

Abstract
The pathophysiological importance of oxidative damage after traumatic brain injury (TBI) has been extensively demonstrated. The transcription factor nuclear factor erythoid related factor 2 (Nrf2) mediates antioxidant and cytoprotective genes by binding to antioxidant response elements (ARE) present in nuclear DNA. In this study, we characterized the time course of Nrf2-ARE–mediated expression in the cortex and hippocampus using a unilateral controlled cortical impact model of focal TBI. Ipsilateral hippocampal and cortical tissue was collected for Western-blot protein analysis (n=6/group) or quantitative reverse transcription-polymerase chain reaction for mRNA (n=3/group) at 3, 6, 12, 24, 48, and 72 h or 1 week post-injury. Multiple genes mediated by Nrf2-ARE were altered post-TBI. Specifically, Nrf2 mRNA increased significantly post-TBI at 48 and 72 h in the cortex and at 48 and 72 h and 1 week in the hippocampus with a coincident increase in glial fibrillary acidic protein mRNA, thereby implying this response is likely occurring in astrocytes. Presumably linked to Nrf2 activation, heme-oxygenase-1, nicotinamide adenine dinucleotide phosphate-quinone-oxidoreductase 1, glutathione reductase, and catalase mRNA overlap throughout the post-injury time course. This study demonstrates the first evidence of such changes during the first week after focal TBI and that increases in expression of some Nrf2-ARE–mediated cytoprotective genes are not observed until 24–48 h post-injury. Unfortunately, this does not precede, but rather coincides with, the occurrence of lipid peroxidative damage. This is the first known comparison between the time course of peroxidative damage and that of Nrf2-ARE activation during the first week post-TBI. These results underscore the necessity to discover pharmacological agents to accelerate and amplify Nrf2-ARE–mediated expression early post-TBI.
Key words: : 4-hydroxy-2-nonenal, gene expression, lipid peroxidation, Nrf2, oxidative damage, traumatic brain injury
Introduction
Traumatic brain injury (TBI) currently signifies a substantial health and socioeconomic dilemma in the United States. Each year, an estimated 1.5 million new TBI-related cases occur; of those 1.5 million, roughly 50,000 cases will result in death.1 As a consequence of this epidemic, approximately 5.3 million persons are living with a permanent or temporary disability as a result of TBI.1 It is well documented that TBI results in a series of secondary biochemical events that occur after the initial primary injury, thereby drastically exacerbating the deleterious effects triggered by the original mechanical trauma itself.2,3
Despite the failures of TBI clinical trials to date, progress has been made in identifying the numerous molecular and cellular mediators of post-TBI pathophysiology, including oxidative stress.2,4 Arguably the most validated component of the post-TBI secondary injury cascade, oxidative stress is thought to involve an imbalance in the ratio of harmful reactive oxygen and nitrogen species (ROS/RNS) and protective endogenous antioxidant defense enzymes.3,5,6
Numerous studies over the past 30 years have continually implicated oxidative damage in TBI pathophysiology.3,6–12 This was first demonstrated by Kontos and colleagues11,12 who found an almost immediate post-injury increase in brain microvascular superoxide radicals in fluid percussion TBI models. More specifically, previous work has demonstrated that free radical mediated lipid peroxidation (LP) plays a critical role in the acute pathophysiology of TBI.2,6,13 Briefly, LP involves the oxidation of polyunsaturated fatty acids (e.g., arachidonic, linoleic, and docosahexaenoic acids) in cells and membrane phospholipids at allylic carbons. These peroxidized polyunsaturated fatty acids then subsequently undergo phospholipase-mediated hydrolysis and thereby cause disruption of the membrane phospholipid architecture, and hence eventual loss of proper functioning phospholipid-dependent enzymes, ion channels, and key structural proteins. Subsequent cellular dysfunction and neuronal cell death then ensues, ultimately resulting in neurological impairment.1,14 Long-lasting behavioral and cognitive deficits, accompanied by motor deficiencies, devastate TBI victims as manifested by impaired working memory, destabilized impulse control, spatial deficits, and loss of spontaneity.1,14
An endogenous cytoprotective defense system does exist to combat the basal and stress or injury-induced imbalance in ROS/RNS and antioxidant defense enzymes. This innate system is primarily under the control of the pleiotropic transcription factor nuclear factor-erythoid2-related factor 2 (Nrf2).13,15 Since its discovery, Nrf2 has been identified as a key mediator of the inducible cytoprotective response via its interaction with the genomic cis-acting enhancer region of protective genes known as the antioxidant response element (ARE).16,17
Under basal conditions, Nrf2 is sequestered in the cytoplasm by the repressor protein Keap1.16,17 This binding interaction between Nrf2 and Keap1 facilitates proteasomal degradation of Nrf2 by recruitment of Cul3 ubiquitin ligase via the BTB domain of Keap1.18 Only under conditions of cell stress (e.g., oxidative stress, ER stress, injury, toxicity, etc.) is Nrf2 released from Keap1 by a proposed hinge-latch mechanism.19,20 This release thereby allows for subsequent Nrf2 translocation into the nucleus21 where it heterodimerizes with small Maf proteins and binds to the ARE of cytoprotective genes,13 inducing transcription and consequent production of defense proteins (e.g., heme-oxygenase 1; HO-1).
Numerous studies in many different neurodegeneration paradigms have indicated that manipulation of the Nrf2-ARE pathway can dramatically attenuate multiple pathophysiological processes, including oxidative stress,22 mitochondrial dysfunction,23,24 and inflammation.25,26 Moreover, recent work has demonstrated that this Nrf2-ARE defense response is inducible by a variety of small molecules, as demonstrated in several different in vitro 23,27 and in vivo28–33 paradigms. More specifically in regard to TBI, it has recently been shown that the Nrf2-ARE pathway is involved after TBI.34,35 Specifically, the prototypical Nrf2-ARE activator sulforaphane has been shown to attenuate post-TBI pathophysiology, including blood–brain barrier dysfunction,36 edema formation,37 and cognitive deficits.38 Another impressive small molecule capable of potently inducing the Nrf2-ARE response is carnosic acid, previously shown to be protective in vivo in a cerebral ischemia paradigm.39
While previous studies have demonstrated some protective effects with compounds that activate the Nrf2-ARE pathway after TBI, it has not previously been determined how the pathway itself responds after TBI. Characterizing the endogenous Nrf2-ARE response is critical for future development and testing of pharmacological agents that can amplify and enhance this pathway. Determining when best to administer a Nrf2-ARE activating drug—based on both the endogenous Nrf2-ARE response and the time course of oxidative damage post-injury—is of utmost priority for future investigation of this promising therapeutic target.
Thus, in the current study, we compared the time course of Nrf2-ARE mediated expression to that of oxidative damage as represented by the lipid peroxidation aldehydic breakdown product 4-hydroxy-2-nonenal (4-HNE). It was hypothesized that the time course of endogenous Nrf2-ARE activation would mimic that of the time course of oxidative damage (4-HNE); however, it was expected that the magnitude and speed of amplification would be insufficient to provide adequate defense against increasing oxidative damage post-TBI.
Methods
Animals
This study used young adult (8 weeks old) male CF-1 mice (Charles River Labs) weighing 28–32 grams at time of surgery. All animals had ad libitum access to food and water and were housed in the Division of Laboratory Animal Resources sector of the University of Kentucky Chandler Medical Center, which is fully accredited by AALAC. All procedures described herein follow protocols approved by the University of Kentucky's Institutional Animal Care and Use Committee, in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
Mouse model of controlled cortical impact (CCI) TBI
Mice were initially anesthetized in a Plexiglas chamber using 4.0% isoflurane, shaved, weighed, and then placed into a stereotaxic frame (David Kopf, Tujunga, CA). Core body temperature was maintained using an underlying heating pad. Throughout the surgical procedure, mice were kept anesthetized by a constant flow of 3.0% isoflurane and oxygen delivered via nose cone. The head was positioned in the horizontal plane with nose bar set at zero. A 2.0-cm sagittal incision was made in the scalp, and the skin was retracted using hemostats to expose the skull. After exposing the skull, a 4.0-mm diameter craniotomy was made using a dental bur (SS White, Lakewood, NJ) mounted on a cordless Dremel (Racine, WI) lateral (left) to the sagittal suture, centered between bregma and lambda, while leaving the dura mater intact.
Sham-operated (control) mice received anesthesia and all surgical procedures (including craniotomy) but without CCI brain injury. Brain-injured mice received cortical impact injury using an electronically controlled pneumatic impacting device (Precision Systems Instrumentation, TBI-0310 Impactor, Fairfax Station, VA) with a 3.0-mm diameter, beveled (flat) impactor tip. The impact velocity was held at 3.50 meters/sec while the depth of cortical deformation was set at 1.0 mm (severe) as described previously.40 Mortality after this severe CCI brain injury is rare (<5.0%).
After injury, the craniotomy was closed by secure placement of a 6.0-mm in diameter disk of dental acrylic cemented in place with quick bonding liquid cyanoacrylate. The mice were then placed in a Hova-Bator Incubator (model 1583; Randall Burkey Co, Boerne, TX) set at 37°C for at least 20–30 min to prevent post-traumatic hypothermia before returning them to their home cage. Consciousness (i.e., return of right reflex and mobility) was regained within 10 min after the surgery.
Brain-injured and sham mice typically show no untoward effects after recovering from anesthesia and resume normal eating, drinking, and grooming patterns thereafter. In this study, animals survived from 3 h to 7 days post-injury, depending on group assignment. Additional details on surgical procedures have been published previously by our laboratory.40
Quantitative reverse transcription-polymerase chain reaction (RT-PCR) analysis
Quantitative real-time PCR (qRT-PCR) was used to determine mRNA levels of Nrf2-ARE mediated gene targets. Briefly, at a given time point post-injury, mice received an overdose of sodium pentobarbital (200.0 mg/kg intraperitoneally [IP]). The ipsilateral cortex (penumbra tissue and the injured core) and hippocampus were then rapidly dissected out on an ice-chilled stage and immediately transferred to a RNAlater® solution (Ambion Inc.) for 24 h at 4°C to prevent RNase activity and sample degradation. Samples were then placed in a −80°C freezer for storage until further analysis. To isolate total RNA, tissue samples were homogenized in TRIzol® reagent (Ambion Inc.) according to manufacturer specifications. Isolated total RNA was precipitated out using isopropanol, washed with ethanol, and then decontaminated of residual genomic DNA by DNase I treatment per manufacturer specifications.
Total RNA concentrations were determined using a Nanodrop,® with 260/280 ratios of 1.8–2.2 considered acceptable. Purified total RNA (1.0 μg) was then reverse transcribed to acquire complementary total DNA (cDNA). Final cDNA samples were then used for quantitative real-time PCR assay. In this study, qRT-PCR was performed using the StepOne-Plus real-Time PCR System (Applied Biosystems, Carlsbad, CA) in conjunction with TaqMan® primer-probe reagent-based chemistry. Commercial, inventoried TaqMan gene expression assays consisting of a gene-specific set of primers and a fluorogenic internal probe were used (Applied Biosystems). The mouse glyceraldehyde-3-phosphate dehydrogenase (GAPDH) endogenous control was used for normalization purposes of all target gene analysis as previously validated in our laboratory.40
PCR reactions were run in triplicate in a 96 well format using a standard (∼2.5 hours) amplification protocol. Each reaction well of the plate contained a total of 25 μl. The PCR reaction for a specific gene (e.g., HO-1, superoxide dismutase [SOD]1, etc.) contained 10.0 μl of 1:10 diluted total cDNA and a total of 15.0 μl of a TaqMan PCR master mix and gene-specific primers and probe. The PCR reaction for GAPDH gene expression assay contained 2.0 μl of 1:10 diluted total cDNA and 23.0 μl of a TaqMan PCR master mix and control gene primers and probe. After PCR reaction, the resulting amplification curves were then further analyzed by the established ΔΔCt method wherein GAPDH was used as the reference gene and the sham group as a control.40
Western blotting of 4-HNE adducted proteins and a Nrf2-ARE mediated target
Western blotting technique was used to detect levels of 4-HNE adducts and the protein levels of the Nrf2-ARE mediated target HO-1. Briefly, at a given time point post-injury, mice received an overdose of sodium pentobarbital (200.0 mg/kg IP). The ipsilateral cortex (penumbra tissue and the injured core) and hippocampus were then rapidly dissected out on an ice-chilled stage and immediately transferred to Triton lysis buffer (1.0% Triton, 20.0 mM Tris HCL, 150.0 mM NaCl, 5.0 mM ethylene glycol tetraacetic acid, 10.0 mM ethylenediaminetetraacetic acid, and 10.0% glycerol) containing protease inhibitors (Complete Mini Protease Inhibitor Cocktail; Roche Diagnostics, Indianapolis, IN). Samples were then sonicated and centrifuged for 30 min (13,000 rpm at 4°C). The supernatants were collected and the remaining pellet was discarded.
Protein concentrations were determined using a BCA Protein Assay (Pierce). An aliquot of each protein sample (15.0 μg for 4-HNE; boiled 25.0 μg for Nrf2-ARE targets) was separated on an SDS–PAGE precast gel (12% Bis-Tris w/v acrylamide; Criterion XT, Bio-Rad) using a XT-MES running buffer system and then transferred to nitrocellulose membranes using a semi-dry electro-transferring unit at 15 volts for 30 min at room temperature. Membranes were then incubated for 1 h at room temperature in a 5.0% milk/Tris buffered solution (TBS) blocking solution. The membranes were then incubated overnight at 4°C in blocking solution with 0.5 mM Tween-20 (TBST) containing the appropriate dilution of primary antibody.
A mouse monoclonal primary antibody was used for detecting 4-HNE bands (1:100 dilution; Japan Institute for Control of Aging, JaICA, Japan). For detection of the Nrf2-ARE target HO-1, the following primary antibody was used: rabbit polyclonal anti-heme-oxygenase-1 (1:100; 32 kD band; Santa Cruz Biotechnology, CA). A goat anti-mouse or anti-rabbit secondary antibody (2 h incubation at room temperature) conjugated to an infrared dye (1:5000, IRdye-800CW, Rockland) was used for detection of the primary labeled bands. Wet membranes were then imaged and quantified using a Li-Cor Odyssey InfraRed Imaging System and accompanying software (Li-Cor Biosciences, Lincoln, NE). For the 4-HNE blot analysis, all bands ranging from 250 kD to 50 kD were quantified for each lane, representing the smear of 4-HNE labeled proteins for each sample. This was then analyzed as percent of control samples.41
Sample size and data analysis
The sample sizes for this study were three per group (qRT-PCR analysis) and six per group (Western blot analysis). All data were expressed as the mean±standard deviation (SD) and were analyzed using GraphPad PRISM version 5.0 statistical graphing software. Both qRT-PCR and immunoblot quantification data were analyzed by appropriately designed analysis of variance followed by Dunnett post-hoc tests as appropriate. A p value <0.05 was considered significant for all statistical analyses.
Results
Lipid peroxidation time course after TBI (cortex and hippocampus)
As shown in Figure 1, CCI-TBI resulted in a progressive increase in both ipsilateral cortical (CTX) and hippocampal (HIPP) levels of 4-HNE modified proteins. In the CTX, there was a significant increase at 48 and 72 h post-injury compared with sham (uninjured mice) with the peak occurring at 72 h post-injury. This increase had dissipated by 1 week post-injury, returning to sham levels. In the HIPP, a similar but delayed time course is present with significant increases in 4-HNE at 72 h and 1 week post-injury compared with sham animals. In both cases, the time course of 4-HNE demonstrates a progressive increase with robust, profound increases present at 48–72 h in the cortex and 72 h–1 week in the hippocampus.
FIG. 1.

Time course of 4-hydroxy-2-nonenal (4-HNE) after focal controlled cortical impact-traumatic brain injury (TBI) in the mouse. Western blot analysis of 4-HNE in ipsilateral cortex (CTX, A) and hippocampus (HIPP, B) revealed a significant increase in 4-HNE at 48 and 72 hours (in CTX) and at 72 hours and 1 week post-injury (in HIPP) after TBI as compared to sham controls. Analyzed by One-way ANOVA followed by Dunnett's post-hoc test. *=p<.05. Error bars represent±standard deviation. Black box in left most lane indicates the molecular weight range (250 kD to 50 kD) that was included in the densitometry analysis.
Nrf2-ARE mediated expression is altered after TBI (cortex)
Real-time qRT-PCR analysis of Nrf2-ARE mediated gene expression in the cortex (Fig. 2) revealed multiple changes after TBI. HO-1 mRNA increased 20-fold at 24 and 72 h post-injury in the cortex (p<0.05). HO-1 protein expression (Fig. 3) was also significantly increased, but not until 72 h post-injury when it was increased by approximately 200-fold. A noticeable, but non-significant, increase developed soon after CCI-TBI, however. Glutathione-s-transferase mu-5 (GST-mu5) mRNA decreased significantly at 24 and 48 h post-injury in the cortex (p<0.05). Conversely, SOD1 mRNA expression did not significantly change in the cortex. Likewise, there were no significant changes in glutathione reductase (GR) mRNA, catalase (CAT) mRNA, or Keap1 mRNA expression in the cortex. No significant change was observed in SOD2 mRNA in either brain region compared with sham controls.
FIG. 2.

Nuclear factor erythoid related factor-2-antioxidant response elements (Nrf2-ARE) mediated mRNA expression is altered in the cortex (CTX) following traumatic brain injury (TBI). Real-time qRT-PCR analysis of Nrf2-ARE mediated gene expression revealed multiple changes following TBI. Nrf2 (see A) mRNA increased at 48 and 72 hours post-injury (p<.05); heme oxygenase-1 (HO-1; see B) mRNA increased by 20 fold at both 24 and 72 hours post-injury (p<.05) and nicotinamide adenine dinucleotide phosphate-quinone oxidoreductase 1 (NQO1; see C) showed a significant increase at both 72 hours and 1 week post-injury in the cortex (p<.05). Interestingly, astrocytic glial fibrillary acidic protein (GFAP; see D) mRNA increased at 24, 48, 72 hours, and 1 week post-injury (p<.05) although GFAP is not directly mediated via Nrf2-ARE. Superoxide dismutase-1 (SOD1; see E), superoxide dismutase-2 (SOD2; see F) glutathione reductase (GR; see G), catalase (CAT; see J), or Keap1 (see K) mRNA expression mRNA expression did not change significantly at any time point. In contrast to the Nrf2-ARE regulated increases in HO-1 (B) and NQO1 (C), glutathione-s-transferase mu-5 (GST-mu5; see H) mRNA decreased significantly at 24 and 48 hours post-injury (p<.05). However, there was a significant increase in glutathione peroxidase-3 (GPx-3; see I) at 1 week post-injury (p<.05). Analyzed by one-way ANOVA followed by Dunnett s post-hoc test. *=p<.05. Error bars represent +/− standard deviation.
FIG. 3.
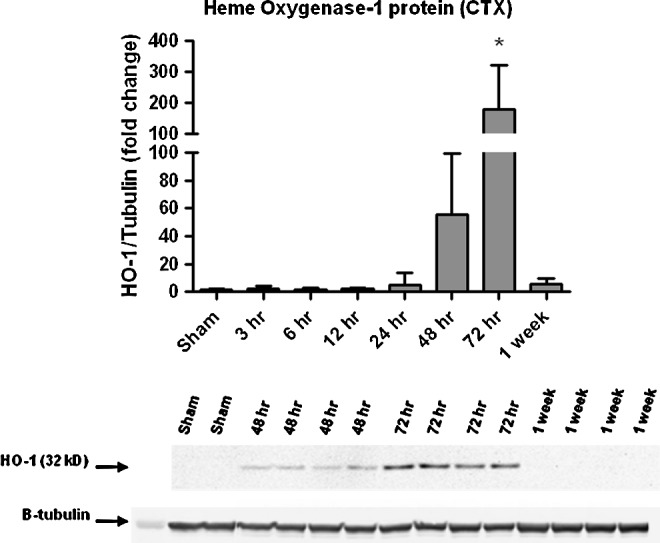
Time course of heme-oxygenase 1 (HO-1) protein expression after focal controlled cortical impact-traumatic brain injury (TBI) in the mouse (cortex). Western blot analysis of HO-1 in ipsilateral cortex (CTX) revealed a robust significant increase in HO-1 protein at 72 h after TBI compared with sham controls. Sample immunoblot for HO-1 presented below the graph illustrates this increased expression. Analyzed by one-way analysis of variance followed by Dunnett post-hoc test. *=p<0.05. Error bars represent±standard deviation.
Nrf2-ARE mediated expression is altered after TBI (hippocampus)
Real-time qRT-PCR analysis of Nrf2-ARE mediated gene expression in the hippocampus (Fig. 4) revealed multiple robust changes after TBI. Similar to what was found for the cortex, HO-1 mRNA increased by nearly 20-fold at 24 and 48 h in the hippocampus (p<0.05). Glial fibrillary acidic protein (GFAP) mRNA increased at 24, 48, and 72 h and 1 week post-injury (p<0.05) in both brain regions. CAT mRNA increased at 72 h in the hippocampus (p<0.05) but did not significantly change in the cortex. GST-mu5 mRNA decreased at 6, 24, and 48 h in the hippocampus (p<0.05). While glutathione peroxidase-3 (GPx-3) mRNA increased at 1 week post-injury in the cortex, it actually increased at both 72 h and 1 week post-injury in the hippocampus (p<0.05) compared with sham controls. SOD1 mRNA was significantly decreased in the hippocampus at 3, 6, 48, and 72 h (p<0.05). GR mRNA, however, increased in the hippocampus at 24 h (p<0.05) but without significant change in corresponding GR protein expression (data not shown).
FIG. 4.

Nuclear factor erythoid related factor-2-antioxidant response element (Nrf2-ARE) mediated mRNA expression is altered in the hippocampus (HIPP) after traumatic brain injury (TBI). Real-time qRT-PCR analysis of Nrf2-ARE mediated gene expression in the hippocampus revealed multiple changes following TBI. Similar to what was found for the cortex (compare to Fig. 2), Nrf2 (see A) mRNA significantly increased at 48 hours, 72 hours, and 1 week post-injury (p<.05); heme-oxygenase 1 (HO-1; see B) mRNA increased by nearly 20 fold at 24 and 48 hours; glutathione peroxidase-3 (GPx-3; see I) mRNA increased at 1 week post-injury in the hippocampus (p<.05) and glial fibrillary acidic protein (GFAP; see D) mRNA increased at 24, 48, 72 hours and 1-week post-injury (p<.05). It should be noted that the astrocyte marker GFAP is not a specific Nrf2-ARE mediated target, but Nrf2-ARE activation does occur abundantly in astrocytes. However, nicotinamide adenine dinucleotide phosphate-quinone oxidoreductase 1 (NQO1; see C) mRNA expression did not significantly change in contrast to the changes seen in cortex (see Fig. 2). Catalase (CAT; see J) mRNA increased significantly at 72 hours in hippocampus (p<.05); a similar, but not significant, increase was seen in cortex at the same time point (see Fig. 2). Similarly, glutathione reductase (GR; see G) mRNA showed a small increase in the hippocampus at 24 hours (p<.05). As in the cortex, glutathione-s-transferase mu-5 (GST-mu5; see H) mRNA decreased at 6, 24, 48 hours in hippocampus (p<.05). Superoxide dismutase-1 (SOD-1; see E) and superoxide dismutase-2 (SOD2; see F) mRNAs were significantly decreased in the hippocampus at 3, 6, 48, and 72 hours (p<.05). Keap1 (see K) mRNA expression did not significantly change at any time points post-injury in the hippocampus or cortex (see Fig. 2). Analyzed by one-way ANOVA followed by Dunnett's post-hoc test. *=p<.05. Error bars represent +/− standard deviation.
Summary of expression changes after focal CCI-TBI in the young adult male mouse
As summarized in Table 1, real-time qRT-PCR analysis of Nrf2-ARE mediated gene expression revealed multiple changes after TBI in both the ipsilateral cortex and hippocampus. Of high importance are findings such as nicotinamide adenine dinucleotide phosphate-quinone-oxidoreductase 1 (NADPH NQO1) mRNA increasing at 72 h and 1 week (p<0.05) post-injury in the cortex, signifying activation of the Nrf2-ARE pathway. There was no change observed in the repressor Keap1 mRNA in either brain region after TBI compared with sham controls, thereby suggesting that all observed Nrf2-ARE mediated changes were not dependent on decreased Keap1 levels but rather dependent on Nrf2-ARE activation itself.
Table 1.
Summary of mRNA Expression Changes after Focal Controlled Cortical Impact in the Mouse
| Time point post-injury | ||||||
|---|---|---|---|---|---|---|
| Gene | 3 h | 6 h | 24 h | 48 h | 72 h | 1 wk |
| HO-1 | – | – | ⇑↑ | ↑ | ⇑ | – |
| SOD1 | ↓ | ↓ | – | ↓ | ↓ | – |
| SOD2 | – | – | – | – | – | – |
| GFAP | – | – | ⇑↑ | ⇑↑ | ⇑↑ | ⇑↑ |
| GR | – | – | ↑ | – | – | – |
| GPx-3 | – | – | – | – | ↑ | ↑ |
| NQO1 | – | – | – | – | ⇑ | ⇑ |
| CAT | – | – | – | – | ↑ | – |
| GST-mu5 | – | ↓ | ↓ | ↓ | – | – |
| Keap1 | – | – | – | – | – | – |
| Nrf2 | – | – | – | ⇑↑ | ⇑↑ | ↑ |
Open arrows, ipsilateral cortical; closed arrows, hippocampal. HO-1, heme-oxygenase 1; SOD, superoxide dismutase; GFAP, glial fibrillary acidic protein; GR, glutathione reductase; GPx-3, glutathione peroxidase 3; NQO1, nicotinamide adenine dinucleotide phosphate-quinone-oxidoreductase 1; CAT, catalase; GST-mu5, glutathione-s-transferase mu 5; Nrf2, nuclear factor erythoid related factor2.
Interestingly, Nrf2 mRNA also increased at 48 and 72 h in the cortex and at 48 and 72 hours and 1 week post-injury in the hippocampus (p<0.05), thereby demonstrating the first evidence of such Nrf2 changes post-TBI. This may be from a positive feedback type activation of Nrf2 itself, because it has been shown previously that ARE enhancer sequences are present in and around the Nrf2 promoter.13
Lastly, GFAP mRNA was significantly increased at 24, 48, and 72 hours and 1 week post-injury (p<0.05) in both brain regions (cortex and hippocampus). While the astrocyte marker GFAP is not truly a specific Nrf2-ARE mediated target, it is important to note that much of the previously documented Nrf2-ARE pathway activation is thought to occur in astrocytes and other glia in vivo.24,28
Discussion
Given the overwhelming impact of TBI on society, it is imperative to discover and translate rational therapies for the clinical management of this growing epidemic. Oxidative stress has consistently been identified as one of the key pathological processes underlying the deleterious secondary injury cascade after TBI.3,6–12 However, although three distinct direct antioxidant therapies (e.g., polyethylene glycol conjugated superoxide dismutase [PEG-SOD], tirilazad, and dexanabinol) have been investigated, none has proven effective in rigorous large-scale clinical trials.42 The Nrf2-ARE pathway may provide a comprehensive, pleiotropic approach to more fully attenuate oxidative damage after TBI.
The current study provides critical information by defining the temporal and spatial profiles of both lipid peroxidation and Nrf2-ARE mediated expression changes post-injury in a mouse model of focal TBI. Although previous studies have investigated compounds that activate the Nrf2-ARE pathway after TBI, it has not yet been determined how the pathway itself responds in a focal TBI model. By defining the temporal and spatial profile of the Nrf2-ARE pathway after TBI, it will be easier to determine the optimal time to administer a potential Nrf2-ARE activating drug.
In contrast to other reports in differing TBI paradigms,43 we did not see a significant increase in HO-1 protein levels until the 72 h post-injury time point. Although a significant increase (approximately 10-20–fold) in HO-1 mRNA was observed at both 24 and 72 h post-injury (cortex), this increase in mRNA did not translate to a significant increase in HO-1 protein except at 72 h post-injury when it was highly elevated in comparison with sham by approximately 100–200 fold.
Previous studies have indicated it may be possible for such extreme differences in the levels of mRNA versus protein of Nrf2-ARE mediated targets to occur.44 Indeed, we previously published a study demonstrating a similar disconnect between mRNA and protein levels of a different protein, inducible nitric oxide synthase (iNOS), after focal CCI in the mouse.40 The delay of 48 h between the peak in cortical HO-1 mRNA at 24 h and the peak of HO-1 protein at 72 h, however, is less precedented but may have multiple explanations related to the well-documented oxidative stress and lipid peroxidative damage occurring during the first 72 h after TBI.42
First, while HO-1 gene transcription in the nucleus may take place relatively uninhibited during the first 24 h post-TBI, the corresponding translation of mRNA in the cytoplasm could be initially suppressed during the first 24 h of intense post-traumatic oxidative stress and rapidly intensifying lipid peroxidation known to occur in that cellular compartment after TBI.2 Second, the post-traumatic oxidative stress and damage will modify cellular proteins, thereby inhibiting their normal function as previously demonstrated for glutamine synthetase in normal brain aging and Alzheimer disease brains45 as well as SOD-1 in familial amyotrophic lateral sclerosis mice during the progression of their motor functional decline.46In addition, the oxidative modification of these proteins can subsequently cause them to be less recognizable by the antibodies used for their quantification.
Hence, in the present study, much of the newly synthesized HO-1 protein may have been oxidatively modified as quickly as it is being produced, thereby rendering it unable to be detected by the antibody based technique (e.g., Western blot) used in the current study. In essence, it is possible that there is indeed an earlier net increase in HO-1 protein—as some other studies43 have reported—but we were unable to detect it with the methods used in the current study until the later 72 h time-point.
The critical issue in relation to the current study, however, is that the Nrf2-ARE pathway is indeed being activated (as evidenced by the increases in various downstream target mRNA levels) post-injury. This activation of the Nrf2-ARE pathway is temporally and spatially related to the preceding and concurrent increase in oxidative damage. This furthers the notion that while the Nrf2-ARE pathway is activated post-injury, it is unable to provide adequate protection and must be amplified and temporally accelerated to antagonize the injury induced deleterious increase in oxidative damage.
In the current study, we demonstrate that the time course of endogenous Nrf2-ARE activation closely mimics that of the time course of oxidative damage (as represented by 4-HNE). This is likely because of the mechanism by which Nrf2-ARE activation is induced—the persistent production of free radicals/reactive species results in a modification to the key cysteine residues present on the Keap1 repressor protein. Such modifications could result in a conformational change in Keap1, thereby liberating Nrf2 to translocate to the nucleus and bind to enhancer ARE sequences in the promoter regions of antioxidant genes as described previously.13 This process may continue until the stimulus (oxidative stress) is mitigated or removed.
It is clear, however, that either the magnitude or the haste with which the Nrf2-ARE mediated amplification occurs is somehow insufficient to provide adequate protection against ongoing oxidative damage during the acute secondary injury phase. Hence, harnessing this pathway for neuroprotection will therefore require modulating this response to increase its magnitude and speed up its activation early enough post-TBI before oxidative damage becomes overpowering. Ongoing studies will determine the pre-clinical utility of amplifying and enhancing this defense pathway via administration of Nrf2-ARE activating pharmacological agents after TBI.
Acknowledgments
The authors would like to thank Kimberly M. Carrico for expert technical assistance. This work was supported by grants NIH-NIDA 1T32 DA022738, NIH-NINDS 1T32 NS077889, NIH-NINDS 2P30 NS051220-01, and funds from the Kentucky Spinal Cord & Head Injury Research Trust.
Author Disclosure Statement
No competing financial interests exist.
References
- 1.Thurman D.J., Alverson C., Dunn K.A., Guerrero J., and Sniezek J.E. (1999). Traumatic brain injury in the United States: a public health perspective. J. Head Trauma Rehabil. 14, 602–615 [DOI] [PubMed] [Google Scholar]
- 2.Deng Y., Thompson B.M., Gao X., and Hall E.D. (2007). Temporal relationship of peroxynitrite-induced oxidative damage, calpain-mediated cytoskeletal degradation and neurodegeneration after traumatic brain injury. Exp. Neurol. 205, 154–165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Singh I.N., Sullivan P.G., Deng Y., Mbye L.H., and Hall E.D. (2006). Time course of post-traumatic mitochondrial oxidative damage and dysfunction in a mouse model of focal traumatic brain injury: implications for neuroprotective therapy. J. Cereb. Blood Flow Metab. 26, 1407–1418 [DOI] [PubMed] [Google Scholar]
- 4.Thompson S.N., Gibson T.R., Thompson B.M., Deng Y., and Hall E.D. (2006). Relationship of calpain-mediated proteolysis to the expression of axonal and synaptic plasticity markers following traumatic brain injury in mice. Exp. Neurol. 201, 253–265 [DOI] [PubMed] [Google Scholar]
- 5.Hall E.D., and Braughler J.M. (1993). Free radicals in CNS injury. Res. Publ. Assoc. Res. Nerv. Ment. Dis. 71, 81–105 [PubMed] [Google Scholar]
- 6.Hall E.D., McCall J.M., and Means E.D. (1994). Therapeutic potential of the lazaroids (21-aminosteroids) in acute central nervous system trauma, ischemia and subarachnoid hemorrhage. Adv. Pharmacol. 28, 221–268 [DOI] [PubMed] [Google Scholar]
- 7.Hall E.D., Andrus P.K., Yonkers P.A., Smith S.L., Zhang J.R., Taylor B.M., and Sun F.F. (1994). Generation and detection of hydroxyl radical following experimental head injury. Ann. N. Y. Acad. Sci. 738, 15–24 [DOI] [PubMed] [Google Scholar]
- 8.Smith S.L., Andrus P.K., Zhang J.R., and Hall E.D. (1994). Direct measurement of hydroxyl radicals, lipid peroxidation, and blood-brain barrier disruption following unilateral cortical impact head injury in the rat. J. Neurotrauma 11, 393–404 [DOI] [PubMed] [Google Scholar]
- 9.Ansari M.A., Roberts K.N., and Scheff S.W. (2008). A time course of contusion-induced oxidative stress and synaptic proteins in cortex in a rat model of TBI. J. Neurotrauma 25, 513–526 [DOI] [PubMed] [Google Scholar]
- 10.Ansari M.A., Roberts K.N., and Scheff S.W. (2008). Oxidative stress and modification of synaptic proteins in hippocampus after traumatic brain injury. Free Radic. Biol. Med. 45, 443–452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kontos H.A., and Povlishock J.T. (1986). Oxygen radicals in brain injury. Cent. Nerv. Syst. Trauma 3, 257–263 [DOI] [PubMed] [Google Scholar]
- 12.Kontos H.A., and Wei E.P. (1986). Superoxide production in experimental brain injury. J. Neurosurg. 64, 803–807 [DOI] [PubMed] [Google Scholar]
- 13.Kensler T.W., Wakabayashi N., and Biswal S. (2007). Cell survival responses to environmental stresses via the Keap1-Nrf2-ARE pathway. Annu. Rev. Pharmacol. Toxicol. 47, 89–116 [DOI] [PubMed] [Google Scholar]
- 14.Lindner M.D., Plone M.A., Cain C.K., Frydel B., Francis J.M., Emerich D.F., and Sutton R.L. (1998). Dissociable long-term cognitive deficits after frontal versus sensorimotor cortical contusions. J. Neurotrauma 15, 199–216 [DOI] [PubMed] [Google Scholar]
- 15.Chan J.Y., Han X.L., and Kan Y.W. (1993). Isolation of cDNA encoding the human NF-E2 protein. Proc. Natl. Acad. Sci. U. S. A. 90, 11366–11370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Itoh K.N., Wakabayashi Y., Katoh Y., Ishii T., Igarashi K., Engel J.D., and Yamamoto M. (1999). Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev. 13, 76–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Moi P., Chan K., Asunis I., Cao A., and Kan Y.W. (1994). Isolation of NF-E2-related factor 2 (Nrf2), a NF-E2-like basic leucine zipper transcriptional activator that binds to the tandem NF-E2/AP1 repeat of the beta-globin locus control region. Proc. Natl. Acad. Sci. U. S. A. 91, 9926–9930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kobayashi A., Kang M.I., Okawa H., Ohtsuji M., Zenke Y., Chiba T., Igarashi K., and Yamamoto M. (2004). Oxidative stress sensor Keap1 functions as an adaptor for Cul3-based E3 ligase to regulate proteasomal degradation of Nrf2. Mol Cell Biol 24:7130–7139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kobayashi A., Kang MI, Watai Y, Tong KI, Shibata T, Uchida K, and Yamamoto M. (2006). Oxidative and electrophilic stresses activate Nrf2 through inhibition of ubiquitination activity of Keap1. Mol. Cell Biol. 26, 221–229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tong K.I., Kobayashi A., Katsuoka F., and Yamamoto M. (2006). Two-site substrate recognition model for the Keap1-Nrf2 system: a hinge and latch mechanism. Biol. Chem. 387, 1311–1320 [DOI] [PubMed] [Google Scholar]
- 21.Jain A.K., Bloom D.A., and Jaiswal A.K. (2005). Nuclear import and export signals in control of Nrf2. J. Biol. Chem. 280, 29158–29168 [DOI] [PubMed] [Google Scholar]
- 22.Johnson J.A., Johnson D.A., Kraft A.D., Calkins M.J., Jakel R.J., Vargas M.R., and Chen P.C. (2008). The Nrf2-ARE pathway: an indicator and modulator of oxidative stress in neurodegeneration. Ann. N. Y. Acad. Sci. 1147, 61–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Calkins M.J., Jakel R.J., Johnson D.A., Chan K., Kan Y.W., and Johnson J.A. (2005). Protection from mitochondrial complex II inhibition in vitro and in vivo by Nrf2-mediated transcription. Proc. Natl. Acad. Sci. U. S. A. 102, 244–249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Calkins M.J., Vargas M.R., Johnson D.A., and Johnson J.A. (2010). Astrocyte-specific overexpression of Nrf2 protects striatal neurons from mitochondrial complex II inhibition. Toxicol. Sci. 115, 557–568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Innamorato N.G., Rojo A.I., Garcia-Yague A.J., Yamamoto M., de Ceballos M.L., and Cuadrado A. (2008). The transcription factor Nrf2 is a therapeutic target against brain inflammation. J. Immunol. 181, 680–689 [DOI] [PubMed] [Google Scholar]
- 26.Rojo A.I, Innamorato N.G, Martin-Moreno A.M., de Ceballos M.L., Yamamoto M., and Cuadrado A. (2010). Nrf2 regulates microglial dynamics and neuroinflammation in experimental Parkinson's disease. Glia 58, 588–598 [DOI] [PubMed] [Google Scholar]
- 27.Li J., Johnson D., Calkins M., Wright L., Svendsen C., and Johnson J. (2005). Stabilization of Nrf2 by tBHQ confers protection against oxidative stress-induced cell death in human neural stem cells. Toxicol. Sci. 83, 313–328 [DOI] [PubMed] [Google Scholar]
- 28.Vargas M.R., Johnson D.A., Sirkis D.W., Messing A., and Johnson J.A. (2008). Nrf2 activation in astrocytes protects against neurodegeneration in mouse models of familial amyotrophic lateral sclerosis. J. Neurosci. 28, 13574–13581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhao J., Kobori N., Aronowski J., and Dash P.K. (2006). Sulforaphane reduces infarct volume following focal cerebral ischemia in rodents. Neurosci. Lett. 393, 108–112 [DOI] [PubMed] [Google Scholar]
- 30.Zhao X., Sun G., Zhang J., Strong R., Dash P.K., Kan Y.W., Grotta J.C., and Aronowski J. (2007). Transcription factor Nrf2 protects the brain from damage produced by intracerebral hemorrhage. Stroke 38, 3280–3286 [DOI] [PubMed] [Google Scholar]
- 31.Innamorato N.G., Jazwa A., Rojo A.I., Garcia C., Fernandez-Ruiz J., Grochot-Przeczek A., Stachurska A., Jozkowicz A., Dulak J., and Cuadrado A. (2010). Different susceptibility to the Parkinson's toxin MPTP in mice lacking the redox master regulator Nrf2 or its target gene heme oxygenase-1. PLoS One 5, e11838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kraft A.D., Resch J.M., Johnson D.A., and Johnson J.A. (2007). Activation of the Nrf2-ARE pathway in muscle and spinal cord during ALS-like pathology in mice expressing mutant SOD1. Exp. Neurol. 20, 107–117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Son T.G., Camandola S., Arumugam T.V., Cutler R.G., Telljohann R.S., Mughal M.R., Moore T.A., Luo W., Yu Q.S., Johnson D.A., Johnson J.A., Greig N.H., and Mattson M.P. (2010). Plumbagin, a novel Nrf2/ARE activator, protects against cerebral ischemia. J. Neurochem. 112, 1316–1326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jin W., Wang H., Yan W., Zhu L., Hu Z., Ding Y., and Tang K. (2009). Role of Nrf2 in protection against traumatic brain injury in mice. J. Neurotrauma 26, 131–139 [DOI] [PubMed] [Google Scholar]
- 35.Jin W., Wang H., Ji Y., Zhu L., Yan W., Qiao L., and Yin H. (2009). Genetic ablation of Nrf2 enhances susceptibility to acute lung injury after traumatic brain injury in mice. Exp. Biol. Med. 234, 181–189 [DOI] [PubMed] [Google Scholar]
- 36.Zhao J., Moore A.N., Redell J.B., and Dash P.K. (2007). Enhancing expression of Nrf2-driven genes protects the blood brain barrier after brain injury. J. Neurosci. 27, 10240–10248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhao J., Moore A.N., Clifton G.L., and Dash P.K. (2005). Sulforaphane enhances aquaporin-4 expression and decreases cerebral edema following traumatic brain injury. J. Neurosci. Res. 82, 499–506 [DOI] [PubMed] [Google Scholar]
- 38.Dash P.K., Zhao J., Orsi S.A., Zhang M., and Moore A.N. (2009). Sulforaphane improves cognitive function administered following traumatic brain injury. Neurosci. Lett. 460, 103–107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Satoh T.K., Kosaka K., Itoh K., Kobayashi A., Yamamoto M., Shimojo Y., Kitajima C., Cui J., Kamins J., Okamoto S., Izumi M., Shirasawa T., and Lipton S.A. (2008). Carnosic acid, a catechol-type electrophilic compound, protects neurons both in vitro and in vivo through activation of the Keap1/Nrf2 pathway via S-alkylation of targeted cysteines on Keap1. J. Neurochem. 104, 1116–1131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hall E.D., Wang J.A., and Miller D.M. (2012). Relationship of nitric oxide synthase induction to peroxynitrite-mediated oxidative damage during the first week after experimental traumatic brain injury. Exp. Neurol. 238, 176–182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Miller D.M., Singh I.N., Wang J.A., and Hall E.D. (2013). Administration of the Nrf2-ARE activators sulforaphane and carnosic acid attenuates 4-hydroxy-2-nonenal-induced mitochondrial dysfunction ex vivo. Free Radic. Biol. Med. 57, 1–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bains M., and Hall E.D. (2012). Antioxidant therapies in traumatic brain and spinal cord injury. Biochim. Biophys. Acta 1822, 675–684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cheng Z.G., Zhang G.D., Shi P.Q., Du B.S. (2013). Expression and antioxidation of Nrf2/ARE pathway in traumatic brain injury. Asian Pac. J. Trop. Med, 6, 305–310 [DOI] [PubMed] [Google Scholar]
- 44.Dowell J.A., and Johnson J.A. (2013). Mechanisms of Nrf2 protection in astrocytes as identified by quantitative proteomics and siRNA screening. PLoS One 8, e70163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Smith C.D., Carney J.M., Starke-Reed P.E., Oliver C.N., Stadtman E.R., Floyd R.A., and Markesbery W.R. (1991). Excess brain protein oxidation and enzyme dysfunction in normal aging and in Alzheimer disease. Proc. Natl. Acad. Sci. U. S. A. 88, 10540–10543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Andrus P.K., Fleck T.J., Gurney M.E., and Hall E.D. (1998). Protein oxidative damage in a transgenic mouse model of familial amyotrophic lateral sclerosis. J. Neurochem. 71, 2041–2048 [DOI] [PubMed] [Google Scholar]