case with tranilast and Aβ40. Alternatively, tranilast may bind to most Aβ monomer conformations with low affinity, although this is unlikely given its significant effects on fibrillation. Because tranilast is not present in the fibrils and does not affect A11-reactive oligomer formation, the effect on fibrillation likely stems from binding of tranilast to Aβ monomer conformations that favor fibril formation, shifting the thermodynamic equilibrium of sampled conformations.
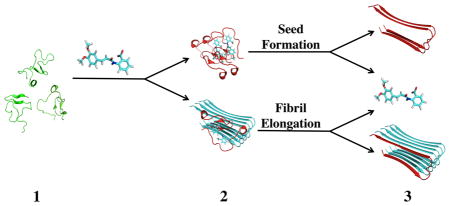
Presumably, tranilast binds to monomer conformations capable of docking onto growing fibrils, but not to actual fibril conformations, the two of which are thought to be different in the dock-and-lock scheme of Aβ fibril growth.46–49 We propose the mechanisms depicted in Figure 6 for tranilast promotion of Aβ fibril formation. Aβ monomers exist in a dynamic equilibrium sampling multiple conformations (step 1). Tranilast binds to Aβ monomers and stabilizes conformations compatible of binding to pre-existing fibrilar aggregates or creating new fibril seeds. These monomers then dock onto growing fibrils or to each other to create new fibril seeds (step 2). This docking leads to a conformational change that causes the monomer to lock onto the fibril or new seed, releasing tranilast for binding to another Aβ monomer (step 3). In this model, tranilast acts by promoting fibril elongation or seed formation with one tranilast molecule able to promote the incorporation of many Aβ monomers into the aggregates. This model can explain how despite having low binding affinity, tranilast has a large effect on Aβ fibril seeding and elongation.
Figure 6.

Proposed mechanism for tranilast binding to Aβ monomers, explaining the increase in fibril formation. (1) Aβ monomers (green) sample many different conformations in solution. (2) Tranilast binds to Aβ monomers and stabilizes conformations (red) capable of docking onto fibrils (cyan) or creating fibril seeds, promoting fibril aggregation. (3) Upon locking onto the fibril or fibril seed, the Aβ monomer adopts a new conformation, releasing tranilast and extending the aggregate.27,49
Supplementary Material
Figure 3.

AFM images of Aβ40 and 42 samples incubated with either 0:1 (−Tranilast) or 5:1 (+Tranilast) molar ratios of tranilast to Aβ. No fibrils were observed at 0 h for any sample. After 12 h, fibrils were observed in both Aβ42 samples, and only a sparse amount of fibrils were observed in the Aβ40 sample with tranilast. After 72 h of incubation at 37 °C, more fibrils were observed in the Aβ40 sample containing tranilast and in both Aβ42 samples. No fibrils were observed in the Aβ40 without tranilast at any point. AFM samples were prepared from 20 μM Aβ in PBS + 5% DMSO, incubated at 310 K without agitation, in either the presence or absence of 100 μM tranilast.
Acknowledgments
Funding
This work was supported in part by University of California-Los Angeles Jim Easton Consortium for Alzheimer’s Drug Discovery and Biomarker Development (G.B.); RJG Foundation Grant 20095024 (G.B.); and a Cure Alzheimer’s Fund Grant (G.B.).
Footnotes
The authors declare no competing financial interest.
The solubility of tranilast under the same buffer conditions used for the CSP titrations was confirmed by measuring a proportional increase in 1H NMR to increasing tranilast concentration. Overlays of spectra from the CSP titrations are also presented, along with the fit curves for the 10 residues in each peptide that were used to determine Kd values. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.2010 Alzheimer’s Disease Facts and Figures, Executive Summary. Alzheimer’s Association; Chicago: 2010. [Google Scholar]
- 2.Chong YH, Shin YJ, Lee EO, Kayed R, Glabe CG, Tenner AJ. ERK1/2 activation mediates Abeta oligomer-induced neurotoxicity via caspase-3 activation and tau cleavage in rat organotypic hippocampal slice cultures. J Biol Chem. 2006;281:20315–20325. doi: 10.1074/jbc.M601016200. [DOI] [PubMed] [Google Scholar]
- 3.Masters CL, Cappai R, Barnham KJ, Villemagne VL. Molecular mechanisms for Alzheimer’s disease: implications for neuroimaging and therapeutics. J Neurochem. 2006;97:1700–1725. doi: 10.1111/j.1471-4159.2006.03989.x. [DOI] [PubMed] [Google Scholar]
- 4.Dahlgren KN, Manelli AM, Stine WB, Jr, Baker LK, Krafft GA, LaDu MJ. Oligomeric and fibrillar species of amyloid-beta peptides differentially affect neuronal viability. J Biol Chem. 2002;277:32046–32053. doi: 10.1074/jbc.M201750200. [DOI] [PubMed] [Google Scholar]
- 5.Pike CJ, Burdick D, Walencewicz AJ, Glabe CG, Cotman CW. Neurodegeneration induced by beta-amyloid peptides in vitro: the role of peptide assembly state. J Neuroscience: Official J Soc Neurosci. 1993;13:1676–1687. doi: 10.1523/JNEUROSCI.13-04-01676.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Luhrs T, Ritter C, Adrian M, Riek-Loher D, Bohrmann B, Dobeli H, Schubert D, Riek R. 3D structure of Alzheimer’s amyloid-beta(1–42) fibrils. Proc Natl Acad Sci U S A. 2005;102:17342–17347. doi: 10.1073/pnas.0506723102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lorenzo A, Yankner BA. Beta-amyloid neurotoxicity requires fibril formation and is inhibited by congo red. Proc Natl Acad Sci U S A. 1994;91:12243–12247. doi: 10.1073/pnas.91.25.12243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Harper JD, Wong SS, Lieber CM, Lansbury PT. Observation of metastable Abeta amyloid protofibrils by atomic force microscopy. Chem Biol. 1997;4:119–125. doi: 10.1016/s1074-5521(97)90255-6. [DOI] [PubMed] [Google Scholar]
- 9.Roher AE, Chaney MO, Kuo YM, Webster SD, Stine WB, Haverkamp LJ, Woods AS, Cotter RJ, Tuohy JM, Krafft GA, Bonnell BS, Emmerling MR. Morphology and toxicity of Abeta-(1–42) dimer derived from neuritic and vascular amyloid deposits of Alzheimer’s disease. J Biol Chem. 1996;271:20631–20635. doi: 10.1074/jbc.271.34.20631. [DOI] [PubMed] [Google Scholar]
- 10.Townsend M, Shankar GM, Mehta T, Walsh DM, Selkoe DJ. Effects of secreted oligomers of amyloid beta-protein on hippocampal synaptic plasticity: a potent role for trimers. J Physiol. 2006;572:477–492. doi: 10.1113/jphysiol.2005.103754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lesne S, Koh MT, Kotilinek L, Kayed R, Glabe CG, Yang A, Gallagher M, Ashe KH. A specific amyloid-beta protein assembly in the brain impairs memory. Nature. 2006;440:352–357. doi: 10.1038/nature04533. [DOI] [PubMed] [Google Scholar]
- 12.Barghorn S, Biernat J, Mandelkow E. Purification of recombinant tau protein and preparation of Alzheimer-paired helical filaments in vitro. Methods Mol Biol. 2005;299:35–51. doi: 10.1385/1-59259-874-9:035. [DOI] [PubMed] [Google Scholar]
- 13.Cummings BJ, Cotman CW. Image analysis of beta-amyloid load in Alzheimer’s disease and relation to dementia severity. Lancet. 1995;346:1524–1528. doi: 10.1016/s0140-6736(95)92053-6. [DOI] [PubMed] [Google Scholar]
- 14.Kuo YM, Emmerling MR, Vigo-Pelfrey C, Kasunic TC, Kirkpatrick JB, Murdoch GH, Ball MJ, Roher AE. Water-soluble Abeta (N-40, N-42) oligomers in normal and Alzheimer disease brains. J Biol Chem. 1996;271:4077–4081. doi: 10.1074/jbc.271.8.4077. [DOI] [PubMed] [Google Scholar]
- 15.McLean CA, Cherny RA, Fraser FW, Fuller SJ, Smith MJ, Beyreuther K, Bush AI, Masters CL. Soluble pool of Abeta amyloid as a determinant of severity of neuro-degeneration in Alzheimer’s disease. Ann Neurol. 1999;46:860–866. doi: 10.1002/1531-8249(199912)46:6<860::aid-ana8>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- 16.Kato T, Tagami H. Successful treatment of cheilitis granulomatosa with tranilast. J Dermatol. 1986;13:402–403. doi: 10.1111/j.1346-8138.1986.tb02965.x. [DOI] [PubMed] [Google Scholar]
- 17.See F, Watanabe M, Kompa AR, Wang BH, Boyle AJ, Kelly DJ, Gilbert RE, Krum H. Early and Delayed Tranilast Treatment Reduces Pathological Fibrosis Following Myocardial Infarction. Heart Lung Circ. 2013;22:122–132. doi: 10.1016/j.hlc.2012.08.054. [DOI] [PubMed] [Google Scholar]
- 18.Subramaniam V, Ace O, Prud’homme GJ, Jothy S. Tranilast treatment decreases cell growth, migration and inhibits colony formation of human breast cancer cells. Exp Mol Pathol. 2011;90:116–122. doi: 10.1016/j.yexmp.2010.10.012. [DOI] [PubMed] [Google Scholar]
- 19.Rogosnitzky M, Danks R, Kardash E. Therapeutic potential of tranilast, an anti-allergy drug, in proliferative disorders. Anticancer Res. 2012;32:2471–2478. [PubMed] [Google Scholar]
- 20.Izumi K, Mizokami A, Li YQ, Narimoto K, Sugimoto K, Kadono Y, Kitagawa Y, Konaka H, Koh E, Keller ET, Namiki M. Tranilast inhibits hormone refractory prostate cancer cell proliferation and suppresses transforming growth factor beta1-associated osteoblastic changes. Prostate. 2009;69:1222–1234. doi: 10.1002/pros.20975. [DOI] [PubMed] [Google Scholar]
- 21.Chakrabarti R, Subramaniam V, Abdalla S, Jothy S, Prud’homme GJ. Tranilast inhibits the growth and metastasis of mammary carcinoma. Anticancer Drugs. 2009;20:334–345. doi: 10.1097/CAD.0b013e328327994e. [DOI] [PubMed] [Google Scholar]
- 22.Hiroi M, Onda M, Uchida E, Aimoto T. Anti-tumor effect of N-[3,4-dimethoxycinnamoyl]-anthranilic acid (trani-last) on experimental pancreatic cancer. J Nippon Med Sch. 2002;69:224–234. doi: 10.1272/jnms.69.224. [DOI] [PubMed] [Google Scholar]
- 23.Schneider A, Moraru A, Krüger C, Laage R, Pitzer C. Use Of Tranilast And Derivatives Thereof For The Therapy Of Neurological Conditions. 20110112187. United States Patent Application. 2011 2008.
- 24.Platten M, Wild-Bode C, Wick W, Leitlein J, Dichgans J, Weller M. N-[3,4-Dimethoxycinnamoyl]-anthranilic acid (tranilast) inhibits transforming growth factor-beta relesase and reduces migration and invasiveness of human malignant glioma cells. Int J Cancer. 2001;93:53–61. doi: 10.1002/ijc.1289. [DOI] [PubMed] [Google Scholar]
- 25.Bitan G, Teplow DB. Preparation of aggregate-free, low molecular weight amyloid-beta for assembly and toxicity assays. Methods Mol Biol. 2005;299:3–9. doi: 10.1385/1-59259-874-9:003. [DOI] [PubMed] [Google Scholar]
- 26.Wang C. Solution NMR studies of Abeta monomer dynamics. Protein Peptide Lett. 2011;18:354–361. doi: 10.2174/092986611794653969. [DOI] [PubMed] [Google Scholar]
- 27.Sgourakis NG, Yan Y, McCallum SA, Wang C, Garcia AE. The Alzheimer’s peptides Abeta40 and 42 adopt distinct conformations in water: a combined MD/NMR study. J Mol Biol. 2007;368:1448–1457. doi: 10.1016/j.jmb.2007.02.093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang Y, Xiao J, Suzek TO, Zhang J, Wang J, Bryant SH. PubChem: a public information system for analyzing bioactivities of small molecules. Nucleic Acids Res. 2009;37:W623–633. doi: 10.1093/nar/gkp456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Molecular Operating Environment (MOE) Chemical Computing Group, I; Montreal, Quebec City, Canada: 2012. p. 10. [Google Scholar]
- 30.Halgren T. MMFF VI. MMFF94s option for energy minimization studies. J Comput Chem. 1999;20:720–729. doi: 10.1002/(SICI)1096-987X(199905)20:7<720::AID-JCC7>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 31.Sobolev V, Sorokine A, Prilusky J, Abola EE, Edelman M. Automated analysis of interatomic contacts in proteins. Bioinformatics. 1999;15:327–332. doi: 10.1093/bioinformatics/15.4.327. [DOI] [PubMed] [Google Scholar]
- 32.Uno K, Iijima K, Koike T, Abe Y, Asano N, Ara N, Shimosegawa T. A pilot study of scheduled endoscopic balloon dilation with oral agent tranilast to improve the efficacy of stricture dilation after endoscopic submucosal dissection of the esophagus. J Clin Gastroenterol. 2012;46:e76–82. doi: 10.1097/MCG.0b013e31824fff76. [DOI] [PubMed] [Google Scholar]
- 33.Prud’homme GJ. Cancer stem cells and novel targets for antitumor strategies. Curr Pharm Des. 2012;18:2838–2849. doi: 10.2174/138161212800626120. [DOI] [PubMed] [Google Scholar]
- 34.Kuba-Miyara M, Agarie K, Sakima R, Imamura S, Tsuha K, Yasumoto T, Gima S, Matsuzaki G, Ikehara T. Inhibitory effects of an ellagic acid glucoside, okicamelliaside, on antigen-mediated degranulation in rat basophilic leukemia RBL-2H3 cells and passive cutaneous anaphylaxis reaction in mice. Int Immunopharmacol. 2012;12:675–681. doi: 10.1016/j.intimp.2012.01.013. [DOI] [PubMed] [Google Scholar]
- 35.Jin UH, Lee SO, Safe S. Aryl hydrocarbon receptor (AHR)-active pharmaceuticals are selective AHR modulators in MDA-MB-468 and BT474 breast cancer cells. J Pharmacol Exp Ther. 2012;343:333–341. doi: 10.1124/jpet.112.195339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Glinka Y, Mohammed N, Subramaniam V, Jothy S, Prud’homme GJ. Neuropilin-1 is expressed by breast cancer stem-like cells and is linked to NF-kappaB activation and tumor sphere formation. Biochem Biophys Res Commun. 2012;425:775–780. doi: 10.1016/j.bbrc.2012.07.151. [DOI] [PubMed] [Google Scholar]
- 37.Subramaniam V, Chakrabarti R, Prud’homme GJ, Jothy S. Tranilast inhibits cell proliferation and migration and promotes apoptosis in murine breast cancer. Anticancer Drugs. 2010;21:351–361. doi: 10.1097/CAD.0b013e328334992c. [DOI] [PubMed] [Google Scholar]
- 38.Sato S, Takahashi S, Asamoto M, Naiki T, Naiki-Ito A, Asai K, Shirai T. Tranilast suppresses prostate cancer growth and osteoclast differentiation in vivo and in vitro. Prostate. 2010;70:229–238. doi: 10.1002/pros.21056. [DOI] [PubMed] [Google Scholar]
- 39.Prud’homme GJ, Glinka Y, Toulina A, Ace O, Subramaniam V, Jothy S. Breast cancer stem-like cells are inhibited by a non-toxic aryl hydrocarbon receptor agonist. PloS One. 2010;5:e13831. doi: 10.1371/journal.pone.0013831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Izumi K, Mizokami A, Shima T, Narimoto K, Sugimoto K, Kobori Y, Maeda Y, Konaka H, Koh E, Namiki M. Preliminary results of tranilast treatment for patients with advanced castration-resistant prostate cancer. Anticancer Res. 2010;30:3077–3081. [PubMed] [Google Scholar]
- 41.Goto T, Nemoto T, Ogura K, Hozumi T, Funata N. Successful treatment of desmoid tumor of the chest wall with tranilast: a case report. J Med Case Rep. 2010;4:384. doi: 10.1186/1752-1947-4-384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nakajima K, Okita Y, Matsuda S. Sensitivity of scirrhous gastric cancer to 5-fluorouracil and the role of cancer cell-stromal fibroblast interaction. Oncol Rep. 2004;12:85–90. [PubMed] [Google Scholar]
- 43.Yashiro M, Murahashi K, Matsuoka T, Nakazawa K, Tanaka H, Osaka H, Koyama T, Ohira M, Chung KH. Tranilast (N-3,4-dimethoxycinamoyl anthranilic acid): a novel inhibitor of invasion-stimulating interaction between gastric cancer cells and orthotopic fibroblasts. Anticancer Res. 2003;23:3899–3904. [PubMed] [Google Scholar]
- 44.Noguchi N, Kawashiri S, Tanaka A, Kato K, Nakaya H. Effects of fibroblast growth inhibitor on proliferation and metastasis of oral squamous cell carcinoma. Oral Oncol. 2003;39:240–247. doi: 10.1016/s1368-8375(02)00092-1. [DOI] [PubMed] [Google Scholar]
- 45.Selkoe DJ. Amyloid beta-protein precursor: new clues to the genesis of Alzheimer’s disease. Curr Opin Neurobiol. 1994;4:708–716. doi: 10.1016/0959-4388(94)90014-0. [DOI] [PubMed] [Google Scholar]
- 46.Krishnamoorthy J, Brender JR, Vivekanandan S, Jahr N, Ramamoorthy A. Side-chain dynamics reveals transient association of Abeta(1–40) monomers with amyloid fibers. J Phys Chem B. 2012;116:13618–13623. doi: 10.1021/jp305279w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Straub JE, Thirumalai D. Principles governing oligomer formation in amyloidogenic peptides. Curr Opin Struct Biol. 2010;20:187–195. doi: 10.1016/j.sbi.2009.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Massi F, Straub JE. Energy landscape theory for Alzheimer’s amyloid beta-peptide fibril elongation. Proteins. 2001;42:217–229. doi: 10.1002/1097-0134(20010201)42:2<217::aid-prot90>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- 49.Esler WP, Stimson ER, Jennings JM, Vinters HV, Ghilardi JR, Lee JP, Mantyh PW, Maggio JE. Alzheimer’s disease amyloid propagation by a template-dependent dock-lock mechanism. Biochemistry. 2000;39:6288–6295. doi: 10.1021/bi992933h. [DOI] [PubMed] [Google Scholar]
- 50.Tolcher AW, Messersmith WA, Mikulski SM, Papadopoulos KP, Kwak EL, Gibbon DG, Patnaik A, Falchook GS, Dasari A, Shapiro GI, Boylan JF, Xu ZX, Wang K, Koehler A, Song J, Middleton SA, Deutsch J, Demario M, Kurzrock R, Wheler JJ. Phase I study of RO4929097, a gamma secretase inhibitor of Notch signaling, in patients with refractory metastatic or locally advanced solid tumors. J Clin Oncol. 2012;30:2348–2353. doi: 10.1200/JCO.2011.36.8282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tong G, Wang JS, Sverdlov O, Huang SP, Slemmon R, Croop R, Castaneda L, Gu H, Wong O, Li H, Berman RM, Smith C, Albright CF, Dockens R. A contrast in safety, pharmacokinetics and pharmacodynamics across age groups after a single 50 mg oral dose of the gamma-secretase inhibitor avagacestat. Br J Clin Pharmacol. 2013;75:136–145. doi: 10.1111/j.1365-2125.2012.04339.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Xia W, Wong ST, Hanlon E, Morin P. gamma-secretase modulator in Alzheimer’s disease: shifting the end. J Alzheimer’s Dis: JAD. 2012;31:685–696. doi: 10.3233/JAD-2012-120751. [DOI] [PubMed] [Google Scholar]
- 53.Chang L, Bakhos L, Wang Z, Venton DL, Klein WL. Femtomole immunodetection of synthetic and endogenous amyloid-beta oligomers and its application to Alzheimer’s disease drug candidate screening. J Mol Neurosci. 2003;20:305–313. doi: 10.1385/JMN:20:3:305. [DOI] [PubMed] [Google Scholar]
- 54.Dovey HF, John V, Anderson JP, Chen LZ, de Saint Andrieu P, Fang LY, Freedman SB, Folmer B, Goldbach E, Holsztynska EJ, Hu KL, Johnson-Wood KL, Kennedy SL, Kholodenko D, Knops JE, Latimer LH, Lee M, Liao Z, Lieberburg IM, Motter RN, Mutter LC, Nietz J, Quinn KP, Sacchi KL, Seubert PA, Shopp GM, Thorsett ED, Tung JS, Wu J, Yang S, Yin CT, Schenk DB, May PC, Altstiel LD, Bender MH, Boggs LN, Britton TC, Clemens JC, Czilli DL, Dieckman-McGinty DK, Droste JJ, Fuson KS, Gitter BD, Hyslop PA, Johnstone EM, Li WY, Little SP, Mabry TE, Miller FD, Audia JE. Functional gamma-secretase inhibitors reduce beta-amyloid peptide levels in brain. J Neurochem. 2001;76:173–181. doi: 10.1046/j.1471-4159.2001.00012.x. [DOI] [PubMed] [Google Scholar]
- 55.Baures PW, Peterson SA, Kelly JW. Discovering transthyretin amyloid fibril inhibitors by limited screening. Bioorg Med Chem. 1998;6:1389–1401. doi: 10.1016/s0968-0896(98)00130-8. [DOI] [PubMed] [Google Scholar]
- 56.Isvoran A, Badel A, Craescu CT, Miron S, Miteva MA. Exploring NMR ensembles of calcium binding proteins: perspectives to design inhibitors of protein-protein interactions. BMC Struct Biol. 2011;11:24. doi: 10.1186/1472-6807-11-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kawatkar S, Moustakas D, Miller M, Joseph-McCarthy D. Virtual fragment screening: exploration of MM-PBSA rescoring. J Comput-Aided Mol Des. 2012;26:921–934. doi: 10.1007/s10822-012-9590-x. [DOI] [PubMed] [Google Scholar]
- 58.Stark JL, Powers R. Application of NMR and molecular docking in structure-based drug discovery. Top Curr Chem. 2012;326:1–34. doi: 10.1007/128_2011_213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ai R, Chang CE. Ligand-specific homology modeling of human cannabinoid (CB1) receptor. J Mol Graphics Modell. 2012;38:155–164. doi: 10.1016/j.jmgm.2012.05.002. [DOI] [PubMed] [Google Scholar]
- 60.Rungsardthong K, Mares-Samano S, Penny J. Virtual screening of ABCC1 transporter nucleotidebinding domains as a therapeutic target in multidrug resistant cancer. Bioinformation. 2012;8:907–911. doi: 10.6026/97320630008907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Skariyachan S, Mahajanakatti AB, Sharma N, Karanth S, Rao S, Rajeswari N. Structure based virtual screening of novel inhibitors against multidrug resistant superbugs. Bioinformation. 2012;8:420–425. doi: 10.6026/97320630008420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Strambi A, Mori M, Rossi M, Colecchia D, Manetti F, Carlomagno F, Botta M, Chiariello M. Structure Prediction and Validation of the ERK8 Kinase Domain. PloS One. 2013;8:e52011. doi: 10.1371/journal.pone.0052011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wadood A, Ulhaq Z. In silico identification of novel inhibitors against Plasmodium falciparum dihydroorate dehydrogenase. J Mol Graphics Modell. 2013;40:40–47. doi: 10.1016/j.jmgm.2012.11.010. [DOI] [PubMed] [Google Scholar]
- 64.Park IH, Li C. Dynamic ligand-induced-fit simulation via enhanced conformational samplings and ensemble dockings: a survivin example. J Phys Chem B. 2010;114:5144–5153. doi: 10.1021/jp911085d. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.