

Abstract
Background
Gastroenteropancreatic neuroendocrine tumors have occasionally been described in association with neurofibromatosis type 1, whereas an association with neurofibromatosis type 2 has never been reported.
Case presentation
This report refers to an Italian 69 year old woman with neurofibromatosis type 2 and a pancreatic gastrinoma. In the past she had encephalic meningiomas, a tongue schwannoma and bilateral acoustic neurinomas. She presented with weight loss and a long-term history of diarrhea, responsive to proton pump inhibitors. Upper gastrointestinal endoscopy revealed peptic ulcer of the duodenal bulb. Blood tests were normal, except for the elevation of plasma gastrin (1031 pg/ml; reference value <108) and chromogranin A (337 U/L; reference value <36). After secretin stimulation testing, the plasma gastrin level rose to 3789 pg/ml. The abdomen magnetic resonance imaging and gallium68-DOTATOC positron emission tomography scan demonstrated the presence of a 1.2 x 2 cm lesion in the pancreatic head and a liver metastatis. Pancreatic endoscopic ultrasound with fine needle aspiration revealed cytomorphologic features suggestive of pancreatic gastrinoma. Brain magnetic resonance showed a pituitary microadenoma. There was no evidence of hyperparathyroidism. The genetic test for multiple endocrine neoplasia type 1 syndrome mutation was negative.
Conclusion
This report focuses on the first case of coexistence of gastrinoma with neurofibromatosis type 2. Although the clinical relevance of this association remains to be determined, our case report will surely give cause for due consideration.
Keywords: Gastrinoma, Neurofibromatosis type 2, Multiple endocrine neoplasia syndrome type 1, Neuroendocrine tumors, Zollinger Ellison syndrome
Background
Both gastroenteropancreatic neuroendocrine tumors (GEP NET) and neurofibromatosis type 2 (NF2) are fairly rare diseases [1,2]. GEP NET have occasionally been described in association with neurofibromatosis type 1 (NF1) [3], whereas an association with NF2 has never been reported.
Gastrinoma is a gastrin-secreting tumor usually located in the duodenal wall or in the pancreas [4]. Chronic hypergastrinemia causes gastric acid hypersecretion with consequent peptic ulcer disease, diarrhea and gastroesophageal reflux disease (Zollinger Ellison syndrome, ZES), usually responsive to proton pump inhibitor (PPI) therapy. With an incidence of 0.5-2/million population/year gastrinoma may be sporadic or may develop in the setting of multiple endocrine neoplasia type 1 (MEN1) syndrome [5-8]. The tumors associated with MEN1 syndrome have a more benign course than sporadic neuroendocrine tumors, although more aggressive tumors have rarely been described in association with MEN1 syndrome [9]. MEN1 is an autosomal dominant inherited disease, due to inactivating mutations of the MEN1 gene, located on the long arm of chromosome 11 (11q13) [10]. Diagnostic criteria for MEN1 include the presence of at least two of the three main MEN1 associated lesions (primary hyperparathyroidism, GEP NET and anterior pituitary tumors) or the association of any typical MEN1-associated neoplasia and a positive familial history [11]. Diagnostic confirmation is achievable through MEN1 genetic testing.
According to available data, an association between GEP NET and NF1 has already been reported and it is well recognized that NF1 patients may develop NET, usually in the duodenum or periampullary region [12-14] and occasionally in the pancreas [15]. Gastric carcinoids have also been described [16].
NF2 is an autosomal dominant inherited disease characterized by the predisposition to develop tumors of the central nervous system (CNS) [2]. NF2 is caused by mutations of the NF2 gene that codes for an oncosuppressor protein named merlin or schwannomin and is located on chromosome 22 (22q12.2) [17]. A common manifestation of the disease is the development of vestibular schwannomas, tumors in other cranial nerves and meningiomas, usually multifocal and intracranial, but sometimes also spinal or intradural [18].
At present an association between GEP NET and NF2 has never been reported. The present case report refers to a woman with NF2 and a pancreatic gastrinoma.
Case presentation
In 2011 an Italian 69 years old woman was referred to our Gastrointestinal Unit due to an 8 years long history of diarrhea, dyspepsia and weight loss. Diarrhea was responsive to PPI. Two years before (2009) she was diagnosed as having NF2 syndrome, according to internationally accepted clinical criteria [19], based on the finding of multiple encephalic meningiomas (partially removed by surgery), a tongue schwannoma and bilateral acoustic neurinomas. In addition, a non-functioning adrenal adenoma, has also been diagnosed. To investigate the cause of diarrhea, the patient had already undergone both a colonoscopy, negative, and an upper gastrointestinal endoscopy, which provided clear cut evidence of a duodenal bulb ulceration; during the endoscopy the measurement of intragastric pH was not performed, since the patient was on PPI. After two weeks of PPI withdrawal laboratory tests revealed a marked increase in plasma gastrin levels (1051 pg/ml; reference value, r.v. <108), chromogranin A (337 U/L; r.v. <36), glucagon (524 pg/ml; r.v. 25–250). The liver, kidney, thyroid and parathyroid function tests were normal. Moreover, secondary causes of hypergastrinemia were excluded.A secretin stimulation test (Secrelux 2U/Kg, Goldham-Bioglan, Zusmarhausen. Germany) was performed and showed a diagnostic increase of plasma gastrin levels (from 1031 to 3789 pg/ml) within ten minutes from the secretin injection (Figure 1). The patient underwent Octreoscan® with the evidence of a hypercaptant pancreatic lesion and a second one in the right frontal area, consistent with a meningiomatous lesion. Other imaging studies included abdomen magnetic resonance (MR) and Gallium-68-DOTATOC positron emission tomography (PET) (Figure 2), which revealed a 1.2 × 2 cm nodular lesion in the pancreatic head, a liver metastatis and, the already known cerebral meningioma. After several months the patient underwent pancreatic endoscopic ultrasound with fine needle aspiration, demonstrating cytomorphologic features suggestive of pancreatic gastrinoma, with well-differentiated cells expressing Chromogranin A, synaptophysin and gastrin at immunohistochemistry. Ki-67 index was 2% and no mitosis was found in the sample.Due to the coexistence of a gastrinoma and an adrenal adenoma, a brain and encephalic trunk MR (Figure 3) was performed to investigate the possible presence of a pituitary neoplasm: this revealed partial empty sella, the residual meningiomatous lesions and a pituitary microadenoma, which resulted non functioning at hormonal blood tests. There was no evidence of hyperparathyroidism, but, given the presence of a pancreatic gastrinoma, a pituitary and an adrenal adenoma, the patient underwent genetic testing for MEN1 mutations, which was negative. Genetic test for the CDKN1B gene was not performed since the patient died because of massive bleeding from a cerebral meningioma.
Figure 1.
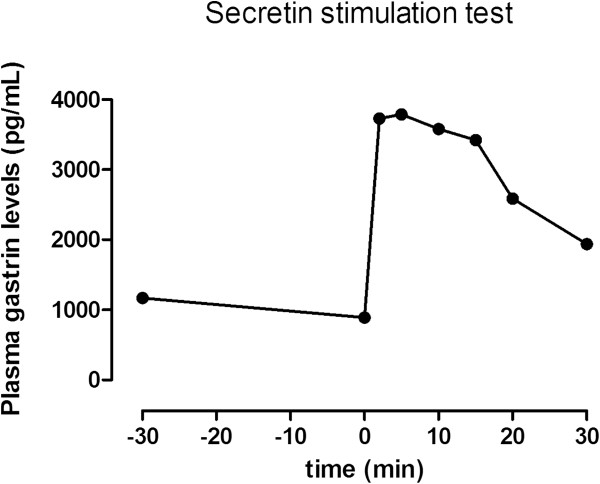
Secretin stimulation test in the patient. Plasma gastrin levels were measured at -30 and 0 time, and 2, 5, 10, 15, 20 and 30 minutes after intravenous secretin infusion (Secrelux 2U/kg, Goldham-Bioglan, Zusmarhausen. Germany). The increase from 1031 to 3789 pg/ml was observed within ten minutes.
Figure 2.
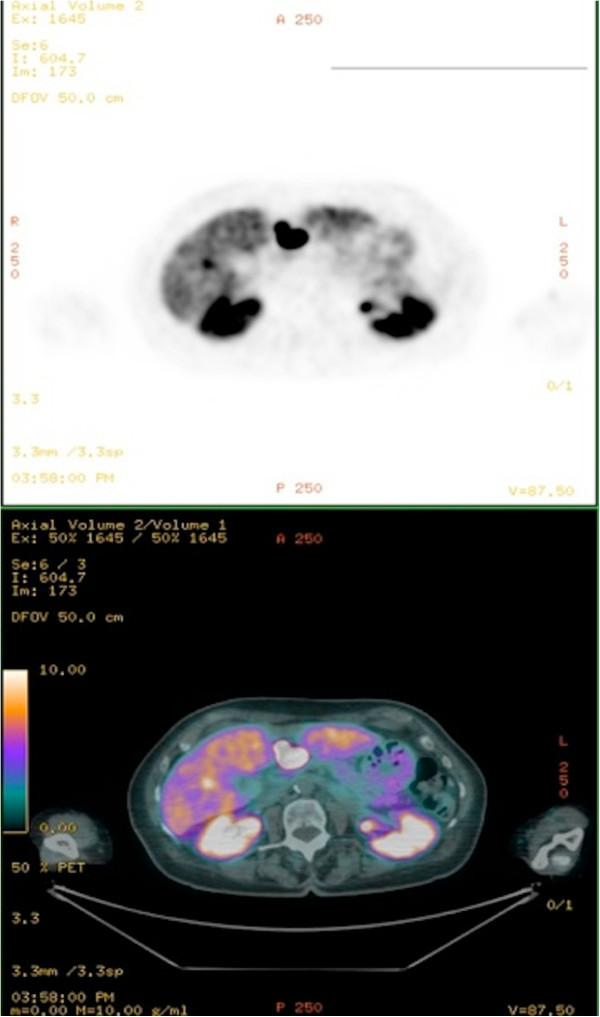
Gallium-68-DOTATOC PET. Gallium68 PET detected the presence of a nodular lesion of cm 1.2 x 2 in the pancreatic head and a suspected liver metastatic lesion.
Figure 3.
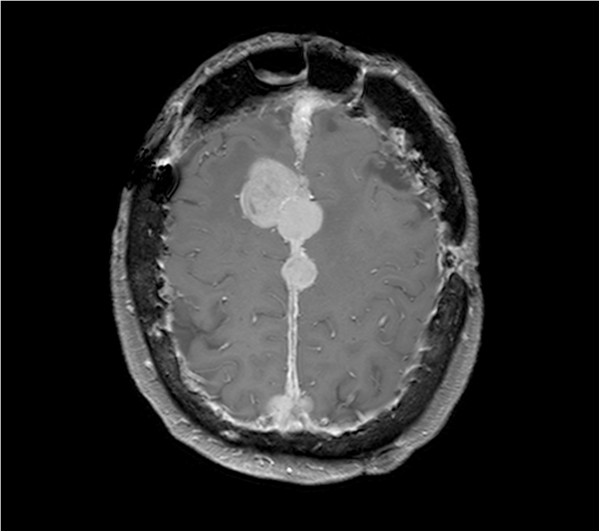
Brain MR with contrast. This T1-weighted image detected multiple meningiomas enhancing intensely and homogeneously after administration of gadolinium.
Conclusions
We have reported the first case report of the association between NF2 and a pancreatic gastrinoma, although it remains to be determined whether this association is merely casual or not. The possible underlying pathogenetic mechanisms need also elucidation.
The association between NF1 and GEP NET has already been established. Several case reports and reviews of the literature describe the coexistence of NF1 and neuroendocrine neoplasms, such as somatostatinomas, gastrinomas [20], insulinomas [21] and gastric carcinoids [8-12]. In patients with NF1 NETs are most commonly localized in the duodenum and in the peri-ampullary region. Although NF1 associated duodenal neuroendocrine tumors are not distinctive, they tend to be pure somatostatinomas [22,23] or less frequently gastrinomas, whereas duodenal tumors not associated with NF1 are frequently multihormonal, but asymptomatic [24]. Other gastrointestinal manifestations can occur in several locations, in the form of gastrointestinal stromal tumors or ganglioneuromas [3]. Also an association between NF1 and MEN2A is described in one case report [25].
As concerns patients with NF2, schwannomas of the intestinal tract are a possible complication [26], whereas there are no data about coexistence with GEP NET and/or MEN1 syndrome.
An increased occurrence of endocrine tumors, as well as of other non endocrine tumors, including collagenomas, ependimomas, schwannomas and meningiomas has been recently reported in patients with MEN1 [27-29]. Meningiomas arising in MEN1 setting are usually silent, thus the diagnosis is often late, with a mean interval of 18 years from the diagnosis of MEN1; they have a higher incidence in patients with ZES in the MEN1 setting than in patients with sporadic ZES [30].
The present report focuses on the first case of gastrinoma in a NF2 patient. This patient might have a MEN1 like phenotype, as she showed the clinical features typical of MEN1 syndrome (pancreatic neuroendocrine tumor, pituitary microadenoma and adrenal adenoma), despite the absence of hyperparathyroidism and MEN1 gene mutations. The phenotype of MEN1 is broad and different combinations of endocrine and non endocrine manifestations have been described. About 30% MEN1 patients do not carry detectable MEN1 gene mutations [31]. Patients without detectable mutations may have mutations in introns or in the regulatory or untranslated regions. Moreover, the polymerase chain reaction does not reveal a large deletion causing the loss of a whole exon. Recently, germ line mutations have been found in the CDKN1B gene, encoding a cyclin dependent kinase inhibitor protein that inhibits cell cycle progression [32,33], even if only 1.6-2.8% of all tested cases of MEN1 have CDKN1B mutations [34]. The possible pathogenic mechanisms underlying the dual development of gastrinoma and NF2 remain to be elucidated. Interestingly, a possible explanation for the high prevalence of neuroendocrine tumors in NF1 might be the loss of neurofibromin, a tumor suppressor protein, representing the main product of the NF1 gene. Again, the mutation of the G protein Ras, often seen in NF1 tumors, could promote tumorigenesis due to a prolonged or persistent arrest in the activated GTP loaded state. Recent studies have clearly shown that also the tumor-suppressor protein merlin, mutated in NF2, controls Ras activity [35]. Furthermore, merlin probably regulates angiogenesis to support schwannoma growth, although the exact mechanisms are still unexplored [36]. Angiogenesis is an important determinant of tumor growth also in neuroendocrine neoplasms, so merlin could influence their development and growth. Another possible explanation of the association between NF2 and GEP NET may be the likely common embryological origin of nervous and neuroendocrine cells from the neural crest, although some authors have recently suggested that the neuroendocrine system might originate from the endoderm [37,38].
Overall, the possible pathogenic mechanisms underlying the occurrence of both gastrinoma and NF2, which would allow to discriminate between an actual connection and a merely casual association, remain to be elucidated.
Consent
Written informed consent was obtained from the patient for publication of this case report and any accompanying images. A copy of the written consent is available for review by the Editor-in-Chief of this journal.
Abbreviations
GEP NET: Gastroenteropancreatic neuroendocrine tumors; NF2: Neurofibromatosis type 2; NF1: Neurofibromatosis type 1; ZES: Zollinger Ellison syndrome; PPI: Proton pump inhibitor; MEN1: Multiple endocrine neoplasia type 1; CNS: Central nervous system; MR: Magnetic resonance; PET: Positron emission tomography.
Competing interests
We have no conflict of interest to declare.
Authors’ contributions
SM planned the work. SM, AZ and RER wrote the paper and subsequently performed its critical review, contributing equally to this work. AZ and FC carried out the literature research; MP contributed to the acquisition of data and their interpretation. DC and MP revised all the materials and manuscript. All authors read and approved the final manuscript.
Pre-publication history
The pre-publication history for this paper can be accessed here:
Contributor Information
Sara Massironi, Email: sara.massironi@policlinico.mi.it.
Alessandra Zilli, Email: zilli.alessandra@libero.it.
Roberta Elisa Rossi, Email: robertaelisa.rossi@gmail.com.
Federica Cavalcoli, Email: federica.cavalcoli@studenti.unimi.it.
Dario Conte, Email: dario.conte@unimi.it.
Maddalena Peracchi, Email: maddalena.peracchi@unimi.it.
Acknowledgments
The authors acknowledge Paolo Giorgio Arcidiacono, MD and Silvia Carrara, MD (Vita-Salute San Raffaele University, San Raffaele Scientific Institute, Milan, Italy) for their help in patient’s clinical management, and Marcello Hinxman-Allegri for the linguistic revision.
References
- Öberg KE. Gastrointestinal neuroendocrine tumors. Ann Oncol. 2010;21:72–80. doi: 10.1093/annonc/mdq290. [DOI] [PubMed] [Google Scholar]
- Evans DG. Neurofibromatosis type 2 (NF2): a clinical and molecular review. Orphanet J Rare Dis. 2009;4:16. doi: 10.1186/1750-1172-4-16. http://www.ncbi.nlm.nih.gov/pubmed/?term=Evans+DG%3A+Neurofibromatosis+type+2+(NF2)%3A+a+clinical+and+molecular+review.+Orphanet+J+Rare+Dis+2009%2C+4%3A16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuller CE, Williams GT. Gastrointestinal manifestations of type 1 neurofibromatosis (von Recklinghausen's disease) Histopathology. 1991;19:1–11. doi: 10.1111/j.1365-2559.1991.tb00888.x. [DOI] [PubMed] [Google Scholar]
- Krampitz JW, Norton JA. Current management of the Zollinger-Ellison syndrome. Adv Surg. 2013;47:59–79. doi: 10.1016/j.yasu.2013.02.004. [DOI] [PubMed] [Google Scholar]
- Ito T, Igarashi H, Jensen RT. Zollinger-Ellison syndrome: recent advances and controversies. Curr Opin Gastroenterol. 2013;29:650–661. doi: 10.1097/MOG.0b013e328365efb1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen RT, Niederle B, Mitry E, Ramage JK, Steinmuller T, Lewington V, Scarpa A, Sundin A, Perren A, Gross D, O'Connor JM, Pauwels S, Kloppel G. Frascati. Consensus Conference; European Neuroendocrine Tumor Society. Gastrinoma (duodenal and pancreatic) Neuroendocrinology. 2006;84:173–182. doi: 10.1159/000098009. [DOI] [PubMed] [Google Scholar]
- Klöppel G, Anlauf M. Gastrinoma–morphological aspects. Wien Klin Wochenschr. 2007;119:579–584. doi: 10.1007/s00508-007-0885-1. [DOI] [PubMed] [Google Scholar]
- Anlauf M, Garbrecht N, Henopp T, Schmitt A, Schlenger R, Raffel A, Krausch M, Gimm O, Eisenberger CF, Knoefel WT, Dralle H, Komminoth P, Heitz PU, Perren A, Klöppel G. Sporadic versus hereditary gastrinomas of the duodenum and pancreas: distinct clinic-pathological and epidemiological features. World J Gastroenterol. 2006;12:5440–5446. doi: 10.3748/wjg.v12.i34.5440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akerström G, Hessman O, Hellman P, Skogseid B. Pancreatic tumours as part of he MEN-1 syndrome. Best Pract Res Clin Gastroenterol. 2005;19:819–830. doi: 10.1016/j.bpg.2005.05.006. [DOI] [PubMed] [Google Scholar]
- Thakker RV, Newey PJ, Walls GV, Bilezikian J, Dralle H, Ebeling PR, Melmed S, Sakurai A, Tonelli F, Brandi ML. Endocrine Society. Clinical practice guidelines for multiple endocrine neoplasia type 1 (MEN1) J Clin Endocrinol Metab. 2012;97:2990–3011. doi: 10.1210/jc.2012-1230. [DOI] [PubMed] [Google Scholar]
- Piecha G, Chudek J, Wiecek A. Multiple Endocrine Neoplasia type 1. Eur J Intern Med. 2008;19:99–103. doi: 10.1016/j.ejim.2007.08.004. [DOI] [PubMed] [Google Scholar]
- Ferner RE, Huson SM, Thomas N, Moss C, Willshaw H, Evans DG, Upadhyaya M, Towers R, Gleeson M, Steiger C, Kirby A. Guidelines for the diagnosis and management of individuals with neurofibromatosis. J Med Genet. 2007;44:81–88. doi: 10.1136/jmg.2007.049122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams VC, Lucas J, Babcock MA, Gutmann DH, Korf B, Maria BL. Neurofibromatosis type 1 revisited. Pediatrics. 2009;23:124–133. doi: 10.1542/peds.2007-3204. [DOI] [PubMed] [Google Scholar]
- Boyd KP, Korf BR, Theos A. Neurofibromatosis type 1. J Am Acad Dermatol. 2009;61:1–16. doi: 10.1016/j.jaad.2008.12.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishi T, Kawabata Y, Hari Y, Imaoka H, Ishikawa N, Yano S, Maruyama R, Tajima Y. A case of pancreatic neuroendocrine tumor in a patient with neurofibromatosis-1. World J Surg Oncol. 2012;10:153. doi: 10.1186/1477-7819-10-153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart W, Traynor JP, Cooke A, Griffiths S, Onen NF, Balsitis M, Shah AA, Upadhyaya M, Tobias ES. Gastric carcinoid: germline and somatic mutation of the neurofibromatosis type 1 gene. Fam Cancer. 2007;6:147–152. doi: 10.1007/s10689-006-9002-2. [DOI] [PubMed] [Google Scholar]
- Seizinger BR, Martuza RL, Gusella JF. Loss of genes on chromosome 22 in tumorigenesis of human acoustic neuroma. Nature. 1986;322:644–647. doi: 10.1038/322644a0. [DOI] [PubMed] [Google Scholar]
- Mautner VF, Lindenau M, Baser ME, Hazim W, Tatagiba M, Haase W, Samii M, Wais R, Pulst SM. The neuroimaging and clinical spectrum of neurofibromatosis 2. Neurosurgery. 1996;38:880–885. doi: 10.1097/00006123-199605000-00004. [DOI] [PubMed] [Google Scholar]
- Hoa M, Slattery WH 3rd. Neurofibromatosis 2. Otolaryngol Clin North Am. 2012;45:315–332. doi: 10.1016/j.otc.2011.12.005. [DOI] [PubMed] [Google Scholar]
- Lee WS, Koh YS, Kim JC, Park CH, Joo YE, Kim HS, Cho CK, Choi SK, Rew JS, Kim SJ. Zollinger-Ellison syndrome associated with neurofibromatosis type 1: a case report. BMC Cancer. 2005;5:85. doi: 10.1186/1471-2407-5-85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fung JW, Lam KS. Neurofibromatosis and insulinoma. Postgrad Med J. 1995;71:485–486. doi: 10.1136/pgmj.71.838.485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao C, Shah A, Hanson DJ, Howard JM. Von Recklinghausen's disease associated with duodenal somatostatinoma: contrast of duodenal versus pancreatic somatostatinomas. J Surg Oncol. 1995;59:67–73. doi: 10.1002/jso.2930590116. [DOI] [PubMed] [Google Scholar]
- Cappelli C, Agosti B, Braga M, Cumetti D, Gandossi E, Rizzoni D, Agabiti Rosei E. Von Recklinghausen's neurofibromatosis associated with duodenal somatostatinoma. A case report and review of the literature. Minerva Endocrinol. 2004;29:19–24. [PubMed] [Google Scholar]
- Dayal Y, Tallberg KA, Nunnemacher G, DeLellis RA, Wolfe HJ. Duodenal carcinoids in patients with and without neurofibromatosis. A comparative study. Am J Surg Pathol. 1986;10:348–357. doi: 10.1097/00000478-198605000-00007. [DOI] [PubMed] [Google Scholar]
- Gkaliagkousi E, Erlic Z, Petidis K, Semertzidis P, Doumas M, Zamboulis C, Neumann HP, Douma S. Neurofibromatosis type 1: should we screen for other genetic syndromes? A case report of co-existence with multiple endocrine neoplasia 2A. Eur J Clin Invest. 2009;39:828–832. doi: 10.1111/j.1365-2362.2009.02174.x. [DOI] [PubMed] [Google Scholar]
- Ogasawara N, Sasaki M, Ishiguro H, Itoh Y, Nojiri S, Kubota E, Wada T, Kataoka H, Kuwabara Y, Joh T. Gastric schwannoma with adjacent external progression harbored aberrant NF2 gene. Dig Endosc. 2009;21:192–195. doi: 10.1111/j.1443-1661.2009.00885.x. [DOI] [PubMed] [Google Scholar]
- Kato H, Uchimura I, Morohoshi M, Fujisawa K, Kobayashi Y, Numano F, Goseki N, Endo M, Tamura A, Nagashima C. Multiple endocrine neoplasia type 1 associated with spinal ependymoma. Intern Med. 1996;35:285–289. doi: 10.2169/internalmedicine.35.285. [DOI] [PubMed] [Google Scholar]
- Chigot JP, Bendib S, Turpin G, Benlian P. Characteristic pathological associations in multiple endocrine neoplasia type 1. Presse Med. 1996;25:1229–1233. [PubMed] [Google Scholar]
- Banik S, Hasleton PS, Lyon RL. An unusual variant of multiple endocrine neoplasia syndrome: a case report. Histopathology. 1984;8:135–144. doi: 10.1111/j.1365-2559.1984.tb02328.x. [DOI] [PubMed] [Google Scholar]
- Asgharian B, Chen YJ, Patronas NJ, Peghini PL, Reynolds JC, Vortmeyer A, Zhuang Z, Venzon DJ, Gibril F, Jensen RT. Meningiomas may be a component tumor of multiple endocrine neoplasia type 1. Clin Cancer Res. 2004;10:869–880. doi: 10.1158/1078-0432.CCR-0938-3. [DOI] [PubMed] [Google Scholar]
- Ozawa A, Agarwal SK, Mateo CM, Burns AL, Rice TS, Kennedy PA, Quigley CM, Simonds WF, Weinstein LS, Chandrasekharappa SC, Collins FS, Spiegel AM, Marx SJ. The parathyroid/pituitary variant of multiple endocrine neoplasia type 1 usually has causes other than p27Kip1 mutations. J Clin Endocrinol Metab. 2007;92:1948–1951. doi: 10.1210/jc.2006-2563. [DOI] [PubMed] [Google Scholar]
- Pellegata NS, Quintanilla-Martinez L, Siggelkow H, Samson E, Bink K, Höfler H, Fend F, Graw J, Atkinson MJ. Germ-line mutations in p27Kip1 cause a multiple endocrine neoplasia syndrome in rats and humans. Proc Natl Acad Sci U S A. 2006;103:15558–15563. doi: 10.1073/pnas.0603877103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agarwal SK, Mateo CM, Marx SJ. Rare germline mutations in cyclin-dependent kinase inhibitor genes in multiple endocrine neoplasia type 1 and related states. J Clin Endocrinol Metab. 2009;94:1826–1834. doi: 10.1210/jc.2008-2083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Georgitsi M, Raitila A, Karhu A, van der Luijt RB, Aalfs CM, Sane T, Vierimaa O, Mäkinen MJ, Tuppurainen K, Paschke R, Gimm O, Koch CA, Gündogdu S, Lucassen A, Tischkowitz M, Izatt L, Aylwin S, Bano G, Hodgson S, De Menis E, Launonen V, Vahteristo P, Aaltonen LA. Germline CDKN1B/p27Kip1 mutation in multiple endocrine neoplasia. J Clin Endocrinol Metab. 2007;92:3321–3325. doi: 10.1210/jc.2006-2843. [DOI] [PubMed] [Google Scholar]
- Morrison H, Sperka T, Manent J, Giovannini M, Ponta H, Herrlich P. Merlin/neurofibromatosis type 2 suppresses growth by inhibiting the activation of Ras and Rac. Cancer Res. 2007;67:520–527. doi: 10.1158/0008-5472.CAN-06-1608. [DOI] [PubMed] [Google Scholar]
- Wong HK, Shimizu A, Kirkpatrick ND, Garkavtsev I, Chan AW, di Tomaso E, Klagsbrun M, Jain RK. Merlin/NF2 regulates angiogenesis in schwannomas through a Rac1/semaphorin 3 F-dependent mechanism. Neoplasia. 2012;14:84–94. doi: 10.1593/neo.111600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rawdon BB, Andrew A. Origin and differentiation of gut endocrine cells. Histol Histopathol. 1993;8:567–580. [PubMed] [Google Scholar]
- Sidhu GS. The endodermal origin of digestive and respiratory tract APUD cells. Histopathologic evidence and a review of the literature. Am J Pathol. 1979;96:5–20. [PMC free article] [PubMed] [Google Scholar]