

Abstract
The aim of this study was to evaluate the potential efficacy of SB-3CT, a matrix metallopeptidase 9 inhibitor, on behavioral and histological outcomes after traumatic brain injury (TBI) in rats. Adult male Sprague-Dawley rats were randomly divided into three groups (n=15/group): TBI with SB-3CT treatment, TBI with saline, and sham injury. The TBI model was induced by a fluid percussion TBI device. SB-3CT (50 mg/kg in 10% dimethyl sulfoxide) was administered intraperitoneally at 30 min, 6 h, and 12 h after the TBI. Motor function (beam-balance/beam-walk tests) and spatial learning/memory (Morris water maze) were assessed on post-operative Days 1−5 and 11–15, respectively. Fluoro-Jade staining, immunofluorescence, and cresyl violet-staining were carried out for histopathological evaluation at 24 h, 72 h, and 15 days after TBI, respectively. It was shown that TBI can result in significant behavioral deficit induced by acute neurodegeneration, increased expression of cleaved caspase-3, and long-term neuronal loss. SB-3CT intervention via the current regime provides robust behavioral protection and hippocampal neurons preservation from the deleterious effects of TBI. Hence, the efficacy of SB-3CT on TBI prognosis could be ascertained. It is believed that the current study adds to the growing literature in identifying SB-3CT as a potential therapy for human brain injury.
Key Words: : hippocampus, neuroprotection, SB-3CT, traumatic brain injury
Introduction
Traumatic brain injury (TBI) is a major cause of morbidity and mortality.1 Studies over the past two decades have demonstrated that as a result of secondary autodestructive insult after TBI, long-term motor and cognitive disabilities occur due to serious damage in the central nervous system (CNS).2–4 Despite advances in preclinical research, as well as improvement in clinical intensive care during recent years, no effective pharmacological therapy is available to promote functional recovery after TBI.5,6 It is therefore important to identify an effective therapeutic agent and its intervention regime to improve functional outcome after TBI.
Matrix metalloproteinases (MMPs) are a family of extracellular zinc- and calcium-dependent endopeptidases that degrade components of extracellular matrix (ECM).7,8 Their elevation was demonstrated after TBI.9,10 More specifically, the elevation of MMP-9 can cause the increase in capillary permeability and will also lead to brain edema, a typical symptom of secondary injury after TBI.11,12 It was demonstrated in our previous study 13 that the amount of MMP-9 in both the severe TBI and moderate TBI groups were significantly increased over that of the sham group within 6 h after TBI. With the increase in time, MMP-9 protein levels in the severe TBI and moderate TBI groups kept on increasing, and the maximal levels of the two groups were all reached at 24 h and 72 h after TBI in the ipsilateral and contralateral hemispheres, respectively. Further, Grossetete and colleagues have shown that the elevation of MMP-3 and MMP-9 after TBI has a negative correlation with the prognosis of patients. Given these findings, we therefore proposed that MMP-9 may be a useful target for rescue therapies after TBI.14
Broad-spectrum MMP inhibitors (MMPIs) have been tested in patients with ischemia. It was shown that these agents were efficient in reducing cell loss and edema15 but they also caused fibrosis of joints with arthritic-like symptoms.16 This has led to a search for highly selective inhibitors that could retain the desired neuroprotective effects without any joint adverse effects. SB-3CT, a highly selective inhibitor known to target only MMP-2 and MMP-9, is one of such substance that is promising in treating a range of pathologies including those of CNS.17 It was shown by Cui and colleagues that SB-3CT treatment for seven days could lessen neuronal laminin degradation and provide protection on neurons from ischemic cell death.18 It was further confirmed by Liu and colleagues that SB-3CT could attenuate occludin protein loss and claudin-5 redistribution, thus attenuating blood–brain barrier damage in early ischemic stroke stage.19 These investigations were all carried out in cerebral ischemia, whereas no information is available on the behavioral and histological outcomes resulting from the subsequent SB-3CT treatment after TBI.
The current study thus was designed to explore whether SB-3CT with a rational intervention regime could facilitate favorable neurobehavioral outcomes and attenuate histological damage after TBI. A fluid percussion TBI model was applied in rats.20 SB-3CT treatment at a dosage of 50 mg/kg was then carried out via repeated intraperitoneal administration at 30 min, 6 h, and 12 h, respectively, after TBI. Both the behavioral and histological outcomes after TBI in rats were assessed to give qualitative and quantitative evaluation on the effect of post-operative intervention using SB-3CT.
Methods
Animals
Forty-five male Sprague-Dawley rats weighing 310–330 g were used in this study. The rats were randomly divided into three groups: TBI with SB-3CT (n=15; TBI+SB group), TBI with saline (n=15; TBI+SA group), and sham injured groups (n=15; briefed as sham group). Animals were housed in individual cages in a temperature (22°C) and humidity-controlled (50% relative) animal facility with a 12-h light/dark cycle. Animals had free access to food and water during the duration of the experiments and were held in the animal facility for at least seven days prior to surgery. The animal procedures were approved by the Animal Care and Experiment Committee of School of Medicine, Shanghai JiaoTong University.
Surgical preparation
Rats were anesthetized with 4% isoflurane in a carrier gas mixture of nitrous oxide/oxygen (2:1 ratio), intubated, and mechanically ventilated with a rodent volume ventilator (Harvard Apparatus model 683, Holliston, MA). A surgical level of anesthesia was maintained with 2% isoflurane. Rats were mounted in a stereotaxic frame, a scalp incision made along the midline, and a 4.8-mm diameter craniectomy was performed on the right parietal bone (centered at −4.5 mm bregma and right lateral 3.0 mm). A rigid plastic injury tube (modified Leur-loc needle hub, 2.6 mm inside diameter) was secured over the exposed, intact dura with cyanoacrylate adhesive. Two skull screws (2.1 mm diameter, 6.0 mm length) were placed into burr holes, 1 mm rostral to bregma and 1 mm caudal to lambda. The injury tube was secured to the skull and screws with cranioplastic cement (Plastics One, Roanoke, VA). Rectal temperature was continuously monitored and maintained within normal ranges during surgical preparation by a feedback temperature controller pad (CWE model TC-1000, Ardmore, PA). Temporalis muscle temperature was measured by insertion of a 29-gauge needle temperature probe (Physitemp unit TH-5, probe MT-29/2, Clifton, NJ) between the skull and temporalis muscle. The mean arterial blood pressure was monitored continuously throughout the whole duration of TBI and post-operational treatment, and blood gases were measured before injury and at 15 min after injury.
Fluid percussion TBI and post-operational treatment with SB-3CT
A fluid percussion device was used to create TBI as described in detail previously.20 Briefly, this device consists of a Plexiglas cylindrical reservoir, 60 cm long and 4.5 cm in diameter, bounded at one end by a rubber-covered Plexiglas piston mounted on O rings. The opposite end is fitted with a transducer housing ended with a 2.6-mm-inside-diameter Luer Lok fitting. The entire system is filled with 37°C isotonic saline. Injury is induced by the descent of a metal pendulum striking the piston, thereby injecting a small volume of saline epidurally into the closed cranial cavity and producing brief displacement and deformation of neuronal tissue. The resulting pressure pulse is measured in air by an extracranial transducer (Statham PA 85–100; Gould, Oxnard, CA) and recorded on a storage oscilloscope (Tektronix 5111; Tektronix, Beaverton, OR). Animals were injured at a moderate magnitude of injury (2.12–2.16 atm), as described in detail previously.20
As to the TBI+SB group, SB-3CT (50 mg/kg in 10% dimethyl sulfoxide; Enzo Life Sciences, Inc, Farmingdale, NY) was injected intraperitoneally starting at 30 min after TBI, followed by second and third injections at 6 h and 12 h. These time points were chosen according to our previous findings that MMP-9 activity was up-induced within the first 24 h following TBI in the same animal model.13 Weight-matched control animals received injections of an equal volume of vehicle (10% dimethyl sulfoxide in saline) only. A final volume of not more than 0.4% of body weight was injected using an ultrafine 30-gauge insulin needle. An intraperitoneal route of delivery was chosen as effective delivery of this inhibitor to the brain had previously been reported in adult rats via the same route.21 Further, similar dosages have been shown to be effective in inhibiting MMP activity in adult rat models of spinal cord injury17 and ischemia21 without any confounding toxic effects. After SB-3CT or saline treatment in the current study, the animals were returned to the normal rat housing rooms (22°C, 50% relative humidity, 12-h light/12- h dark cycle).
Behavioral monitoring
Motor function (beam-balance/beam-walk tests) and spatial learning/memory (Morris water maze; MWM) were assessed as the behavioral index on post-operative Days 1−5 and 11−15, respectively.
Gross vestibulomotor function was assessed on a beam balance task: the time duration that animals could remain on an elevated, 1.5-cm-wide wooden beam was recorded (up to a maximum of 60 sec).22 Three trials were performed per animal per day. Training/pre-assessment was completed on the day prior to surgery/injury. Spinning on the beam was counted as a fall. Animals fell onto a cushioned pad to avoid injury.
Finer components of vestibulomotor function and coordination were assessed using a modified beam-walking task.23 On the day prior to injury, the rats were trained to escape from a bright light and loud white noise (model #15800C; Lafayette Instruments, Inc., Lafayette, IN) by traversing a narrow wooden beam (2.5×100 cm) and entering a darkened goal box at the opposite end. The noise and light were terminated when the rat entered the goal box. Four pegs (3 mm diameter and 4 cm high) were equally spaced along the center of the beam to increase the difficulty of the task. Performance was assessed by measuring the latency to traverse the beam. The rats were kept in the goal box for 30 sec between trials. If the rats did not cross the beam in 60 sec or fell off, the light and noise were stopped and the rats were placed in the goal box. Data for each session are the mean of three trials.
Spatial learning was assessed in a MWM,24 which has been shown to be sensitive to assess cognitive function after TBI.25–28 To this end, we utilized a protocol that replicated one previously reported by Kline and colleagues.27 Briefly, the maze consisted of a plastic pool (180 cm diameter; 60 cm high) filled with tap water (26±1°C) to a depth of 28 cm and was situated in a room with salient visual cues that remained constant throughout the study. The platform was a clear acrylic glass stand (10 cm diameter, 26 cm high) that was positioned at 26 cm from the maze wall in the southwest quadrant and held constant for each rat. Spatial learning began on post-operative day 11 and consisted of providing a block of four daily trials (4-min inter-trial interval) for five consecutive days (11–15) to locate the platform when it was submerged 2 cm below the water's surface (i.e., invisible to the rat). On Day 15, the platform was raised 2 cm above the water's surface (i.e., visible to the rat) as a control procedure to determine the contributions of non-spatial factors (e.g., sensorimotor performance, motivation, and visual acuity) on cognitive performance. For each daily block of trials, the rats were placed in the pool facing the wall at each of four possible start locations (north, east, south, or west) in a randomized manner. Each trial lasted until the rat climbed onto the platform or until 120 sec had elapsed, whichever occurred first. Rats that failed to locate the goal within the allotted time were manually guided to it. All rats remained on the platform for 30 sec before being placed in a heated incubator between trials. The times and swim distances of the four daily trials for each rat were averaged and used in the statistical analyses.
Tissue collection and sectioning
Rats were euthanized 24 h (acute histology experiment), 72 h (apoptotic effector cleaved caspase-3 expression), or on Day 15 (chronic histology examination) after TBI, by deep sodium pentobarbital anesthesia (100 mg/kg, intraperitoneally) followed by transcardial perfusion with 100 mL of 0.1 M sodium phosphate buffer (PB; pH=7.4) followed by 350 mL of 4% paraformaldehyde (pH, 7.4). Brains were removed and post-fixed for 1 h in 4% paraformaldehyde at 4°C. Brains were next cryoprotected in 10% sucrose solution for one day followed by two days in a 30% sucrose solution, and then frozen on powdered dry ice. Using a sliding microtome (American Optical, Model 860), 45 μm coronal sections were cut. Every serial section starting at −2.12 mm bregma and ending at −4.80 mm bregma was saved in 24-well cell culture plates. Systematic random sampling techniques were used for selecting tissue sections for staining and stereological analysis.
Histology
Acute histology
Neuronal degeneration was assessed at 24 h after TBI using histofluorescent staining, Fluoro-Jade B (FJ-B). Briefly, tissue sections were mounted onto gelatin-coated slides in a 1:1 ratio of 0.1M PB and distilled H2O (dH2O), air-dried overnight, and subsequently immersed in 100% alcohol (3 min), 70% alcohol (1 min), dH2O (1 min), and 0.006% potassium permanganate (15 min). After that, sections were rinsed with dH2O (1 min), incubated in 0.001% FJ-B (Histo-Chem Inc., Pine Bluff, AK) staining solution in 0.1% acetic acid for 30 min, rinsed again in dH2O (3 min), and air-dried. Finally, the sections were immersed in xylene and cover-slipped with DePeX mounting medium (Electron Microscopy Sciences, Fort Washington, PA). Cell counting of CA2-3 and hilus was done by an investigator blinded to the groups as previously described.29 The degeneration cell counts were made on a microscope (Nikon E600, Nikon, Tokyo) using a computer software (IMAGEJ 1.43, National Institute of Health, USA).
Immunofluorescence
An apoptotic effector cleaved caspase-3 expression was assessed on Day 3 post-injury using immunofluorescent staining. Briefly, free-floating sections were permeabilized for 1 h in 0.1% Triton X-100 (PBT) buffer to reduce non-specific immunolabelling. Double immunostaining was performed by co-incubation of primary antibodies overnight at 4°C. Primary antibodies and dilutions used were anti-Caspase-3 (1:500, abcam), anti-NeuN (1:1000, Chemicon; NeuN, Neuronal Nuclei specific antibody). After rinsing, sample sections were then incubated with goat anti-rabbit conjugated with Alexa 568 (Molecular Probes, 1:1000), goat anti-mouse conjugated with Alexa 488 (Molecular Probes, 1:1000), and 4′,6-diamidino-2-phenylindole (DAPI; Thermo, 1:1000). Tissue sections were mounted onto gelatin-coated slides in a 1:1 ratio of 0.1M PB and distilled H2O (dH2O) and cover-slipped with Fluoromount G mounting medium (Electron Microscopy Sciences, Fort Washington, PA). The number of viable neuronal cells in hippocampal CA2-3 and hilus was counted on a microscope (Nikon E600, Nikon, Tokyo) using a computer software (Image Plus 2.0, Motic, Xiamen, China). The mean number of stained cells was determined by three researchers who were blinded to the groups as previously described.30
Chronic histology
Long-term neuronal survival was assessed on Day 15 post-injury. Surviving neurons were stained with cresyl violet and further quantified using stereological techniques.31 Briefly, tissue sections were mounted on gelatin-coated slides and dried overnight before staining. The sections were dehydrated at room temperature by a series of ethanol immersions: 70% (2 min×1), 95% (2 min×2), and 100% (2 min×2), followed by immersion in xylene (16 min). The sections were then rehydrated in a series of ethanol immersions: 100% (2 min×2), 95% (2 min×2), and 75% (2 min×1), then rinsed with dH2O (30 sec×2). The sections were next stained with cresyl violet acetate (0.1%) for 6 min, followed by rinsing in dH2O (15 sec×2), differentiated by immersion in 95% ethanol with 0.15% acetic acid (8 min), and dehydrated in a series of ethanol immersions (95% [30 sec×2] and 100% [30 sec×2]) and cleared by immersion in xylene (5 min×2). The sections were cover-slipped with Permount (Fisher Scientific, Hampton, NH). Neuronal survival was assessed in the CA2-3 and hilus of hippocampal fields. The cresyl violet cell counts were made on a microscope (Nikon E600, Nikon, Tokyo) using a computer software (Stereo Investigator™ 8.0, Microbrightfield, Inc., Williston, VT) by a blinded examiner.
Statistical analysis
Data analysis was performed using SPSS software (Version 17, Chicago, IL) which adheres to a general linear model. Alpha level for Type I error was set at 0.05 for rejecting null hypotheses. Data are presented as mean±standard deviation. A one-way analysis of variance (ANOVA) was used to compare physiological variables measured between groups. Degenerating neuronal counts from FJ-B staining, cleaved caspase-3 expression and surviving neuronal counts from cresyl violet staining were each analyzed using a one-way ANOVA and a Dunnett post hoc analysis comparing each group to the sham. Behavioral data for beam walk and MWM latency were analyzed using repeated measures ANOVA with assessment days as the repeated variable within subjects followed by Dunnett post hoc analysis.
Results
Physiological data
Physiological parameters (pH, partial pressure of carbon dioxide, partial pressure of oxygen, mean arterial blood pressure, brain temperature, and rectal temperature) were assessed for each group at 15 min before injury and 15 min after TBI or sham procedures as shown in Table 1. All physiologic variables were within normal ranges for both the TBI+SB group and the TBI+SA group.
Table 1.
Physiological Variables
| 24 h | 72 h | 15 days | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Groups | Sham | TBI+SAline | TBI+SB-3CT | Sham | TBI+SAline | TBI+SB-3CT | Sham | TBI+SAline | TBI+SB-3CT |
| Pre-trauma 15 min | |||||||||
| pH | 7.43±0.06 | 7.44±0.13 | 7.43±0.04 | 7.45±0.08 | 7.43±0.06 | 7.42±0.03 | 7.46±0.05 | 7.47±0.06 | 7.45±0.07 |
| PCO2 (mm Hg) | 38.70±1.43 | 39.01±2.17 | 38.53±1.68 | 37.77±0.53 | 38.80±1.47 | 38.20±1.33 | 39.30±1.61 | 38.30±1.81 | 37.90±2.01 |
| PO2 (mm Hg) | 138.17±5.49 | 135.26±2.48 | 136.23±1.28 | 139.19±6.64 | 139.17±7.21 | 138.35±5.66 | 135.57±3.45 | 136.18±4.89 | 138.60±3.47 |
| MABP (mm Hg) | 118.21±4.11 | 117.48±4.34 | 119.23±4.12 | 117.63±3.73 | 119.11±3.25 | 120.25±4.71 | 116.66±4.59 | 115.59±4.33 | 120.31±2.92 |
| Brain t(°C) | 36.89±0.11 | 36.91±0.09 | 36.72±0.18 | 36.87±0.21 | 36.79±0.11 | 36.81±0.15 | 36.79±0.22 | 36.61±0.27 | 36.79±0.13 |
| Rectal t(°C) | 37.02±0.13 | 36.89±0.20 | 36.93±0.11 | 36.76±0.18 | 36.84±0.12 | 36.71±0.21 | 36.81±0.09 | 36.89±0.18 | 36.78±0.14 |
| Post-trauma 15 min | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| PH | 7.45±0.06 | 7.46±0.07 | 7.45±0.12 | 7.43±0.08 | 7.43±0.03 | 7.44±0.15 | 7.43±0.06 | 7.45±0.05 | 7.43±0.06 |
| PCO2 (mm Hg) | 38.23±1.82 | 39.26±1.78 | 38.77±2.12 | 38.11±0.92 | 38.55±1.47 | 39.81±1.56 | 38.86±1.51 | 38.89±1.98 | 38.27±1.43 |
| PO2 (mm Hg) | 140.23±6.34 | 139.64±3.71 | 138.63±3.72 | 138.72±2.47 | 137.50±7.23 | 138.98±6.47 | 135.57±3.62 | 136.18±4.92 | 138.60±3.59 |
| MABP (mm Hg) | 120.31±3.08 | 118.20±4.08 | 119.23±4.08 | 117.63±3.77 | 116.66±4.79 | 121.01±2.11 | 117.73±4.34 | 118.21±4.34 | 119.00±3.81 |
| Brain t (°C) | 36.91±0.18 | 36.83±0.15 | 36.89±0.09 | 36.67±0.18 | 36.81±0.12 | 36.83±0.15 | 36.72±0.17 | 36.81±0.21 | 36.88±0.15 |
| Rectal t (°C) | 37.11±0.09 | 36.78±0.14 | 36.78±0.20 | 36.82±0.16 | 36.80±0.09 | 36.91±0.20 | 36.93±0.18 | 36.67±0.19 | 36.79±0.18 |
TBI, traumatic brain injury; PCO2, partial pressure of carbon dioxide; PO2, partial pressure of oxygen; MABP, mean arterial blood pressure; t, temperature.
Behavioral outcome: Beam walk performance
No pre-surgical differences were observed among groups, as all rats were capable of balancing on the beam for the allotted 60 sec (Fig. 1A). The TBI injured groups were all markedly impaired relative to the sham group, which was still able to maintain pre-surgery balancing ability during the whole testing period. At Day 1 after TBI, the TBI+SA group exhibited statistically lower beam-balance latencies (p<0.01, TBI+SA vs. sham; Fig. 1A), higher beam-walk latencies (p<0.01, TBI+SA vs. sham; Fig. 1B), compared with those of the sham group. Such kind of phenomena peaked at Day 1 after TBI, and after that time point, the beam balance and beam walk latencies slowly returned back to the normal ranges. However, even at Day 5 after TBI, a significant difference could still be observed in beam balance latency (p<0.01, TBI+SA vs. sham, Fig.1A) and beam walk latency (p<0.01, TBI+SA vs. sham, Fig.1B) between the TBI+SA and sham groups. Upon SB-3CT treatment, such behavioral deficit was attenuated obviously since Day 2 after TBI. For example, at Day 2 after TBI, the beam balance latency of TBI+SB group was 30.08 sec—approximately 53.17% of that of the sham group (p<0.01), higher than the relative latency of the TBI+SA group (23.39%; TBI+SA over sham, p<0.01; Fig. 1A). At Day 5 after TBI, the beam balance latency of the TBI+SB group approached about 92.47% of that of the sham group (p<0.01), whereas the TBI+SA group only reached about 67.22% of that of the sham group (p<0.01; Fig. 1A). As to the beam walk latency, at Day 2 after TBI, the TBI+SB group was 44.9 sec—approximately nine times that of the sham group (p<0.01), lower than the latency of the TBI+SA group (56.5 sec, 11.3 times of that of the sham group; p<0.01; Fig. 1B). At Day 5 after TBI, the beam walk latency of the TBI+SB group was 17.5 sec—approximately 3.5 times of that of the sham group (p<0.01), lower than the one of the TBI+SA group (42.5 sec, 8.5 times of that of the sham group, p<0.01; Fig. 1B).
FIG. 1.

Motor testing. In the beam balance test (A) or beam walk test (B), rats in the traumatic brain injury (TBI)+SB-3CT (SB) group exhibited significantly improved performance compared to the TBI+SAline (SA) group. (*p<0.01, TBI+SA group versus sham group at d1, d2, d3, d4, and d5, respectively;≠p<0.01, TBI+SB group versus sham group at d1, d2, d3, d4, and d5, respectively; #p<0.01, TBI+SB group versus TBI+SA group at d1, d2, d3, d4, and d5, respectively).
Behavioral outcome: MWM performance
During the spatial memory examination, a statistically significant between-group effect on the latency to reach the submerged platform was noted in the MWM test. As shown in Figure 2, the TBI+SA group exhibited statistically higher latencies, compared with the sham group, at each time points evaluated (p<0.01, TBI+SA vs. sham for all time points). The post-hoc analysis revealed that the rats in TBI+SB group were able to locate the escape platform significantly quicker than TBI+SA group. At day 11, it took 98.1 sec for the rats in TBI+SB group to locate the escape platform, whereas the one in TBI+SA group needed 112.1 sec (p<0.01, TBI+SB vs. TBI+SA). However, the rats in the sham group only needed around 84.9 sec for doing this (p<0.01, TBI+SB vs. sham; p<0.01, TBI+SA vs. sham). At Day 15 after TBI, it seems that the rats in the TBI+SB group recovered quite well as their latency approached closely to that of the sham group. However, a significant difference could still be observed between these two groups (p<0.01, TBI+SB vs. sham).
FIG. 2.

Morris water maze testing. Rats in the traumatic brain injury (TBI)+SB-3CT (SB) group exhibited significantly improved performance on the MWM task, compared with the TBI+SA group (open circles; *p<0.01, TBI+SA group versus sham group at d11, d12, d13, d14, and d15, respectively;≠p<0.01, TBI+SB group versus sham group at d11, d12, d13, d14, and d15, respectively; #p<0.01, TBI+SB group versus TBI+SA group at d11, d12, d13, d14, and d15, respectively).
Histological outcome: Fluoro-Jade staining
This experiment measured at 24 h post-injury was designed to evaluate the effect of SB-3CT administered just post-injury on TBI induced neuronal degeneration. Neuronal degeneration was detected using FJ-B at 24 h after TBI (Fig. 3) and further quantified in the stratum pyramidal of the injured CA2-3 and hilus region (Fig. 4). Pyramidal cells staining positive for FJ-B fluoresced brightly with prominent somas and extensive dendritic arborization along the stratum radiatum. More degenerating neurons in the injured hippocampus of the TBI+SA group could be observed than the TBI+SB groups (Fig. 3). Post hoc Dunnett test revealed that, compared with the TBI+SA group, there was a significant reduction in degeneration neurons in the SB-3CT treated group both in the CA2-3 (p<0.01, Fig. 4) and hilus regions (p<0.01, Fig. 4). Upon SB-3CT treatment, a reduction of 40.7% in the degenerating neurons was achieved in the injured CA2-3 region of the hippocampus after TBI and a reduction of 50.7% in the hilus region (Fig. 4).
FIG. 3.
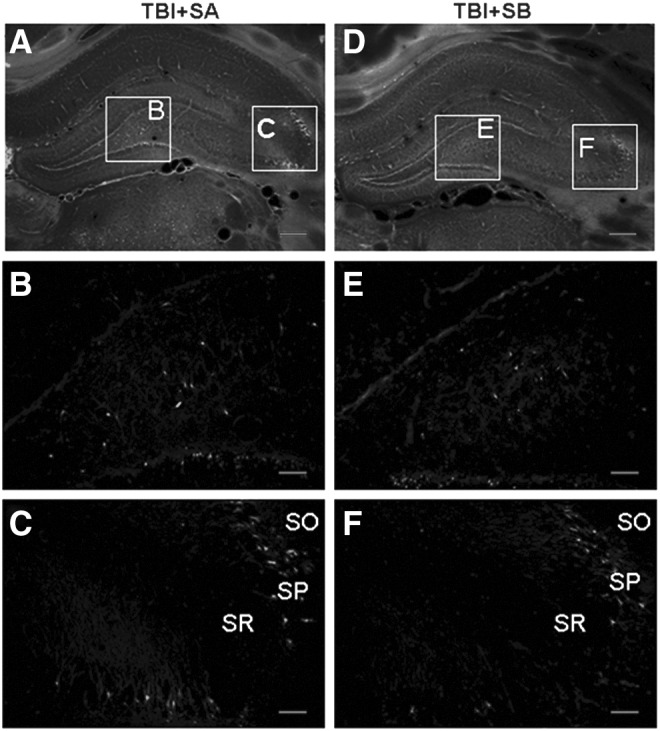
Fluoro-Jade B histofluorescence of degenerating neurons at 12 h post-traumatic brain injury (TBI) in the injured hippocampus. The micrographs are representative images of coronal sections (−3.6 mm bregma) of the injured hippocampus of TBI+SAline (SA; A) and TBI+SB-3CT (SB; B). (B, C, E, and F) Magnified images of the lined rectangles in A and B. In the injured hippocampus, degenerating neurons were found in CA2-3 (C, F) and hilus (B,E). A significant increase in the number of degenerating neurons was observed in both CA2-3 and hilus in TBI+SB group (E and F), compared with TBI+SA (B and C). (Scale bars=25μm (A and D) and 100 μm (B, C, E, and F). SO, stratum oriens; SP, stratum pyramidal; SR, stratum radiatum).
FIG. 4.

Quantitative analysis on Fluoro-Jade B positive neurons in the injured hippocampus of traumatic brain injury (TBI)+saline (SA) and TBI+SB-3CT (SB) groups at 12h post-TBI. *p<0.01 for the comparison between TBI+SB and TBI+SA groups.
Histological outcome: Cleaved caspase-3 expression
The pyramidal cells in CA2-3 and hilus regions of the injured hippocampus were co-stained with DAPI, NeuN, and cleaved caspase 3 (Fig. 5 and Fig. 6), respectively. It was shown that TBI induced the cleaved caspase-3 expression as its positive staining was localized in both TBI+SA and TBI+SB groups in both CA2-3 (Fig. 5E, H, F, and I) and hilus (Fig. 6E, H, F, and I), compared with the sham group (Fig. 5B and C; Fig. 6B and C), respectively. However, it seems that such an up-expression in cleaved caspase-3 could be moderated with the treatment of SB-3CT as relatively lower number of such cells could be located. The above staining was further quantified (Fig. 7). It was shown that at 72 h the NeuN staining of visible cells in the injured CA2-3 and hilus in TBI+SA rats was significantly reduced to 70.6±5.8% (p<0.01) and 79.8±7.1% (p<0.01) relative to those of the sham group. However, upon SB-3CT treatment on the rats after TBI (TBI+SB group), the NeuN stained cells were increased to 85.5±7.2% (p<0.01) and 88.6±7.8% (p<0.01), respectively, of those of the sham group. A significant difference in the relative amounts of NeuN stained cells was observed between the TBI+SA and TBI+SB groups in both the CA2-3 (p<0.01) and hilus (p<0.01) regions. Conversely, cleaved caspase-3 stained percentage in the injured CA2-3 and hilus of TBI+SA group is 698.6% (p<0.01) and 456.6% (p<0.01), respectively, of those of the sham group. However, upon SB-3CT treatment on the rats after TBI (TBI+SB group), the cleaved caspase−3 positive cells were decreased to 439.6% (p<0.01) and 238.2% (p<0.01), respectively, of those of the sham group. A significant difference in the relative amounts of cleaved caspase−3 positive stained cells was observed between the TBI+SA and TBI+SB groups in both the CA2-3 (p<0.01) and hilus (p<0.01) regions, respectively. However, even though the cleaved caspase-3 was down-regulated with the administration of SB-3CT, its expression was still far beyond the normal range. On the other hand, cleaved-caspase-3 labeling also could be located randomly and sporadically in other cells in addition to neurons as shown in Figure 5E, H, F, and I, and Figure 6E, H, F, and I.
FIG. 5.

Representative morphology stained with NeuN, cleaved-caspase-3, and 4′,6-diamidino-2-phenylindole (DAPI) in the CA2-3 hippocampal region from the sham (A, B and C), traumatic brain injury (TBI)+saline (D, E and F), and TBI+SB-3CT (G, H, and I) groups at 72 h after TBI. Merge1 showed NeuN colocalized with cleaved caspase-3; Merge2 showed DAPI colocalized with NeuN and cleaved caspase-3. The inset in each sub-figure is a representative localized image of high magnification. Scale bars=25 μm for A-I; 100 μm for insets.
FIG. 6.

Representative morphology stained with NeuN, cleaved-caspase-3, and 4′,6-diamidino-2-phenylindole in the hilus hippocampal region from the sham (A, B and C), traumatic brain injury (TBI)+saline (SA; D, E, and F), and TBI+SB-3CT (SB; G, H, and I) groups at 72 h after TBI. Merge1 showed NeuN colocalized with cleaved caspase-3; Merge2 showed DAPI colocalized with NeuN and cleaved caspase-3. The inset in each sub-figure is a representative localized image of high magnification. Scale bars=25 μm for A-I; 100 μm for insets.
FIG. 7.

Quantitative analysis on the NeuN-positive cells and cleaved caspase−3 positive cells in both the CA2-3 and hilus regions of three groups. (*p<0.01, traumatic brain injury (TBI)+saline (SA) group vs. sham group in the CA2-3 and hilus regions, respectively;≠p<0.01, TBI+SB-3CT (SB) group versus sham group in the CA2-3 and hilus regions, respectively; #p<0.01, TBI+SB group versus TBI+SA group in the CA2-3 and hilus regions, respectively).
Histological outcome: Cresyl violet staining
Rats were euthanized and brain sections were stained with cresyl violet. As shown in Figure 8, cellular-level injuries could be identified after TBI as the density of positive cells was reduced in both the TBI+SA (Fig. 8C) and TBI+SB (Fig. 8D) groups, compared with that of the sham group (Fig. 8B). Consistent with the above findings, upon SB-3CT treatment, such reduction in viable neural density obviously was inhibited as more positive cells could be observed in Figure 8D than in Figure 8C. Moreover, the CA2-3 stratum pyramidale in TBI animals (Fig. 8C and 8D) consistently appeared narrower than in sham TBI animals (Fig. 8B). Pyramidal neurons in the injured ipsilateral CA2-3 region were quantified further using stereological techniques (Fig. 8E). Post hoc Dunnett test indicated that both TBI injured groups with (p<0.01, TBI+SB vs. sham) or without SB-3CT treatment (p<0.01, TBI+SA vs. sham) had significantly fewer surviving neurons, compared with that of the sham group (Fig. 8E). However, SB-3CT treatment can significantly attenuate such a reduction induced by TBI as a significant difference in the number of viable neuronal was observed between the TBI+SA and TBI+SB groups (p<0.01; Fig. 8E).
FIG. 8.

Cresyl violet staining of the injured hippocampus at 15 d after traumatic brain injury (TBI). (A) Cresyl violet staining of the injured hippocampus in TBI+SB-3CT (SB) group, and outlined area is CA2-3; (B-D) Cresyl violet staining of localized areas of the injured hippocampus in sham, TBI+SAline (SA) and TBI+SB groups at 15 days after TBI, respectively. A significant increase in the number of surviving neurons was observed in CA2-3 in TBI+SB group (D), compared with TBI+SA group (C). Scale bars=100 μm (A) and 10 μm (B, C, and D). Quantitative analysis on cresyl violet positive cells in the injured CA2-3 region of hippocampus was shown in E. Data represent mean±SEM. (*p<0.01, TBI+SA group versus sham group;≠p<0.01, TBI+SB group versus sham group; #p<0.01, TBI+SB group versus TBI+SA group.
Discussion
It was demonstrated in this study that SB-3T, a selective inhibitor of MMP-9, can be an effective therapeutic strategy for TBI. When administered at 30 min, 6 h, and 12 h after TBI in rats, SB-3CT could provide both acute and chronic neuroprotection from the deleterious effects from TBI. The improvement in both motor and cognitive outcomes correlates well with significant hippocampal tissue preservation. In behavioral experiments, SB-3CT improved sensorimotor function (beam-balance/beam-walk tests) and spatial learning/memory retention (MWM) as assessed on Days 1–5 and 11–15 post injury, respectively. In histological experiments, SB-3CT attenuated the acute neuronal degeneration of 41.4% and 40.1% induced by TBI in CA2-3 and hilus, respectively, and decreased chronic neuronal loss of 45.3% in CA2-3 induced by TBI. At 72 h after TBI, SB-3CT not only decreased the neuron loss but also attenuated the up-expression of an apoptotic effector, cleaved caspase-3 by 34.7% and 45.8% in CA2-3 and hilus, respectively. The above results correlate well with other studies that shows that SB-3CT can protect against a variety of pathological conditions, such as brain ischemia.18,19 It is well understood that secondary injury in TBI is caused by autodestructive insults such as ischemia, a phenomenon commonly seen with an injured hippocampus.
Broad-spectrum pharmacological MMPIs significantly reduce brain damage after insults in animal models.32,33 For example, MMPIs can block the disruption of the blood–brain barrier and also were also proposed to treat neuroinflammation.34 However, previous human clinical trials with MMPIs, representing hydroxamate derivatives, failed because of adverse effects attributed, at least in part, to their lack of specificity.21,35 Further, it was shown by Nelson and colleagues that broad-spectrum MMPIs, tested in patients with ischemia, caused fibrosis of joints with arthritic-like symptoms.16 On the other hand, MMPs have been implicated to play beneficial roles in recovery following stroke.18 Shortly after secondary injury such as ischemia, a cascade of events is initiated in attempt to repair the damage, a process similar to that found in wound healing.36,37 Blood vessels also are dependent on the plasminogen-activator system and on MMPs for their regeneration.38 A suitable balanced level of MMP-9 activity is important for vascular remodeling after spinal cord injury.39 Delayed or extended inhibition of MMPs, especially through the use of broad-spectrum inhibitors, might prove deleterious, as confirmed by Zhao and colleagues.40 Accounting for these concerns, SB-3CT was chosen in this study as it is a selective inhibitor. Repeated intraperitoneal administration of SB-3CT was carried out at 30 min, 6 h, and 12 h after TBI as an intervention regime which included both acute and delayed inhibition. Such design might offer the following advantages: 1) Selective inhibition of MMP-9/MMP-2 gelatinases to minimize the undesired adverse effects usually caused by broad-spectrum MMPIs as mentioned above and 2) A realistic time window in which injured TBI patients could be escorted to a hospital setting to begin treatment.
The postulated therapeutic effects of MMPIs are linked to various pathological mechanisms including suppression of the activity of MMPs induced by brain injury,41,42 the decreasing of tenascin-C production,43 which attenuates anti-Thy1.1 nephritis,44 and the blocking of tumor necrosis factors α–converting enzyme and Fas Ligand–converting enzyme,45,46 preventing the delayed-type hypersensitivity lesions in the central nervous system47 and blocking edema in intracerebral hemorrhage in the rat.15 Recently, it was shown by Cui and colleagues that treatment with SB-3CT in the embolus-induced ischemia model had an unexpected outcome, compared with the control group. Expression of inactive pro-MMP-9 increased significantly during ischemia—a small portion of which was processed to active MMP-9, which commonly manifests as pathology associated with ischemia. Down regulation of pro-MMP-9 expression was observed when ischemic animals were treated with SB-3CT. Collectively, it is believed that most probably, the neuroprotective effect of SB-3CT works through not only binding MMP-9 to block pathological cascade of secondary brain injury but also through inhibiting the expression of pro-MMP-9.18 This also was indicated by an investigation carried out by Ranasinghe and colleagues.48 In their study, significant inhibition of brain pro-MMP-9 activity after hypoxic ischemia was observed by SB-3CT. Similar phenomena was observed in TBI models that increase in the pro-MMP-9 level was also observed in the lesioned cortex within 24 h after TBI in addition to the up-regulation of activated MMP-9.49 Moreover, the pro-MMP-9 levels in the lesioned cortex remained elevated for even 10 d after TBI. Early investigation also revealed that pro-MMP-9 levels are elevated as early as 3 h after trauma.9 It was further demonstrated that SB-3CT treatment could attenuate both the levels of pro-MMP-9 and active MMP-9.49
Our previous study demonstrated that moderate TBI induced significant increases in both MMP-9 mRNA and protein levels in the ipsilateral hemisphere (cortex and hippocampus) from 6 h to 1 week after TBI.13 We also found that the peak of early cell death and expression of apoptosis related protein caspase-3 after TBI is at 24 h and 72 h post-TBI.50 Both studies confirmed that expression of MMP-9 and caspse-3 is synchronous. The spatio-temporal pattern of MMP-9 and caspase-3 up-regulation strongly correlated with the variation in delayed neuronal death in the same model of TBI used in the present study.50,51 Hence, it can be postulated that SB-3CT, a selective inhibitor of MMP-9, may provide protection against secondary insults and improve behavior deficit by reducing cellular loss associated with a reduction in caspase-3. With regard to the intervention regime, its proposed target to the delayed neuronal death might mainly benefit from its dosages at 6 h and 12 h after the TBI designed in this work. Interestingly, this time window is of a clinical therapeutic interest.
On the other hand, acute SB-3CT administration (carried out at 30 min after TBI in this study) does not directly match the secondary insult time course and functional recovery period. However, it is interesting to find that significant loss of behavioral deficit caused by acute neuro-degeneration and neuronal loss also was lessened by SB-3CT post-treatment. Similar administration was shown to be effective in inhibiting MMP activity in adult rat models of spinal cord injury52 and ischemia17 without any confounding toxic effects. Moreover, it was also demonstrated that apoptotic cell death within the hippocampus was significantly reduced, and the presence of immature neurons was significantly increased if acute post-intervention was carried out after TBI.53 According to the early neuropathological findings after TBI,54 and the beneficial effect of early post-traumatic mild hypothermia on hippocampal cell death after TBI,50 it is possible that early or acute intervention may exert positive effects on the protection of neurons or behavioral recovery.
Future studies are still required to elucidate the underlying mechanism of how SB-3CT can result in decreased cellular loss in the injured ipsilateral hippocampus and the optimal dosage and intervention regime, such as the detailed cellular and molecular signaling pathways. Moreover, the elucidation of both individual acute and delayed intervention effects of SB-3CT after TBI is important for clinical reference. Thus, these investigations are ongoing in our group.
Acknowledgment
This work was supported by grants from 973 project (No. 2012CB518100, No. 2010CB944804), National Key Basic Research Project (No. 81271381), National Science and Nature Grant (No. 81000829), and the Fundamental Research Funds for the Central Universities (No. 11QB1402200).
Author Disclosure Statement
No competing financial interests exist.
References
- 1.Thurman D.J., Alverson C., Dunn K.A., Guerrero J., and Sniezek J.E. (1999). Traumatic brain injury in the United States: a public health perspective. J. Head Trauma Rehabil. 14, 602–615 [DOI] [PubMed] [Google Scholar]
- 2.Hayes R. L., Jenkins L.W., and Lyeth B.G. (1992). Neurotransmittermediated mechanisms of traumatic brain injury: acetylcholine and excitatory amino acids. J. Neurotrauma 9, 173–187 [PubMed] [Google Scholar]
- 3.Faden A.I. (1996). Pharmacologic treatment of acute traumatic brain injury. J. Am. Med. Assoc. 276, 569–570 [PubMed] [Google Scholar]
- 4.McIntosh T. K., Saatman K.E., Raghupathi R., Graham D.I., Smith D.H., Lee V.M., and Trojanowski J.Q. (1998). The Dorothy Russell Memorial Lecture. The molecular and cellular sequelae of experimental traumatic brain injury: pathogenetic mechanisms. Neuropathol. Appl. Neurobiol. 24, 251–267 [DOI] [PubMed] [Google Scholar]
- 5.Narayan R.K., Michel M.E., Ansell B., Baethmann A., Biegon A., and Bracken M.B. (2002) Clinical trials in head injury. J. Neurotrauma 19,503–557, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Xiong Y., Zhang Y., Mahmood A., Meng Y., Zhang. Z.G., Morris D.C., and Chopp M. (2012) Neuroprotective and neurorestorative effects of thymosin β4 treatment initiated 6 hours after traumatic brain injury in rats. J. Neurosurg. 116, 1081–1092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rosenberg G.A. (2002). Matrix metalloproteinases in neuroinflammation. Glia 39, 279–291 [DOI] [PubMed] [Google Scholar]
- 8.Wang J., and Tsirka S. (2005). Contribution of extracellular proteolysis and microglia to intracerebral hemorrhage. Neurocrit. Care 3, 77–85 [DOI] [PubMed] [Google Scholar]
- 9.Wang X., Jung J.C., Asahi M., Chwang. W., Russo L., Moskowitz. M.A., Dixon C.E., Fini E., and Lo E.H. (2000) Effects of matrix metalloproteinase-9 gene knock-out on morphological and motor outcomes after traumatic brain injury. J. Neurosci. 20, 7037–7042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Noble L.J., Donovan F., Igarashi T., Goussev S., and Werb Z. (2002). Matrix metalloproteinases limit functional recovery after spinal cord injury by modulation of early vascular events. J. Neurosci. 22, 5726–5735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Higashida T., Kreipke C.W., Rafols J.A., Peng C., Schafer S., Schafer P., Ding JY, and Dornbos D., 3rd, Li X, Guthikonda M., Rossi N.F., Ding Y. (2002).The role of hypoxia-inducible factor-1α, aquaporin-4, and matrix metalloproteinase-9 in blood-brain barrier disruption and brain edema after traumatic brain injury. J Neurosurg. 114, 92–101 [DOI] [PubMed] [Google Scholar]
- 12.Ralay R.H., Zunich S.M., Choi N., Hodge J.N., and Wainwright M.S. (2011). Mild stretch-induced injury increases susceptibility to interleukin-1β-induced release of matrix metalloproteinase-9 from astrocytes. J. Neurotrauma 28,1757–1766 [DOI] [PubMed] [Google Scholar]
- 13.Jia F., Mao Q., Liang Y.M., and Jiang J.Y. (2010). Matrix metalloproteinase-9 expression and protein levels after fluid percussion injury in rats: the effect of injury severity and brain temperature. J. Neurotrauma 27,1059–1068 [DOI] [PubMed] [Google Scholar]
- 14.Grossetete M., Phelps J., Arko L., Yonas H., and Rosenberg G.A.(2009). Elevation of matrix metalloproteinases 3 and 9 in cerebrospinal fluid and blood in patients with severe traumatic brain injury. Neurosurgery 65,702–708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rosenberg G. and Navratil M. (1997). Metalloproteinase inhibition blocks edema in intracerebral hemorrhage in the rat. Neurology 48, 921–926 [DOI] [PubMed] [Google Scholar]
- 16.Nelson A.R., Fingleton B., Rothenberg M.L., and Matrisian L.M. (2000). Matrix metalloproteinases: biologic activity and clinical implications. J. Clin. Oncol. 18, 1135–1149 [DOI] [PubMed] [Google Scholar]
- 17.Ranasinghe H.S., Scheepens A., Sirimanne E., Mitchell M.D., Williams C.E., and Fraser M. (2012) Inhibition of MMP-9 activity following hypoxic ischemia in the developing brain using a highly specific inhibitor. Dev. Neurosci. 34, 417–427 [DOI] [PubMed] [Google Scholar]
- 18.Cui J., Chen. S, Zhang C., Meng F., Wu W., Hu R., Hadass O., Lehmidi T., Blair G.J., Lee M., Chang M., Mobashery S., Sun G.Y., and Gu Z. (2012) Inhibition of MMP-9 by a selective gelatinase inhibitor protects neurovasculature from embolic focal cerebral ischemia. Mol. Neurodegener. 15, 1186–1191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu J., Jin X., Liu K.J., and Liu W. (2012) Matrix metalloproteinase-2-mediated occludin degradation and caveolin-1-mediated claudin-5 redistribution contribute to blood-brain barrier damage in early ischemic stroke stage. J. Neurosci. 32, 3044–3057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dixon C.E., Lyeth B.G., Povlishock J.T., Findling R.L, Hamm R.J., Marmarou A., Young H.F., and Hayes R.L. (1987). A fluid percussion model of experimental brain injury in the rat. J. Neurosurg. 67, 110–119 [DOI] [PubMed] [Google Scholar]
- 21.Gu Z., Cui J., Brown S., Fridman R., Mobashery S., Strongin A., and Lipton S. (2005) A highly specific inhibitor of matrix metalloproteinase-9 rescues laminin from proteolysis and neurons from apoptosis in transient focal cerebral ischemia. J Neurosci. 25, 6401–6408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Singleton R.H., Yan H.Q., Fellows-Mayle W., and Dixon C.E. (2010) Resveratrol attenuates behavioral impairments and reduces cortical and hippocampal loss in a rat controlled cortical impact model of traumatic brain injury. J Neurotrauma 27, 1091–1099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Feeney D.M., Gonzalez A., and Law W.A. (1982). Amphetamine, haloperidol, and experience interact to affect rate of recovery after motor cortex injury. Science 217, 855–857 [DOI] [PubMed] [Google Scholar]
- 24.Morris R. (1984). Developments of a water-maze procedure for studying spatial learning in the rat. J. Neurosci. Meth. 11, 47–60 [DOI] [PubMed] [Google Scholar]
- 25.Hamm R.J., Dixon C.E., Gbadebo D.M., Singha A.K., Jenkins L.W., Lyeth B.G., and Hayes R.L. (1992). Cognitive deficits following traumatic brain injury produced by controlled cortical impact. J. Neurotrauma 9, 11–20 [DOI] [PubMed] [Google Scholar]
- 26.Kline A.E., Massucci J.L., Marion D.W., and Dixon C.E. (2002). Attenuation of working memory and spatial acquisition deficits after a delayed and chronic bromocriptine treatment regimen in rats subjected to traumatic brain injury by controlled cortical impact. J. Neurotrauma 19, 415–425 [DOI] [PubMed] [Google Scholar]
- 27.Kline A.E., McAloon R.L., Henderson K.A., Bansal U.K., Ganti B.M., Ahmed R.H., Gibbs R.B., and Sozda C.N. (2010). Evaluation of a combined therapeutic regimen of 8-OH-DPAT and environmental enrichment after experimental traumatic brain injury. J. Neurotrauma 27, 2021–2032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Scheff S.W., Baldwin S.A., Brown R.W., and Kraemer P.J. (1997). Morris water maze deficits in rats following traumatic brain injury: lateral controlled cortical impact. J. Neurotrauma 14, 615–627 [DOI] [PubMed] [Google Scholar]
- 29.Eadie B.D., Redila V.A., and Christie B.R.(2005) Voluntary exercise alters the cytoarchitecture of the adult dentate gyrus by increasing cellular proliferation, dendritic complexity, and spine density. J. Comp. Neurol. 23, 39–47 [DOI] [PubMed] [Google Scholar]
- 30.Kang S.W., Choi S.K., Park E., Chae S.J., Choi S., Jin J.H., Lee G.J., and Park H.K.(2011) Neuroprotective effects of magnesium-sulfate on ischemic injury mediated by modulating the release of glutamate and reduced of hyperreperfusion. Brain Res. 31, 121–128 [DOI] [PubMed] [Google Scholar]
- 31.Feng J.F., Van K.C., Gurkoff G.G., Kopriva C., Olszewski R.T., Song M., Sun S., Xu. M., Neale J.H., Yuen P.W., Lowe D.A., Zhou J., and Lyeth B.G. (2011) Post-injury administration of NAAG peptidase inhibitor prodrug, PGI-02776, in experimental TBI. Brain Res. 1395, 62–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Romanic A.M., White R.F., Arleth A.J., Ohlstein E.H., and Barone F.C. (1998) Matrix metalloproteinase expression increases after cerebral focal ischemia in rats: inhibition of matrix metalloproteinase-9 reduces infarct size. Stroke 29, 1020–1030 [DOI] [PubMed] [Google Scholar]
- 33.Asahi M., Asahi K., Jung J.C., del G.J., Fini M.E., and Lo E.H. (2000) Role for matrix metalloproteinase 9 after focal cerebral ischemia: effects of gene knockout and enzyme inhibition with BB-94. J. Cereb. Blood Flow Metab. 20, 1681–1689 [DOI] [PubMed] [Google Scholar]
- 34.Yong V.W., Power C., Forsyth P., and Edwards D.R. (2001) Metalloproteinases in biology and pathology of the nervous system. Nat. Rev. Neurosci. 2, 502–511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Coussens L.M., Fingleton B., and Matrisian L.M. (2002) Matrix metalloproteinase inhibitors and cancer: trials and tribulations. Science 295, 2387–2392 [DOI] [PubMed] [Google Scholar]
- 36.Yong V.W. (2005) Metalloproteinases: mediators of pathology and regeneration in the CNS. Nat. Rev. Neurosci. 6, 931–944 [DOI] [PubMed] [Google Scholar]
- 37.Rosenberg G.A. (2009) Matrix metalloproteinases and their multiple roles in neurodegenerative diseases. Lancet Neurol. 8, 205–216 [DOI] [PubMed] [Google Scholar]
- 38.Vu T.H., Shipley J.M., Bergers G., Berger J.E., Helms J.A., Hanahan D., Shapiro S.D., Senior R.M., and Werb Z. (1998)MMP-9/gelatinase B is a key regulator of growth plate angiogenesis and apoptosis of hypertrophic chondrocytes. Cell 93, 411–422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nagy V., Bozdagi O., Matynia A., Balcerzyk M., Okulski P., Dzwonek J., Costa R.M., Silva A.J., Kaczmarek L., and Huntley G.W. (2006) Matrix metalloproteinase-9 is required for hippocampal late-phase long-term potentiation and memory. J. Neurosci. 26, 1923–1934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhao B.Q., Wang S., Kim H.Y., Storrie H., Rosen B.R., Mooney D.J., Wang X., and Lo E.H. (2006) Role of matrix metalloproteinases in delayed cortical responses after stroke. Nat. Med. 12, 441–445 [DOI] [PubMed] [Google Scholar]
- 41.Demestre M., Wells G.M., Miller K.M., Smith K.J., Hughes R.A., Gearing A.J., and Gregson N.A.(2004) Characterisation of matrix metalloproteinases and the effects of a broad-spectrum inhibitor (BB-1101) in peripheral nerve regeneration. Neuroscience 124, 767–779 [DOI] [PubMed] [Google Scholar]
- 42.Sood R.R., Taheri S., Candelario J.E., Estrada E.Y., and Rosenberg G.A.(2008) Early beneficial effect of matrix metalloproteinase inhibition on blood-brain barrier permeability as measured by magnetic resonance imaging countered by impaired long-term recovery after stroke in rat brain. J. Cereb. Blood Flow Metab. 28, 431–438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vyavahare N., Jones P.L., Tallapragada S., and Levy R.J. (2000) Inhibition of matrix metalloproteinase activity attenuates tenascin-C production and calcification of implanted purified elastin in rats. Am. J. Pathol. 157, 885–893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Steinmann N.K., Ziswiler R., Küng M., and Marti H.P. (1998) Inhibition of matrix metalloproteinases attenuates anti-Thy1.1 nephritis. J. Am. Soc. Nephrol. 9, 397–407 [DOI] [PubMed] [Google Scholar]
- 45.Liedtke W., Cannella B., Mazzaccaro R.J., Clements J.M., Miller K.M., Wu K.W., Gearing A.J., and Raine C.S. (1998) Effective treatment of models of multiple sclerosis by matrix metalloproteinase inhibitors. Ann. Neurol. 44, 35–46 [DOI] [PubMed] [Google Scholar]
- 46.Redford E.J., Smith K.J., Gregson N.A., Davies M., Hughes P., Gearing A.J., Miller K., and Hughes R.A. (1997) A combined inhibitor of matrix metalloproteinase activity and tumour necrosis factor-alpha processing attenuates experimental autoimmune neuritis. Brain 120, 1895–1905 [DOI] [PubMed] [Google Scholar]
- 47.Matyszak M.K. and Perry V.H. (1996) Delayed-type hypersensitivity lesions in the central nervous system are prevented by inhibitors of matrix metalloproteinases. J. Neuroimmunol. 69, 141–149 [DOI] [PubMed] [Google Scholar]
- 48.Ranasinghe H.S., Scheepens A., Sirimanne E., Mitchell M.D., Williams C.E., and Fraser M. (2012) Inhibition of MMP-9 activity following hypoxic ischemia in the developing brain using a highly specific inhibitor. Dev. Neurosci. 34, 417–427 [DOI] [PubMed] [Google Scholar]
- 49.Hadass O., Tomlinson BN., Gooyit M., Chen S., Purdy J.J., Walker J.M., Zhang C., Giritharan A.B, Purnell W., Robinson C.R., Shin D., Schroeder V.A., Suckow M.A., Simonyi A.Y., Sun G., Mobashery S., Cui J., Chang M., and Gu Z. (2013) Selective inhibition of matrix metalloproteinase-9 attenuates secondary damage resulting from severe traumatic brain injury. PLoS One. 8, e76904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jia F., Mao Q., Liang Y.M., and Jiang J.Y. (2009) Effect of post-traumatic mild hypothermia on hippocampal cell death after traumatic brain injury in rats. J. Neurotrauma. 26, 243–252 [DOI] [PubMed] [Google Scholar]
- 51.Jia F., Mao Q., Liang Y.M., and Jiang J.Y. (2014) The effect of hypothermia on the expression of TIMP-3 after traumatic brain injury in rats. J. Neurotrauma 31, 387–394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yu F., Kamada H., Niizuma K., Endo H., and Chan P.H. (2008) Induction of MMP-9 expression and endothelial injury by oxidative stress after spinal cord injury. J. Neurotrauma 25, 184–195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rau T.F., Kothiwal A.S., Rova A.R., Brooks D.M., and Poulsen D.J. (2012) Treatment with low-dose methamphetamine improves behavioral and cognitive function after severe traumatic brain injury. J. Trauma Acute Care Surg. 73, 165–172 [DOI] [PubMed] [Google Scholar]
- 54.Clark R.S., Kochanek P.M., Dixon C.E., Chen M., Marion D.W., Heineman S., DeKosky S.T., and Graham S.H. (1997) Early neuropathologic effects of mild or moderate hypoxemia after controlled cortical impact injury in rats. J. Neurotrauma 14, 179–189 [DOI] [PubMed] [Google Scholar]