

Abstract
Isoprenoids constitute the largest class of natural products with greater than 55,000 identified members. They play essential roles in maintaining proper cellular function leading to maintenance of human health, plant defense mechanisms against predators, and are often exploited for their beneficial properties in the pharmaceutical and nutraceutical industries. Most impressively, all known isoprenoids are derived from one of two C5-precursors, isopentenyl diphosphate (IPP) or dimethylallyl diphosphate (DMAPP). In order to study the enzyme transformations leading to the extensive structural diversity found within this class of compounds there must be access to the substrates. Sometimes, intermediates within a biological pathway can be isolated and used directly to study enzyme/pathway function. However, the primary route to most of the isoprenoid intermediates is through chemical catalysis. As such, this review provides the first exhaustive examination of synthetic routes to isoprenoid and isoprenoid precursors with particular emphasis on the syntheses of intermediates found as part of the 2C-methylerythritol 4-phosphate (MEP) pathway. In addition, representative syntheses are presented for the monoterpenes (C10), sesquiterpenes (C15), diterpenes (C20), triterpenes (C30) and tetraterpenes (C40). Finally, in some instances, the synthetic routes to substrate analogs found both within the MEP pathway and downstream isoprenoids are examined.
Keywords: Enzyme mechanism, isoprenoids, terpenes, MEP pathway.
INTRODUCTION
With greater than 55,000 representative compounds, isoprenoids (also referred to as terpenes) are the largest and most chemically diverse family [1]. Isoprenoids are classified into groups according to the number of carbon atoms they contain, and include: hemiterpenes (C5), monoterpenes (C10), sesquiterpenes (C15), diterpenes (C20), triterpenes (C30), tetraterpenes (C40) and so forth. They play numerous functional roles in primary metabolic processes that are necessary for cell survival including cell wall and membrane biosynthesis (phytosterols), electron transport (plastoquinones), photosynthetic pigments (carotenoids), and hormones in plants (gibberellins) [2, 3]. In addition, microbial production of isoprenoids has found their way into industry as a source for nutraceuticals, pharmaceuticals and, most recently, in biofuel applications [4]. Some examples include fragrances and essential oils (menthol, eucalyptol, limonene), cosmetics (squalene), disinfectants (pinene and camphor), antimicrobials (taxol and artimesinen), food supplements (Vitamins A, E and K), food colorants (xanthanins), fuels (farnesene and botryococcenes) and dozens more of related industrial applications.
The chemistry of isoprenoid biosynthesis allows for a tremendous structural diversity within its members, both in terms of the carbon length of the molecule (e.g. monoterpenes, diterpenes) and in the connectivity observed in the isoprenoid chain after biosynthetic steps which process diphosphates (e.g. cyclization of a triterpene, condensation of two sesquiterpene diphosphates). Understandably, this chemical diversity has enabled members of the isoprenoid family to play a wide range of important biological roles throughout nature as previously mentioned. However, the biology of isoprenoid molecules is beyond the scope of this review. Generally speaking, research into isoprenoid chemistry includes work that aims to understand their role in endogenous biological pathways of relevance to human health (two such examples are the enzymology of cholesterol biosynthesis and the role of protein farnesylation in cancer progression), as well as work which is designed to isolate and characterize isoprenoid secondary metabolites of value to society (e.g. anticancer agents such as Taxol, or potential renewable fuels such as isoprene, farnesene, or botryococcenes), human health, plant and fungal biochemistry. Thus, the development of chemical routes to isoprenoid compounds aids the synthesis of some pharmacologically-valuable compounds and intermediates (many of which themselves represent challenging synthetic targets) as well as enables enzymological characterization and inhibition studies. In this review, we will focus primarily on synthetic routes that have been developed for isoprenoid intermediates for the purpose of enzyme discovery and characterization.
The chemical diversity of all isoprenoids is derived from one of two simple C5 building blocks: isopentenyl diphosphate (IPP) and its isomer dimethylallyl diphosphate (DMAPP). These universal precursors are produced by either of two routes: the mevalonate or the 2C-methyl-D-erythritol 4-phosphate (MEP) (also known as the non-mevalonate or 1-deoxy-D-xylulose 5-phosphate, DXP) pathway (Scheme 1). Both biosynthetic routes are distributed throughout nature, and as a general rule of thumb the mevalonate pathway is prevalent in eukaryotes and archaea, whereas the MEP pathway is widespread in eubacteria. For this review, the synthetic intermediates found as part of the mevalonate pathway will not be discussed. Briefly, the MEP pathway begins with condensation of glyceraldehyde-3-phosphate (G3P) and pyruvate to generate DXP through the action of deoxyxylulose phosphate synthase (DXS) [5]. DXP then undergoes a carbon skeleton rearrangement and a NADPH-dependent reduction catalyzed by MEP synthase, which represents the first committed step in the MEP pathway [6]. Addition of cytidyl monophosphate (CMP) to MEP via the action of a CTP-dependent (CDPME) synthase [7], followed by activation of the tertiary alcohol to the tertiary phosphate (CDPME2P) with an ATP-dependent kinase [8] results in the cyclized product, methylerythritol-2,4-cyclodiphosphate (cMEPP), catalyzed by the similarly named synthase [9]. The final two steps each involve the reductive elimination of water, first through the action of cMEPP reductase to provide hydroxydimethylallyl disphosphate (HDMAPP) followed by HDMAPP reductase that result in the formation of both IPP and DMAPP [10]. IPP isomerase (IDI) interconverts IPP and DMAPP to provide the appropriate ratios for normal cellular function [11]. Condensation of the two C5-building blocks catalyzed by geranyl diphosphate synthase (GPPS) provides the monoterpene, GPP. This C10 subunit is the building block for the thousands of monoterpenes discovered thus far, a few of which are represented in this review. Addition of another IPP to the GPP framework by farnesyl diphosphate synthase (FPPS) results in the C15 foundation from which thousands of sesquiterpenes and triterpenes are found in Nature. Finally, geranylgeranyl diphosphate synthase (GGPPS) catalyzes the condensation of another IPP subunit to the FPP framework to provide GGPP, which is a precursor for many naturally occurring antioxidants, for example β-carotene. While not exhaustive, for there are scores of synthetic routes to varying chain-length isoprenoids and isoprenoid precursors, this review will focus on the syntheses of MEP pathway intermediates to the universal precursors, IPP and DMAPP, followed by representative synthetic routes to longer branched, unsaturated hydrocarbons that may be of interest to the reader.
Scheme. (1).

The MEP pathway to isoprenoids.
Isoprenoid Precursors-Methylerythritol Phosphate (MEP) Pathway
Since the MEP pathway discovery to isoprenoids in the early 90s, considerable effort toward elucidating the enzymes responsible for their biosynthesis ensued [6, 12, 13]. Early studies revealed the MEP pathway was distributed in most bacteria, the chloroplasts of photosynthetic organisms, unicellular eukaryotes (e.g. green algae) and the malaria parasite [14]. Most importantly, the MEP pathway enzymes are excellent targets for the development of antimicrobial agents for there are no known human orthologs [15]. The first enzyme in the MEP pathway to isoprenoids, deoxyxylulose phosphate synthase, converts pyruvate and glyceraldehyde-3-phosphate to (–)-1-deoxy-D-xylulose 5-phosphate (DXP). Conversion of DXP to MEP through MEP synthase represents the first committed step to all isoprenoids where the mevalonate pathway is not present. Therefore, a fundamental understanding of the biological role of MEP synthase was exhaustively pursued, largely through the use of synthetic (isotopically-labeled) DX(P) substrates and corresponding substrate analogs.
Many examples for the enzyme-assisted synthesis of both DX and DXP and the corresponding isotopomers are reported in the literature [16-19]. However, preparation of large quantities of these deoxyxyluloses is not the preferred route largely due to purification problems, the requirement for expensive labeled precursors and scalability. In addition, biosynthetic production of DX(P) analogs is largely unattainable due to the stringent substrate specificity of the corresponding enzymes responsible for their biosynthesis. Therefore, synthetic preparations to these compounds are preferred due to the versatility of the chemical transformations from relatively cheap starting material, high-yielding reactions, and simple purifications.
Syntheses of 1-deoxy-D-xylulose 4 (DX)-The first examples for the synthesis of the D-enantiomer of DX were reported by Estramareix et al. in the early 80s. Following protection and oxidative cleavage of D-arabinitol 1, condensation of isopropylidene-protected D-glyceraldehye and the acetaldehyde dithioacetal anion provided a mixture of the protected threose and erythro-isomers. Separation of the threose enantiomer from the erythro-form followed by deprotection provided D-deoxylulose 4 over three steps in 15% yield [20]. The same group improved upon the synthesis to deoxyxylulose and the 1-2H3-isotopically-labeled derivativethrough condensation of aldehyde 2 with methylmagnesium iodide to provide the 2,4-O-benzylidene-D-threose intermediate 3. Following oxidation and deprotection, the free sugar 4 was obtained by brominolysis of the distannylidene derivative (Scheme 2) [21, 22].
Scheme. (2).

Estramareix synthetic route to DX.
Importantly, the Grignard reaction for introduction of the methyl group is now the most widely used method for incorporation of isotopes into the C-1 position of deoxyxylulose (phosphate). Further, Estramareix et al. provided the foundation for future synthetic efforts through exploitation of the D-threose scaffold in order to set the correct stereochemistry in the final product. As such, multiple independent routes to the free sugar using either D-threose or its oxidized derivative, D-tartrate, were employed and overall yields were much improved [23-25], with the highest reported yield of 68% over five steps from dimethyl-D-tartrate [26]. A generalized reaction scheme for DX 4 syntheses from multiple laboratories starting from either the D-tartrate 5 or D-threitol 6 derivatives are outlined below (Scheme 3).
Scheme. (3).

Generalized synthetic routes to enantiomerically pure DX.
The previously outlined synthetic routes to DX provide a convenient platform for incorporation of a stable isotope (2H or 13C) into the C-1 position of the free sugar via an aldehyde intermediate. However, incorporation of an isotopic tag at other positions along the polyol backbone is not feasible using these approaches. An alternative strategy was adopted by Giner et al. that enabled insertion of deuterium at the C-3 and/or C-4 positions of DX. Briefly, deuterium-hydrogen exchange of the phosphonium ylide 7 with a 10:1:1 mixture of CH2Cl2:D2O:CD3OD followed by a Wittig reaction with the benzyl-protected glycoaldehyde 8 provided the conjugated 5-carbon enone 9 with 85% deuterium incorporation at the C-3 position. Subsequent Sharpless asymmetric dihydroxylation and debenzylation via hydrogenolysis provided the enantiomerically pure DX 4 in good yields [27-29]. Conversely, the deuterium was conveniently inserted into the C-4 position (~70% deuterium) by reduction of the propargyl alcohol 10 with lithium aluminum deuteride, followed by oxidation, asymmetric dihydroxylation then deprotection, although yields were not reported for the reduction step [24]. The authors also demonstrated incorporation of a 13C-label into the C-1 position of aldehyde 11 to provide 1-13C-DX 4 in 52% yield over four steps through enone intermediate 12 (Scheme 4).
Scheme. (4).

Giner’s syntheses of C-1 isotopically labeled DX.
A convenient chemoenzymatic synthesis for 1-deoxy-5,5-2H2-D-xylulose was outlined by Piel and Boland starting from 2,3-O-isopropylide-dimethyl-D-tartrate. Briefly, pig liver esterase was used to convert the starting material to the monoester. Following reduction of the ester to the alcohol with LiBEt3D, methylation of the acid and deprotection of the isopropylidene moiety, the C-5 dideuterated DX was synthesized over four steps in 37% yield [30]. However, scalability was an issue since the first step required the use of the esterase.
Syntheses of 1-deoxy-D-xylulose 5-phosphate 16 (DXP), isotopomers, and fluorinated analogs-The first report for the synthesis of enantiomerically pure DXP was outlined by Begley et al. in 1998 [31]. The key step in this synthesis was desymmetrization of the C2-symmetric dibenzylated-D-threitol by monophosphorylation with dibenzylchlorophosorochloridate in 40% yield. The primary alcohol was converted to the methyl ketone using modified procedures previously outlined then debenzylation via hydrogenolysis provided DXP over seven steps in 5% yield from diethyl-D-tartrate. Shortly thereafter, Blagg and Poulter reported a more efficient chemical synthesis for DXP by exploiting (–)-2,3-O-isopropylidene-D-threitol 13 as a more suitable starting material for the chemical transformations (Scheme 5). The triisopropylsilyloxy-protected DX 14 proved to be a versatile intermediate in the synthesis. Simple deprotection under acidic conditions provided DX 4 over five steps in 69% yield. Alternatively, following ketolization of the ketone with ethylene glycol and removal of the triisopropylsilyloxy-protecting group, the primary alcohol 15 was phosphorylated with trimethylated phosphite in the presence tellurium (IV) chloride. Phosphate deprotection in the presence of TMSBr followed by acidic workup and purification by cellulose chromatography provided pure DXP 16 in four steps from the fully protected DX in 71% yield [26].
Scheme. (5).

Blagg and Poulter’s synthetic routes to both DX and DXP from D-threitol.
MEP synthase is generally recognized as an excellent target for the development of novel antimicrobial agents. However, the enzyme-catalyzed reaction mechanism for conversion of DXP to MEP remained elusive. In order to address this gap, multiple isotopically-labeled DXP and DXP analogs were synthesized to determine the stereochemical course for the transformation. In 2003, Cox et al. reported a synthetic route to DXP from propargyl diol 17 where deuterium was incorporated into either the C3-18 or C4-19 positions, respectively, using a modified protocol reported by Giner et al. [32]. Silyl deprotection and selective phosphorylation of the primary alcohol over the secondary alcohol provided 20 and 21, respectively. However, the overall yield and enantiopurity was suboptimal (~20%, 84% e.e.) and no demonstration for isotopic label incorporation was discussed [33]. The same authors amended their synthesis for incorporation of the deuterium label into either the [3-2H]-16 or [4-2H]-16 position of DXP over eight steps in 15% or 7% yield (Scheme 6), respectively, and corroborated previous reports that MEP synthase converts DXP to MEP through a retroaldol/aldol mechanism [34-36]. Shortly thereafter, Liu et al. reported two independent routes to the identical compounds with similar yields (~8%, 95% e.e.) [37].
Scheme. (6).

Cox’s synthetic routes to DXP from the propargyl alcohol.
There are countless reports in the literature for the synthesis of DXP analogs that extend beyond the scope of this review. However, one class of DXP analogs deserves discussion since they were designed for three reasons: potential as novel inhibitors, determination of the enzyme-catalyzed reaction mechanism, and substrate specificity. Specifically, the fluorinated analogs of either DX or DXP were chemically synthesized for this purpose. The first report of a fluorinated deoxyxylulose analog was reported by Bouvet and O’Hagan for the synthesis of both 1-fluoro and 1,1-difluoro-1-deoxy-D-xylulose [38]. In their synthesis of the mono-fluorinated analog 24, the authors utilized a Wittig reaction for coupling of the silyl-protected glycoaldehyde 22 with the mono-fluorinated phosphorous ylide 23 in a reaction analogous to previous reports for the [3- and [4-2H]-DX syntheses [29]. However, Sharpless asymmetric dihydroxylation was unsuccessful thus necessitating standard dihydroxylation with osmium tetraoxide to provide the racemic fluorinated diol 25. Deprotection of the silyl group provided the (±) C-1 monofluorinated DX in 30% yield over three steps as a 3:2 mixture of the α- and β-cyclic fluorinated anomers 26 and 27 (Scheme 7).
Scheme. (7).

O’Hagan’s synthetic route to CF-DX.
Using O’Hagan’s syntheses as an inspiration, Fox and Poulter synthesized the 1-fluoro-, 1,1-difluoro- and 1,1,1-trifluoromethyl-1-deoxy-D-xylulose 5-phosphate analogs for mechanistic studies with MEP synthase [39]. Upon installation of the oxirane moiety into aldehyde 28, the key step in the synthesis of the 1-fluoromethyl-DXP (CF-DXP) was the regioselective ring opening of the intermediate oxirane 29 with diisopropylamine trihydrogen fluoride [40] at the C-1 position to provide the monofluorinated alcohol 30 in 45-55% yield. Debenzylation followed by regioselective phosphorylation of the primary alcohol with dibenzyl phosphoroiodidate and subsequent Dess-Martin oxidation provided the fully protected CF-DXP. Debenzylation of the phosphate by hydrogenolysis in tert-butanol instead of methanol or ethanol was necessary to avoid formation of the stable hemiketal due to the presence of the adjacent fluorine. The free acid effectively removed the isopropylidene moiety to provide CF-DXP 31 in 10% yield, with greater than 95% purity, over nine steps as a mixture of the ketone and hydrate forms (77:23) (Scheme 8). A much lengthier synthesis of the identical compound was independently reported by Liu and coworkers. In their synthesis of 1-fluoromethyl-DXP, fluorination of the primary alcohol, which was derived from D-arabinose in three steps, with triflate and tetrabutylammonium fluoride (TBAF) provided the monofluorinated sugar. Deprotection of the 1,2-diol, benzylation and reductive ring opening with sodium borohydride provided the open chain-polyol. Regioselective benzyolation of the primary alcohol and Swern oxidation resulted in the fully protected monofluorinated deoxyxylulose. A series of protection and deprotection steps allowed for the introduction of the phosphate group with trimethyl phosphite in the presence of tellurium chloride and 2,6-lutidene to afford the protected CF-DXP. Deprotection of the phosphate methyl groups with TMSBr followed by debenzylation by hydrogenolysis provided the free acid of CF-DXP 31. This compound was estimated to be 80% pure as judged by 19F NMR spectroscopy and was obtained in less than 3% yield over 14 steps [36]. In the same manuscript, Liu et al. described the synthesis of either the C-3 or C-4 monofluorinated DXP. These synthetic routes were a modified procedure as outlined for the C-1 monofluorinated DXP, required over 15 steps and the final material was obtained in low yields.
Scheme. (8).

Fox and Poulter’s synthesis to CF-DXP from a D-threitol derivative.
Simple modification of O’Hagan’s synthesis for the 1,1-difluoromethyl deoxyxylulose, via the identical difluoromethylenephosphonate intermediate 33, provided 1,1-difluoro-1-deoxy-D-xylulose 5-phosphate 35 over five steps from the isopropylidene-protected D-tartrate 5. The key difference was installation of the dibenzyl phosphate into the tartrate monoester 32 prior to reaction with the lithiated diethyl(difluoromethyl)phosphonate. C-P bond cleavage of the phosphonate sugar 33, after azeotropic removal of water, with sodium methoxide provided the fully protected CF2-DXP 34 in 83% yield. Deprotection provided the difluorinated sugar phosphate 35 in 53% yield over five steps from the tartrate monoester as a mixture of the ketone/hydrate (2:98) as judged by 19F NMR spectroscopy (Scheme 9).
Scheme. (9).

Fox and Poulter’s synthesis of CF2-DXP from the D-tartrate derivative.
The key step in the synthesis of 1,1,1-trifluoromethyl-1-deoxy-D-xylulose 5-phosphate (CF3-DXP) was insertion of the trifluormethyl moiety into the identical TIPS-protected aldehyde intermediate found in Blagg’s synthesis of DXP [26] using (trifluoromethyl)trimethylsilane and a catalytic amount of tert-butoxide, following a procedure outlined by Olah et al. [41], to provide the bissilyl ether as a 3:1 mixture of diastereomers. Following removal of the silyl ethers with two equivalents of TBAF, regioselective phosphorylation of the primary alcohol was carried out analogous to the synthesis for the monofluorinated DXP derivative. Deprotection using the same strategy as for the mono- and difluorinated DXP analogs afforded the trifluorinated sugar phosphate exclusively as the ketone hydrate in 46% yield over eight steps and greater than 95% purity as judged by 19F NMR spectroscopy.
Syntheses of 2C-methyl-D-erythritol 40 (ME)-The five carbon phosphosugar 2C-methyl-D-erythritol 4-phosphate is the first committed intermediate in the mevalonate-independent route, or MEP pathway, to isoprenoid compounds. The methylerythritol sugar is also known to be recognized, imported and incorporated into (after in vivo phosphorylation to form MEP) the isoprenoid products of various prokaryotes [42, 43]. Thus, synthetic preparations of ME and isotopically-enriched ME proved crucial to the efforts to identify the enzymes responsible for IPP and DMAPP biosynthesis through the MEP pathway [44, 45]. More recently, preparations of ME were instrumental in the discovery of its ability to act as a small-molecule stimulant of isoprenoid biosynthesis [46], and in studies of its influence upon plant development [47]. The identification of MEP synthase (which makes MEP from DXP) and downstream enzymes provided an impetus for synthetic preparations of MEP itself [48].
Although ME is produced in the leaves and flowers of some plants [49], and abiotically through the atmospheric oxidation of isoprene [50-52], discovery efforts in the MEP pathway necessitated access to multi-milligram quantities of the sugar. Prior to these efforts, no dedicated chemical syntheses of the sugar existed, although some syntheses of derivatives of ME had been reported [53]. A facile route to ME was reported by Duvold et al. in 1997, wherein ME was produced in 85% yield (80% e.e.) from 3-methylfuran-2(5H)-one 36 or 25% from citraconic anhydride 37 in four steps [54].
Sharpless asymmetric dihydroxylation was used to introduce the C-2 and C-3 hydroxyl groups with the correct stereochemistry into cis-olefin 38 to provide the diacetate 39 protected ME in excellent yield. Deprotection provided ME 40 in 80% yield over three steps from the furan 36 (Scheme 10) [55]. This route also provided for the incorporation of heavy atom labels into the backbone of ME (as [1,1,4,4-2H]-ME) through reduction of the starting material with LiAlD4. Fontana and coworkers also reported a route to ME from dimethylfumarate (31% yield, 84% e.e., 6 steps) [56].
Scheme. (10).

Rohmer’s synthesis of ME from 3-methylfuran-2(5H)-one or citraconic anhydride.
In this case, establishment of the correct stereochemistry of the hydroxyl groups was afforded through asymmetric epoxidation of the trans-alkene 44 to give epoxide 45 followed by ring opening with sodium hydroxide to provide the benzyl-protected ME 46. Reduction of 46 via hydrogenolysis over palladium gave ME 40 in moderate yield over eight steps (Scheme 11). Heavy atom incorporation is also possible using this route, either in the form of deuterium (reduction with LiAlD4 or similar reagent) or 13C (use of U-13C-dimethylfumarate). Asymmetric hydroxylation or epoxidation of 3-methylbut-2-enyl-1,4-diols to introduce the stereochemistry of the C-2 and -3 hydroxyl groups proved to be a facile and useful means that also found utility in the synthesis of MEP itself and a trifluorinated-derivative of ME [57]. Even more recently, a strategy to ME and its stereoisomers was published reliant upon this means to introduce hydroxyl functionalities [58], although the scheme largely utilizes methods previously reported in the synthesis of MEP [59, 60]. In another compelling application of the asymmetric dihydroxylation reaction in the context of branched carbohydrate synthesis, Taylor and coworkers described a two-step synthesis to ME 40 from 4-methyl 1,2-dioxine 47 (Scheme 12) [61].
Scheme. (11).

Fontana’s synthesis of ME from dimethylfumarate.
Scheme. (12).

Taylor’s synthesis of ME from 4-methyl 1,2-dioxine.
Central to this approach was generation of the dioxine intermediate diol 48 with the correct stereochemistry through photooxidation of a 1,3 butadiene precursor. Although the enantiomeric excess for the synthesis of ME using this method was somewhat low, the authors were able to produce a number of ME analogs with good yield and high enantioselectivity.
There are several reports for the enantiopure synthesis of ME. The use of protected carbohydrates as precursors to ME is a particularly effective means to ensure enantiopurity. Kis et al. were among the first to report such a strategy in their synthesis of ME and MEP in 2000, wherein 1,2:5,6-di-O-isopropylidene-D-mannitol was converted to ME in 10 steps or MEP in 13 steps [62]. In 2003, Hoeffler et al. reported the synthesis of enantiopure ME in 43% yield over 8 steps from 1,2-O-isopropylidene-α-D-xylofuranose 49 [63, 64]. In the authors’ strategy, the 2-methyl functionality in the final product was introduced into a TBDPS-protected 1,2-O-isopropylidene-α-D-xylofuranose 50 by oxidation of the secondary alcohol to ketone 51, followed by methyl-Grignard addition to the less hindered face of the carbohydrate ring to provide the carbon backbone 52 in the correct stereochemistry. Multiple high yielding deprotection/protection steps (53-55) followed by periodate cleavage and aldehyde 56 reduction then debenzylation of the penultimate intermediate 57 resulted in the final ME 40 product (Scheme 13).
Scheme. (13).

Hoeffler’s synthesis of ME from 1,2-O-isopropylidene-α-D-xylofuranose.
In this manner, the authors were able to exclusively obtain the desired diastereomer of the branched chain sugar. As with similar reports, incorporation of deuterium or tritium labels into the final product was enabled in the strategy.
Coates and coworkers have reported a versatile synthesis of ME, MEP and cMEPP from D-arabitol as the starting material [65, 66]. In the authors’ strategy, benzylidene-protected D-arabitol 58 was first converted to 1,3-benzylidene-D-threitol 59 and subsequently transformed to (2S,4R)-cis-2-phenyl-4-tert-butyldimethyl-silyloxy-1,3-dioxan-5-one 61 in two steps. The dioxanone intermediate was selectively alkylated on the axial face (20:1) with methylmagnesium bromide to form the methyltetrol 62 core of ME and related metabolites. The final product is generated by silyl-deprotection to provide 62 then hydrogenolysis to enantiopure ME 40 (8% overall yield from D-arabitol in 9 steps), or alternatively 62 may be further modified to form MEP or cMEPP (Scheme 14). Furthermore, the novel methylerythritol analogue, 1-amino-1-deoxy-2C-methylerythritol was also produced using this approach [65]. Lai and coworkers have reported modifications to this dioxanone strategy which resulted in a shorter synthesis with a slightly higher yield (7 steps, 11.5% overall yield) [67]. A similar strategy of diastereocontrolled addition of an alkyl Grignard reagent into a protected ketone was used by Chattopadhyay and coworkers to provide a straightforward and economical route to ME and other methyltetrol isomers [68].
Scheme. (14).

Coates’ synthesis of ME from D-arabitol.
Syntheses of 2C-methyl-D-erythritol 4-phosphate 71 (MEP)-Similar to other MEP pathway intermediates, MEP was previously synthesized using an enzymatic approach through condensation of glyceraldehyde-3-phosphate and pyruvate [7, 55]. As was observed in the synthesis of ME, several chemical routes to the 4-phosphate MEP are reported, and similarly involve asymmetric dihydroxylation of substituted diols or utilization of carbohydrate starting materials. Poulter and coworkers demonstrated the synthesis of MEP using the former approach in 2000 and 2002 in their synthesis of MEP as a monosodium salt and free acid, respectively [60, 69].
The sodium salt of MEP was synthesized from 1,2-propane diol 64 in 7 steps with an overall yield of 32% (78% e.e.). Following silyl-protection 65 of the primary alcohol and oxidation to provide ketone 66, the cis-configuration was installed through a Wittig-Horner olefination reaction to give cis-olefin 67 in excellent yield. Reduction of the ester and phosphorylation of primary alcohol 68 provided the phosphotriester 69 in 84% yield over the two steps. Dihydroxylation of the olefin 69 was conducted using the method of Sharpless after installation of the phosphotriester, which necessitated buffering of the standard reaction conditions with NaHCO3, to provide the protected MEP 70 in moderate yield [55, 70]. Conversion of the phosphomethylesters to the corresponding acids was accomplished with TMS-Br under acidic conditions, and MEP 71 as the monosodium salt was formed upon neutralization (Scheme 15). Formation of MEP as the free acid is accomplished by introducing the phosphate functionality as a benzyl-protected triester, which is ultimately removed via hydrogenation over Pd/C. Hoeffler et al. used this strategy to install the phosphate moiety into MEP ultimately from an 1,2-O-isopropylidene-α-D-xylofuranose starting material using an approach inspired from the authors earlier work toward the preparation of ME (as discussed above) [64]. Similarly, both Fontana [71] and Koppisch and Poulter [69] utilized benzyl ester protected phosphate groups to form the free acid of MEP from approaches that were inspired by previous efforts of their respective teams.
Scheme. (15).

Koppisch and Poulter’s synthesis of MEP from 1, 2-propane diol.
Synthesis of MEP from various protected carbohydrates, as noted above, has been reported by a number of teams. Enantiopurity of the final product is ensured by utilization of the carbohydrate starting material, and their use also provides a convenient means to introduce isotopic labels. Enantiopure preparations of MEP as the sodium salt from 1,2:5,6-di-O-isopropylidene-D-mannitol proceeded in 9% yield over 14 steps (as compared to 32% over 7 steps) [72] and preparation of the free acid from 1,2-O-isopropylidene-α-D-xylofuranose proceeded in 32% yield over 7 steps (compared to 27% yield over 5 steps) [64]. More recent syntheses from carbohydrate-based starting material have also provided facile access to other metabolites based on an ME backbone (such as MEP or cMEPP) [65, 66] or in the synthesis of derivatives of MEP [73]. As mentioned earlier, a deprotected intermediate of the methylated dioxanone 72 in Coates and coworkers synthesis of ME from D-arabitol is readily phosphorylated by lithiation with n-BuLi followed by reaction with dibenzyl chlorophosphoridate to provide the benzyl-protected MEP 73. Removal of the benzyl protecting groups via hydrogenation followed by treatment with ammonia provided MEP as the ammonium salt in good yield 71 (Scheme 16) [65].
Scheme. (16).

Coates’synthesis of MEP from D-arabitol.
In 2007, Koumbis and coworkers devised a synthesis of MEP from D-arabinose acetonide 74 [74]. In this case, the authors converted the starting material into an erythritol acetonide by tosylation of the free hydroxyls followed by LiAlH4 reduction to form the 1, 4 diol 75. Monoderivitization of the diol with various protecting groups was performed using a tin-mediated procedure developed for 1,3- and vicinal 1,2-diols resulted in a high ratio (5:1) of the desired monoprotected alcohol 76 [75]. Following benzylation of the free alcohol to to provide the fully protected 77, silyl-deprotection 78 and phosphorylation as its benzylated derivative 79 was accomplished in over 90% yield. Deprotection via hydrogenation ultimately provided MEP 71 as its free acid in 37% yield over 7 steps (Scheme 17). Crick and coworkers used a similar approach to synthesize enantiopure MEP as its free acid from 1,2-O-isopropylidene-α-D-xylofuranose [76, 77]. Similar to the syntheses described earlier, a dibenzyl protected phosphate moiety was key to formation of MEP as its corresponding free acid.
Scheme. (17).

Koumbis’ synthesis of MEP from D-arabinose acetonide.
Syntheses of 4-diphosphocytidyl-2C-methyl-D-erythritol 83 (CDPME)-Relatively fewer synthetic procedures to CDPME 83 exist, and with the exception of a fully enzymatic approach [7], most depend significantly upon prior routes toward the synthesis of the free acid of MEP. Koppisch and Poulter reported the first synthetic preparation of CDPME in 2002 (Scheme 18) [60]. In the authors’ synthesis, the key step was coupling of MEP 82 (as a tributylammonium salt) with cytidine monophosphate 81 (as a triethylammonium salt) via activation of the phosphate 80 with trifluoroacetic anhydride and methylimidazole, respectively. This phosphoramidite based approach was originally developed in a synthesis of UDP-galactofuranose, but has been incorporated into most reported syntheses of CDPME as well [78].
Scheme. (18).

Koppisch and Poulter’s synthesis of CDPME from MEP and CMP.
Upon workup and purification using size exclusion chromatography, the ammonium form of CDPME 83 was synthesized from MEP in a single pot with an overall yield of 40%. This phosphoramidite based coupling strategy has been employed in most CDPME syntheses reported to date, with the most notable exception being an enzymatic coupling strategy to form CDPME with recombinant CDPME synthase (IspD) and synthetic MEP prepared from 1,2-O-isopropylidene-α-D-xylofuranose [77]. In a similar fashion, D-arabitol was used in the synthesis of CDPME by Lai and coworkers through phosphoramidite coupling of alkylammonium salts of MEP and CMP [67].
Syntheses of 4-diphosphocytidyl-2C-methy-D-erythritol 2-phosphate 90 (CDP-ME2P)-In this synthesis, the hydroxyl groups of benzylidene diol 84 (from a previous synthesis [66]) were phosphorylated, in a stepwise manner, with first dibenzyl phosphochloridate 85 then phosphorous trichloride in the presence of ethanol to provide the double phosphotriester 86 in good yield. Benzyl deprotection was performed by hydrogenolysis in one step to provide the diethylphosphate protected ME2P 87 in near quantitative yield. Triethylammonium cytidine monophosphate 88 was then activated to the phosphoramidite and coupled with the phosphoramidite of 87 to provide the phosphate-protected CDP-ME2P 89 in moderate yield. The diethyl phosphate moiety was deprotected using TMSI to provide CDP-ME2P 90 in 16% overall yield over five steps (Scheme 19) [76].
Scheme. (19).

Coates’synthesis of CDP-ME2P.
Crick and coworkers also reported a route to the MEP intermediate CDP-ME2P from D-arabitol. In this work, the authors extended the synthetic route to ME/MEP developed by Urban sky et al. to form several benzylphosphoester or methylphosphoester derivatives of the aforementioned authors’ key dioxanone intermediate. In one case, the tertiary hydroxyl of the dioxanone intermediate was converted to a dibenzylphosphoester with PCl3 and benzyl alcohol, while its primary hydroxyl was transformed into a methylphosphoester with dimethyl phosphochloridate/n-BuLi. Coupling of this product to CMP through formation of the CMP phosphoramidite was accomplished in the manner of previous syntheses albeit in low yield. A second strategy, wherein the bisphosphorylated dioxanone intermediate was formed as a benzylphosphoester (at the primary hydroxyl group) and ethylphosphoester (at the tertiary hydroxyl) was formed first, and upon hydrogenation over Pd/C, 2,4-bisphosphomethylerythritol was formed as the 2-ethylphos-phoester. Phosphoramidite coupling of this material with CMP proceeded smoothly, and upon deprotection yielded CDP-ME2P in good yield [76].
Syntheses of 2C-methylerythritol 2,4-cyclodisphophate 95 (cMEPP)- Cyclization of 4-diphosphocytidyl-2C-methyl-D-eryth-ritol 2-phosphate by cMEPP synthase generates 2C-methyl-D-erythritol 2,4-cylodiphosphate (MECDP) [9]. An efficient synthesis of cMEPP was reported by Giner and Ferris using 2C-methyl-D-erythritol 1,3-diacetate 91 as the starting material. The authors reported simultaneous phosphorylation of both the primary and tertiary alcohol through diisopropyl dibenzyl phosphite under basic conditions and in situ oxidation to provide the dibenzyl protected phosphate 92 in 75% yield. Hydrogenation proceeded cleanly to provide the deprotected phosphate 93 followed by phosphate coupling via carbodiimide cyclization 94 and acetate removal under careful basic conditions to provide the cyclic diphosphate 95 in four steps with 42% overall yield from the diacetate (Scheme 20) [79].
Scheme. (20).

Giner and Ferris synthesis of cMEPP.
A second, lengthier synthesis began with 1,3-benzylidene-D-threitol 96. Following periodate cleavage, 97 was selectively protected with TBSCl at the primary alcohol to provide the monoether 98. A phase-transfer-promoted RuO4 oxidation provided ketone 99 followed by reduction with methylmagnesium bromide 100 and removal of the protecting group yielding the benzylidene protected ME 101. Double phosphorylation using phosphoramidite coupling instilled the two benzyl-protected phosphate esters 102 and selective debenzylation of the phosphate esters via hydrogenation provided the bis-phosphomonoester 103, which was subsequently cyclized using 1, 1’-carbonyl diimidazole to provide the penultimate intermediate 104 [65, 66]. The final product, cMEPP 95, was generated by simple hydrogenation over palladium hydroxide (Scheme 21). This method required 9 steps as opposed to the 4 steps required in the previous synthesis and was also lower yielding (10% vs. 42%).
Scheme. (21).

Coates’ synthesis of cMEPP from the protected D-threitol.
Syntheses of (E)-4-hydroxydimethylallyl diphosphate 107 (HDMAPP)-This straightforward synthesis was reported independently by several research groups in rapid succession upon its initial discovery in 2002 [80-86]. Of the seven, three utilized a strategy to incorporate an isotopic tag in either penultimate or final step of the synthesis. The most efficient synthesis of non-radiolabeled HDMAPP was obtained in two steps from the commercially available 2-methyl-2-vinyloxirane 105 in 72% overall yield (Scheme 22). Importantly, the (E)-geometry of the chloroalcohol 106 was preferred (97:3) upon ring opening in the presence of titanium (IV) tetrachloride as determined from nuclear Overhauser effects spectroscopy (NOESY) followed by diphosphorylation to provide HDMAPP 107. From the identical starting material 105, ring opening in the presence of cupric (II) chloride provided the chloroaldehyde 108 in moderate yield. Following conversion of the aldehyde to the methylacetal 109, either a deuterium or tritium was introduced in the final step to provide labeled HDMAPP in 23% or 15% yield, respectively [85].
Scheme. (22).

Hecht’s synthesis of HDMAPP and isotopomers from the vinyloxirane.
Fox and Poulter demonstrated the (E)-chloroaldehyde as a versatile intermediate for insertion of an isotopic label to C-1 in the penultimate step of the synthesis (Scheme 23). The overall yield for reduction of the intermediate chloroaldehyde 108 to the chloroalcohol 106 followed by phosphorylation provided HDMAPP 107, with purification of the intermediate olefin, were 66% [87].
Scheme. (23).
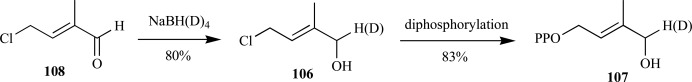
Fox and Poulter’s synthesis of HDMAPP via the chloroaldehyde intermediate.
Isopentenyl diphosphate and dimethylallyl disphosphate (IPP and DMAPP)-Both IPP and DMAPP are derived from HDMAPP, where the MEP pathway is operative, by reductive elimination of water [88] and represent the requisite building blocks to all isoprenoids. The synthesis of both compounds was optimized in the mid-80s by Davisson and Poulter during their investigations for improving the diphosphorylation step of the allylic 110 or non-allylic 111 prenyl alcohols over previously published reports [89]. Briefly, the authors found that when pyrophosphoric acid was treated with tetra-n-butyl ammonium hydroxide and immediately lyophilized, tris(tetra-n-butyl) ammonium hydrogen pyrophospate was obtained on a multigram scale at near quantitative yields for the displacement reaction with the corresponding activated allylic 112 and nonallylic 113 prenyl moieties. Following cation exchange and cellulose chromatography, the ammonium form for prenyl disphosphates, 114 and 115, were typically obtained in excess of 75% yield on gram scales and could be stored for long durations under cool temperatures with minimal decomposition (Scheme 24) [90, 91].
Scheme. (24).

Davisson’s synthesis of both IPP and DMAPP.
ISOPRENOIDS
Monoterpenes
Monoterpenes, which comprise the C10 class of isoprenoids, are largely derived from condensation of IPP to DMAPP in either a ‘head-to-tail’ or ‘non-head-to-tail’ fashion or, alternatively, through condensation of two DMAPP monomers. These enzyme-catalyzed reactions ultimately result in the extremely large structural diversity observed within this class of compounds [92-94]. They are often found in pesticides, essential oils, fragrances, for use in homeopathic medicine, and are commonly used building blocks to higher-value chemicals, thus demonstrating their utility for industrial applications [95, 96]. There are countless reports in the literature for the chemical syntheses of the acyclic, mono- and bicyclic monoterpenes and the reader is directed to several sources for reviewing synthetic approaches to these industrially useful compounds [97-101]. Some common monoterpenes are found in (Fig. 1) and a few representative examples for their syntheses are presented.
Fig. (1).

Examples of naturally occurring acyclic, monocylic and bicyclic monoterpenes.
The cyclic monoterpene, (+)-limonene, is derived from cyclization of GPP and has proven to be an essential building block toward the synthesis of dozens of closely related compounds. Further, limonene is primarily accessed through cold-pressed orange oil on large scales, making this monoterpene a rather inexpensive chiral starting material suitable for chemical synthesis, and is therefore utilized as building blocks to dozens of related compounds. Representative examples of these chemical transformation are found in a review by Thomas and coworkers and some basic chemical transformations from limonene found in (Fig. 2) [102]. Another common bicyclic monoterpene, camphor, is found in the wood of camphor trees and is most often utilized as a plasticizer in certain materials, for therapeutics, as a wax, and as an aromatic fragrance in essential oils. The synthesis of camphor from α-pinene was first demonstrated in 1922 and served as a building block for hundreds of chemical transformations to related isoprenoid derivatives [103].
Fig. (2).

Common chemical transformations from (+)-limonene and (+)-camphor.
Among bicyclic monoterpenes, α- and β-pinenes are among the most widely distributed of the isoprenoid family and are found in the essential oils of most trees. They are currently of industrial importance as solvents and for the commercial preparation of camphor and α-terpinol [104]. However, the bicylic system of pinene presented a synthetic challenge. Fallis and coworkers addressed this through utilization of a bicyclic heptane system common to many monoterpenes within this structural class of isoprenoids. Following several high-yielding steps to convert ester 116 to the well-known Hagemann’s ester 117, transformation to the dimethyl keto acetate 118 then tosylation to 119 provided a convenient leaving group for base-assisted cyclization to provide the bicyclic core 120. A Wittig reaction with methylenetriphenylphosphorane and 120 yielded β-pinene 121, which was subsequently isomerized to α-pinene 122 by hydrogenation over palladium/carbon (Scheme 25) [105]. All chemi-cal transformations were in excess of 70% yield, making this strat-egy to this class of compounds useful for large scale production.
Scheme. (25).

Fallis’ synthesis of both β-pinene (121) and α-pinene (122).
Another, more facile synthesis to β-pinene was developed by Snider et al. using a stereospecific [2+2] cycloaddition of ketenes to alkenes. In this synthesis, the authors converted geranic acid 123 to the corresponding acid chloride 124. This was a versatile intermediate since the presence of the electron-withdrawing halogen enabled formation of the ketene 125 with the necessary regiochemistry for subsequent cyclization to the vinyl ketones. β-Pinene 121 was generated from the ketene through a Wolff-Kishner reduction or to the monoterpene, chrysanthenone 126, through simple isomerization (Scheme 26) [106].
Scheme. (26).

Snider’s synthesis of both β-pinene and chrysanthenone.
The bicyclic monoterpene, carene, is one of the major constituents in turpentine [107]. In one synthesis, the cyclic hexene group was constructed from isoprene 127 and ketone 128 by a Diels-Alder reaction to form the methylated cyclohexene regioisomers 129 and 130. Following methyl addition to the ketone to provide a racemic mixture of tertiary alcohols 131 and 132 the bicyclic product, 3-carene 133, and the methyl regioisomer 134 were obtained using thionyl chloride (Scheme 27) [108]. This synthesis is useful since 3-carene is used for the construction of other monoterpenes and cyclopropanes. A detailed account of synthetic approaches to new monoterpenes using carene as the building block are found in a recent review [109].
Scheme. (27).

Johnson’s synthesis of a stereoisomeric mixture of 3-carene.
Whereas there are countless numbers of chemical syntheses to monoterpenes and their structural derivatives, many are traced back to a few, basic mono- and bicyclic compounds. There are dozens of reviews, manuscripts and books readily available that enable a detailed understanding of the evolution for chemical approaches to a myriad of monoterpenes and those presented in this review are meant to serve as a nucleation point for future synthetic efforts.
Sesquiterpenes
Sesquiterpenes, which comprise the C15 class of isoprenoids, are predominantly derived from the condensation of an additional IPP monomer to the C10 monoterpene framework to provide farnesyl diphosphate (FPP). They are a structurally diverse group where all are derived from FPP via differing enzyme-catalyzed cyclization reactions and carbon skeleton rearrangements. They are commonly found as essential oils in plants in the mono-, bi- and tricyclic forms and have been shown to have a wide range of biological activities [110]. However, in spite of the expansive structural diversity, it is the acyclic form that has garnered the most attention from a synthetic chemistry perspective. Briefly, previous studies have demonstrated that mammalian Ras proteins, when farnesylated, result in cellular signal transduction leading to normal cell growth, differentiation and survival. However, mutant Ras-proteins that are always activated result in undifferentiated cell growth and are now implicated in greater than 30% of human cancers [111, 112]. The enzyme responsible for farnesylation (prenylation) of Ras, protein farnesyltransferase (PFTase), when inhibited resulted in reversion of the undifferentiated cell growth to a normal cell. Hence, synthesis of FPP analogs as anti-cancer therapeutics is an area of intense research [113]. Primary to this goal was the synthesis of suicide inhibitors against PFTase to prevent farnesylation through covalent attachment of the FPP analog to the enzyme by trapping a carbocation intermediate within the enzyme active site [114].
A primary concern in the synthesis of farnesyl diphosphate analogs is maintenance of the requisite all trans-isomer in the final product. Gibbs and co-workers designed multiple synthetic routes to both the 3- and 7-vinyl substituted farnesyl diphosphate (VFPP) analogs through the vinyl triflate intermediate 136 via a ketone ester 135 to provide the E-isomer 137 with high stereoselectivity (16:1) and in good yields [115]. Reduction, alcohol activation and diphosphorylation provided the all E-isomer of 3-VFPP 138 (Scheme 28). This synthetic approach was also implemented in the synthesis of the all E-7-VFPP analog; albeit, in much lower yields since two coupling steps using the vinyl triflate strategy were necessary, required 11 steps and less than 10% overall yield [116].
Scheme. (28).

Gibbs’ synthesis of 3-VFPP.
In a recent report, Gibbs et al. improved on their synthesis of 7-VFPP by coupling of 5-lithio-2, 3-dihydrofuran 139 with an isoprenylorganocuprate 140 to provide the corresponding alkenylcuprate intermediate 141. This compound proved to be a versatile intermediate since this affords the opportunity to introduce multiple functional groups into ultimately the 7-position of FPP. Following conversion of 141 into the corresponding organozinc intermediate 142 over four steps in 25% yield, the vinyl-substituted farnesyl skeleton was completed via coupling to the silyl-protected vinyl iodide 143 through palladium triphenylphosphine and removal of the silyl-protecting group to provide 7-vinylfarnesol 144. Diphosphorylation provided 7-VFPP 145 over six steps in 12% overall yield (Scheme 29) [117].
Scheme. (29).

Gibbs’ alternate synthetic route to 7-VFPP.
Diterpenes
Diterpenes, which comprise the C20 class of isoprenoids, are derived from condensation of IPP to FPP to form geranylgeranyl diphosphate (GGPP). Some representative examples include, dolichols, ubiquinones (plastoquinones), phytyl derivatives and geranylgeranylated proteins [118]. The structural diversity is amplified through multivariant cyclizations via carbocation intermediates and alkyl shifts to provide mono-, di-, and tricyclic compounds, which are further processed depending on organismal need. As a result of this diversity, it is not surprising that there are greater than 11,000 diterpenes that have been identified [119]. Therefore, in a similar vein as with the sesquiterpenes, this section will provide some representative examples for the chemical syntheses of GGPP analogs since there is ongoing interest for identifying novel compounds for medicinal purposes.
One class of inhibitors consists of the nitrogen-containing bisphosphonates, which are used in treating bone resorption diseases such as osteoporosis and metastatic bone disease [120]. These bisphosphonates work by depleting cells of GGPP, leading to inhibition of protein isoprenylation [121]. In order to determine the exact mechanism by which bisphosphonates inhibit isoprenoid metabolism, a GGPP analog was synthesized using a synthetic scaffold that is amenable to varying isoprenoid chain lengths. A facile synthesis for one of the bisphosphonates containing the geranyl substituent is described below.
Treatment of tetraethyl methylenebisphosphonate 146 with sodium hydride, 15-crown-5 ether, and farnesyl bromide 147 provided the dialkylated bisphosphonate 148 product harboring the geranyl moieties. Although not high yielding, the mono- or dialkylation product could be regulated through careful titration with the base [122]. Deprotection of 148 with TMS-Br provided the GGPP analog 149 in 95% yield (Scheme 30). This strategy has proven successful for addition of alkylation reagents of varying chain length.
Scheme. (30).

Schull’s synthesis of digeranyl-substituted bisphosphonate.
In addition to the bisphosphonates, another strategy using GGPP analogs was hypothesized for inhibition of GGPP synthase. One of the reasons for developing this alternative synthetic route was the observation that bisphosphonates tend to be poorly absorbed into the body through oral delivery. Creating compounds that have increased hydrophobicity was postulated to have better absorption properties [120]. Based upon biochemical evidence that a developing carbocation intermediate was formed within the enzyme active site of GGPP synthase the aza analog of GGPP was necessarily synthesized.
In the synthesis of an aza-GGPP 155, alkylation of farnesyl chloride 150 with α-lithio formamidine 151 generated an amidine, which was subsequently hydrolyzed to a farnesyl-N-methylamine 152. Alkylation of the amine with t-butylbromoacetate resulted in the β-aminoester 153 in excellent yield. Following reduction of 153 with lithium aluminum hydride, the resulting geranylgeraniol N-methylamine 154 was converted to the corresponding diphosphate with methanesulfonyl chloride and tris(tetrabutylammonium) hydrogen pyrophosphate to provide aza-GGPP 155 over four steps in 30% yield from commercially available farnesyl chloride (Scheme 31) [123].
Scheme. (31).

Coates’synthesis of aza-GGPP.
As described previously, several FPP analogs were synthesized by Gibbs and Wiemer using a novel palladium, copper-catalyst coupling approach. This concept was expanded by Coates et al. to synthesize new GGPP analogs thought to act as inhibitors of PGGTase I, which attaches a geranylgeranyl moiety from GGPP to a cysteine in a CAAX-type box, where leucine is the carboxyl terminal residue [124]. Three new GGPP analogs were synthesized containing phenyl, tert-butyl and cyclopropyl substituents. The phenyl-substituted GGPP analog was prepared from an ethyl acetoacetate dianion 156 coupled with farnesyl bromide 157 to provide the β-ketoester 158. The potassium enolate was then transformed into the vinyl triflate 159. Coupling of the triflate with phenylboronic acid and silver oxide as the base yielded the desired phenyl ester 160. Reduction of the ester with DIBALH and subsequent phosphorylation provided 3-PhGGPP 162 over six steps in 11% yield (Scheme 32) [125].
Scheme. (32).

Mu’s synthesis of 3-PhGGPP.
Triterpenes
Triterpenes comprise the C30 class of isoprenoids and are biosynthesized through the condensation of two farnesyl diphosphate (FPP) units followed by reduction. The majority of triterpenes in biological systems result from ‘head-to-head’ condensations of FPP, which result in a linkage between the first carbons in each respective farnesyl group (a 1’-1 linkage). There are other examples of triterpenes with varied linkages in nature and one such triterpene, botryococcene (1’-3 linkage), will be discussed later in this section.
The linear triterpene, squalene, is a key precursor in the biosynthesis of cholesterol and other steroids. During the condensation of FPP monomers in the biosynthesis of squalene, a cyclopropyl diphosphate intermediate (presqualene diphosphate or PSPP) is formed and subsequently reduced by NADPH to form the final product [126]. Squalene itself then is transformed into the fused tetracyclic core characteristic of sterols and steroids. The synthesis of sterols, steroids, and other cyclic triterpenes represents a large body of information and is beyond the scope of this review. In this section, we will summarize approaches to construction of several representative linear triterpenes. Readers with an interest in cyclic triterpenes are directed to other comprehensive reviews.
An early synthesis of squalene was based on systematic olefin synthesis. In step 1, The five-carbon and six-carbon building blocks for the external segments of squalene were derived from α-acetyl-α-chlorobutyrolactone 163 to generate homoallylic chloride 164 and 3,5-dichloropentan-2-one 165 over four steps. Coupling of the C-5 and C-6 fragments with n-BuLi provided the homogeranyl backbone 166. Following epoxidation and elimination, the resulting homogeranyl chloride 167 was ready for squalene synthesis.
Step 2 entailed utilizing ketone 165 as the starting material for the synthesis of the internal segment of squalene. Briefly, following ketalization of 165 to generate 1, 3-dioxolane 168, coupling with t-butyl acetoacetate 169 and deprotection provided the central segment, 3,6-dichlorooctane-2,7-dione 170, for squalene assembly. Condensation of the products from steps 1 and 2 were accomplished by treatment of two equivalents of homogeranyl chloride 167 with two equivalents of n-BuLi and one equivalent of dione 170 to provide the dicholorosqualene intermediate 171. Subsequent epoxidation and oxygen elimination as employed in the construction of homogeranyl chloride provided squalene 172 in 15 steps (Scheme 33); however, yields for this synthesis were not estimated [127].
Scheme. (33).

Cornforth’s synthesis of squalene.
One challenge in the chemical synthesis of squalene is stereoselectivity, as all bonds are found in the trans-configuration. In a later synthesis, a stereoselective Claisen rearrangement was employed. Starting with succinaldehyde 173, treatment of the resulting alcohol with iPrMgBr yielded bis-allylic alcohol 174 in good yield. The alcohol was subject to a Claisen rearrangement with 3-methoxyisoprene 175 to the C-20 tetraenedione 176, which was subsequently reduced with sodium borohydride to provide the tetraenediol 177. Repetition of this sequence of reactions provided the C-30 squalene 178 backbone. Rearrangement of the terminal double bonds and elimination of the alcohol groups were accomplished through the use of thionyl chloride in a substitution reaction to provide the terminal halides 179, whereupon reduction with lithium aluminum hydride yielded squalene 172 over eight steps in 7% yield (Scheme 34) [128].
Scheme. (34).

Petersen’s synthesis of squalene using a Claisen rearrangement.
As mentioned previously, linear, branched triterpenes (such as those with 1’-3 isoprenoid linkages) are also found in nature. The best example of a triterpene of this nature is botryococcene, which is produced and accumulated by the B race of the green algae from which its name is derived, Botryococcus braunii [129-131]. The most abundant of the hydrocarbons in B. braunii Race B is the tetramethylated derivative of the triterpene, C-34 botryococcene [132]. This compound is of special interest since it was demonstrated the botryococcenes are compatible with existing oil infrastructures and, following hydrocracking, can be used directly as a ‘drop-in’ fuel [133].
The chemical synthesis of the tetramethylated botryococcene is challenging since there are six stereocenters on the carbon framework. To the best of our knowledge, there is only a single published report for the total synthesis of this isoprenoid with the correct stereochemistry in the final product, although there are several reports of the C-30 botryococcene and related analogs chemical synthesis [134-137]. Certainly, considering the interest in botryococcenes as a source of biofuel, novel synthetic approaches to the methylated versions may prove fruitful in order to study the enzyme(s) responsible for methylated botryococcene biosynthesis. Briefly, White et al. recognized the symmetry elements present in the structure and thus devised a convergent strategy for condensing the two symmetric fragments with the core segment to provide the correct stereochemistry in the final product. Specifically, as outlined in (Scheme 35), Step 1, the synthesis of the symmetric fragment began with a five step approach from methyl (2S)-3-hydroxy-2-methylpropionate 180 to the homoallylic alcohol 181 in 29% yield. Head-to-tail union of 181 proceeded through coupling of the dilithio species 182 with the tosylated alcohol 183 to provide the enantiomerically pure C12 segment 184 in excellent yield.
Scheme. (35).

White’s synthesis of C34 botrycoccene.
The synthesis of the central subunit proved to be much more challenging. The primary concern was introduction of a quaternary carbon center with the requisite R-configuration in the core fragment. Starting with R-methyl ester 185, conversion to the pivotal intermediate methoxymethyl ester 186 was accomplished in six steps in good yields. Installation of the quaternary center with the correct stereochemistry was accomplished by γ-deprotonation and 1,4-elimination of the methoxymethyl ester 187 to provide a 3:2 diastereomeric mixture of ester 188. The authors then used an enantioconvergent strategy to convert both diastereomeric intermediates to the central fragment through alternating protection and deprotection strategies to provide the enantiomerically pure diiodide central subunit 190 over thirteen steps in 7% yield. Activation of homoallylic alcohol 184 required four steps to the cuprate 191 intermediate and enabled coupling of the core fragment 190 to provide (–)-C34-botryococcene 192 over five steps in 8% yield (Scheme 35) [138].
Tetraterpenes
Tetraterpenes, which comprise the C40 class of isoprenoids, are derived from two C20 geranylgeranyl disphosphates in a head-to-head condensation reaction. Representative examples include the carotenoids and xanthophylls, which have strong anti-oxidant properties and are essential components in photosynthetic machinery [139]. Further, carotenoids are used extensively in the food and cosmetic industries as food colorants, protection against UV-irradiation, and as nutritional supplements [140, 141]. Although the majority of tetraterpenes are isolated from biological sources, the structural complexity of these isoprenoids proved to be a synthetic challenge largely due to the extensive double bond network and introduction of the correct stereochemistry at remote positions along the carbon framework. Two representative examples for achieving this end are presented.
Lycopene is an acyclic tetraterpene and the immediate precursor to both α- and β-carotene that consists of 13 double bonds all in the trans-configuration. This compound’s high radical scavenging ability in addition to being synthetically challenging were major drivers toward lycopene synthesis [142]. A favorable synthetic route to the all trans-lycopene exploited two Wittig-Horner reactions to introduce double bonds under mild reaction conditions. The starting material, 4,4-dimethoxy-3-methylbutanal 193, was condensed with methylene bisphosphonic acid tetraethyl ester 194 in the presence of NaH to provide the C6 propenyl phosphonate 195. Wittig-Horner condensation with the methyl heptenone 196 yielded the dimethoxytriene 197 in the correct double bond configuration. Following deprotection under acidic conditions, aldehyde 198 was condensed with the identical biphosphonic acid 194 to provide the all-E-tetraene 199. Another Wittig-Horner reaction with diketone 200 provided the all trans-lycopene 201 over five steps in 27% yield (Scheme 36) [143].
Scheme. (36).

Shen’s synthesis of lycopene.
Lutein is a xanthophyll found in plants for non-photochemi-cal quenching of singlet excited state chlorophyll, which occurs at high light intensities, thus prevents photoinhibition. In addition, lutein is a major dietary carotenoid abundant in fruits and vegetables. The predominant stereoisomer is (3R,3’R,6’R)-β,ε-carotene-3,3’-diol [144]. The strategy used in one synthesis to make this isomer (and others) was to couple C-15 and C-10 building blocks, then elongate the resulting C-15 hydroxyaldehyde through a Wittig salt intermediate. Commercially available α-ionone 202 was the starting material for the synthesis of nitrile 203, which enabled facile chain elongation. Selective oxidation of 203 to the corresponding conjugated ketone 204 was accomplished in the presence of peroxide and bleach. Careful reduction of the ketone in the presence of borohydride provided racemic alcohol 205 in excellent yield. Reduction of the nitrile moiety to the aldehyde 206 using DIBAL-H resulted in the racemic mixture of hydroxyaldehydes. To isolate the desired (3R, 6R) configuration, kinetic resolution of 206 was employed using an enzyme-mediated acylation through Lipase AK. Fortunately, the desired stereoisomer 207 remained unreactive and was readily purified from the acylated enantiomer. The aldehyde was subjected to olefination using the protected Wittig salt 208 harboring the C10 internal portion of the lutein carbon framework resulting in a mixture of cis- and trans-configured 209 products. Following deprotection of the acetal to provide aldehyde 210, coupling with a second Wittig salt reagent 211 appended to the remaining lutein framework provided a mixture of cis- and trans-lutein 212. The diastereomeric lutein was isomerized under refluxing conditions and isolated via column chromatography and crystallization resulting in stereomerically pure (3R,3’R,6’R) lutein 213 in 6% yield over nine steps (Scheme 37) [145].
Scheme. (37).

Khachik and Chang’s synthesis of lutein.
SUMMARY
Isoprenoids are a tremendous illustration of the cornucopia of chemical diversity and unique biochemical roles that are possible within members of a single molecular family. A detailed understanding of these structures, and of their roles, is empowered by the development of synthetic methodologies to produce the corresponding target molecules, and as substrates or products for detailed enzymological studies. In nature, the biosynthesis of isoprenoids affects the conversion of small molecule acids (such as those used in the mevalonate pathway) or carbohydrates (such as those used in the methylerythritol phosphate pathway) into molecules that are largely aliphatic hydrocarbons. Similarly, as is outlined in our review, the synthetic methodologies to produce a given isoprenoid may draw from and utilize chemical reactions associated with the construction of hydrocarbons, carbohydrates, or diphosphate based bioconjugates.
Whereas the biological production of isoprenoids will likely be the major route to these compounds, chemical synthesis of isoprenoids and their corresponding precursors will always be necessary. This is especially relevant when probing enzyme activity thus requiring the synthesis of substrate analogs. Emphasis on the chemical syntheses of MEP pathway intermediates to isoprenoids was a result of the exhaustive efforts required to probe the enzyme function responsible for tens of thousands of natural products that are found in all corners of life. It is also important to highlight the potential of longer chain isoprenoids for use in industrial applications, including nutracueticals, pharmaceuticals, cosmetics and as a source for biofuels due to the energy rich composition of these important compounds.
ACKNOWLEDGEMENTS
We thank Mr. Lucas Harrington for critical review of the manuscript. Funding for this review was supplied in part by the US Department of Energy under Contract DE-EE0003046 awarded to the National Alliance for Advanced Biofuels and Bioproducts (NAABB) and the Office of Energy Efficiency and Renewable Energy and Bioenergy Technologies (EERE-BETO). Funding was also provided by the Los Alamos National Laboratory Directed Research and Development Grant #LDRD20130091DR.
CONFLICT OF INTEREST
The authors confirm that this article content has no conflict of interest.
REFERENCES
- 1.Christianson DW. Unearthing the roots of the terpenome. Curr. Opin. Chem. Biol. 2008;12:141–150. doi: 10.1016/j.cbpa.2007.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bach TJ, Rohmer M. Isoprenoid Synthesis in Plants and Microorganisms. Springer: New York. 2012:505. [Google Scholar]
- 3.Chappell J. Biochemistry and molecular biology of the isoprenoid biosynthetic pathway in plants. Annu. Rev. Plant. Phys. 1995;46:521–547. [Google Scholar]
- 4.Kirby J, Keasling JD. Biosynthesis of plant isoprenoids: Perspectives for microbial engineering. Annu. Rev. Plant. Biol. 2009;60:335–355. doi: 10.1146/annurev.arplant.043008.091955. [DOI] [PubMed] [Google Scholar]
- 5.Eubanks LM, Poulter CD. Rhodobacter capsulatus DXP synthase: Steady-state kinetics and substrate binding. Biochemistry. 2001;40:8613–8614. doi: 10.1021/bi0205303. [DOI] [PubMed] [Google Scholar]
- 6.Koppisch AT, Fox DT, Blagg BS, Poulter CD. E.coli MEP synthase: steady-state kinetic analysis and substrate binding. Biochemistry. 2002;41:236–243. doi: 10.1021/bi0118207. [DOI] [PubMed] [Google Scholar]
- 7.Rohdich F, Schuhr CA, Hecht S, Herz S, Wungsintaweekul J, Eisenreich W, Zenk MH, Bacher A. Biosynthesis of isoprenoids.A rapid method for the preparation of isotope-labeled 4-diphosphocytidyl-2C-methyl-D-erythritol. J. Am. Chem. Soc. 2000;122:9571–9574. [Google Scholar]
- 8.Miallau L, Alphey MS, Kemp LE, Leonard GA, McSweeney SM, Hecht S, Bacher A, Eisenreich W, Rohdich F, Hunter WN. Biosynthesis of isoprenoids: crystal structure of 4-diphosphocytidyl-2C-methyl-D-erythritol kinase. Proc. Natl. Acad. Sci. USA. 2003;100:9173–9178. doi: 10.1073/pnas.1533425100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Herz S, Wungsintaweekul J, Schuhr CA, Hecht S, Luttgen H, Sagner S, Fellermeier M, Eisenreich W, Zenk MH, Bacher A, Rohdich F. Biosynthesis of terpenoids: YgbB protein converts 4-diphosphocytidyl-2C-methyl-D-erythritol 2-phosphate to 2C-methyl-D-erythritol 2, 4-cyclo-diphosphate. Proc. Natl. Acad. Sci. USA. 2000;97:2486–2490. doi: 10.1073/pnas.040554697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Seemann M, Rohmer M. Isoprenoid biosynthesis via the methylerythritol phosphate pathway: GcpE and LytB, two novel iron-sulphur proteins. Cr. Chim. 2007;10:748–755. [Google Scholar]
- 11.Zhou CF, Li ZR, Wiberley-Bradford AE, Weise SE, Sharkey TD. Isopentenyl diphosphate and dimethylallyl diphosphate/isopentenyl diphosphate ratio measured with recombinant isopentenyl diphosphate isomerase and isoprene synthase. Anal. Biochem. 2013;440:130–136. doi: 10.1016/j.ab.2013.05.028. [DOI] [PubMed] [Google Scholar]
- 12.Eisenreich W, Schwarz M, Cartayrade A, Arigoni D, Zenk MH, Bacher A. The deoxyxylulose phosphate pathway of terpenoid biosynthesis in plants and microorganisms. Chem. Biol. 1998;5:R221–233. doi: 10.1016/s1074-5521(98)90002-3. [DOI] [PubMed] [Google Scholar]
- 13.Rohmer M. The discovery of a mevalonate-independent pathway for isoprenoid biosynthesis in bacteria, algae and higher plants. Nat. Prod. Rep. 1999;16:565–74. doi: 10.1039/a709175c. [DOI] [PubMed] [Google Scholar]
- 14.Jomaa H, Wiesner J, Sanderbrand S, Altincicek B, Weidemeyer C, Hintz M, Turbachova I, Eberl M, Zeidler J, Lichtenthaler HK, Soldati D, Beck E. Inhibitors of the nonmevalonate pathway of isoprenoid biosynthesis as antimalarial drugs. Science. 1999;285:1573–1576. doi: 10.1126/science.285.5433.1573. [DOI] [PubMed] [Google Scholar]
- 15.Testa CA, Brown MJ. The methylerythritol phosphate pathway and its significance as a novel drug target. Curr. Pharm. Biotech. 2003;4:248–259. doi: 10.2174/1389201033489784. [DOI] [PubMed] [Google Scholar]
- 16.Putra SR, Lois LM, Campos N, Boronat A, Rohmer M. Incorporation of [2,3-C-13(2)]- and [2,4-C-13(2)]-D-1-deoxyxylulose into ubiquinone of Escherichia coli via the mevalonate-independent pathway for isoprenoid biosynthesis. Tetrahedron Lett. 1998;39:23–26. [Google Scholar]
- 17.Shabat D, List B, Lerner RA, Barbas CF. A short enantioselective synthesis of 1-deoxy-L-xylulose by antibody catalysis. Tetrahedron Lett. 1999;40:1437–1440. [Google Scholar]
- 18.Hecht S, Kis F, Eisenreich W, Amslinger S, Wungsintaweekul J, Herz S, Rohdich F, Bacher A. Enzyme-assisted preparation of isotope-labeled 1-deoxy-D-xylulose 5-phosphate. J. Org. Chem. 2001;66:3948–3952. doi: 10.1021/jo0100300. [DOI] [PubMed] [Google Scholar]
- 19.Zhou YF, Cui Z, Li H, Tian J, Gao WY. Optimized enzymatic preparation of 1-deoxy-D-xylulose 5-phosphate. Bioorg. Chem. 2010;38:120–123. doi: 10.1016/j.bioorg.2010.01.003. [DOI] [PubMed] [Google Scholar]
- 20.Therisod M, Fischer JC, Estramareix B. The origin of the carbon chain in the thiazole moiety of thiamine in Escherichia coli - Incorporation of deuterated 1-deoxy-D-threo-2-pentulose. Biochem. Biophys. Res. Commun. 1981;98:374–379. doi: 10.1016/0006-291x(81)90850-0. [DOI] [PubMed] [Google Scholar]
- 21.David S, Estramareix B, Fischer JC, Therisod M. 1-Deoxy-D-threo-2-pentulose - the precursor of the 5-carbon chain of the thiazole of thiamine. J. Am. Chem. Soc. 1981;103:7341–7342. [Google Scholar]
- 22.David S, Estramareix B, Fischer JC, Therisod M. The biosynthesis of thiamine - syntheses of [1,1,1,5-(H4)-H-2]-1-deoxy-D-threo-2-pentulose and incorporation of this sugar in biosynthesis of thiazole by Escherichia coli cells. J. Chem. Soc. Perk. T 1. 1982:2131–2137. [Google Scholar]
- 23.Kennedy IA, Hill RE, Pauloski RM, Sayer BG, Spenser ID. Biosynthesis of vitamin B-6 - Origin of pyridoxine by the union of 2 acyclic precursors, 1-deoxy-D-xylulose and 4-hydroxy-L-threonine. J. Am. Chem. Soc. 1995;117:1661–1662. [Google Scholar]
- 24.Giner JL, Jaun B. Biosynthesis of isoprenoids in Escherichia coli: Retention of the methyl H-atoms of 1-deoxy-D-xylulose. Tetrahedron Lett. 1998;39:8021–8022. [Google Scholar]
- 25.Meyer O, Hoeffler JF, Grosdemange-Billiard C, Rohmer M. Practical synthesis of 1-deoxy-D-xylulose and 1-deoxy-D-xylulose 5-phosphate allowing deuterium labelling. Tetrahedron. 2004;60:12153–12162. [Google Scholar]
- 26.Blagg BSJ, Poulter CD. Synthesis of 1-deoxy-D-xylulose and 1-deoxy-D-xylulose-5-phosphate. J. Org. Chem. 1999;64:1508–1511. doi: 10.1021/jo981966k. [DOI] [PubMed] [Google Scholar]
- 27.Crispino GA, Ho PT, Sharpless KB. Selective perhydroxylation of squalene - taming the arithmetic demon. Science. 1993;259:64–66. doi: 10.1126/science.8418495. [DOI] [PubMed] [Google Scholar]
- 28.Wang ZM, Sharpless KB. Asymmetric dihydroxylation of alpha-substituted styrene derivatives. Synlett (11): 1993:603–604. [Google Scholar]
- 29.Giner JL, Jaun B, Arigoni D. Biosynthesis of isoprenoids in Escherichia coli: The fate of the 3-H and 4-H atoms of 1-deoxy-d-xylulose. Chem. Commun. (Camb) 1998:1857–1858. [Google Scholar]
- 30.Piel J, Boland W. Highly efficient and versatile synthesis of isotopically labelled 1-deoxy-D-xylulose. Tetrahedron Lett. 1997;38:6387–6390. [Google Scholar]
- 31.Taylor SV, Vu LD, Begley TP, Schorken U, Grolle S, Sprenger GA, Bringer-Meyer S, Sahm H. Chemical and enzymatic synthesis of 1-Deoxy-D-xylulose-5-phosphate. J. Org. Chem. 1998;63:2375–2377. [Google Scholar]
- 32.Giner JL. New and efficient synthetic routes to 1-deoxy-D-xylulose. Tetrahedron Lett. 1998;39:2479–2482. [Google Scholar]
- 33.Cox RJ, de Andres-Gomez A, Godfrey CRA. Rapid and flexible synthesis of 1-deoxy-D-xylulose-5-phosphate, the substrate for 1-deoxy-D-xylulose-5-phosphate reductoisomerase. Org. Biomol. Chem. 2003;1:3173–3177. doi: 10.1039/b307314a. [DOI] [PubMed] [Google Scholar]
- 34.Wong U, Cox RJ. The chemical mechanism of D-1-deoxyxylulose-5-phosphate reductoisomerase from Escherichia coli. Angew. Chem. Int. Ed. Engl. 2007;46:4926–4929. doi: 10.1002/anie.200700647. [DOI] [PubMed] [Google Scholar]
- 35.Fox DT, Poulter CD. Mechanistic studies with 2-C-methyl-D-erythritol 4-phosphate synthase from Escherichia coli. Biochemistry. 2005;44:8360–8368. doi: 10.1021/bi047312p. [DOI] [PubMed] [Google Scholar]
- 36.Wong A, Munos JW, Devasthali V, Johnson KA, Liu HW. Study of 1-deoxy-D-xylulose-5-phosphate reductoisomerase: Synthesis and evaluation of fluorinated substrate analogues. Org. Lett. 2004;6:3625–3628. doi: 10.1021/ol048459b. [DOI] [PubMed] [Google Scholar]
- 37.Munos JW, Pu XT, Mansoorabadi SO, Kim HJ, Liu HW. A secondary kinetic isotope effect study of the 1-deoxy-D-xylulose-5-phosphate reductoisomerase-catalyzed reaction: Evidence for a retroaldol-aldol rearrangement. J. Am. Chem. Soc. 2009;131:2048–2049. doi: 10.1021/ja807987h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bouvet D, O'Hagan D. The synthesis of 1-fluoro- and 1,1-difluoro- analogues of 1-deoxy-D-xylulose. Tetrahedron. 1999;55:10481–10486. [Google Scholar]
- 39.Fox DT, Poulter CD. Synthesis and evaluation of 1-deoxy-D-xylulose 5-phosphoric acid analogues as alternate substrates for methylerythritol phosphate synthase. J. Org. Chem. 2005;70:1978–1985. doi: 10.1021/jo048022h. [DOI] [PubMed] [Google Scholar]
- 40.Muehlbacher M, Poulter CD. Isopentenyl-diphosphate isomerase: inactivation of the enzyme with active-site-directed irreversible inhibitors and transition-state analogues. Biochemistry. 1988;27:7315–7328. doi: 10.1021/bi00419a021. [DOI] [PubMed] [Google Scholar]
- 41.Prakash GKS, Krishnamurti R, Olah GA. Synthetic methods and reactions.141. Fluoride-induced trifluoromethylation of carbonyl-compounds with trifluoromethyltrimethylsilane (TMS-CF3) - a trifluoro-methide equivalent. J. Am. Chem. Soc. 1989;111:393–395. [Google Scholar]
- 42.Testa CA, Cornish RA, Poulter CD. The sorbitol phosphotransferase system is responsible for transport of 2-C-methyl-D-erythritol into Salmonella enterica serovar typhimurium. J. Bacteriol. 2004;186:473–480. doi: 10.1128/JB.186.2.473-480.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Duvold T, Cali P, Bravo JM, Rohmer M. Incorporation of 2-C-methyl-D-erythritol, a putative isoprenoid precursor in the mevalonate-independent pathway, into ubiquinone and menaquinone of Escherichia coli. Tetrahedron Lett. 1997;38:6181–6184. [Google Scholar]
- 44.Charon L, Hoeffler JF, Pale-Grosdemange C, Lois LM, Campos N, Boronat A, Rohmer M. Deuterium-labelled isotopomers of 2-C-methyl-D-erythritol as tools for the elucidation of the 2-C-methyl-D-erythritol 4-phosphate pathway for isoprenoid biosynthesis. Biochem. J. 2000;346 (Pt 3):737–42. [PMC free article] [PubMed] [Google Scholar]
- 45.Charon L, Hoeffler JF, Pale-Grosdemange C, Rohmer M. Synthesis of [3,5,5,5-H-2(4)]-2-C-methyl-D-erythritol, a substrate designed for the elucidation of the mevalonate independent route for isoprenoid biosynthesis. Tetrahedron Lett. 1999;40:8369–8373. [Google Scholar]
- 46.Bitok JK, Meyers CF. 2C-Methyl-D-erythritol 4-phosphate enhances and sustains cyclodiphosphate synthase IspF activity. ACS Chem. Biol. 2012;7:1702–1710. doi: 10.1021/cb300243w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hemmerlin A, Hoeffler JF, Meyer O, Tritsch D, Kagan IA, Grosdemange-Billiard C, Rohmer M, Bach TJ. Cross-talk between the cytosolic mevalonate and the plastidial methylerythritol phosphate pathways in Tobacco Bright Yellow-2 cells. J. Biol. Chem. 2003;278:26666–26676. doi: 10.1074/jbc.M302526200. [DOI] [PubMed] [Google Scholar]
- 48.Kuzuyama T, Takahashi S, Watanabe H, Seto H. Direct formation of 2-C-methyl-D-erythritol 4-phosphate from 1-deoxy-D-xylulose 5-phosphate by 1-deoxy-D-xylulose 5-phosphate reductoisomerase, a new enzyme in the non-mevalonate pathway to isopentenyl diphosphate. Tetrahedron Lett. 1998;39:4509–4512. [Google Scholar]
- 49.Sagner S, Eisenreich W, Fellermeier M, Latzel C, Bacher A, Zenk MH. Biosynthesis of 2-C-methyl-D-erythritol in plants by rearrangement of the terpenoid precursor, 1-deoxy-D-xylulose 5-phosphate. Tetrahedron Lett. 1998;39:2091–2094. [Google Scholar]
- 50.Ding X, Zheng M, Yu LP, Zhang XL, Weber RJ, Yan B, Russell AG, Edgerton ES, Wang XM. Spatial and seasonal trends in biogenic secondary organic aerosol tracers and water-soluble organic carbon in the southeastern United States. Environ. Sci. Technol. 2008;42:5171–5176. doi: 10.1021/es7032636. [DOI] [PubMed] [Google Scholar]
- 51.Li QA, Wang W, Zhang HW, Wang YJ, Wang B, Li L, Li HJ, Wang BJ, Zhan J, Wu M, Bi XH. Development of a compound-specific carbon isotope analysis method for 2-methyltetrols, biomarkers for secondary organic aerosols from atmospheric isoprene. Anal. Chem. 2010;82:6764–6769. doi: 10.1021/ac100214p. [DOI] [PubMed] [Google Scholar]
- 52.Xia X, Hopke PK. Seasonal variation of 2-methyltetrols in ambient air samples. Environ. Sci. Technol. 2006;40:6934–6937. doi: 10.1021/es060988i. [DOI] [PubMed] [Google Scholar]
- 53.Witczak ZJ, Whistler RL. Synthesis of 3-C-(hydroxymethyl)erythritol and 3-C-methylerythritol. Carbohydr. Res. 1984;133: 235–245. doi: 10.1016/0008-6215(84)85201-5. [DOI] [PubMed] [Google Scholar]
- 54.Duvold T, Bravo JM, PaleGrosdemange C, Rohmer M. Biosynthesis of 2-C-methyl-D-erythritol, a putative C-5 intermediate in the mevalonate independent pathway for isoprenoid biosynthesis. Tetrahedron Lett. 1997;38:4769–4772. [Google Scholar]
- 55.Kolb HC, Vannieuwenhze MS, Sharpless KB. Catalytic asymmetric dihydroxylation. Chem. Rev. 1994;94:2483–2547. [Google Scholar]
- 56.Fontana A, Messina R, Spinella A, Cimino G. Simple and versatile synthesis of branched polyols: (+)-2-C-methylerythritol and (+)-2-C-methylthreitol. Tetrahedron Lett. 2000;41:7559–7562. [Google Scholar]
- 57.Wang H, Zhao XM, Li YH, Lu L. Asymmetric synthesis of four isomers of 2-C-trifluoromethylerythritol. J. Org. Chem. 2006;71:3278–3281. doi: 10.1021/jo052282x. [DOI] [PubMed] [Google Scholar]
- 58.Ghosh SK, Butler MS, Lear MJ. Synthesis of 2-C-methylerythritols and 2-C-methylthreitols via enantiodivergent sharp less dihydroxylation of trisubstituted olefins. Tetrahedron Lett. 2012;53:2706–2708. [Google Scholar]
- 59.Koppisch AT, Blagg BSJ, Poulter CD. Synthesis of 2-C-methyl-D-erythritol 4-phosphate: The first pathway-specific intermediate in the methylerythritol phosphate route to isoprenoids. Org. Lett. 2000;2:215–217. doi: 10.1021/ol991299x. [DOI] [PubMed] [Google Scholar]
- 60.Koppisch AT, Poulter CD. Synthesis of 4-diphosphocytidyl-2-C-methyl-D-erythritol and 2-C-methyl-D-erythritol-4-phosphate. J. Org. Chem. 2002;67:5416–5418. doi: 10.1021/jo025736o. [DOI] [PubMed] [Google Scholar]
- 61.Robinson TV, Pedersen DS, Taylor DK, Tiekink ERT. Dihydroxylation of 4-substituted 1,2-dioxines: A concise route to branched erythro sugars. J. Org. Chem. 2009;74:5093–5096. doi: 10.1021/jo900669u. [DOI] [PubMed] [Google Scholar]
- 62.Kis K, Wungsintaweekul J, Eisenreich W, Zenk MH, Bacher A. An efficient preparation of 2-C-methyl-D-erythritol 4-phosphoric acid and its derivatives. J. Org. Chem. 2000;65:587–592. doi: 10.1021/jo9905118. [DOI] [PubMed] [Google Scholar]
- 63.Hoeffler JF, Grosdemange-Billiard C, Rohmer M. Synthesis of tritium labelled 2-C-methyl-D-erythritol, a useful substrate for the elucidation of the methylerythritol phosphate pathway for isoprenoid biosynthesis. Tetrahedron Lett. 2000;41:4885–4889. [Google Scholar]
- 64.Hoeffler JF, Pale-Grosdemange C, Rohmer M. Chemical synthesis of enantiopure 2-C-methyl-D-erythritol 4-phosphate, the key intermediate in the mevalonate-independent pathway for isoprenoid biosynthesis. Tetrahedron. 2000;56:1485–1489. [Google Scholar]
- 65.Lagisetti C, Urbansky M, Coates RM. The dioxanone approach to (2S 3R)-2-C-methylerythritol 4-phosphate and 2-4-cyclodiphosphate and various MEP analogues. J. Org Chem. 2007;72:9886–9895. doi: 10.1021/jo0711900. [DOI] [PubMed] [Google Scholar]
- 66.Urbansky M, Davis CE, Surjan JD, Coates RM. Synthesis of enantiopure 2-C-methyl-D-erythritol 4-phosphate and 2-4-cyclodiphosphate from D-arabitol. Org. Lett. 2004;6:135–138. doi: 10.1021/ol0362562. [DOI] [PubMed] [Google Scholar]
- 67.Odejinmi SI, Rascon RG, Chen W, Lai K. Formal synthesis of 4-diphosphocytidyl-2-C-methyl D-erythritol from D-(+)-arabitol. Tetrahedron. 2012;68:8937–8941. doi: 10.1016/j.tet.2012.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sharma A, Das P, Chattopadhyay S. Concise asymmetric syntheses of the (+)-2-C-methyltetritol isomers. Tetrahedron Asymmetry. 2008;19:02167–2170. [Google Scholar]
- 69.Koppisch AT, Blagg BS, Poulter CD. Synthesis of 2-C-methyl-D-erythritol 4-phosphate the first pathway-specific intermediate in the methylerythritol phosphate route to isoprenoids. Org Lett. 2000;2:215–217. doi: 10.1021/ol991299x. [DOI] [PubMed] [Google Scholar]
- 70.Bennani YL, Sharpless KB. Asymmetric dihydroxylation (Ad) of NN-DIALKYL and N-methoxy-N-methyl alpha beta-unsaturated and beta gamma-unsaturated amides. Tetrahedron Lett. 1993;34:2079–2082. [Google Scholar]
- 71.Fontana A. Concise synthesis of (+)-2-C-methyl-D-erythritol-4-phosphate. J. Org. Chem. 2001;66:2506–2508. doi: 10.1021/jo005732o. [DOI] [PubMed] [Google Scholar]
- 72.Kis K, Wungsintaweekul J, Eisenreich W, Zenk MH, Bacher A. An efficient preparation of 2-C-methyl-D-erythritol 4-phosphoric acid and its derivatives. J. Org. Chem. 2000;65:587–592. doi: 10.1021/jo9905118. [DOI] [PubMed] [Google Scholar]
- 73.Hirsch G, Grosdemange-Billiard C, Tritsch D, Rohmer M. (3R 4S)-3 4 5-trihydroxy-4-methylpentylphosphonic acid, an isosteric phosphonate analogue of 2-C-methyl-D-erythritol 4-phosphate, a key intermediate in the new pathway for isoprenoid biosynthesis. Tetrahedron Lett. 2004;45:519–521. [Google Scholar]
- 74.Koumbis AE, Kotoulas SS, Gallos JK. A convenient synthesis of 2-C-methyl-D-erythritol 4-phosphate and isotopomers of its precursor. Tetrahedron. 2007;63:2235–2243. [Google Scholar]
- 75.David S, Hanessian S. Regioselective manipulation of hydroxyl groups via organotin derivatives. Tetrahedron. 1985;41:0643–663. [Google Scholar]
- 76.Narayanasamy P, Eoh H, Brennan PJ, Crick DC. Synthesis of 4-diphosphocytidyl-2-C-methyl-D-erythritol 2-phosphate and kinetic studies of Mycobacterium tuberculosis IspF. Chem. Biol. 2010;17:117–122. doi: 10.1016/j.chembiol.2010.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Narayanasamy P, Eoh H, Crick DC. Chemoenzymatic synthesis of 4-diphosphocytidyl-2-C-methyl-D-erythritol: a substrate for IspE. Tetrahedron Lett. 2008;49:4461–4463. doi: 10.1016/j.tetlet.2008.05.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Marlow AL, Kiessling LL. Improved chemical synthesis of UDP-galactofuranose. Org. Lett. 2001;3:2517–2519. doi: 10.1021/ol016170d. [DOI] [PubMed] [Google Scholar]
- 79.Giner JL, Ferris WV. Synthesis of 2-C-methyl-D-erythritol 2,4-cyclopyrophosphate. Org. Lett. 2002;4:1225–1226. doi: 10.1021/ol025661a. [DOI] [PubMed] [Google Scholar]
- 80.Fox DT, Poulter CD. Synthesis of (E)-4-hydroxydimethylallyl diphosphate.An intermediate in the methyl erythritol phosphate branch of the isoprenoid pathway. J. Org. Chem. 2002;67:5009–5010. doi: 10.1021/jo0258453. [DOI] [PubMed] [Google Scholar]
- 81.Amslinger S, Kis K, Hecht S, Adam P, Rohdich F, Arigoni D, Bacher A, Eisenreich W. Biosynthesis of terpenes.Preparation of (E)-1-Hydroxy-2-methyl-but-2-enyl 4-diphoshate an intermediate of the deoxyxylulose phosphate pathway. J. Org. Chem . 2002; 67:4590–4594. doi: 10.1021/jo025705t. [DOI] [PubMed] [Google Scholar]
- 82.Ward JL, Beale MH. Synthesis of (2E)-4-hydroxy-3-methylbut-2-enyl diphosphate, a key intermediate in the biosynthesis of isoprenoids. J. Chem. Soc. Perk. T. 1. 2002:710–712. [Google Scholar]
- 83.Giner JL. A convenient synthesis of (E)-4-hydroxy-3-methyl-2-butenyl pyro-phosphate and its [4-C-13]-labeled form. Tetrahedron Lett. 2002;43:5457–5459. [Google Scholar]
- 84.Gao WY, Loeser R, Raschke M, Dessoy MA, Fulhorst M, Alpermann H, Wessjohann LA, Zenk MH. (E)-4-hydroxy-3-methylbut-2-enyl diphosphate: An intermediate in the formation of terpenoids in plant chromoplasts. Angew. Chem. Int. Ed. Engl. 2002;41:2604–2607. doi: 10.1002/1521-3773(20020715)41:14<2604::AID-ANIE2604>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- 85.Hecht S, Amslinger S, Jauch J, Kis K, Trentinaglia V, Adam P, Eisenreich W, Bacher A, Rohdich F. Studies on the non-mevalonate isoprenoid biosynthetic pathway.Simple methods for preparation of isotope-labeled (E)-1-hydroxy-2-methylbut-2-enyl 4-diphosphate. Tetrahedron Lett. 2002;43:8929–8933. [Google Scholar]
- 86.Wolff M, Seemann M, Grosdemange-Billiard C, Tritsch D, Campos N, Rodriguez-Concepcion M, Boronat A, Rohmer M. Isoprenoid biosynthesis via the methylerythritol phosphate pathway.(E)-4-hydroxy-3-methylbut-2-enyl diphosphate: chemical synthesis and formation from methylerythritol cyclodiphosphate by a cell-free system from Escherichia coli. Tetrahedron Lett. 2002;43:2555–2559. [Google Scholar]
- 87.Fox DT, Poulter CD. Synthesis of (E)-4-hydroxydimethylallyl diphosphate.An intermediate in the methyl erythritol phosphate branch of the isoprenoid pathway. J. Org. Chem. 2002;67:5009–5010. doi: 10.1021/jo0258453. [DOI] [PubMed] [Google Scholar]
- 88.Rohdich F, Hecht S, Gartner K, Adam P, Krieger C, Amslinger S, Arigoni D, Bacher A, Eisenreich W. Studies on the nonmevalonate terpene biosynthetic pathway: Metabolic role of IspH (LytB) protein. Proc. Natl. Acad. Sci. U S A. 2002;99:1158–1163. doi: 10.1073/pnas.032658999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Cramer F, Bohm W. Synthese von geranyl-pyrophospat und farnesyl-pyrophosphat. Angew. Chem. Int. Edit. 1959;71:775–775. [Google Scholar]
- 90.Davisson VJ, Woodside AB, Neal TR, Stremler KE, Muehlbacher M, Poulter CD. Phosphorylation of isoprenoid alcohols. J. Org. Chem. 1986;51:4768–4779. [Google Scholar]
- 91.Davisson VJ, Woodside AB, Poulter CD. Synthesis of allylic and homoallylic isoprenoid pyrophosphates. Method. Enzymol. 1985;110:130–144. doi: 10.1016/s0076-6879(85)10068-6. [DOI] [PubMed] [Google Scholar]
- 92.Walsh CT. Revealing coupling patterns in isoprenoid alkylation biocatalysis. ACS Chem. Biol. 2007;2:296–298. doi: 10.1021/cb700094s. [DOI] [PubMed] [Google Scholar]
- 93.Thulasiram HV, Erickson HK, Poulter CD. Chimeras of two isoprenoid synthases catalyze all four coupling reactions in isoprenoid biosynthesis. Science. 2007;316:73–76. doi: 10.1126/science.1137786. [DOI] [PubMed] [Google Scholar]
- 94.Thulasiram HV, Erickson HK, Poulter CD. A common mechanism for branching, cyclopropanation, and cyclobutanation reactions in the isoprenoid biosynthetic pathway. J. Am. Chem. Soc. 2008;130:1966–1971. doi: 10.1021/ja0771282. [DOI] [PubMed] [Google Scholar]
- 95.Mcgarvey DJ, Croteau R. Terpenoid metabolism. Plant Cell. 1995;7:1015–1026. doi: 10.1105/tpc.7.7.1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Lange BM, Ahkami A. Metabolic engineering of plant monoterpenes, sesquiterpenes and diterpenes-current status and future opportunities. Plant Biotechnol. J. 2013;11:169–196. doi: 10.1111/pbi.12022. [DOI] [PubMed] [Google Scholar]
- 97.Thomas AF, Bessiere Y, ApSimon J, editors. 7 pp. John Wiley & Sons Inc Hoboken: In: The Total Synthesis of Natural Products ; 1988. The synthesis of monoterpenes 1980-1986. pp. 274–454. [Google Scholar]
- 98.Borschberg HR. New strategies for the synthesis of monoterpene indole alkaloids. Curr. Org. Chem. 2005;9:1465–1491. [Google Scholar]
- 99.Takacs JM, Boito SC, Myoung YC. Recent applications of catalytic metal-mediated carbocyclizations in asymmetric synthesis. Curr. Org. Chem. 1998;2:233–254. [Google Scholar]
- 100.Frauenfelder C, Schmid GA, Vogelsang T, Borschberg HJ. Flexible synthetic approaches to monoterpene indole alkaloids. Chimia. 2001;55:828–830. [Google Scholar]
- 101.Bettadaiah BK, editor. University of Msore Mysore: 2003. Studies on new synthetic strategies for O- and S-derivatives of monoterpenes. [Google Scholar]
- 102.Thomas AF, Bessiere Y. Limonene. Nat. Prod. Rep. 1989;6:291–309. [Google Scholar]
- 103.Money T. Camphor - A chiral starting material in natural product synthesis. Nat. Prod. Rep. 1985;2:253–289. doi: 10.1039/np9850200253. [DOI] [PubMed] [Google Scholar]
- 104.de Carvalho C, da Fonseca MMR. Biotransformation of terpenes. Biotechnol. Adv. 2006;24:134–142. doi: 10.1016/j.biotechadv.2005.08.004. [DOI] [PubMed] [Google Scholar]
- 105.Thomas MT, Fallis AG. Total synthesis of (+/-) alpha-pinene and (+/-) beta-pinene - general route to bicyclo[3..1]heptanes. . Tetrahedron Lett. 1973:4687–4690. [Google Scholar]
- 106.Kulkarni YS, Snider BB. Intramolecular [2 + 2] cyclo-additions of ketenes2. Synthesis of chrysanth.none beta-pinene, beta-cis-bergamotene, and beta-trans-Bergamotene. J. Org. Chem . 1985;50:2809–2810. [Google Scholar]
- 107.Cocker W. Review of some investigations of chemistry of carene. J. Soc. Cosmet. Chem. 1971;22:249–284. [Google Scholar]
- 108.Kadow JF, Johnson CR. Synthesis of fused cyclopropanes from gamma-stannyl alcohols. Tetrahedron Lett. 1984;25:5255–5258. [Google Scholar]
- 109.Macaev FZ, Malkov AV. Use of monoterpenes, 3-carene and 2-carene, as synthons in the stereoselective synthesis of 2,2-dimethyl-1,3-disubstituted cyclopropanes. Tetrahedron. 2006;62:9–29. [Google Scholar]
- 110.Merfort I. Perspectives on sesquiterpene lactones in inflammation and cancer. Curr. Drug Targets. 2011;12:1560–1573. doi: 10.2174/138945011798109437. [DOI] [PubMed] [Google Scholar]
- 111.Bos JL. Ras oncogenes in human cancer - a review. Cancer Res. 1989;49:4682–4689. [PubMed] [Google Scholar]
- 112.Harris CM, Poulter CD. Recent studies of the mechanism of protein prenylation. Nat. Prod. Rep. 2000;17:137–144. doi: 10.1039/a904110i. [DOI] [PubMed] [Google Scholar]
- 113.Ohkanda J, Knowles DB, Blaskovich MA, Sebti SM, Hamilton AD. Inhibitors of protein farnesyltransferase as novel anticancer agents. Curr. Top. Med. Chem. 2002;2:303–323. doi: 10.2174/1568026023394281. [DOI] [PubMed] [Google Scholar]
- 114.Mu YQ, Gibbs RA, Eubanks LM, Poulter CD. Cuprate-mediated synthesis and biological evaluation of cyclopropyl- and tert-butylfarnesyl diphosphate analogs. J. Org. Chem. 1996;61:8010–8015. doi: 10.1021/jo9614203. [DOI] [PubMed] [Google Scholar]
- 115.Gibbs RA, Krishnan U, Dolence JM, Poulter CD. A stereoselective palladium copper-catalyzed route to isoprenoids - synthesis and biological evaluation of 13-methylidenefarnesyl diphosphate. J. Org. Chem. 1995;60:7821–7829. [Google Scholar]
- 116.Rawat DS, Gibbs RA. Synthesis of 7-substituted farnesyl diphosphate analogues. Org. Lett. 2002;4:3027–3030. doi: 10.1021/ol026176i. [DOI] [PubMed] [Google Scholar]
- 117.Placzek AT, Gibbs RA. New synthetic methodology for the construction of 7-substituted farnesyl diphosphate analogs. Org. Lett. 2011;13:3576–3579. doi: 10.1021/ol201069x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Wiemer AJ, Wiemer DF, Hohl RJ. Geranylgeranyl diphosphate synthase: An emerging therapeutic target. Clin. Pharmacol. Ther. 2011;90:804–812. doi: 10.1038/clpt.2011.215. [DOI] [PubMed] [Google Scholar]
- 119.Boca Raton Florida: 2003. Dictionary of Natural Products Chapman & Hall through CRC Press Version 11. [Google Scholar]
- 120.Szabo CM, Matsumura Y, Fukura S, Martin MB, Sanders JM, Sengupta S, Cieslak JA, Loftus TC, Lea CR, Lee HJ, Koohang A, Coates RM, Sagami H, Oldfield E. Inhibition of geranylgeranyl diphosphate synthase by bisphosphonates and diphosphates: A potential route to new bone antiresorption and antiparasitic agents. J. Med. Chem. 2002;45:2185–2196. doi: 10.1021/jm010412y. [DOI] [PubMed] [Google Scholar]
- 121.Wiemer AJ, Yu JS, Lamb KM, Hohl RJ, Wiemer DF. Mono- and dialkyl isoprenoid bisphosphonates as geranylgeranyl diphosphate synthase inhibitors. Bioorgan. Med. Chem. 2008;16:390–399. doi: 10.1016/j.bmc.2007.09.029. [DOI] [PubMed] [Google Scholar]
- 122.Shull LW, Wiemer AJ, Hohl RJ, Wiemer DF. Synthesis and biological activity of isoprenoid bisphosphonates. Bioorg. Med. Chem. 2006;14:4130–4136. doi: 10.1016/j.bmc.2006.02.010. [DOI] [PubMed] [Google Scholar]
- 123.Steiger A, Pyun HJ, Coates RM. Synthesis and characterization of aza analog inhibitors of squalene and geranylgeranyl diphosphate synthases. J. Org. Chem. 1992;57:3444–3449. [Google Scholar]
- 124.Casey PJ, Seabra MC. Protein prenyltransferases. J. Biol. Chem. 1996;271:5289–5292. doi: 10.1074/jbc.271.10.5289. [DOI] [PubMed] [Google Scholar]
- 125.Mu YQ, Eubanks LM, Poulter CD, Gibbs RA. Coupling of isoprenoid triflates with organoboron nucleophiles: Synthesis and biological evaluation of geranylgeranyl diphosphate analogues. Bioorg. Med. Chem. 2002;10:1207–1219. doi: 10.1016/s0968-0896(01)00390-x. [DOI] [PubMed] [Google Scholar]
- 126.Dewar MJS, Ruiz JM. Mechanism of the biosynthesis of squalene from farnesyl pyrophosphate. Tetrahedron. 1987;43:2661–2674. [Google Scholar]
- 127.Cornforth JW, Cornforth RH, Mathew KK. A stereoselective synthesis of squalene. J. Chem. Soc. 1959:2539–2547. [Google Scholar]
- 128.Faulkner DJ, Petersen MR. Application of claisen rearrangement to synthesis of trans trisubstituted olefinic bonds - synthesis of squalene and insect juvenile-hormone. J. Am. Chem. Soc. 1973;95:553–563. doi: 10.1021/ja00783a040. [DOI] [PubMed] [Google Scholar]
- 129.Weiss TL, Roth R, Goodson C, Vitha S, Black I, Azadi P, Rusch J, Holzenburg A, Devarenne TP, Goodenough U. Colony organization in the green alga Botryococcus braunii (Race B) is specified by a complex extracellular matrix. Eukaryot. Cell. 2012;11:1424–1440. doi: 10.1128/EC.00184-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Eroglu E, Okada S, Melis A. Hydrocarbon productivities in different Botryococcus strains: comparative methods in product quantification. J. Appl. Phycol. 2011;23:763–775. doi: 10.1007/s10811-010-9577-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Lovejoy KS, Davis LE, McClellan LM, Lillo AM, Welsh JD, Schmidt EN, Sanders CK, Lou AJ, Fox DT, Koppisch AT, Del Sesto RE. Evaluation of ionic liquids on phototrophic microbes and their use in biofuel extraction and isolation. J. Appl. Phycol. 2013;25:973–981. [Google Scholar]
- 132.Niehaus TD, Kinison S, Okada S, Yeo YS, Bell SA, Cui P, Devarenne TP, Chappell J. Functional identification of triterpene methyltransferases from Botryococcus braunii Race B. J. Biol. Chem. 2012;287:8163–8173. doi: 10.1074/jbc.M111.316059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Hillen LW, Pollard G, Wake LV, White N. Hydrocracking of the oils of Botryococcus braunii to transport fuels. Biotechnol. Bioeng. 1982;24:193–205. doi: 10.1002/bit.260240116. [DOI] [PubMed] [Google Scholar]
- 134.Pulis AP, Aggarwal VK. Synthesis of enantioenriched tertiary boronic esters from secondary allylic carbamates.Application to the synthesis of C30 botryococcene. J. Am. Chem. Soc. 2012;134:7570–7574. doi: 10.1021/ja303022d. [DOI] [PubMed] [Google Scholar]
- 135.Davies JJ, Krulle TM, Burton JW. Total synthesis of 7, 11-cyclobotryococca-5, 12, 26-triene using an oxidative radical cyclization as a key step. Org. Lett. 2010;120:2738–2741. doi: 10.1021/ol100794k. [DOI] [PubMed] [Google Scholar]
- 136.Hird NW, Lee TV, Leigh AJ, Maxwell JR, Peakman TM. The total synthesis of 10-(R,S)-C-30 botryococcene and botryococcane and a new synthesis of a general intermediate to the botryococcene family. Tetrahedron Lett. 1989;30:4867–4870. [Google Scholar]
- 137.White JD, Reddy GN, Spessard GO. Total synthesis of (-)-botryococcene. J. Am. Chem. Soc. 1988;110:1624–1626. [Google Scholar]
- 138.White JD, Reddy GN, Spessard GO. Total synthesis of (-)-C-34-botryococcene, the principal triterpenoid hydrocarbon of the freshwater alga Botryococcus braunii. J. Chem. Soc. Perk. T 1. 1993:759–767. [Google Scholar]
- 139.Dall'Osto L, Holt NE, Kaligotla S, Fuciman M, Cazzaniga S, Carbonera D, Frank HA, Alric J, Bassi R. Zeaxanthin protects plant photosynthesis by modulating chlorophyll triplet yield in specific light-harvesting antenna subunits. J. Biol. Chem. 2012;287:41820–41834. doi: 10.1074/jbc.M112.405498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Shegokar R, Mitri K. Carotenoid lutein: a promising candidate for pharmaceutical and nutraceutical applications. J. Diet. Suppl. 2012;9:183–210. doi: 10.3109/19390211.2012.708716. [DOI] [PubMed] [Google Scholar]
- 141.Yamaguchi M. Role of carotenoid beta-cryptoxanthin in bone homeostasis. J. Biomed. Sci. 2012;19 doi: 10.1186/1423-0127-19-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Di Mascio P, Kaiser S, Sies H. Lycopene as the most efficient biological carotenoid singlet oxygen quencher. Arch. Biochem. Biophys. 1989;274:532–538. doi: 10.1016/0003-9861(89)90467-0. [DOI] [PubMed] [Google Scholar]
- 143.Shen RP, Jiang XY, Ye WD, Song XH, Liu L, Lao XJ, Wu CL. A novel and practical synthetic route for the total synthesis of lycopene. Tetrahedron. 2011;67:5610–5614. [Google Scholar]
- 144.Landrum JT, Bone RA. Lutein, zeaxanthin, and the macular pigment. Arch. Biochem. Biophys. 2001;385:28–40. doi: 10.1006/abbi.2000.2171. [DOI] [PubMed] [Google Scholar]
- 145.Khachik F, Chang AN. Total synthesis of (3R,3 ' R,6 ' R)-Lutein and its stereoisomers. J. Org. Chem. 2009;74:3875–3885. doi: 10.1021/jo900432r. [DOI] [PubMed] [Google Scholar]