

Summary
An analogue of the anticancer drug cisplatin (mtPt) was delivered to mitochondria of human cells using a peptide specifically targeting this organelle. mtPt induces apoptosis without damaging nuclear DNA, indicating that mtDNA damage is sufficient to mediate the activity of a platinum-based chemotherapeutic. This study is the first to demonstrate specific delivery of a platinum drug to mitochondria and to investigate the effects of directing this agent outside the nucleus.
Introduction
cis-Diamminedichloroplatinum(II), or cisplatin, is an effective chemotherapeutic agent that is used in nearly 50% of all cancer patients (Galanski et al., 2005). The second generation platinum-based drugs, carboplatin and oxaliplatin, also play an important role in modern chemotherapy. These compounds bind to nuclear DNA, resulting primarily in intrastrand DNA cross-links (Todd et al., 2009). The lesions they cause inhibit transcription, ultimately triggering apoptosis and cell death (Todd et al., 2009). It is important, however, to understand whether alternative cellular targets besides nuclear DNA can potentiate the activity of platinum-based drugs because they offer the opportunity to treat resistant tumors. Furthermore, a greater understanding of other platinum drug targets might allow treatment-limiting side effects to be mitigated.
Owing to their central role in facilitating apoptosis, mitochondria are being actively explored as potential anticancer drug targets (Fulda et al., 2010). Mitochondria contain their own circular DNA (mtDNA), the potential importance of which as a target during platinum-based chemotherapy has not been fully evaluated. Previous studies have proposed mitochondrial and not nuclear DNA as the critical target of cisplatin in potentiating its anticancer activity (Cullen et al., 2007), and under certain conditions higher levels of cisplatin adducts are observed in mitochondrial relative to nuclear DNA (Murata et al., 1990; Olivero et al., 2005). Mitochondria also appear to play a role in mediating cellular resistance to cisplatin. Cisplatin-resistant cell lines have elevated mitochondrial membrane potentials (Andrews et al., 1992; Isonishi et al., 2001), sustain less damage to mtDNA when treated with the drug (Hirama et al., 2006), and exhibit substantially less mitochondrial uptake of cisplatin (Groessl et al., 2011) compared to non-resistant parent lines.
To investigate more precisely the effects of mitochondrial targeting by a potential platinum chemotherapeutic, we designed such a complex that would selectively localize to this organelle. A mitochondria-penetrating peptide (MPP) was appended to the cis-{Pt(NH3)2}2+ DNA-binding unit of cisplatin and carboplatin. MPPs are short, cell-permeable peptide sequences comprising alternating lipophilic and cationic residues that exhibit minimal toxicity towards human cells (Horton et al., 2008). Here, we describe the synthesis and biological properties of a platinum(II) complex conjugated to the N-terminus of an MPP to determine the effect of mitochondrial targeting on the activity of a platinum-based agent. This study is the first to probe the consequences of platinum directed specifically to mitochondria in a cancer cell.
Platinum-peptide conjugates reported previously use a variety of different linking strategies. Such conjugates have been prepared by attaching the peptide to the non-leaving group ligand (amine) of a platinum(II) complex (Robillard et al., 2000; Barragan et al., 2009; Damian et al., 2010), the leaving group ligand (carboxylate) of a platinum(II) complex (Ndinguri et al., 2009), or through axial ligands of a platinum(IV) prodrug (Mukhopadhyay et al., 2008; Graf et al., 2012). Here we began with the novel platinum(II) complex [Pt(succac)(NH3)2](NO3), where succac = succinylacetonate, as detailed in the Online Methods. Structural and spectroscopic characterization data for this complex are shown in Supplementary Figure S1. The succac ligand contains both a β-diketonate group for coordination to platinum as the leaving group ligand with a dangling carboxylic acid functionality for amide-bond formation. [Pt(succac)(NH3)2](NO3) was conjugated to the N-terminus of the MPP r(Fxr)3, where r and Fx are the unnatural amino acid residues d-arginine and l-cyclohexylalanine, respectively. This peptide was selected for conjugation because it exhibits no toxicity towards human cells (Horton et al., 2012) and is composed of artificial amino acids and is therefore not degraded by proteases (Fonseca et al., 2011). This peptide/platinum conjugate is referred to as mtPt (Figure 1A). A fluorophore-labeled analogue, mtPt-TAMRA (Supplementary Figure S2), was also prepared featuring attachment of carboxytetramethylrhodamine (TAMRA) on the amino side-chain of a C-terminal lysine. For both compounds, the platinum unit was attached while these peptides remained on the solid-phase support. The peptides were then cleaved from the resin with neat TFA and purified by reverse-phase HPLC. The purified Pt-peptide conjugates were characterized by ESI-MS and 195Pt NMR spectroscopy (Supplementary Notes 2-3 and Supplementary Figure S1 and S2). The ability to tether cis-diammineplatinum(II) groups to peptides on the solid-phase via linkages in their leaving groups using [Pt(succac)(NH3)2](NO3) represents a general strategy for the design of platinum-peptide conjugates.
Figure 1. mtPt localizes to mitochondria and is active against a cisplatin-resistant cell line.
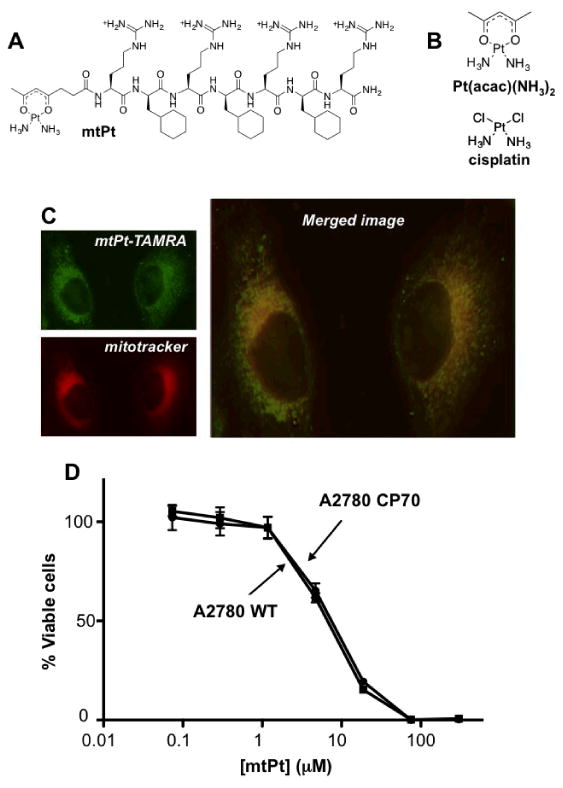
(A) Structure of mtPt. (B) Structures of cisplatin and Pt(acac)(NH3)2. (C) Intracellular localization of mtPt-TAMRA in HeLa cells by fluorescence microscopy. (D) Cytotoxicity of mtPt. Viability of A2780 WT and cisplatin-resistant A2780CP70 cells treated for 72 h with mtPt. Mean values plotted, n ≥ 3, error bars indicate SEM.
In order to assess the intracellular localization of mtPt, fluorescence imaging studies were carried out with the TAMRA-labeled analogue. Localization was first assessed in HeLa cells, for they have a well-defined mitochondrial network that can be readily imaged. Representative images revealing colocalization of the TAMRA conjugate with MitoTracker deep red are shown in Figure 1C. Imaging was also performed in WT A2780 and cisplatin-resistant A2780 CP70 ovarian cancer cell lines. Pearson's correlation coefficients (PCCs) quantitatively describing colocalization of the two dyes in these cell lines are given in Supplementary Table 2. For both cell lines, mtPt(TAMRA) localizes specifically to the mitochondria with PCCs ranging from 0.36 to 0.53. These results also clearly indicate that mtPt(TAMRA) does not localize in the nucleus; Pearson's correlation coefficients for overlap with the nuclear stain are all effectively zero.
The cytoxicity of mtPt was then studied using the MTT assay in A2780 WT and A2780CP70 ovarian cancer cell lines. Cisplatin, [Pt(acac)(NH3)2](SO4)0.5, and [Pt(succac)(NH3)2](NO3) were included as controls. The results are summarized in Supplementary Table S1 and Supplementary Figure S3, and representative dose-response curves for mtPt are depicted in Figure 1D. Cisplatin is toxic to A2780 cells at submicromolar concentrations, with an IC50 of 0.60 ± 0.08 μM. In the resistant CP70 line, however, the IC50 value (5.2 ± 1.4 μM) increases by almost an order of magnitude. The calculation of resistance factors (R.F.s), defined as the ratios of IC50 values of resistant to wild-type cell lines, provides an estimate of differential toxicity. The R.F. for cisplatin is 8.7, indicating that it is susceptible to resistance factors expressed in A2780 CP70 cells. [Pt(acac)(NH3)2](SO4)0.5 and [Pt(succac)(NH3)2](NO3) are both less toxic than cisplatin (IC50 values of 13.9 ± 1.9 and 220 ± 40 μM, respectively), but exhibit similar resistance factors. The lower toxicity for the latter two compounds is expected, given that chelating β-diketonate ligands decrease the rate of departure of the leaving group from the platinum coordination sphere, which leads to lower biological activity (Wilson et al., 2012). The toxicity of mtPt is greater than that of the precursor diketonate compounds, with an IC50 of 7.5 ± 0.3 μM, but importantly, the activity is unaffected in the resistant cell line (Figure 1D). The mechanisms used by this cell line to evade the action of platinum drugs, including increased nucleotide excision repair and drug efflux (Parker et al., 1991), are presumably less able to interfere with the action of a drug that targets mitochondria, allowing mtPt to retain activity in both cell lines.
Further links between the activity of mtPt and events occurring within mitochondria were sought by assessing DNA damage, effects on cell cycle, and the generation of mitochondrial reactive oxygen species. Damage to nuclear and mitochondrial DNA was assessed by monitoring the efficiency of PCR amplification of long segments of isolated nuclear and mitochondrial DNA. A2780 cells were exposed to mtPt, [Pt(acac)(NH3)2](SO4)0.5, or cisplatin for 6 h. PCR amplification of an 8.9-kb mitochondrial genomic segment and a 12.2-kb nuclear genomic segment was analyzed and compared to results for untreated controls. mtPt caused a statistically significant reduction in the amplification of the mitochondrial but not the nuclear DNA segment (Figure 2A). The reverse effect was observed for cisplatin (Figure 2A). These results indicate that mtPt damages mitochondrial DNA without causing nuclear lesions on DNA. This conclusion was corroborated by flow cytometry studies, which revealed that mtPt does not induce cell cycle arrest in A2780 cells (Figure 2B and Supplementary Figure S9). The genomic DNA-targeting compounds cisplatin and [Pt(acac)(NH3)2](SO4)0.5, in contrast, both induced significant cell cycle arrest. Although the mechanism of action for mtPt is clearly different from that of platinum drugs acting in the nucleus, apoptotic cell death still occurs, as judged by Annexin V/Sytox staining and flow cytometry (Figure 2D and Supplementary Figure S5).
Figure 2. The cellular effects of mtPt treatment.
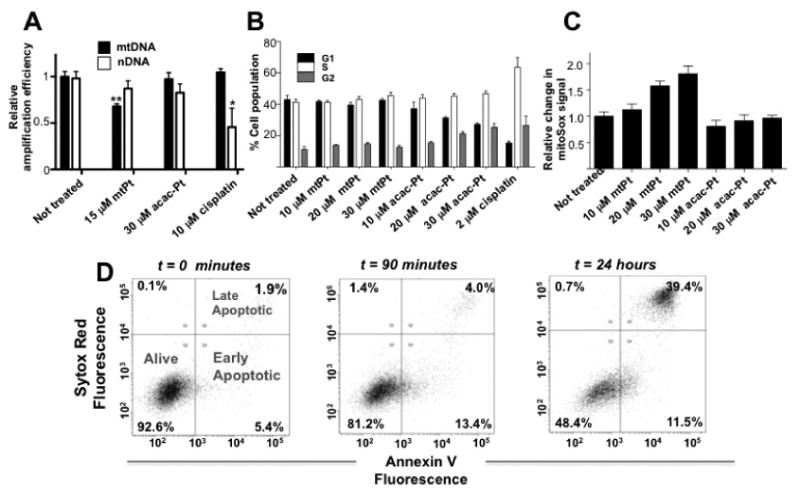
(A) DNA damage caused by platinum drugs. Relative amplification of an 8.9 kb mitochondrial specific gene segment is reduced by 6 h of treatment with 15 μM mtPt but not by 30 μM treatment with [Pt(acac)(NH3)2](SO4)0.5 (acac-Pt) or 15 μM treatment with cisplatin. Mean values plotted, n = 4, all values normalized to non-treated control. p < 0.05 = * p < 0.01 = **. (B) Cell cycle arrest caused by platinum drugs. In contrast to cisplatin and the parent compound, mtPt does not induce cell cycle arrest. Quantitative analysis of distribution into G1, S and G2 cell cycle phases was assessed by propidium iodide staining and flow cytometry. Mean values plotted, n = 3. (C) Assessment of mitochondrial ROS production by MitoSox™ staining of A2780 cells treated with the indicated concentrations of mtPt and [Pt(acac)(NH3)2](SO4)0.5 (acac-Pt) for 24 h. Mean values plotted, n = 3, all values normalized to non-treated control. (D) mtPt induces slow-onset apoptosis. Annexin V/Sytox Red staining of A2780 cells treated with 30 μM mtPt at indicated time points.
Damage to mitochondrial DNA can produce reactive oxygen species (ROS) that lead to mitochondrial dysfunction and cell death (Santos et al., 2003). We therefore assessed whether mtPt treatment increases the levels of superoxide in mitochondria. mtPt induced significant increases in mitochondrial superoxide in A2780 cells following 24 hours of treatment, as judged by studies using a fluorogenic dye that is specific for O2‒ as a ROS. The [Pt(acac)(NH3)2](SO4)0.5 parent compound, in contrast, caused minimal superoxide production (Figure 2C). These results support the hypothesis that mtPt generates mitochondrial dysfunction.
To further probe a potential connection between damage to mitochondrial DNA and mtPt cytotoxicity, we assessed mtPt activity in mouse embryonic fibroblasts (MEFs) expressing a form of mitochondrial DNA polymerase gamma (PolG) lacking functional 3′- to 5′-exonuclease activity. The PolG m/m line is proofreading deficient and accumulates mtDNA point mutations at a 3–5-fold higher rate than their WT counterparts (Kujoth et al., 2005). We therefore hypothesized that they might be highly sensitive to agents that damage mtDNA. Indeed, we observed that PolG m/m cells are more sensitive to mtPt than wild type cells, with greater levels of apoptosis being generated at an equivalent dose (Figure 3). This potentiation of toxicity was not observed for cisplatin or [Pt(acac)(NH3)2](SO4)0.5 (Figure 3 and Supplementary Figure S6). These data suggest that mtPt-induced apoptosis results from damage specifically to mtDNA.
Figure 3. Cells with defective DNA Pol G are sensitized to mtPt.
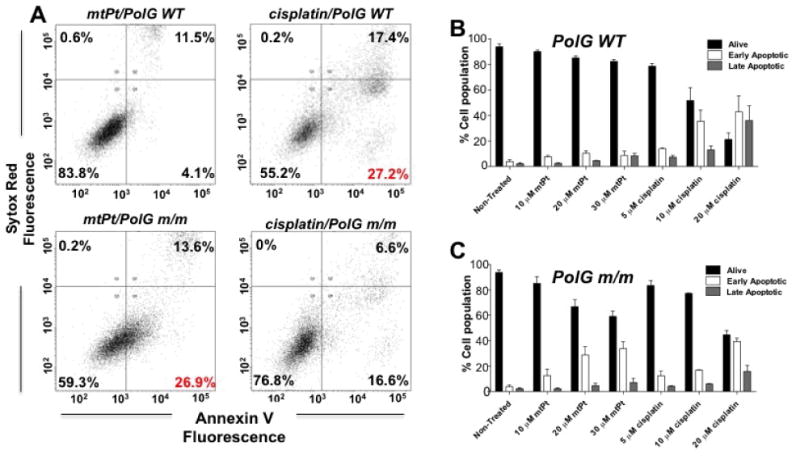
(A) Representative distribution of PolG WT and PolG m/m cells into alive, early apoptotic and late apoptotic populations after 24 h. In these trials, 30 μM mtPt, and 10 μM cisplatin were used. Populations assessed by Annexin V/Sytox staining and flow cytometry. (B) and (C) Quantitation of apoptotic cell death in Pol G WT and mutant m/m cells. Mean values plotted, n = 3. All error bars indicate SEM.
Significance
In summary, we have prepared and characterized a mitochondrially localized conjugate of cisplatin using chemistry that can be readily applied to access a wide range of platinum(II)-peptide conjugates targeted to subcellular locales. Although less cytotoxic than cisplatin, presumably owing to slower ligand exchange kinetics, mtPt is equally effective at killing wild-type and cisplatin-resistant ovarian cancer cells. We find that mtPt is delivered to mitochondria and damages mtDNA. Moreover, we present evidence that a cell line with reduced mitochondrial genomic integrity is highly sensitive to treatment with mtPt, linking its activity directly to mtDNA damage. Nuclear DNA damage and cell cycle arrest, both well-characterized aspects of the cisplatin mechanism of action, were not observed for the mitochondrially-targeted compound. These findings provide the first direct evidence that damage to mtDNA by platinum chemotherapeutics is toxic to cancer cells and indicate that mitochondria are potential targets for anticancer therapy.
Experimental Methods
General Materials and Methods
Succinylacetone (Levenson, 2009) and [Pt(acac)(NH3)](SO4)0.5 (Wilson et al., 2012) were synthesized as previously described. Peptides were also prepared as reported (Mourtada et al., 2013) and purified by reverse-phase HPLC. Other reagents were purchased from commercial vendors.
Physical Measurements
NMR spectra were acquired on a Bruker DPX-400 spectrometer in the MIT Department of Chemistry Instrumentation Facility (DCIF). For FTIR spectra, samples were prepared as KBr disks and data were recorded with a ThermoNicolet Avatar 360 spectrophotometer running the OMNIC software. Electrospray ionization mass spectrometry (ESI-MS) measurements were acquired on an Agilent Technologies 1100 series LC-MSD trap. Solutions used for biological studies were dissolved in MilliQ water (or PBS for cisplatin) and sterile-filtered. The platinum concentrations of the solutions were determined by graphite-furnace atomic absorption spectroscopy (GFAAS) using a Perkin-Elmer AAnalyst600 spectrometer.
Synthesis of [Pt(succac)(NH3)2](NO3)
Cisplatin (500 mg, 1.67 mmol) and AgNO3 (552 mg, 3.25 mmol) were stirred in 10 mL H2O in the absence of light at room temperature for 16 h. The resulting mixture was filtered to remove AgCl. To the filtrate, a solution of NaOH (67 mg, 1.68 mmol) and succinylacetone (269 mg, 1.70 mmol) in 5 mL of H2O was added dropwise. After stirring at rt for 5 h, the resulting solution was concentrated to dryness at 60 °C under reduced pressure to afford an orange oil. This oil was dissolved in 3 mL of H2O and acidified with three drops of 25% HNO3. Acetone (50 mL) was added, and the resulting turbid white suspension was stirred for approximately 3 min, resulting in the deposition of an oily orange-brown residue. The turbid supernatant was decanted and mixed with 50 mL of a 1:1 (v/v) mixture of acetone and diethyl ether. Upon stirring at rt for approximately 5 min, an orange-brown residue deposited again. The cloudy supernatant was decanted and poured directly into 150 mL of diethyl ether. The mixture was stored at ‒40 °C for 1.5 h and filtered to collect a white solid. The white solid was washed with 2 × 10 mL diethyl ether and then dried in vacuo. Yield: 203 mg (28%). Characterization data are given in Supplementary Note 1.
Synthesis of mtPt
A 50-μmol portion of FMOC-r(Fxr)3 on Rink amide resin was placed in a fritted 2.5-mL Torviq disposable syringe and swelled with 2 mL of anhydrous DMF for 1 h. The N-terminal FMOC group was removed by treating the resin with a solution of 25% 4-methylpiperidine in DMF (v/v) for 30 min. The deprotected resin was washed with 5 × 1.5 mL DMF. A mixture of 90 mg (200 μmol) [Pt(NH3)2(succac)]NO3 and 76 mg (200 μmol) HATU was dissolved in 1.5 mL of 10% N,N-diisopropylethylamine (DIPEA) in DMF (v/v) and added to the deprotected resin. The reaction vessel was shaken at rt for 60 min, then washed sequentially with 5 × 1.5 mL of DMF and 5 × 1.5 mL CH2Cl2 and dried in vacuo. The dry resin was treated with a TFA/water/triisopropylsilane 95/2.5/2.5% (v/v) solution for 90 min to detach mtPt. mtPt was precipitated from the cleavage solution with diethyl ether. The resulting solid mtPt was separated by centrifugation and purified by semi-preparative HPLC with a C18 reverse-phase column (9.4 mm × 250 mm). Fractions from sequential runs containing mtPt were pooled and lyophilized. The purity of the final product was assessed via analytical HPLC. Purification conditions and characterization data is given in Supplementary Note 2.
Synthesis of mtPt-TAMRA
A 50-μmol portion of FMOC-r(Fxr)3K-MTT was deprotected in a 3% solution of TFA in DCM for 30 min and coupled to (5,6)-TAMRA using 4 equiv (5,6)-TAMRA, 4 equiv HBTU, and 8 equiv DIPEA in DMF for 2 h. The resulting resin was labeled with 200 μmol (90 mg) of [Pt(NH3)2(succac)]NO3 using the procedure described above for the preparation of mtPt. The purity of the two isomers (5 and 6) of mtPt-TAMRA was assessed by analytical HPLC. The 5-isomer (tr = 26.5 min), ≥ 95% pure based on the integrated chromatogram, was carried forward for biological studies. The distinction between the 5- and 6-isomers was made based on 1H NMR spectroscopy (Supplementary Figure S15). Characterization is given in Supplementary Note 3.
X-ray Crystallography
Single crystals of succinylacetone and [Pt(succac)(NH3)2](NO3) were grown from hexanes/Et2O and MeOH/Et2O, respectively. Data were obtained, solved, and refined as previously described (Wilson et al., 2012). Refinement data and thermal ellipsoid plots are given in Supplementary Table 3 and Supplementary Figure S1. These data have been deposited in the Cambridge Crystallographic Data Centre (CCDC) under the accession codes of CCDC 938791 and 938792 for succinylacetone and [Pt(succac)(NH3)2](NO3), respectively.
Cell Culture
A2780 and A2780CP70 (wild-type and cisplatin–resistant ovarian cancer) cell lines were obtained from Fox Chase Cancer Center and cultured in RPMI-1640 medium supplemented with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin. PolG WT and m/m MEFs were cultured in high glucose DMEM supplemented with 10% FBS. The cells were incubated in a humidified atmosphere containing 5% CO2 at 37 °C.
Cytotoxicity Assays
The colorimetric MTT assay was used to assess cytotoxicity of the compounds using a previously described protocol (Wilson et al., 2012). From the resulting dose-response curves, 50% growth inhibitory concentration (IC50) values were determined by interpolation.
Cell Imaging Studies
A2780 and A2780CP70 cells were seeded in an imaging dish in 2 mL of growth medium at 60% confluence. The growth medium was swapped with premixed medium containing 1 or 10 μM of mtPt-TAMRA, and the cells were allowed to incubate with the dye for 1 h. MitoTracker Green and Hoechst 33258, were added to the cells at 1.25–2.5 μM concentrations and allowed to incubate for 30 min. At the end of the incubation period, the medium was aspirated, and the cells were washed with 3 × 1 mL PBS and submerged in 2 mL of dye-free DMEM. The imaging experiments were performed using a Zeiss Axiovert 200M inverted epifluorescence microscope equipped with an EM-CCD digital camera (Hamamatsu) and a MS200 XY Piezo Z stage (Applied Scientific Instruments). The light source was an X-Cite 120 metal-halide lamp (EXFO) and the fluorescence images were obtained with an oil-immersion objective at 63× magnification. The microscope was operated by the Volocity software program of Perkin-Elmer. Colocalization of the dyes was quantitated using the program ImageJ (Schneider et al., 2012), using a previously described protocol (French et al., 2008). HeLa cells were imaged using a previously described protocol (Fonseca et al., 2011).
Analysis of Mitochondrial Superoxide Levels
A2780 cells were plated in 12-well plates at a density of 105 cells/mL and allowed to attach overnight. Cells were then treated with the indicated concentrations of mtPt and [Pt(acac)(NH3)](SO4)0.5 and incubated for 24 or 48 h. The medium was removed and cells were washed with Hank's Buffered Saline (HBS) and then incubated with 5 μM MitoSox reagent (Invitrogen) in HBS for 30 min in the absence of light. Cells were washed three times with HBS, harvested by trypsinization and analyzed via flow cytometry with FACSCanto. At least 104 cells were analyzed for each sample.
Annexin V Apoptosis Assay
A2780 cells were plated in a 12-well dish at a density of 105 cells/mL and allowed to adhere overnight. PolG WT and m/m cells were plated at a density of 5 × 104 cells/well. A2780 Cells were treated with the indicated concentrations of mtPt for 90 min, 8 h and 24 h. PolG WT and m/m cells were incubated for 24 h. Following incubation cells were stained at room temperature with Annexin V-FITC for 15 min and 5 nM Sytox Red for an additional 15 min. Analysis was performed via flow cytometry with FACSCanto. At least 104 cells were analyzed for each sample.
Cell Cycle Analysis
A2780 cells were seeded in a 12-well dish at a density of 105 cells/mL and allowed to adhere overnight. Cells were treated with the indicated concentrations of mtPt and [Pt(acac)(NH3)](SO4)0.5 and incubated for 24 h. Cells were harvested by trypsinization and fixed at 4 °C in 70% ethanol for 2 h. Cells were centrifuged, ethanol was removed, and the remaining cell pellet was washed once with PBS. Cells were resuspended in PBS and incubated with 0.2 mg/mL RNAse A for 1 h at 37 °C, then stained with 10 μg/mL propidium iodide and analyzed immediately via flow cytometry with FACSCanto. Quantitation of data was performed using FlowJo. At least 104 cells were analyzed for each sample.
Determination of DNA Damage by qPCR
A2780 cells were seeded at 2 × 105 cells/well and allowed to adhere overnight. Cells were treated with either 15 μM mtPt, 30 μM [Pt(acac)(NH3)](SO4)0.5 or 15 μM cisplatin for 6 h and harvested after trypsinization. DNA was isolated from cell pellets using the Sigma GenElute mammalian genomic DNA miniprep kit according to the manufacturer's instructions. Amplification of an 8.9 kb segment of mitochondrial DNA or a 12.2 kb segment of genomic DNA was performed using the Elongase long range PCR enzyme kit (Invitrogen) as described previously (Santos et al., 2006). Quantitation of amplified product was performed by Pico Green staining and normalized to non-treated value.
Supplementary Material
Acknowledgments
Work in the lab of S.J.L. was supported by the National Cancer Institute under grant CA034992. J.J.W. is supported by a David H. Koch Graduate Fellowship. Work in the lab of S.O.K. was supported by the Canadian Institute of Health. Dr. Tomas Prolla (University of Wisconsin-Madison) provided PolgD257A mice from which mouse embryonic fibroblasts were generated.
Footnotes
Potential competing financial interests: S.J.L. declares a financial interest in Blend Therapeutics.
Additional information: Supplemental Information can be found with this article online at http://dx.doi.org/10.1016/j.chembiol.
References
- Andrews PA, Albright KD. Mitochondrial defects in cis-diamminedichloroplatinum (II)-resistant human ovarian carcinoma cells. Cancer Res. 1992;52:1895–1901. [PubMed] [Google Scholar]
- Barragan F, Moreno V, Marchan V. Solid-phase synthesis and DNA binding studies of dichloroplatinum(II) conjugates of dicarba analogues of octreotide as new anticancer drugs. Chem Commun. 2009;31:4705–4707. doi: 10.1039/b909698a. [DOI] [PubMed] [Google Scholar]
- Cullen K, Yang Z, Schumaker L, Guo ZJ. Mitochondria as a critical target of the chemotheraputic agent cisplatin in head and neck cancer. J Bioenerg Biomembr. 2007;39:43–50. doi: 10.1007/s10863-006-9059-5. [DOI] [PubMed] [Google Scholar]
- Damian MS, Hedman HK, Elmroth SKC, Diederichsen U. Synthesis and DNA interaction of platinum complex/peptide chimera as potential drug candidates Eur. J Org Chem. 2010;32:6161–6170. [Google Scholar]
- Fonseca SB, et al. Rerouting chlorambucil to mitochondria combats drug deactivation and resistance in cancer cells. Chem Biol. 2011;18:445–453. doi: 10.1016/j.chembiol.2011.02.010. [DOI] [PubMed] [Google Scholar]
- Mourtada R, et al. Re-directing an alkylating agent to mitochondria alters drug target and cell death mechanism. PLOS One. 2013;8:e60253. doi: 10.1371/journal.pone.0060253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- French AP, Mills S, Swarup R, Bennett MJ, Pridmore TP. Colocalization of fluorescent markers in confocal microscope images of plant cells Nat. Protoc. 2008;3:619–628. doi: 10.1038/nprot.2008.31. [DOI] [PubMed] [Google Scholar]
- Fulda S, Galluzzi L, Kroemer G. Targeting mitochondria for cancer therapy. Nat Rev Drug Discov. 2010;9:447–464. doi: 10.1038/nrd3137. [DOI] [PubMed] [Google Scholar]
- Galanski M, Jakupec MA, Keppler BK. Update of the preclinical situation of anticancer platinum complexes: novel design strategies and innovative analytical approaches. Curr Med Chem. 2005;12:2075–2094. doi: 10.2174/0929867054637626. [DOI] [PubMed] [Google Scholar]
- Graf N, Mokhtari TE, Papayannopoulos IA, Lippard SJ. Platinum(IV)-chlorotoxin (CTX) conjugates for targeting cancer cells. J Inorg Biochem. 2012;110:58–63. doi: 10.1016/j.jinorgbio.2012.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groessl M, Zava O, Dyson PJ. Cellular uptake and subcellular distribution of ruthenium-based metallodrugs under clinical investigation versus cisplatin. Metallomics. 2011;3:591–599. doi: 10.1039/c0mt00101e. [DOI] [PubMed] [Google Scholar]
- Hirama M, Isonishi S, Yasuda M, Ishikawa H. Characterization of mitochondria in cisplatin-resistant human ovarian carcinoma cells. Oncol Rep. 2006;16:997–1002. 2006. [PubMed] [Google Scholar]
- Horton KL, Stewart KM, Fonseca SB, Guo Q, Kelley SO. Mitochondria-penetrating peptides. Chem Biol. 2008;15:375–382. doi: 10.1016/j.chembiol.2008.03.015. [DOI] [PubMed] [Google Scholar]
- Horton KL, Pereira MP, Stewart KM, Fonseca SB, Kelley SO. Tuning the activity of mitochondria-penetrating peptides for delivery or disruption. Chembiochem. 2012;13:476–485. doi: 10.1002/cbic.201100415. [DOI] [PubMed] [Google Scholar]
- Isonishi S, Saitou M, Yasuda M, Tanaka T. Mitochondria in platinum resistant cells. Hum Cell. 2001;14:203–210. [PubMed] [Google Scholar]
- Kujoth GC, Hiona A, Pugh TD, Someya S, Panzer K, Wohlgemuth SE, et al. Mitochondrial DNA mutations, oxidative stress, and apoptosis in mammalian aging. Science. 2005;309:481–484. doi: 10.1126/science.1112125. [DOI] [PubMed] [Google Scholar]
- Levenson CH. Process for making succinyl acetone. U S Patent 5,276,180. 1994
- Mourtada R, Fonseca SB, Wisnovsky SP, Pereira MP, Wang X, Hurren R, et al. Re-Directing an alkylating agent to mitochondria alters drug target and cell death mechanism. PLoS One. 2013;8:e60253. doi: 10.1371/journal.pone.0060253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukhopadhyay S, Barnés CM, Haskel A, Short SM, Barnes KR, Lippard SJ. Conjugated platinum (IV)–peptide complexes for targeting angiogenic tumor vasculature. Bioconjugate Chem. 2008;19:39–49. doi: 10.1021/bc070031k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murata T, Hibasami H, Maekawa S, Tagawa T, Nakashima K. Biochem Int. 1990;20:949–955. [PubMed] [Google Scholar]
- Ndinguri MW, Solipuram R, Gambrell RP, Aggarwal S, Hammer RP. Peptide targeting of platinum anti-cancer drugs. Bioconjugate Chem. 2009;20:1869–1878. doi: 10.1021/bc900065r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olivero OA, Semino C, Kassim A, Lopez-Larraza DM, Poirier MC. Preferential binding of cisplatin to mitochondrial DNA of Chinese hamster ovary cells. Mutat Res Lett. 1995;346:221–230. doi: 10.1016/0165-7992(95)90039-x. [DOI] [PubMed] [Google Scholar]
- Parker RJ, Eastman A, Bostick-Bruton F, Reed E. Acquired cisplatin resistance in human ovarian cancer cells is associated with enhanced repair of cisplatin-DNA lesions and reduced drug accumulation. J Clin Invest. 1991;87:772–777. doi: 10.1172/JCI115080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robillard MS, Valentijn A, Meeuwenoord NJ, van der Marel GA, van Boom JH, Reedijk J. The first solid-phase synthesis of a peptide-tethered platinum (ii) Complex. Angew Chem, Int Ed. 2000;39:3096–3099. doi: 10.1002/1521-3773(20000901)39:17<3096::aid-anie3096>3.0.co;2-d. [DOI] [PubMed] [Google Scholar]
- Santos JH, Hunakova LU, Chen Y, Bortner C, Van Houten BJ. Cell sorting experiments link persistent mitochondrial dna damage with loss of mitochondrial membrane potential and apoptotic cell death. J Biol Chem. 2003;278:1728–1734. doi: 10.1074/jbc.M208752200. [DOI] [PubMed] [Google Scholar]
- Santos JH, Meyer JN, Mandavilli BS, Van Houten B. Quantitative PCR-based measurement of nuclear and mitochondrial DNA damage and repair in mammalian cells. Methods Mol Biol. 2006;314:183–199. doi: 10.1385/1-59259-973-7:183. [DOI] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todd RC, Lippard SJ. Inhibition of transcription by platinum antitumor compounds. Metallomics. 2009;1:280–291. doi: 10.1039/b907567d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson JJ, Lippard SJ. In vitro anticancer activity of cis-diammineplatinum(ii) complexes with β-diketonate leaving group ligands. J Med Chem. 2012;55:5326–5336. doi: 10.1021/jm3002857. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.