

Abstract
Despite a conserved role for histones as general DNA packaging agents, it is now clear that another key function of these proteins is to confer variations in chromatin structure to ensure dynamic patterns of transcriptional regulation in eukaryotes. The incorporation of histone variants is particularly important to this process. Recent knockdown and knockout studies in various cellular systems, as well as direct mutational evidence from human cancers, now suggest a crucial role for histone variant regulation in processes as diverse as differentiation and proliferation, meiosis and nuclear reprogramming. In this Review, we provide an overview of histone variants in the context of their unique functions during mammalian germ cell and embryonic development, and examine the consequences of aberrant histone variant regulation in human disease.
Since their discovery, the existence of histone variants has suggested an alternative mechanism for introducing small variations into the eukaryotic epigenome (FIG. 1), which is now known to govern fundamental aspects of chromatin structural organization, nucleosomal dynamics and transcription (see REFS 1–3 for recent reviews on histone variants). An emerging finding is that mutations in specific histone variants and their associated chaperone machinery contribute to human disease, which suggests an essential function for regulation of histone variants during crucial periods of cellular development (TABLES 1,2). For decades, nucleosomal histones were considered to be highly stable proteins that have predicted half-lives of several months to years in non-dividing cells. Recent analyses of histone H3 and H4 dynamics in living cells have challenged this notion by showing that newly synthesized histone variants are rapidly incorporated at promoters, enhancers, regulatory elements and coding regions of highly transcribed genes, in most cases, independent of DNA replication. With recent progress in identifying genome-wide enrichment profiles and functions for distinct histone variants, these proteins are now gaining increased attention in chromatin biology, partly because of the disease links that are discussed here.
Figure 1. Human core and linker histone variants.

a | Variants of the core histones H2A (yellow), H2B (red), H3 (blue) and H4 (green) are shown. Unstructured amino- terminal tails are shown as black lines. Specific amino acid residues are depicted at key differences among variants of a common histone protein family (for example, H2A.X, H2BE and H3.3). Different shades of colour are used to indicate protein sequences that are highly divergent between canonical histones (which are listed first) and their variants (for example, H2A.Bbd, histone H2B type W-T (H2BFWT) and histone H3-like centromeric protein A (CENP-A)). b | Human linker histone variants are shown. Owing to high sequence divergence between variants, specific amino acid differences are not shown. Unstructured amino- and carboxy-terminal domains are shown in light grey. Globular domains are shown in brown. Canonical serine/threonine PXK phosphorylation sites that are targeted by cyclin-dependent kinases are indicated in magenta. Alternative names of variants are given in parentheses. aa, amino acid; mH2A1, macroH2A1. Part a is adapted, with permission, from REF. 74 © (2006) Canadian Science Publishing and has been updated to reflect current data on histone variant structures.
Table 1.
Core histone variants in vertebrate development
| Histone | Number of gene copies | Cell-cycle expression | Location | Function | Knockout phenotype | Refs |
|---|---|---|---|---|---|---|
| H2A | 15 | RD | Throughout the genome | Core histone | ND | 153,154 |
| H2A.X | 1 | RI | Throughout the genome | DNA repair (mediated by the phosphorylated form γH2A.X) and genome integrity | Male infertility (that is, defects in sperm meiosis) in mice | 27 |
| H2A.Z | 2 | RI | Throughout the genome | Gene activation, gene silencing and chromosome segregation | Embryonic lethality in H2A.Z.1-knockout mice at E4.5–E7.5 | 48,50,52 |
| MacroH2A | 2 | Possibly RI | Inactive X chromosome | Gene silencing | Brain malformation in zebrafish | 57–59 |
| H2A.Bbd | 3 | RI | Active X chromosome and euchromatin | Active transcription | ND | 18,22 |
| H2B | 17 | RD | Throughout the genome | Core histone | ND | 153,154 |
| TSH2B | 1 | Possibly RI | Throughout the sperm genome and in telomeres of somatic cells | Chromatin-to-nucleoprotamine transition | ND | 19,21 |
| H2BFWT | 1 | Possibly RI | Primate telomeres in sperm | ND | ND, as there is no gene product in mice | 153,154 |
| H2BE | 1 | RI | Throughout the genome of olfactory neurons | ND | Overexpression of olfactory receptor in mice | 79 |
| H3.1 | 10 | RD | Throughout the genome | Core histone | ND | 87,153,154 |
| H3.2 | 3 | RD | Throughout the genome | Core histone | ND | 87,153,154 |
| H3.3 | 2 | RD and RI | Throughout the genome | Gene activation, silencing and chromosome segregation | ND; adult infertility in H3.3A-gene-trap and H3.3B-knockout mice | 42–44 |
| H3.4 | 1 | RI | Sperm genome and nucleolus of somatic cells | ND | ND | 20 |
| H3.5 | 1 | Possibly RI | Euchromatin in hominid testis | ND | ND, as there is no gene product in mice | 15 |
| H3.X and H3.Y | 1 of each | Possibly RI | Euchromatin in primates | ND | ND, as there is no gene product in mice | 53 |
| CENP-A | 1 | RI | Centromeres | Chromosome segregation | Embryonic lethality at E3.5–E8.5 in mice | 113 |
| H4 | 14 | RD | Throughout the genome | Core histone | ND | 153,154 |
CENP-A, histone H3-like centromeric protein A; E, embryonic day; H2BFWT, histone H2B type W-T; ND, not determined; RD, replication dependent; RI, replication independent.
Table 2.
Core histone variants in human disease
| Histone | Number of gene copies | Cell-cycle expression | Mutation and expression pattern | Tumorigenic consequences | Refs |
|---|---|---|---|---|---|
| H2A.X | 1 | RI | Reduced expression | Increased cancer progression in p53-knockout mice | 60,61 |
| H2A.Z | 2 | RI | Over-expression; oncogene | Numerous cancers | 121–123 |
| MacroH2A | 2 | Possibly RI | Reduced expression; tumour suppressor | Melanoma and other cancers | 141 |
| H3.1 | 10 | RD | K27M in H3.1B | Adult and paediatric gliomas, including GBMs and DIPGs, respectively | 83 |
| H3.3 | 2 | RD and RI | K27M, G34R and G34V in H3.3A | Adult and paediatric gliomas, including GBMs and DIPGs, respectively | 82,83 |
| K36M in H3.3B | Chondroblastoma | 101 | |||
| G34W and G34L in H3.3A | Giant cell tumours in bone | ||||
| CENP-A | 1 | RI | Over-expression; oncogene | Numerous cancers | 115–119 |
CENP-A, histone H3-like centromeric protein A; DIPG, diffuse intrinsic pontine glioma; GBM, adult glioblastoma; ND, not determined; RD, replication dependent; RI, replication independent.
In this Review, we provide a brief overview of histone variants in the context of their evolutionary history, followed by more detailed discussions of our current understanding of the roles of these proteins during germ cell and embryonic development in mammals. We further discuss potentially distinct roles for histone variants in post-replicative cells. Finally, we end with a general summary of data that indicate important contributions of histone variants to human disease, with an emphasis on recent evidence that histones themselves are mutated in specific forms of cancer, and provide thoughts on the future of this ever-expanding field.
An overview of histone variants
Four types of histones make up the nucleosome particle and are referred to as core histones. Dimerized pairs of core histone proteins (that is, H2A–H2B and H3–H4) seem to have evolved together to allow exquisite specificity in their partnerships. Only one H4 isoform (that is, canonical H4) has so far been identified, and H3 variants are generally less diverse than those arising from the H2A and H2B families. This is probably due to the role of H3 and H4 in the formation of core histone tetramers, which ultimately dictate nucleosomal assembly4. H2A variants are generally the most diverse family of the core histone proteins and are accompanied by substantial variation in their amino- and carboxy-terminal tail regions. H2A variants are also divergent both throughout regions that are typically engaged in intranucleosomal histone–histone contacts and at surface residues, which can markedly affect chromatin structure and nucleosomal stability. More minor sequence differences are observed in the H3 and H2B variants, which typically involve only a few amino acid substitutions. However, many of these substitutions occur in the globular core of these proteins, thereby influencing histone–histone interactions in a similar way to that of the H2A variants. The fifth histone type binds to DNA that is positioned between nucleosomes and is termed the linker histone H1. It is the most diverse of all histones and is represented by at least 11 isoforms in mice and 10 isoforms in humans, including 7 somatic variants and 3 germline-specific variants. Recent studies indicate that histone variants confer unique properties on chromatin structure by promoting differential interactions with various associated complex proteins such as chaperones (for example, association between H3.3 and death domain-associated protein 6 (DAXX)5) (FIG. 2), as well as through alterations in the activity of chromatin binders such as ATP-dependent chromatin remodelling complexes (for example, association between H2A.Z and SWR complex (SWR-C; also known as SWR1 (REFS 6,7)), which act in conjunction with histone variants to mediate nucleosomal dynamics and gene expression.
Figure 2. Structural characterization of H3.3 in complex with DAXX: implications for chaperone- specific histone variant deposition.

a | Amino acid sequence substitutions of the histone variant H3.3 relative to the canonical replication-dependent histones H3.1 and H3.2 are shown. H3.3 differs from H3.1 and H3.2 at five and four amino acid positions, respectively. b | The crystal structures of the histone-binding domain of death domain-associated protein 6 (DAXX) in complex with a histone H3.3–H4 dimer have recently been solved5, which elucidates the principles that underlie the specificity of DAXX-mediated H3.3 recognition. Further functional studies indicated a crucial role for G90 in H3.3 (which forms part of the substituted H3.3 core AAIG motif (FIG. 1)) in DAXX-mediated H3.3 recognition5. Data from REF. 5.
Crucial roles in mammalian germ cells
Mammalian histone variants are classified into two major categories: those enriched in somatic cells, which can be further divided into replication-dependent and replication-independent histones; and another group of testis-specific histones that are generally enriched in developing spermatocytes8. During spermatogenesis (that is, when spermatogonia are initially converted into spermatocytes and subsequently into spermatids through meiosis), canonical histones undergo rapid exchange with their variant counterparts, which suggests a role for histone variants during important periods of sperm development. Once sperm cells have reached a mature haploid state, protamines (that is, small, arginine- rich nuclear proteins) replace most, but not all, of the histones in male germ cells. However, before protamine replacement, most somatic histones will probably have been replaced with testis-specific variants, including linker histones H1t9 and H1t2 (also known as H1FNT)10, and core histones H2A.Bbd11, H2AL1, H2AL2 (REF. 12), TSH2B13, H3.4 (also known as H3.1t and H3t)14 and H3.5 (also known as H3.3C)15.
Incorporation of testis-specific histone variants generally leads to decreased stability of DNA–protein interactions, which allows global transitions in chromosomal architecture and ultimately facilitates protamine replacement during sperm maturation. For example, incorporation of H2ABbd, H2A.Lap1 (that is, the mouse H2A.Bbd-like protein) and H2AL2, all of which are enriched in testis, is associated with reduced nucleosomal stability16–18, as all three variants express truncated carboxyl termini and lack sufficient acidic surfaces that are required for tethering H2A–H2B dimers to the core H3–H4 heterotetramer. Similarly, histone octamers that contain TSH2B and H3.4 exhibit low stabilities19 and have reduced H2A–H2B nucleosomal associations20. Therefore, enhanced dynamicity of nucleosomes that contain testis-specific histone variants may be required for appropriate chromatin reorganization during spermatogenesis. With the exception of recent evidence21 that shows a specific role for the H2B variant TSH2B in chromatin-to-nucleoprotamine transitions, functional roles for most variants during mammalian spermatogenesis beyond those of their biophysical properties remain unknown. However, an increasing amount of evidence indicates that active incorporation of these variants might directly affect transcription. For example, depletion of H2A.Bbd results in widespread reduction in basal gene expression and disruptions to alternative splicing22. Furthermore, human cells in which H2A.Bbd has been ectopically expressed show an enrichment for this variant throughout active regions of the genome22,23, which collectively indicates a putative role for this testis-specific variant in the regulation of gene expression.
In addition to testis-specific variants, somatic histone variants have been shown to be involved in meiosis when the X and Y chromosomes condense together to form XY bodies. These XY bodies are transcriptionally silenced through a process known as meiotic sex chromosome inactivation (MSCI) and are considered to be recombinantly inactive, thus protecting sex chromosomes from illicit recombination events, which are hallmarks of active meiosis. The XY body is enriched in specific H2A variants (including macroH2A (mH2A)24 and H2A.X25) and H3.3 (REF. 26). The phosphorylated form of H2A.X (γH2A.X) also has a crucial role in male germ cells, as H2A.X-null and transgenic mice fail to properly form XY bodies and undergo MSCI, which results in male sterility27. Another variant, H2A.Z, is also enriched at heterochromatin domains of postmeiotic haploid spermatids, including at the sex chromosomes28, although its functional role during spermatogenesis remains unknown.
On the basis of recent studies that use genome-wide sequencing, following protamine replacement a small proportion of the sperm genome (~4% in humans29 and ~1% in mice30) retains nucleosomes, most of which are clustered at specific developmental gene promoters and at imprinted loci. Large blocks of these remaining nucleosomes carry the ‘active mark’ of histone H3 trimethylated at lysine 4 (H3K4me3), including those at various developmental promoters, HOX gene clusters, microRNA- encoding genes and paternally expressed imprinted loci. Sperm nucleosomes that contain the repressive H3K27me3 modification are also highly enriched at developmental promoters, many of which are repressed in early embryos, including at various bivalent promoters that have been identified in embryonic stem cells (ESCs)29,31. A recent study in sperm has now provided direct evidence for associations between these active H3K4me3-enriched loci and the histone variant H3.3; loci with H3K27me3 are found to be generally occupied by canonical H3.1 and/or H3.2 (REF. 30). However, it remains to be determined whether nucleosomes that originate from sperm have functional roles following fertilization (for example, the transmission of specific chromatin states to preserve transcriptional and cellular ‘memories’), as well as which other histone variants constitute these nucleosomes at specific gene promoters and/or imprinted loci to convey instructive epigenetic information to the zygote. Although the clinical relevance of such findings remains unknown, studies of testis-specific histone variants in the context of human infertility32 give rise to future avenues for chromatin-based diagnostics.
In contrast to various testis-specific variants, only one oocyte-specific histone variant, histone H1oo, is known, and it is the most divergent of all linker histones. Although the specific role of H1oo in oogenesis remains unknown, its high conservation throughout the animal kingdom suggests a vital function during oogenesis and early embryonic development33. In addition, numerous somatic histone variants are abundant in oocytes. For example, H3.3 is highly expressed in ovaries, which suggests a role for this variant during female gametogenesis34. H3.3 is a well-established replication-independent H3 variant, which evolved partly in order to participate in active transcription. Oocytes also express mH2A, which has been implicated in transcriptional repression and is eliminated from the zygote shortly after fertilization. During mouse oogenesis, H3.3 and mH2A are incorporated into euchromatin and heterochromatin, respectively, and are maintained throughout meiosis35,36. Little is known about the genome-wide profiles and functional roles of these variants during mammalian oogenesis. However, recent studies indicate that extensive histone variant dynamics (for example, those of H3.3) are crucial for the establishment of the de novo chromatin landscape after fertilization, which provides a possible mechanism to ‘reset’ chromatin states during the transition from highly specialized gametes to totipotent zygotes.
Roles in the zygote
In mice, immediately following fertilization, sperm nuclear decondensation and the formation of the paternal pronucleus begin when protamines are globally exchanged by H3.3-containing nucleosomes37. As shown in Drosophila melanogaster, with the exception of previously deposited sperm nucleosomes, H3.3 is exclusively contributed by the pre-fertilized oocyte, in the absence of transcriptional activity, by the H3.3-specific chaperone HIRA38,39. Functionally, the paternal deposition of H3.3 seems to be important for the establishment of normal heterochromatin and therefore embryonic development37. Mutating H3.3K27, but not H3.1K27, to arginine decreases levels of both H3K27me3 and H3K27me1, and leads to the induction of aberrant pericentromeric transcription, abnormal chromosomal segregation and developmental arrest in the mouse embryo.
In contrast to paternal deposition of H3.3, fertilization also triggers the removal of accumulated maternal H3.3 and mH2A from the female pronucleus. Such global histone removal is restored during late pronuclear stages, during which H3.2 and H3.3 (but not H3.1 or mH2A) are incorporated into chromatin35. mH2A (specifically mH2A1) is later incorporated in pre-implanted embryos at the eight-cell stage and ultimately associates with the inactive X chromosome in females36,40, which suggests a role in transcriptional repression. Although currently unclear, such data call into question whether a small proportion of the H3.3 or H3.2 pools remain in the early female pronucleus following initial nucleosomal removal to act as transmitted ‘memories’ of prior chromatin states in a similar way to that hypothesized with sperm nucleosomes. It also remains unknown whether specific histone modifications in the female pronucleus are linked to H3 variant turnover. Nevertheless, removal of epigenetic information harboured by prior histone variants in the maternal genome might participate in the initiation of new patterns of gene expression in the totipotent zygote.
Roles in embryonic development
Studies of early embryonic development, especially those using somatic-cell nuclear transfer (SCNT) (BOX 1), imply that much of the molecular basis of tissue-specific gene expression profiles and developmental potential might be deeply rooted in the intricacies of chromatin structure, as determined by alterations in histone variant deposition and function. Genetic approaches to define the functional importance of histone variants have been used, and such approaches are made possible by the unique properties of histone variants, which are often encoded as single or double gene copies throughout the genome (TABLES 1,2). This is in contrast to canonical histones, which are often expressed from numerous clustered genes, and their functions are therefore more difficult to study using genetic approaches.
Box 1. Roles in somatic-cell nuclear transfer embryos.
In both mice and humans, the genomes of differentiated somatic nuclei are remodelled to a totipotent state upon somatic-cell nuclear transfer (SCNT; that is, transplantation into enucleated oocytes). During this remodelling, both linker146 and core histones147 are rapidly turned over and replaced by a new set of histones that are stored in the oocytes. Such events are consistent with the hypothesis that removal of the previous epigenetic information is necessary for proper embryonic development. Whereas H3.2 and H3.3 are incorporated into chromatin following both fertilization and SCNT, H3.1 is only incorporated after SCNT147, which highlights a fundamental difference between these two processes. Although a functional role for these differential deposition patterns remains elusive, it seems that such a phenomenon might contribute to the lower success of somatic nuclear reprogramming. In addition, with regards to H2A variants, canonical H2A, H2A.Z and macroH2A (mH2A) are all removed from chromatin following SCNT, but H2A.X is maintained and actively incorporated during this process147,148. The selective incorporation (H2A.X and H3.2 or H3.3) and elimination (H2A.Z and mH2A) of particular histone variants are believed to contribute to unique chromatin structures that are observed in the nucleus following SCNT, thereby facilitating chromosomal remodelling to promote phenotypic outcomes that are associated with reprogramming. Similarly, studies in <italic>Xenopus laevis</italic> have shown that the homologue of the linker histone H1oo is required for pluripotency gene reactivation during SCNT146. Interestingly, a highly divergent linker histone variant dBigH1, which has recently been identified in <italic>Drosophila melanogaster</italic>, seems to function as a cell-type-specific timer for zygotic genome activation in early embryogenesis149. Currently, a considerable challenge to the research field is to completely characterize the molecular basis of histone variant dynamics following SCNT and during zygotic development in humans. However, studies of such processes during periods of <italic>in vitro</italic> cellular reprogramming, as observed with human induced pluripotent stem cells (iPSCs), provide hope that mechanistic functionalities of such phenomena during early embryonic development will soon be revealed. In fact, recent studies of mH2A in both human150 and mouse151,152 iPSCs show that, during lineage differentiation, mH2A provides perpetual repression at pluripotency genes in association with specific changes in histone modification (for example, increased trimethylation at histone H3 lysine 27 (H3K27me3) and reduced H3K4me2 levels), thereby acting as an epigenetic barrier to reprogramming.
H3.3
Although phenotypes that are associated with complete knockout of genes encoding both H3.3 variants (H3f3a and H3f3b) have not yet been described41, mice with a hypomorphic mutation in H3.3A (which was produced by a retroviral gene trap insertion into H3f3a)42 and H3f3b-knockout mice 43 were both shown to result in the postnatal death of ~50% of homozygous mice and significant defects in fertility in surviving animals. Viable H3.3A mutants showed reduced growth rates, and H3f3a mutants also exhibited neuromuscular deficits. Although disruptions of either H3f3a or H3f3b seem to result in similar phenotypes, mouse embryonic fibroblasts from H3f3b-knockout animals show unique characteristics of pericentric heterochromatin spreading and abnormal karyotypes, which suggest a functional role for H3.3 in the maintenance of genomic integrity. Such a function is further supported by knockdown studies in fertilized mouse zygotes, in which reduced H3.3 accumulation resulted in chromosomal over-condensation and mis-segregation, and subsequently developmental arrest during early embryogenesis44. This phenotype was fully rescued by exogenous expression of H3.3 (but not H3.1); however, replacement with H3.3 harbouring a mutation at K36 (H3.3-K36R) was unable to compensate for H3.3 knockdown. Our group recently showed that H3.3 is required for the proper establishment of H3K27me3 at promoters of developmentally regulated genes in ESCs. H3.3 depletion by both knockdown and knockout was shown to disrupt nucleosomal turnover and binding of the Polycomb repressive complex PRC2 at bivalent promoters. Knockdown of H3.3 in ESCs promoted abnormal differentiation towards the typically restricted trophectoderm lineage, which indicates a crucial role for H3.3 in maintaining proper patterns of embryonic germ layer development45. These data show an essential combinatorial role for histone variant exchange and the subsequent accumulation of necessary histone post-translational modifications during early developmental processes.
Notably, in D. melanogaster, knockout of both H3.3-encoding genes (His3.3A and His3.3B) does not result in lethality but induces male infertility46,47. However, a more recent study has challenged the notion that H3.3 has intrinsic and specific functions in comparison with its canonical counterparts, which showed that replacement of H3.3 with H3.2 at the endogenous His3.3 locus in D. melanogaster results in normal fertility48. These results argue that, at least in the case of D. melanogaster, H3.2 and H3.3 can functionally compensate for one another and are seemingly redundant as long as their expression patterns remain consistent. However, these findings have yet to be verified in a mammalian system.
H2A.Z
Two protein isoforms exist for H2A.Z, which have been re-named H2A.Z.1 and H2A.Z.2 to avoid ambiguity caused by their previous names H2A.Z and H2A.V. The isoforms are encoded by two independent genes H2A.Z.1 (also known as H2AFZ) and H2A.Z.2 (also known as H2AFV), respectively, and they differ only by three amino acids49. H2A.Z.1 has been shown to be essential for animal survival; in mice, knockout of H2AFZ results in early embryonic lethality, indicating that H2A.Z.2 cannot compensate for the loss of H2A.Z.1 (REF. 50). Development of the blastocyst from embryonic day 4.5 (E4.5) requires active cell proliferation, complex differentiation and reorganization of the inner cell mass to form an epithelial monolayer (that is, the primitive ectoderm) followed by gastrulation. During this period, development of H2A.Z.1-null embryos begin to fail; at E6.5, they are significantly underrepresented and show obvious patterns of degeneration50. Similarly, H2A.Z.1-knockout ESCs are unable establish in culture, and knockdown studies further indicate that H2A.Z is required for efficient self-renewal and optimal differentiation of ESCs51,52. Specifically, binding of the core transcription factor octamer-binding protein 4 (OCT4; also known as POU5F1) to genomic target sites is decreased in H2A.Z-knockdown ESCs, which leads to reduced recruitment of mixed-lineage leukaemia (MLL) complexes, decreased H3K4 methylation and concomitant repression of genes that are involved in ESC differentiation. Knockdown of H2A.Z also disrupts appropriate targeting of PRC2 to Polycomb-associated genes, which results in reduced H3K27me3 levels, subsequent activation of normally repressed genes in ESCs and premature differentiation. These data indicate that H2A.Z acts as a critical factor that mediates the activation of differentiation-associated genes and the repression of ESC-specific genes during differentiation.
A growing body of evidence indicates further genetic variation of H2A.Z and H3.3. As described above, two non-allelic H2A.Z-encoding genes have been identified in mice and humans. Genetic variations of H3 variants have been identified in humans and close evolutionary relatives, and all of these variants originate primarily from H3.3 pseudogenes (for example, H3.X, H3.Y and H3.5)15,53. As H2A.Z and H3.3 are enriched at specific genomic loci, which ultimately dictate their distinct functions, future studies will need to independently tag or develop specific antibodies against these newly defined variants to better distinguish between their unique localizations throughout the genome and any differential functions that may exist.
A recent study has indicated that dual incorporation of H2A.Z and H3.3 into nucleosomes results in unstable chromatin environments both in chromatin arrays and in vivo, but not at the level of mononucleosomes54. Both H2A.Z and H3.3 have been found in association with various transcription factor complexes at active transcription start sites (TSSs), bivalent TSSs and enhancers. H2A.Z and H3.3 seem to facilitate active transcription in combination with a variety of other functional factors and associated transcriptional complexes. A wide range of studies has now emphasized a role for H3.3 during early metazoan development and in the maintenance of stem cell pluripotency, but much more remains to be understood regarding its functions during later stages of development, as well as in fully differentiated postmitotic cells. With this in mind, it will be important to consider how H3.3 cooperates with other histone variants, such as H2A.Z, some of which are already known to associate with H3.3-containing nucleosomes throughout highly dynamic regulatory regions of the genome55,56.
mH2A and H2A.X
Although mH2A has long been known to associate with the inactive X chromosome in female mammals57, relatively recent studies have indicated roles for mH2A when incorporated in autosomes58,59. Chromatin immunoprecipitation (ChIP) and genomic tiling microarrays have demonstrated negative correlations between mH2A occupancy and gene expression, which suggests a role for mH2A in transcriptional repression that is similar to its function during X chromosomal inactivation. Interestingly, however, ~12% of active genes are enriched for mH2A, which indicates a potentially more diverse role for mH2A in the regulation of active gene expression22,59. Although complete mammalian knockout of the two mH2A-encoding genes has not yet been reported, mH2A-deficient zebrafish show severe developmental defects, including deformed body structures and malformations in brain anatomy58.
In mice, knockout of H2A.X results in growth retardation, male-specific infertility and meiotic arrest60. Although DNA damage responses can be elicited in the absence of H2A.X, H2A.X-knockout ESCs and mutant mouse embryonic fibroblasts (H2A.XS136A/S136A), which are deficient in H2A.X phosphorylation) show alterations in the efficiency of DNA repair by non-homologous end joining and homologous recombination61–63. As a result of such deficits in DNA damage repair, these cells are more sensitive to DNA damage and have reduced proliferation capacity. As far as we are aware, these groundbreaking studies that link H2A.X and its phosphorylation to genomic instability and cancer were among the first to connect histone variants directly to disease in mouse models and probably humans (see below).
H1 variants
Although the canonical models suggest that H1 incorporation promotes chromatin condensation and thus facilitates the formation of repressive chromatin environments, recent studies show that H1 variants may have more specific nuclear functions. Genome-wide mapping of somatic H1 variants revealed non-uniform genomic distribution and incorporation at both active and repressed loci64. Studies in mouse and D. melanogaster models have identified a role for H1 in the maintenance of repeat regions and global chromosomal architecture65,66. Furthermore, high frequencies of missense mutations have recently been identified in various linker histones across several haematological malignancies67,68. One such mutation was identified in follicular lymphoma and was shown to reduce H1 affinity and residency time on chromatin69. Together, these results indicate a previously unrecognized role for histone H1 variants in human disease.
Challenges
As evidenced from previous discussions, gene targeting is a useful tool for examining unique functions of histone variants in vivo; however, such approaches have been slow to provide compelling biological information about these variants because simultaneous targeting of multiple gene copies is required to achieve complete knockout of any given variant. Various novel bioengineered tools have recently been developed to allow systematic characterization of gene function by means of deletion, alteration or overexpression. These tools include engineered DNA-binding proteins, such as targeted zinc-finger nucleases, transcription activator-like effector nucleases (TALENs) and the clustered regularly interspaced palindromic repeat (CRISPR) system, all of which can efficiently target multiple genes at once. We expect that these new genomic tools will greatly advance future studies of histone variant function in living cells.
Roles in post-replicative cells
It is becoming increasingly clear that functional alterations in histone variant biology — regardless of whether they are due to aberrant expression patterns, disrupted or enhanced interactions with associated enzymatic complexes, or altered genomic deposition — result in devastating phenotypic outcomes that lead to human disease. One particularly important question is whether histone variants, which are expressed and deposited into chromatin throughout the cell cycle, function differently in post-replicative cells in comparison with dividing cells. In postmitotic cells, canonical histones can no longer actively incorporate into chromatin, which results in a ‘shift’ in the balance between histone variants and non-variant ones in chromatin.
One example of this is H3.3, which has been observed to progressively accumulate over the course of 150 days in the rodent brain, with concomitant reductions in levels of canonical H3.1 and H3.2 (REF. 70). Interestingly, however, embryonically derived canonical H3.1 was recently shown to persist for up to a year in rodent brain neurons, which indicates a potential role for such stability in adult neuronal function71. Given the dynamic nature of H3.3, such a phenomenon raises the question of the specific roles of H3.3 in the developing and adult central nervous systems in comparison to its canonical counterparts. Previous biochemical studies in proliferating cells have suggested that H3.3 is preferentially marked by post-translational modification ‘signatures’ that are typically associated with active transcription72, thereby contributing to a genomic ‘barcode’ (REF. 73) or nucleosomal code74 that might result in distinct chromosomal territories or domains, which then influence chromatin states during cellular differentiation and development. However, the mechanisms of such modification enrichment are poorly understood and are unlikely to be reflected in cellular systems, such as postmitotic neurons, that exclusively rely on a single histone H3 for global replacement during development. Although it is currently unclear whether H3.3 indeed accumulates in the brain throughout development and into adulthood, one might expect this variant to become more globally distributed throughout the epigenome and become subsequently marked by post-translational modifications that are previously ‘reserved’ for canonical H3 proteins. If this is true, then such phenomena would directly challenge the notion of histone barcoding in non-dividing cells. One recent study provided evidence to support a role for calcium-dependent DAXX dephosphorylation in mediating H3.3 deposition into chromatin in response to neuronal activity, particularly at regulatory elements that are associated with the expression of immediate early genes75 (FIG. 3). Although such data indicate an extraordinarily dynamic nature for H3.3 in the central nervous system during periods of cellular activity, much more work is needed to fully understand the role of H3 variant exchange and accumulation in the brain during periods of neural development and plasticity.
Figure 3. Histone variant exchange in post-replicative cells.
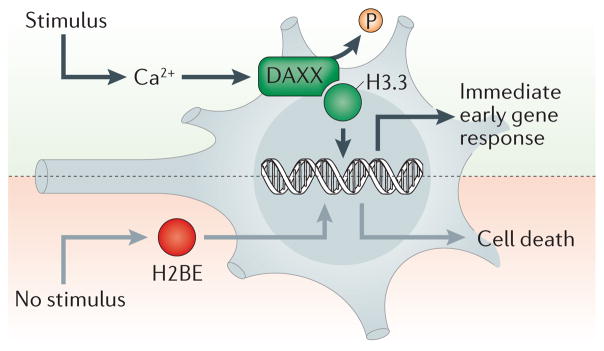
Recent findings have suggested distinct mechanisms of histone variant regulation in postmitotic neurons during periods of heightened or reduced cellular activity. In the presence of a stimulus, calcium-dependent dephosphorylation of death domain-associated protein 6 (DAXX) in embryonic cortical neurons leads to an increase in H3.3 deposition at regulatory elements that are associated with activity-dependent immediate early gene responses. Conversely, during periods of reduced sensory experience, expression of H2BE (which is enriched exclusively in olfactory chemosensory neurons) is enhanced, which results in cell death.
Although it is tempting to assign specific functionalities to H3.3 owing to its ‘behaviour’ in proliferating cells, one must proceed with caution and instead frame questions of histone variant biology within the context of the specific cellular systems examined. Perhaps more appropriate questions should revolve around the consequence of not having options for histone replacement and the effect of exclusively introducing a histone that is thought to be thermodynamically less stable and more dynamic than its canonical counterparts. Analogous to recent discoveries in diffuse intrinsic pontine gliomas (DIPGs) that mutations in histones themselves can result in human disease, mutations in genes that encode H4 (K9fs, where fs denotes frameshift) and H3 (R130C) have been reported76, suggesting direct links between histone protein mutations and mental retardation and intellectual disabilities. Furthermore, following a mutational screen in zebrafish, H3.3 was recently implicated in the process of neuronal differentiation, in which a missense mutation (D123N) was shown to result in the abnormal expression of cranial neural crest specific markers and, eventually, impairments in ectomesenchyme formation77.
Similarly, numerous histone variants of the H2A and H2B families have now been found to be differentially expressed in post-replicative neurons compared with mitotic cells, in both normal and diseased individuals, which suggests unique functions in non-dividing cells. For example, H2A.Z.2.2 (which is the spliced isoform of H2A.Z.2) was recently shown to be enriched in the brain, and was shown to severely destabilize nucleosomes owing partly to its unique carboxyl terminus78. Although the function of H2A.Z.2.2 in postmitotic neurons is unknown, it is probably involved in mediating the dynamic nature of neuronal transcription in response to cellular activity, which is necessary for establishing and maintaining various aspects of synaptic development and plasticity. Interestingly, unlike canonical histones — many of which are encoded by gene clusters and lack both functional promoters and 3′ untranslated regions — most histone variants are encoded by one or two genes and have upstream promoter regulatory elements, which indicate a potential for activity-dependent regulation. Recent data from olfactory chemosensory neurons showed the existence of an exclusively expressed variant of the H2B family, H2BE, the expression of which was shown to be negatively regulated by sensory activity to prevent neuronal cell death during periods of heightened olfactory-mediated sensory experiences79 (FIG. 3). Although such findings represent the first demonstration of an activity-dependent mechanism for histone variant exchange in post-replicative neurons, this is likely to represent a common regulatory phenomenon for these proteins and is sure to gain substantial attention in the future.
Roles in human disease
H3.3: direct mutational evidence that ‘every amino acid matters’
An emerging body of literature has identified increasing numbers of mutations that directly affect chromatin-modifying enzymes in human cancer, which suggests that aberrant regulation of the chromatin modification landscape can lead to oncogenesis (see REF. 80 for a recent review on histone variants and cancer). In particular, genes that affect post-translational modifications of H3 are frequently mutated. Furthermore, the H3.3 variant and its associated chaperones have recently been implicated in cancer. Although H3.3 has previously been reported to be overexpressed in subsets of human tumours81, recent exome sequencing of paediatric gliomas identified missense mutations at K27 and G34 of H3F3A-encoded H3.3 (REFS 82–84) that directly affect post-translational modifications of K27 (REFS 85,86) and K36, respectively85. Both of these residues have vital roles in the regulation of various essential gene expression programmes in mammalian cells. The discovery of H3K27 and H3G34 mutations in paediatric gliomas represents ‘game-changing’ evidence that genetic alterations of histones themselves at or near important and differentially modified residues can lead to cancer in mouse models and humans. It remains unclear how these mutations in histone proteins lead to oncogenesis with such remarkable age and anatomical precision, and the underlying timing and cell-of-origin issues will merit future investigation.
Paradoxically, H3.3 is genomically localized both to euchromatin and to pericentric and telomeric hetero-chromatin87. The histone chaperone HIRA mediates H3.3 deposition at gene bodies and promoters through transcriptionally linked histone replacement88. It is important to note that HIRA-mediated H3.3 deposition is directly linked to cellular signalling mechanisms and is partly regulated by the activity of serine/threonine-protein kinase PAK2 (REF. 89). At heterochromatin, the ATRX–DAXX chaperone complex mediates H3.3 deposition90,91. Recently, mutually exclusive inactivating mutations of both DAXX and ATRX were discovered in pancreatic neuroendocrine tumours (PanNETs), myelodysplastic syndromes, relapsed acute myeloid leukaemia, neuroblastomas and glioblastomas92–94. ATRX and DAXX are required for proper heterochromatin formation and telomere function, and their loss results in aneuploidy and telomere dysfunction95–98. In PanNETs and neuroblastomas, mutations in both ATRX and DAXX correlate with altered telomeres99. Histone H3 mutations have now been identified in paediatric gliomas76, insulinomas100, chondroblastomas and giant cell tumours of bone101, and the co-occurrence of these fairly rare mutational events in malignancy provides strong evidence of a critical role for H3.3 function in the maintenance of cellular proliferation and in cancer progression.
High-grade paediatric gliomas are among the leading causes of terminal cancer in children. Adult glioblastomas (GBMs) are supratentorial and are often found in the cerebral cortex, whereas DIPGs occur in the paediatric brainstem102 and harbour distinct molecular lesions in comparison with adult GBMs103–106. Our group and others recently revealed novel mutations in H3.3A (for example, K27M, G34R and G34V) and canonical H3.1B (K27M)82–85 in both GBMs and DIPGs. Concurrently, inactivating mutations in ATRX and DAXX were also identified in DIPGs82,84. These point mutations in H3.3 map at or close to well-known sites of post-translational modifications, and proteins that are known to ‘write’ or ‘read’ these modifications are also commonly mutated in a wide variety of malignancies107,108. Post-translational modifications of the amino-terminal tail of histone H3 are crucial for multiple chromatin-templated processes. Notably, H3K27 is the target of methylation by Polycomb proteins, which are generally regarded as transcriptional repressors. H3K36 methylation can mediate transcriptional elongation, splicing and DNA damage repair depending on the context. Multiple genes encoding proteins that regulate these modifications are frequently mutated in cancer. Importantly, disruptions of chromatin regulators are highly specific to tumour type; for example, the H3K27 methyltransferase EZH2 is overexpressed in various epithelial malignancies and is lost or mutated in subsets of leukaemia and lymphomas109. These observations suggest that mis-regulation of H3K27 and H3K36 modifications are common in tumorigenesis but that such mis-regulation only occurs in specific cell lineages and developmental contexts.
We and others recently investigated the functional outcome of mutations that affect H3 methylation at K27 (for example, H3-K27M) in human tumours in order to better understand the molecular and biochemical consequences of this substitution, which is found in nearly 80% of human DIPGs. In accordance with results obtained from exome sequencing, quantitative mass spectrometry confirmed that H3.1-K27M and H3.3-K27M constituted up to ~18% of total H3 in DIPG samples regardless of tumour genotype82,83. Furthermore, using a platelet-derived growth factor-induced brain stem glioma mouse model, transgenic H3.3-K27M expression was found to be sufficient to reduce global levels of H3K27me3 with concomitant induction of abnormal proliferating ectopic cell clusters in vivo. Further biochemical characterization of this mutation in cell lines suggests that H3.3-K27M imparts unique gain-of-function properties both to the mutant histone itself and to wild-type H3 in heterotypic oligonucleosomal configurations. Interestingly, converting K27 to all other amino acids, with the modest exception of isoleucine, had little effect on global levels of H3K27me3. H3.3-K27M mutations were later found to directly inhibit allosteric interactions, both in cis and in trans, between PRC2 and endogenous H3 in heterotypic nucleosomal substrates, and this has not been observed with mononucleosomes that contain K27A, K27R or K27Q, which indicates that H3.3-K27M might directly interfere with PRC2 activity that is normally stimulated by H3K27me3. This interference of PRC2 activity was shown to result from direct interactions between K27M and the EZH2 active site, thereby inhibiting its H3K27me3 methyltransferase activity91 (FIG. 4).
Figure 4. H3.3 variant mutations in brain gliomas: direct evidence that ‘every amino acid matters’.

The wild-type histone H3 recruits Polycomb repressive complex 2 (PRC2) and stimulates methyltransferase activity of its catalytic subunit EZH2 (green box),which trimethylates histone H3 at lysine 27 (H3K27me3). The replication-independent histone variant H3.3 mutant that contains the K27M substitution (red box) was recently identified in many diffuse intrinsic pontine gliomas (red circle, bottom left) and supratentorial glioblastomas (red circle, top left). This mutation leads to dominant inhibition of EZH2 in both cis and trans and to concomitant global loss of H3K27me3. These data provide the first direct evidence that mutations in histone variants themselves contribute to human disease.
Supporting this notion, a more recent analysis of genome-wide patterns of H3K27me3 distribution between neural stem cells and H3.3-K27M-expressing DIPG cell lines revealed global reductions in H3K27me3 peak distribution; however, levels of both H3K27me3 and EZH2 were also found to be markedly increased locally at hundreds of loci in H3.3-K27M-expressing cells from patients86. These data suggest that alterations of H3K27me2 and H3K27me3 in chromatin, in terms of the loss and gain of these marks by the H3.3-K27M mutation, are likely to contribute to formation of paediatric DIPGs through altered expression of genes that are associated with tumorigenesis.
Although not directly linked to cancer, recent work has shown a novel role for the H3.3-specific chaperone HIRA in promoting transcriptional ‘restart’ following ultraviolet C (UVC) radiation-mediated DNA damage (a form of genotoxic stress that results in reduced chromosomal integrity and possibly cancer)110. HIRA was found to accumulate before repair at genomic UVC radiation sites to deposit newly synthesized H3.3 histones, thereby priming chromatin for later reactivation of transcription following DNA repair110. It is therefore possible that specific H3.3 mutations identified in cancer (for example, H3.3-G34R and H3.3-G34V) might affect such processes by dampening the ability of H3.3 to promote transcription reactivation following instances of genomic insult.
CENP-A and genomic stability
Recently, histone H3-like centromeric protein A (CENP-A) has also been implicated in cancer. Although CENP-A is present in lower abundance than canonical H3, it is essential for mitosis and cell survival, and is thought to propagate and maintain centromere identity during cell division111,112. CENP-A-null mice fail to survive beyond 6.5 days post-conception and show severe mitotic deficits, nuclear blebbing and bridging, and chromatin hypercondensation and fragmentation113. Altered CENP-A expression results in disrupted stoichiometries between CENP-A and its chaperone Holliday junction recognition protein (HJURP)114, which leads to CENP-A mis-targeting, chromosomal instability and cancer115. CENP-A is upregulated in numerous cancers, including breast cancer, lung adenocarcinoma, colorectal cancers and hepatocellular carcinoma115–119. In addition, overexpression of CENP-A has been demonstrated to result in apoptotic inhibition and cellular proliferation through direct modulation of both cell cycle-associated genes and apoptosis-associated genes120. Such overexpression was suggested to promote instances of CENP-A mis-localization and lead to alterations in chromosomal dynamics and/or gene expression profiles.
H2A variants and cancer
Similar to CENP-A, H2A.Z has been shown to be overexpressed in various human cancers, including breast, lung, colorectal and bladder cancers121–124. The molecular consequences of such overexpression have been elucidated and collectively suggest H2A.Z as a bona fide oncoprotein80. Upregulation of H2A.Z leads to alterations in its incorporation in chromatin, which might be directly regulated by changes in the expression and/or activity of its deposition machinery. H2A.Z, similarly to H3.3 with which it associates within the nucleosome, localizes to active regions of the genome and has been suggested to ‘fine-tune’ gene expression patterns and modulate other non-coding or regulatory regions of the genome that are involved in cancer progression. Additionally, H2A.Z has been implicated in cellular senescence and DNA double-strand break (DSB) repair125, thereby coordinating the maintenance of telomere integrity, chromosomal segregation and genomic stability126–128, all of which are candidate mechanisms that underlie cancer progression.
H2A.X (encoded by H2AFX) is best known for its heavily studied role in the DNA damage response and has been coined the ‘histone guardian’ of the genome129,130. H2A.X is subject to numerous post-translational modifications, including rapid phosphorylation of S139 upon DNA damage. γH2A.X localizes to nuclear foci that are associated with DNA DSBs and is regulated by several phosphoinositide 3-kinase-related protein kinases, such as ATM (ataxia telangiectasia mutated), ATR (ATM and Rad3-related) and DNA-dependent protein kinases, many of which are mutated in human disease. H2A.X can also be phosphorylated (for example, at S16, T136 and Y142), acetylated and ubiquitylated at numerous other residues, all of which confer differential effects on nucleosomal structure and function131–133. Owing to its role in repairing DNA DSBs, both deficiencies in H2A.X and H2A.X haploinsufficiency lead to genomic instability and increased cancer incidence when coupled with p53 disruptions62,63. Furthermore, chromosomal deletions of band 11q23 (REFS 134–136), where H2AFX maps, and alterations in H2AFX copy number137,138 have been reported at high frequencies in various human haematological malignancies and solid tumours, which indicates that aberrant regulation of H2A.X might contribute to cancer initiation and progression.
mH2A, which is the most structurally distinct of the H2A variants, has also been implicated in cancer initiation and progression, and particular isoforms of mH2A have reduced expression in several tumour types (for example, melanoma, and lung, testicular, bladder, colon and ovarian cancers)139–143, which suggests an integral role for mH2A as a tumour suppressor. For example, it was recently reported that transcriptional loss of both mH2A.1 and mH2A.2 isoforms in melanoma promotes progressive malignancy through deregulation of cyclin-dependent kinase 8 (CDK8)141, which has previously been implicated as an oncoprotein in colorectal carcinoma. Similar to H2A.Z, mH2A has also been described as a marker of senescence144, and its loss potentially contributes to malignant transformation. Finally, although a direct link has yet to be established, the H3.3 chaperone ATRX has recently been reported to be a negative regulator of mH2A deposition in chromatin145. Given recent findings that connect gain- and loss-of-function H3 mutations with deletions in ATRX in a range of tumours, it is possible that deregulated mH2A deposition may lead to tumorigenesis.
Conclusions and future perspectives
It is now clear that histone variants represent an important component of the nuclear transcriptional machinery in terms of their distinct deposition profiles as well as structural and biophysical properties. The replication-independent expression and deposition of histone variants differ from those of canonical histones and are important for maintaining various aspects of chromatin integrity following turnover of nucleosomes. Besides simply buffering nucleosomal density, such specialized characteristics of histone variants provide additional complexity to the mammalian epigenome to allow tighter transcriptional control and enhanced heterochromatic maintenance through intrinsic structural differences, differential recruitment of trans-associated factors and, in some cases, altered post-translational modification landscapes.
The introduction of these minor sequence variants into chromatin is physiologically relevant and fundamental to eukaryotic cellular plasticity. However, with the exception of substitutions towards modification-permissive amino acids (for example, both H3.1A31 and H3.2A31 to H3.3S31, which can be phosphorylated), it remains unclear how histone variants acquire and/or differentially enrich for specific chemical modifications that are distinct from their canonical counterparts. For example, given that all lysine residues are conserved across the three H3 species, it seems remarkable that the variant H3.3 is preferentially marked in many cell types by ‘active’ post-translational modifications, whereas canonical H3.1 and H3.2 are more repressively modified. The underlying mechanisms that control such differences have not been fully elucidated; however, differences in deposition machinery are likely to contribute to this phenomenon.
Although recent structural data have uncovered some of the biophysical interactions that control specific variant– chaperone interactions, the molecular mechanisms that target these complexes to distinct epigenomic loci have not been completely characterized. As mentioned above, such targeting mechanisms are probably different in post-replicative cells, such as neurons, in which choices for histone incorporation are limited. Perhaps alternative chaperone complexes and deposition strategies exist in these non-dividing cells to accommodate such limitations, but little is currently known. Given the dynamic nature of histone variants and their ability to be regulated at the level of transcription through upstream signalling, it is likely that the incorporation of these proteins, often intranucleosomally with variants of other histone families (for example, H3.3 and H2A.Z), represents a central mechanism of transcriptional plasticity in eukaryotic cells. Identification of the upstream mediators of such regulation, as well as the consequences of combinatorial variant deposition throughout the genome, will require further study.
Finally, it will be essential to gain a better understanding of the processes that control cell-type-specific expression and deposition of histone variants in future studies of histone variant function. As discussed throughout this Review, numerous histone variants seem to be restricted to specific cell lineages or tissue types, yet it remains unclear how such expression patterns are maintained and what the consequences are of increasing or reducing combinatorial variant deposition across cell types. Aberrations in these processes result in detrimental phenotypic outcomes across numerous mammalian systems, including humans. Although we are clearly still in the infancy of this ever-expanding and diverse field, we imagine that future endeavours related to histone variant biology will hold great promise for human health and disease.
Acknowledgments
The authors thank members of C.D.A.’s laboratory for providing critiques of the manuscript. Given the broad scope of this Review, they apologize for any relevant citations that might have been omitted. I.M., K.-M.N. and C.D.A. are partly supported by grants from the US National Institute of Mental Health (RO1 MH094698-01, P50 MH096890-01) and the Leukemia & Lymphoma Society (LLS-SCOR 7132–08). A.A.S. is supported by a Howard Hughes Medical Institute Damon Runyon Cancer Research Foundation fellowship (DRG-2185-14).
Glossary
- Nucleosomal
Pertaining to the nucleosome, which is the basic unit of chromatin that contains ~147 bp of DNA wrapped around a histone octamer (which is composed of two copies each of histone H3, H4, H2A and H2B, or variants thereof)
- Canonical histones
Archetypical histones of a given family to which all other histone proteins of that same family are compared
- Imprinted loci
Genes that are expressed from only one of the two parental copies, the choice of which is dependent on the sex of the parent from which the gene was derived
- Bivalent promoters
Regions of chromatin that have co-occurrence of histone H3 trimethylated at lysine 27 (H3K27me3) and H3K4me2 or H3K4me3 during embryonic development
- Totipotent
Pertaining to the capacity of an undifferentiated cell to develop into any type of cell
- Pronucleus
The haploid nucleus from a gamete
- Hypomorphic
Pertaining to a mutant allele that does not completely eliminate the wild-type function of a gene and that gives a less severe phenotype than a loss-of-function mutant
- Polycomb repressive complex
(PRC). An epigenetic regulator of gene expression that silences target genes by establishing a repressive chromatin state. PRC2 trimethylates histone H3 at lysine 27. This repressive histone modification is recognized by PRC1, which has ubiquitylating activity. As PRCs can maintain states of gene expression, they have key roles in cell fate maintenance and transitions during development
- Trophectoderm
The outer layer of the blastocyst-stage embryo that gives rise to the trophoblast after implantation and that will provide the bulk of the extra-embryonic lineages of the placenta
- Blastocyst
A pre-implantation embryonic stage that is characterized by the first definitive lineages. It contains a fluid-filled cavity (that is, the blastocoel), a focal cluster of cells from which the embryo develops (that is, the inner cell mass) and peripheral trophoblast cells (which form the placenta)
- Inner cell mass
A cluster of undifferentiated cells in the blastocyst, which give rise to the entire fetus and to some of its extra-embryonic (that is, placental) tissues
- Gastrulation
The process by which the three primitive germ layers are formed in the early embryo; it is one of the first major differentiation events in development
- Core transcription factor
A transcription factor that controls the expression of key pluripotency-related genes. Pluripotent cells are highly responsive to levels of these transcriptional regulators
- Aneuploidy
The presence of an abnormal number of chromosomes
- Blebbing
Formation of protrusions from a membrane. Nuclear blebs are caused by localized separation of fibres from the lamin meshwork
- Bridging
Formation of abnormal connections between two nuclei, in which the nuclear membrane extends across two poorly separated or non-separated chromatin masses. Nuclear bridges are associated with failed and regressed cytokinesis
- Haploinsufficiency
A genetic condition in a diploid organism in which a single functional copy of a gene fails to generate sufficient gene product, leading to an abnormal or diseased state
Footnotes
Competing interests statement
The authors declare no competing interests.
References
- 1.Skene PJ, Henikoff S. Histone variants in pluripotency and disease. Development. 2013;140:2513–2524. doi: 10.1242/dev.091439. [DOI] [PubMed] [Google Scholar]
- 2.Chen P, Zhao J, Li G. Histone variants in development and diseases. J Genet Genom. 2013;40:355–365. doi: 10.1016/j.jgg.2013.05.001. [DOI] [PubMed] [Google Scholar]
- 3.Filipescu D, Szenker E, Almouzni G. Developmental roles of histone H3 variants and their chaperones. Trends Genet. 2013;29:630–640. doi: 10.1016/j.tig.2013.06.002. [DOI] [PubMed] [Google Scholar]
- 4.Talbert PB, Henikoff S. Histone variants — ancient wrap artists of the epigenome. Nature Rev Mol Cell Biol. 2010;11:264–275. doi: 10.1038/nrm2861. [DOI] [PubMed] [Google Scholar]
- 5.Elsasser SJ, et al. DAXX envelops a histone H3.3–H4 dimer for H3.3-specific recognition. Nature. 2012;491:560–565. doi: 10.1038/nature11608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ranjan A, et al. Nucleosome-free region dominates histone acetylation in targeting SWR1 to promoters for H2A.Z replacement. Cell. 2013;154:1232–1245. doi: 10.1016/j.cell.2013.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yen K, Vinayachandran V, Pugh BF. SWR-C and INO80 chromatin remodelers recognize nucleosome-free regions near +1 nucleosomes. Cell. 2013;154:1246–1256. doi: 10.1016/j.cell.2013.08.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Banaszynski LA, Allis CD, Lewis PW. Histone variants in metazoan development. Dev Cell. 2010;19:662–674. doi: 10.1016/j.devcel.2010.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Drabent B, Bode C, Bramlage B, Doenecke D. Expression of the mouse testicular histone gene H1t during spermatogenesis. Histochem Cell Biol. 1996;106:247–251. doi: 10.1007/BF02484408. [DOI] [PubMed] [Google Scholar]
- 10.Martianov I, et al. Polar nuclear localization of H1T2, a histone H1 variant, required for spermatid elongation and DNA condensation during spermiogenesis. Proc Natl Acad Sci USA. 2005;102:2808–2813. doi: 10.1073/pnas.0406060102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ishibashi T, et al. H2A.Bbd: an X-chromosome-encoded histone involved in mammalian spermiogenesis. Nucleic Acids Res. 2010;38:1780–1789. doi: 10.1093/nar/gkp1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Govin J, et al. Pericentric heterochromatin reprogramming by new histone variants during mouse spermiogenesis. J Cell Biol. 2007;176:283–294. doi: 10.1083/jcb.200604141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zalensky AO, et al. Human testis/sperm-specific histone H2B (hTSH2B): molecular cloning and characterization. J Biol Chem. 2002;277:43474–43480. doi: 10.1074/jbc.M206065200. [DOI] [PubMed] [Google Scholar]
- 14.Witt O, Albig W, Doenecke D. Testis-specific expression of a novel human H3 histone gene. Exp Cell Res. 1996;229:301–306. doi: 10.1006/excr.1996.0375. [DOI] [PubMed] [Google Scholar]
- 15.Schenk R, Jenke A, Zilbauer M, Wirth S, Postberg J. H3.5 is a novel hominid-specific histone H3 variant that is specifically expressed in the seminiferous tubules of human testes. Chromosoma. 2011;120:275–285. doi: 10.1007/s00412-011-0310-4. [DOI] [PubMed] [Google Scholar]
- 16.Bao Y, et al. Nucleosomes containing the histone variant H2A.Bbd organize only 118 base pairs of DNA. EMBO J. 2004;23:3314–3324. doi: 10.1038/sj.emboj.7600316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Syed SH, et al. The incorporation of the novel histone variant H2AL2 confers unusual structural and functional properties of the nucleosome. Nucleic Acids Res. 2009;37:4684–4695. doi: 10.1093/nar/gkp473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Soboleva TA, et al. A unique H2A histone variant occupies the transcriptional start site of active genes. Nature Struct Mol Biol. 2012;19:25–30. doi: 10.1038/nsmb.2161. [DOI] [PubMed] [Google Scholar]
- 19.Li A, et al. Characterization of nucleosomes consisting of the human testis/sperm-specific histone H2B variant (hTSH2B) Biochemistry. 2005;44:2529–2535. doi: 10.1021/bi048061n. [DOI] [PubMed] [Google Scholar]
- 20.Tachiwana H, et al. Structural basis of instability of the nucleosome containing a testis-specific histone variant, human H3T. Proc Natl Acad Sci USA. 2010;107:10454–10459. doi: 10.1073/pnas.1003064107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Montellier E, et al. Chromatin-to-nucleoprotamine transition is controlled by the histone H2B variant TH2B. Genes Dev. 2013;27:1680–1692. doi: 10.1101/gad.220095.113. This study is the first to show that TSH2B exchange is required for protamine replacement during spermatogenesis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tolstorukov MY, et al. Histone variant H2A.Bbd is associated with active transcription and mRNA processing in human cells. Mol Cell. 2012;47:596–607. doi: 10.1016/j.molcel.2012.06.011. This study illustrated that H2A.Bbd is enriched throughout active regions of the genome and forms a complex with both elongating RNA polymerase II and spliceosomal elements when stably expressed in HeLa cells. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ioudinkova ES, et al. Distinct distribution of ectopically expressed histone variants H2A.Bbd and macroH2A in open and closed chromatin domains. PLoS ONE. 2012;7:e47157. doi: 10.1371/journal.pone.0047157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hoyer-Fender S, Costanzi C, Pehrson JR. Histone macroH2A1.2 is concentrated in the XY-body by the early pachytene stage of spermatogenesis. Exp Cell Res. 2000;258:254–260. doi: 10.1006/excr.2000.4951. [DOI] [PubMed] [Google Scholar]
- 25.Mahadevaiah SK, et al. Recombinational DNA double-strand breaks in mice precede synapsis. Nature Genet. 2001;27:271–276. doi: 10.1038/85830. [DOI] [PubMed] [Google Scholar]
- 26.van der Heijden GW, et al. Chromosome-wide nucleosome replacement and H3.3 incorporation during mammalian meiotic sex chromosome inactivation. Nature Genet. 2007;39:251–258. doi: 10.1038/ng1949. [DOI] [PubMed] [Google Scholar]
- 27.Fernandez-Capetillo O, et al. H2AX is required for chromatin remodeling and inactivation of sex chromosomes in male mouse meiosis. Dev Cell. 2003;4:497–508. doi: 10.1016/s1534-5807(03)00093-5. [DOI] [PubMed] [Google Scholar]
- 28.Greaves IK, Rangasamy D, Devoy M, Marshall Graves JA, Tremethick DJ. The X and Y chromosomes assemble into H2A.Z-containing facultative heterochromatin following meiosis. Mol Cell Biol. 2006;26:5394–5405. doi: 10.1128/MCB.00519-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hammoud SS, et al. Distinctive chromatin in human sperm packages genes for embryo development. Nature. 2009;460:473–478. doi: 10.1038/nature08162. This paper is the first to use genome-wide sequencing to identify targets of histone retention in human sperm, with a specific emphasis on histone modifications. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Erkek S, et al. Molecular determinants of nucleosome retention at CpG-rich sequences in mouse spermatozoa. Nature Struct Mol Biol. 2013;20:868–875. doi: 10.1038/nsmb.2599. [DOI] [PubMed] [Google Scholar]
- 31.Brykczynska U, et al. Repressive and active histone methylation mark distinct promoters in human and mouse spermatozoa. Nature Struct Mol Biol. 2010;17:679–687. doi: 10.1038/nsmb.1821. [DOI] [PubMed] [Google Scholar]
- 32.Hammoud SS, et al. Genome-wide analysis identifies changes in histone retention and epigenetic modifications at developmental and imprinted gene loci in the sperm of infertile men. Hum Reprod. 2011;26:2558–2569. doi: 10.1093/humrep/der192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tanaka M, Hennebold JD, Macfarlane J, Adashi EY. A mammalian oocyte-specific linker histone gene H1oo: homology with the genes for the oocyte-specific cleavage stage histone (cs-H1) of sea urchin and the B4/H1M histone of the frog. Development. 2001;128:655–664. doi: 10.1242/dev.128.5.655. [DOI] [PubMed] [Google Scholar]
- 34.Torres-Padilla ME, Bannister AJ, Hurd PJ, Kouzarides T, Zernicka-Goetz M. Dynamic distribution of the replacement histone variant H3.3 in the mouse oocyte and preimplantation embryos. Int J Dev Biol. 2006;50:455–461. doi: 10.1387/ijdb.052073mt. [DOI] [PubMed] [Google Scholar]
- 35.Akiyama T, Suzuki O, Matsuda J, Aoki F. Dynamic replacement of histone H3 variants reprograms epigenetic marks in early mouse embryos. PLoS Genet. 2011;7:e1002279. doi: 10.1371/journal.pgen.1002279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chang CC, et al. A maternal store of macroH2A is removed from pronuclei prior to onset of somatic macroH2A expression in preimplantation embryos. Dev Biol. 2005;278:367–380. doi: 10.1016/j.ydbio.2004.11.032. [DOI] [PubMed] [Google Scholar]
- 37.Santenard A, et al. Heterochromatin formation in the mouse embryo requires critical residues of the histone variant H3.3. Nature Cell Biol. 2010;12:853–862. doi: 10.1038/ncb2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Orsi GA, et al. Drosophila Yemanuclein and HIRA cooperate for de novo assembly of H3.3-containing nucleosomes in the male pronucleus. PLoS Genet. 2013;9:e1003285. doi: 10.1371/journal.pgen.1003285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Loppin B, et al. The histone H3.3 chaperone HIRA is essential for chromatin assembly in the male pronucleus. Nature. 2005;437:1386–1390. doi: 10.1038/nature04059. This work is the first to show that HIRA specifically incorporates H3.3 into paternal zygotic chromatin prior to the first round of DNA replication. [DOI] [PubMed] [Google Scholar]
- 40.Costanzi C, Stein P, Worrad DM, Schultz RM, Pehrson JR. Histone macroH2A1 is concentrated in the inactive X chromosome of female preimplantation mouse embryos. Development. 2000;127:2283–2289. doi: 10.1242/dev.127.11.2283. [DOI] [PubMed] [Google Scholar]
- 41.Tang MC, Jacobs SA, Wong LH, Mann JR. Conditional allelic replacement applied to genes encoding the histone variant H3.3 in the mouse. Genesis. 2013;51:142–146. doi: 10.1002/dvg.22366. [DOI] [PubMed] [Google Scholar]
- 42.Couldrey C, Carlton MB, Nolan PM, Colledge WH, Evans MJ. A retroviral gene trap insertion into the histone 3.3A gene causes partial neonatal lethality, stunted growth, neuromuscular deficits and male sub-fertility in transgenic mice. Hum Mol Genet. 1999;8:2489–2495. doi: 10.1093/hmg/8.13.2489. [DOI] [PubMed] [Google Scholar]
- 43.Bush KM, et al. Endogenous mammalian histone H3.3 exhibits chromatin-related functions during development. Epigenetics Chromatin. 2013;6:7. doi: 10.1186/1756-8935-6-7. This paper provides the first evidence that complete knockout of one gene copy encoding H3.3 (H3f3b) results in severe developmental deficits in mice. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lin CJ, Conti M, Ramalho-Santos M. Histone variant H3.3 maintains a decondensed chromatin state essential for mouse preimplantation development. Development. 2013;140:3624–3634. doi: 10.1242/dev.095513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Banaszynski LA, et al. Hira-dependent histone H3.3 deposition facilitates PRC2 recruitment at developmental loci in ES cells. Cell. 2013;155:107–120. doi: 10.1016/j.cell.2013.08.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hodl M, Basler K. Transcription in the absence of histone H3.3. Curr Biol. 2009;19:1221–1226. doi: 10.1016/j.cub.2009.05.048. This study reveals that canonical H3.2 can fully compensate for H3.3 in D. melanogaster. These findings need to be verified in a mammalian model system. [DOI] [PubMed] [Google Scholar]
- 47.Sakai A, Schwartz BE, Goldstein S, Ahmad K. Transcriptional and developmental functions of the H3.3 histone variant in Drosophila. Curr Biol. 2009;19:1816–1820. doi: 10.1016/j.cub.2009.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hodl M, Basler K. Transcription in the absence of histone H3.2 and H3K4 methylation. Curr Biol. 2012;22:2253–2257. doi: 10.1016/j.cub.2012.10.008. [DOI] [PubMed] [Google Scholar]
- 49.Eirin-Lopez JM, Gonzalez-Romero R, Dryhurst D, Ishibashi T, Ausio J. The evolutionary differentiation of two histone H2A.Z variants in chordates (H2A.Z-1 and H2A.Z-2) is mediated by a stepwise mutation process that affects three amino acid residues. BMC Evol Biol. 2009;9:31. doi: 10.1186/1471-2148-9-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Faast R, et al. Histone variant H2A.Z is required for early mammalian development. Curr Biol. 2001;11:1183–1187. doi: 10.1016/s0960-9822(01)00329-3. [DOI] [PubMed] [Google Scholar]
- 51.Hu G, et al. H2A.Z facilitates access of active and repressive complexes to chromatin in embryonic stem cell self-renewal and differentiation. Cell Stem Cell. 2013;12:180–192. doi: 10.1016/j.stem.2012.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Creyghton MP, et al. H2AZ is enriched at Polycomb complex target genes in ES cells and is necessary for lineage commitment. Cell. 2008;135:649–661. doi: 10.1016/j.cell.2008.09.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wiedemann SM, et al. Identification and characterization of two novel primate-specific histone H3 variants, H3. X and H3.Y. J Cell Biol. 2010;190:777–791. doi: 10.1083/jcb.201002043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chen P, et al. H3.3 actively marks enhancers and primes gene transcription via opening higher-ordered chromatin. Genes Dev. 2013;27:2109–2124. doi: 10.1101/gad.222174.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jin C, et al. H3.3/H2A.Z double variant-containing nucleosomes mark ‘nucleosome-free regions’ of active promoters and other regulatory regions. Nature Genet. 2009;41:941–945. doi: 10.1038/ng.409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nekrasov M, et al. Histone H2A.Z inheritance during the cell cycle and its impact on promoter organization and dynamics. Nature Struct Mol Biol. 2012;19:1076–1083. doi: 10.1038/nsmb.2424. This paper shows that unstable nucleosomes located at the TSS are heterotypic with respect to H2A (that is, H2A.Z–H2A), which increases instability to an already unstable environment that is occupied by two variants (for example, H2A.Z–H3.3) [DOI] [PubMed] [Google Scholar]
- 57.Costanzi C, Pehrson JR. Histone macroH2A1 is concentrated in the inactive X chromosome of female mammals. Nature. 1998;393:599–601. doi: 10.1038/31275. [DOI] [PubMed] [Google Scholar]
- 58.Buschbeck M, et al. The histone variant macroH2A is an epigenetic regulator of key developmental genes. Nature Struct Mol Biol. 2009;16:1074–1079. doi: 10.1038/nsmb.1665. [DOI] [PubMed] [Google Scholar]
- 59.Gamble MJ, Frizzell KM, Yang C, Krishnakumar R, Kraus WL. The histone variant macroH2A1 marks repressed autosomal chromatin, but protects a subset of its target genes from silencing. Genes Dev. 2010;24:21–32. doi: 10.1101/gad.1876110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Celeste A, et al. Genomic instability in mice lacking histone H2AX. Science. 2002;296:922–927. doi: 10.1126/science.1069398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bassing CH, et al. Increased ionizing radiation sensitivity and genomic instability in the absence of histone H2AX. Proc Natl Acad Sci USA. 2002;99:8173–8178. doi: 10.1073/pnas.122228699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Celeste A, et al. H2AX haploinsufficiency modifies genomic stability and tumor susceptibility. Cell. 2003;114:371–383. doi: 10.1016/s0092-8674(03)00567-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bassing CH, et al. Histone H2AX: a dosage-dependent suppressor of oncogenic translocations and tumors. Cell. 2003;114:359–370. doi: 10.1016/s0092-8674(03)00566-x. [DOI] [PubMed] [Google Scholar]
- 64.Izzo A, et al. The genomic landscape of the somatic linker histone subtypes H1.1 to H1.5 in human cells. Cell Rep. 2013;3:2142–2154. doi: 10.1016/j.celrep.2013.05.003. [DOI] [PubMed] [Google Scholar]
- 65.Lu X, et al. Drosophila H1 regulates the genetic activity of heterochromatin by recruitment of Su(var)3-9. Science. 2013;340:78–81. doi: 10.1126/science.1234654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Cao K, et al. High-resolution mapping of H1 linker histone variants in embryonic stem cells. PLoS Genet. 2013;9:e1003417. doi: 10.1371/journal.pgen.1003417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Morin RD, et al. Mutational and structural analysis of diffuse large B-cell lymphoma using whole-genome sequencing. Blood. 2013;122:1256–1265. doi: 10.1182/blood-2013-02-483727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Morin RD, et al. Frequent mutation of histone-modifying genes in non-Hodgkin lymphoma. Nature. 2011;476:298–303. doi: 10.1038/nature10351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Okosun J, et al. Integrated genomic analysis identifies recurrent mutations and evolution patterns driving the initiation and progression of follicular lymphoma. Nature Genet. 2014;46:176–181. doi: 10.1038/ng.2856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Pina B, Suau P. Changes in histones H2A and H3 variant composition in differentiating and mature rat brain cortical neurons. Dev Biol. 1987;123:51–58. doi: 10.1016/0012-1606(87)90426-x. [DOI] [PubMed] [Google Scholar]
- 71.Toyama BH, et al. Identification of long-lived proteins reveals exceptional stability of essential cellular structures. Cell. 2013;154:971–982. doi: 10.1016/j.cell.2013.07.037. This study uses metabolic pulse-chase labelling in rats to show that canonical H3 proteins (for example, H3.1) are highly stable in adult neural chromatin. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hake SB, et al. Expression patterns and post-translational modifications associated with mammalian histone H3 variants. J Biol Chem. 2006;281:559–568. doi: 10.1074/jbc.M509266200. [DOI] [PubMed] [Google Scholar]
- 73.Hake SB, Allis CD. Histone H3 variants and their potential role in indexing mammalian genomes: the “H3 barcode hypothesis”. Proc Natl Acad Sci USA. 2006;103:6428–6435. doi: 10.1073/pnas.0600803103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Bernstein E, Hake SB. The nucleosome: a little variation goes a long way. Biochem Cell Biol. 2006;84:505–517. doi: 10.1139/o06-085. [DOI] [PubMed] [Google Scholar]
- 75.Michod D, et al. Calcium-dependent dephosphorylation of the histone chaperone DAXX regulates H3.3 loading and transcription upon neuronal activation. Neuron. 2012;74:122–135. doi: 10.1016/j.neuron.2012.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Najmabadi H, et al. Deep sequencing reveals 50 novel genes for recessive cognitive disorders. Nature. 2011;478:57–63. doi: 10.1038/nature10423. [DOI] [PubMed] [Google Scholar]
- 77.Cox SG, et al. An essential role of variant histone H3.3 for ectomesenchyme potential of the cranial neural crest. PLoS Genet. 2012;8:e1002938. doi: 10.1371/journal.pgen.1002938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Bonisch C, et al. H2A.Z.2.2 is an alternatively spliced histone H2A.Z variant that causes severe nucleosome destabilization. Nucleic Acids Res. 2012;40:5951–5964. doi: 10.1093/nar/gks267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Santoro SW, Dulac C. The activity-dependent histone variant H2BE modulates the life span of olfactory neurons. eLife. 2012;1:e00070. doi: 10.7554/eLife.00070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Vardabasso C, et al. Histone variants: emerging players in cancer biology. Cell Mol Life Sci. 2013;71:379–404. doi: 10.1007/s00018-013-1343-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Graber MW, Schweinfest CW, Reed CE, Papas TS, Baron PL. Isolation of differentially expressed genes in carcinoma of the esophagus. Ann Surg Oncol. 1996;3:192–197. doi: 10.1007/BF02305800. [DOI] [PubMed] [Google Scholar]
- 82.Schwartzentruber J, et al. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature. 2012;482:226–231. doi: 10.1038/nature10833. This work is the first to identify driver mutations that are specific to the histone variant H3.3 in human cancer. [DOI] [PubMed] [Google Scholar]
- 83.Wu G, et al. Somatic histone H3 alterations in pediatric diffuse intrinsic pontine gliomas and non-brainstem glioblastomas. Nature Genet. 2012;44:251–253. doi: 10.1038/ng.1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Khuong-Quang DA, et al. K27M mutation in histone H3.3 defines clinically and biologically distinct subgroups of pediatric diffuse intrinsic pontine gliomas. Acta Neuropathol. 2012;124:439–447. doi: 10.1007/s00401-012-0998-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Lewis PW, et al. Inhibition of PRC2 activity by a gain-of-function H3 mutation found in pediatric glioblastoma. Science. 2013;340:857–861. doi: 10.1126/science.1232245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Chan KM, et al. The histone H3.3K27M mutation in pediatric glioma reprograms H3K27 methylation and gene expression. Genes Dev. 2013;27:985–990. doi: 10.1101/gad.217778.113. References 85 and 86 show the dominant negative effects of histone H3 mutations on the activity of PRC2 methyltransferase activity in paediatric gliomagenesis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Goldberg AD, et al. Distinct factors control histone variant H3.3 localization at specific genomic regions. Cell. 2010;140:678–691. doi: 10.1016/j.cell.2010.01.003. This work provides the first genome-wide profiles of a histone variant (H3.3) in a mammalian system. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Elsaesser SJ, Goldberg AD, Allis CD. New functions for an old variant: no substitute for histone H3.3. Curr Opin Genet Dev. 2010;20:110–117. doi: 10.1016/j.gde.2010.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kang B, et al. Phosphorylation of H4 Ser47 promotes HIRA-mediated nucleosome assembly. Genes Dev. 2011;25:1359–1364. doi: 10.1101/gad.2055511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Drane P, Ouararhni K, Depaux A, Shuaib M, Hamiche A. The death-associated protein DAXX is a novel histone chaperone involved in the replication-independent deposition of H3.3. Genes Dev. 2010;24:1253–1265. doi: 10.1101/gad.566910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Lewis PW, Elsaesser SJ, Noh KM, Stadler SC, Allis CD. DAXX is an H3.3-specific histone chaperone and cooperates with ATRX in replication-independent chromatin assembly at telomeres. Proc Natl Acad Sci USA. 2010;107:14075–14080. doi: 10.1073/pnas.1008850107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Jiao Y, et al. DAXX/ATRX, MEN1, and mTOR pathway genes are frequently altered in pancreatic neuroendocrine tumors. Science. 2011;331:1199–1203. doi: 10.1126/science.1200609. This paper is the first to show that mutations in histone variant-associated chaperones are linked to human PanNETs. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Cheung NK, et al. Association of age at diagnosis and genetic mutations in patients with neuroblastoma. J Am Med Associ. 2012;307:1062–1071. doi: 10.1001/jama.2012.228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Ding L, et al. Clonal evolution in relapsed acute myeloid leukaemia revealed by whole-genome sequencing. Nature. 2012;481:506–510. doi: 10.1038/nature10738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Garrick D, et al. Loss of Atrx affects trophoblast development and the pattern of X-inactivation in extraembryonic tissues. PLoS Genet. 2006;2:e58. doi: 10.1371/journal.pgen.0020058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Michaelson JS, Bader D, Kuo F, Kozak C, Leder P. Loss of Daxx, a promiscuously interacting protein, results in extensive apoptosis in early mouse development. Genes Dev. 1999;13:1918–1923. doi: 10.1101/gad.13.15.1918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Baumann C, Viveiros MM, De La Fuente R. Loss of maternal ATRX results in centromere instability and aneuploidy in the mammalian oocyte and pre-implantation embryo. PLoS Genet. 2010;6:e1001137. doi: 10.1371/journal.pgen.1001137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Wong LH, et al. ATRX interacts with H3.3 in maintaining telomere structural integrity in pluripotent embryonic stem cells. Genome Res. 2010;20:351–360. doi: 10.1101/gr.101477.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Heaphy CM, et al. Altered telomeres in tumors with ATRX and DAXX mutations. Science. 2011;333:425. doi: 10.1126/science.1207313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Cao Y, et al. Whole exome sequencing of insulinoma reveals recurrent T372R mutations in YY1. Nature Commun. 2013;4:2810. doi: 10.1038/ncomms3810. [DOI] [PubMed] [Google Scholar]
- 101.Behjati S, et al. Distinct H3F3A and H3F3B driver mutations define chondroblastoma and giant cell tumor of bone. Nature Genet. 2013;45:1479–1482. doi: 10.1038/ng.2814. This recent paper identifies H3.3-specific driver mutations in chondroblastoma and giant cell tumours of bone, which indicates that H3.3 mutations probably result in a large variety of human cancers. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Broniscer A, Gajjar A. Supratentorial high-grade astrocytoma and diffuse brainstem glioma: two challenges for the pediatric oncologist. Oncologist. 2004;9:197–206. doi: 10.1634/theoncologist.9-2-197. [DOI] [PubMed] [Google Scholar]
- 103.Barrow J, et al. Homozygous loss of ADAM3A revealed by genome-wide analysis of pediatric high-grade glioma and diffuse intrinsic pontine gliomas. Neuro Oncol. 2011;13:212–222. doi: 10.1093/neuonc/noq158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Bax DA, et al. A distinct spectrum of copy number aberrations in pediatric high-grade gliomas. Clin Cancer Res. 2010;16:3368–3377. doi: 10.1158/1078-0432.CCR-10-0438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Zarghooni M, et al. Whole-genome profiling of pediatric diffuse intrinsic pontine gliomas highlights platelet-derived growth factor receptor α and poly (ADP-ribose) polymerase as potential therapeutic targets. J Clin Oncol. 2010;28:1337–1344. doi: 10.1200/JCO.2009.25.5463. [DOI] [PubMed] [Google Scholar]
- 106.Paugh BS, et al. Integrated molecular genetic profiling of pediatric high-grade gliomas reveals key differences with the adult disease. J Clin Oncol. 2010;28:3061–3068. doi: 10.1200/JCO.2009.26.7252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Margueron R, Reinberg D. The Polycomb complex PRC2 and its mark in life. Nature. 2011;469:343–349. doi: 10.1038/nature09784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Wagner EJ, Carpenter PB. Understanding the language of Lys 6 methylation at histone H3. Nature Rev Mol Cell Biol. 2012;13:115–126. doi: 10.1038/nrm3274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Hock H. A complex Polycomb issue: the two faces of EZH2 in cancer. Genes Dev. 2012;26:751–755. doi: 10.1101/gad.191163.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Adam S, Polo SE, Almouzni G. Transcription recovery after DNA damage requires chromatin priming by the H3.3 histone chaperone HIRA. Cell. 2013;155:94–106. doi: 10.1016/j.cell.2013.08.029. This paper is the first to demonstrate an essential role for HIRA, which is an H3.3-specific chaperone, during periods of transcriptional recovery after DNA damage. [DOI] [PubMed] [Google Scholar]
- 111.Black BE, Cleveland DW. Epigenetic centromere propagation and the nature of CENP-A nucleosomes. Cell. 2011;144:471–479. doi: 10.1016/j.cell.2011.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Warburton PE. Chromosomal dynamics of human neocentromere formation. Chromosome Res. 2004;12:617–626. doi: 10.1023/B:CHRO.0000036585.44138.4b. [DOI] [PubMed] [Google Scholar]
- 113.Howman EV, et al. Early disruption of centromeric chromatin organization in centromere protein A (Cenpa) null mice. Proc Natl Acad Sci USA. 2000;97:1148–1153. doi: 10.1073/pnas.97.3.1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Mishra PK, et al. Misregulation of Scm3p/HJURP causes chromosome instability in Saccharomyces cerevisiae and human cells. PLoS Genet. 2011;7:e1002303. doi: 10.1371/journal.pgen.1002303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Tomonaga T, et al. Overexpression and mistargeting of centromere protein-A in human primary colorectal cancer. Cancer Res. 2003;63:3511–3516. [PubMed] [Google Scholar]
- 116.Biermann K, et al. Gene expression profiling identifies new biological markers of neoplastic germ cells. Anticancer Res. 2007;27:3091–3100. [PubMed] [Google Scholar]
- 117.Wu Q, et al. Expression and prognostic significance of centromere protein A in human lung adenocarcinoma. Lung Cancer. 2012;77:407–414. doi: 10.1016/j.lungcan.2012.04.007. [DOI] [PubMed] [Google Scholar]
- 118.McGovern SL, Qi Y, Pusztai L, Symmans WF, Buchholz TA. Centromere protein-A, an essential centromere protein, is a prognostic marker for relapse in estrogen receptor-positive breast cancer. Breast Cancer Res. 2012;14:R72. doi: 10.1186/bcr3181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Li YM, Liu XH, Cao XZ, Wang L, Zhu MH. Expression of centromere protein A in hepatocellular carcinoma. Zhonghua Bing Li Xue Za Zhi. 2007;36:175–178. (in Chinese) [PubMed] [Google Scholar]
- 120.Li Y, et al. shRNA-targeted centromere protein A inhibits hepatocellular carcinoma growth. PLoS ONE. 2011;6:e17794. doi: 10.1371/journal.pone.0017794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Hua S, et al. Genomic analysis of estrogen cascade reveals histone variant H2A.Z associated with breast cancer progression. Mol Systems Biol. 2008;4:188. doi: 10.1038/msb.2008.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Dunican DS, McWilliam P, Tighe O, Parle-McDermott A, Croke DT. Gene expression differences between the microsatellite instability (MIN) and chromosomal instability (CIN) phenotypes in colorectal cancer revealed by high-density cDNA array hybridization. Oncogene. 2002;21:3253–3257. doi: 10.1038/sj.onc.1205431. [DOI] [PubMed] [Google Scholar]
- 123.Rhodes DR, et al. Large-scale meta-analysis of cancer microarray data identifies common transcriptional profiles of neoplastic transformation and progression. Proc Natl Acad Sci USA. 2004;101:9309–9314. doi: 10.1073/pnas.0401994101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Zucchi I, et al. Gene expression profiles of epithelial cells microscopically isolated from a breast-invasive ductal carcinoma and a nodal metastasis. Proc Natl Acad Sci USA. 2004;101:18147–18152. doi: 10.1073/pnas.0408260101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Xu Y, et al. Histone H2A.Z controls a critical chromatin remodeling step required for DNA double-strand break repair. Mol Cell. 2012;48:723–733. doi: 10.1016/j.molcel.2012.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Greaves IK, Rangasamy D, Ridgway P, Tremethick DJ. H2A.Z contributes to the unique 3D structure of the centromere. Proc Natl Acad Sci USA. 2007;104:525–530. doi: 10.1073/pnas.0607870104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Rangasamy D, Berven L, Ridgway P, Tremethick DJ. Pericentric heterochromatin becomes enriched with H2A.Z during early mammalian development. EMBO J. 2003;22:1599–1607. doi: 10.1093/emboj/cdg160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Shia WJ, Li B, Workman JL. SAS-mediated acetylation of histone H4 Lys16 is required for H2A.Z incorporation at subtelomeric regions in Saccharomyces cerevisiae. Genes Dev. 2006;20:2507–2512. doi: 10.1101/gad.1439206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Rogakou EP, Pilch DR, Orr AH, Ivanova VS, Bonner WM. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J Biol Chem. 1998;273:5858–5868. doi: 10.1074/jbc.273.10.5858. [DOI] [PubMed] [Google Scholar]
- 130.Fernandez-Capetillo O, Lee A, Nussenzweig M, Nussenzweig A. H2AX: the histone guardian of the genome. DNA Repair. 2004;3:959–967. doi: 10.1016/j.dnarep.2004.03.024. [DOI] [PubMed] [Google Scholar]
- 131.Li A, et al. Phosphorylation of histone H2A.X by DNA-dependent protein kinase is not affected by core histone acetylation, but it alters nucleosome stability and histone H1 binding. J Biol Chem. 2010;285:17778–17788. doi: 10.1074/jbc.M110.116426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Xiao A, et al. WSTF regulates the H2A.X DNA damage response via a novel tyrosine kinase activity. Nature. 2009;457:57–62. doi: 10.1038/nature07668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Zhu F, et al. Phosphorylation of H2AX at Ser139 and a new phosphorylation site Ser16 by RSK2 decreases H2AX ubiquitination and inhibits cell transformation. Cancer Res. 2011;71:393–403. doi: 10.1158/0008-5472.CAN-10-2012. [DOI] [PubMed] [Google Scholar]
- 134.Monni O, Knuutila S. 11q deletions in hematological malignancies. Leuk Lymphoma. 2001;40:259–266. doi: 10.3109/10428190109057924. [DOI] [PubMed] [Google Scholar]
- 135.Thirman MJ, et al. Rearrangement of the MLL gene in acute lymphoblastic and acute myeloid leukemias with 11q23 chromosomal translocations. New Engl J Med. 1993;329:909–914. doi: 10.1056/NEJM199309233291302. [DOI] [PubMed] [Google Scholar]
- 136.Stankovic T, et al. ATM mutations in sporadic lymphoid tumours. Leuk Lymphoma. 2002;43:1563–1571. doi: 10.1080/1042819021000002884. [DOI] [PubMed] [Google Scholar]
- 137.Parikh RA, et al. Loss of distal 11q is associated with DNA repair deficiency and reduced sensitivity to ionizing radiation in head and neck squamous cell carcinoma. Genes Chromosomes Cancer. 2007;46:761–775. doi: 10.1002/gcc.20462. [DOI] [PubMed] [Google Scholar]
- 138.Srivastava N, Gochhait S, Gupta P, Bamezai RN. Copy number alterations of the H2AFX gene in sporadic breast cancer patients. Cancer Genet Cytogenet. 2008;180:121–128. doi: 10.1016/j.cancergencyto.2007.09.024. [DOI] [PubMed] [Google Scholar]
- 139.Sporn JC, Jung B. Differential regulation and predictive potential of macroH2A1 isoforms in colon cancer. Am J Pathol. 2012;180:2516–2526. doi: 10.1016/j.ajpath.2012.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Sporn JC, et al. Histone macroH2A isoforms predict the risk of lung cancer recurrence. Oncogene. 2009;28:3423–3428. doi: 10.1038/onc.2009.26. [DOI] [PubMed] [Google Scholar]
- 141.Kapoor A, et al. The histone variant macroH2A suppresses melanoma progression through regulation of CDK8. Nature. 2010;468:1105–1109. doi: 10.1038/nature09590. This study shows that mH2A is a critical chromatin factor that is associated with suppression of malignant melanoma development. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Novikov L, et al. QKI-mediated alternative splicing of the histone variant macroH2A1 regulates cancer cell proliferation. Mol Cell Biol. 2011;31:4244–4255. doi: 10.1128/MCB.05244-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Li X, et al. The atypical histone macroH2A1.2 interacts with HER-2 protein in cancer cells. J Biol Chem. 2012;287:23171–23183. doi: 10.1074/jbc.M112.379412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Zhang R, et al. Formation of macroH2A-containing senescence-associated heterochromatin foci and senescence driven by ASF1a and HIRA. Dev Cell. 2005;8:19–30. doi: 10.1016/j.devcel.2004.10.019. [DOI] [PubMed] [Google Scholar]
- 145.Ratnakumar K, et al. ATRX-mediated chromatin association of histone variant macroH2A1 regulates α-globin expression. Genes Dev. 2012;26:433–438. doi: 10.1101/gad.179416.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Jullien J, Astrand C, Halley-Stott RP, Garrett N, Gurdon JB. Characterization of somatic cell nuclear reprogramming by oocytes in which a linker histone is required for pluripotency gene reactivation. Proc Natl Acad Sci USA. 2010;107:5483–5488. doi: 10.1073/pnas.1000599107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Nashun B, Akiyama T, Suzuki MG, Aoki F. Dramatic replacement of histone variants during genome remodeling in nuclear-transferred embryos. Epigenet. 2011;6:1489–1497. doi: 10.4161/epi.6.12.18206. [DOI] [PubMed] [Google Scholar]
- 148.Chang CC, et al. Rapid elimination of the histone variant MacroH2A from somatic cell heterochromatin after nuclear transfer. Cell Reprogramm. 2010;12:43–53. doi: 10.1089/cell.2009.0043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Perez-Montero S, Carbonell A, Moran T, Vaquero A, Azorin F. The embryonic linker histone H1 variant of Drosophila, dBigH1, regulates zygotic genome activation. Dev Cell. 2013;26:578–590. doi: 10.1016/j.devcel.2013.08.011. [DOI] [PubMed] [Google Scholar]
- 150.Barrero MJ, et al. Macrohistone variants preserve cell identity by preventing the gain of H3K4me2 during reprogramming to pluripotency. Cell Rep. 2013;3:1005–1011. doi: 10.1016/j.celrep.2013.02.029. [DOI] [PubMed] [Google Scholar]
- 151.Pasque V, et al. Histone variant macroH2A marks embryonic differentiation in vivo and acts as an epigenetic barrier to induced pluripotency. J Cell Sci. 2012;125:6094–6104. doi: 10.1242/jcs.113019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Gaspar-Maia A, et al. MacroH2A histone variants act as a barrier upon reprogramming towards pluripotency. Nature Commun. 2013;4:1565. doi: 10.1038/ncomms2582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Schones DE, et al. Dynamic regulation of nucleosome positioning in the human genome. Cell. 2008;132:887–898. doi: 10.1016/j.cell.2008.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Luger K, et al. Crystal structure of the nucleosome core particle at 2.8 Å resolution. Nature. 1997;389:251–260. doi: 10.1038/38444. [DOI] [PubMed] [Google Scholar]