

Abstract
We describe the characterization of a gene for mild nonsyndromic autosomal recessive intellectual disability (ID) in two unrelated families, one from Austria, the other from Pakistan. Genome-wide single nucleotide polymorphism microarray analysis enabled us to define a region of homozygosity by descent on chromosome 17q25. Whole-exome sequencing and analysis of this region in an affected individual from the Austrian family identified a 5 bp frameshifting deletion in the METTL23 gene. By means of Sanger sequencing of METTL23, a nonsense mutation was detected in a consanguineous ID family from Pakistan for which homozygosity-by-descent mapping had identified a region on 17q25. Both changes lead to truncation of the putative METTL23 protein, which disrupts the predicted catalytic domain and alters the cellular localization. 3D-modelling of the protein indicates that METTL23 is strongly predicted to function as an S-adenosyl-methionine (SAM)-dependent methyltransferase. Expression analysis of METTL23 indicated a strong association with heat shock proteins, which suggests that these may act as a putative substrate for methylation by METTL23. A number of methyltransferases have been described recently in association with ID. Disruption of METTL23 presented here supports the importance of methylation processes for intact neuronal function and brain development.
INTRODUCTION
About 1% of children worldwide are affected by intellectual disability (ID) (1), which can have a devastating impact on many aspects of the lives of the affected individuals, their families and communities, and is a major challenge at the clinical level. The clinical presentation and etiology of ID is complex and has a high degree of heterogeneity, which leads to a poor rate of molecular diagnosis resulting in below satisfactory clinical management. Although recent advances in sequencing technology have accelerated the rate of gene discovery for ID, even where family sizes are small, the majority of ID genes remain undetected. ID can be divided into two groups: nonsyndromic ID is characterized as the only clinical feature in patients, whereas syndromic ID occurs in combination with one or more additional clinical features (2). A recent review suggested that ∼40 genes for nonsyndromic autosomal recessive ID (NS-ARID) have been identified to date, but estimates there may be in excess of 2500 autosomal ID genes in total, with the majority being recessive (3). Although the physiological roles of ID genes are heterogeneous, increasing numbers of methyltransferases are being implicated in ID, stressing the essential role that methylation plays in the development and function of the central nervous system. For instance, ID has been linked to mutations in the methyltransferases NSUN2 (4,5) EHMT1 (6), FTSJ1, (7) and TRMT1 (8), which have diverse molecular substrates. Here, we report the identification of methyltransferase-like 23 gene (METTL23) truncating mutations in two nonsyndromic ID families, one Austrian and the other Pakistani family, and discuss potential substrates for this protein.
RESULTS
The Austrian ID family (LFKK1) has five children. In four affected siblings—two male and two female—a delay in developmental milestones was noted after the first year of life, and ID was diagnosed, whereas one sister was unaffected. All four affected siblings have a relatively high level of functioning, and a detailed clinical description is given in Supplementary Material.
The phenotypes and the degree of ID in the two affected girls of the Pakistani family (PK31) had a striking similarity to the Austrian family (see Supplementary Material for detailed clinical descriptions). Photographs for the two affected Pakistani girls are shown in Figure 1. Magnetic resonance imaging (MRI) was available for individual PK31-II:1 and II:22. For II:2, MRI showed increased volume of the subcallosal gray matter, and decreased delineation of the basal ganglia-regions strongly implicated in affect regulation, which may correlate with the aggression documented for this individual (Fig. 1).
Figure 1.

Magnetic resonance images: Two independent experts in neuroimaging (Christian Enzinger, Franz Fazekas) inspected the MRI scans blinded to clinical information and detected increased volume of the subcallosal area (SA), which was particularly evident in patient PK31 II:2 (A: red circles; in comparison with patient II:1 in D: green circles). On axial T2-weighted and FLAIR sequences, this led to decreased identifiability of the basal ganglia (B, C red arrows (II:2); compared with E and F (II:1), green arrows). The SA is commonly attributed to the limbic system, belongs to a phylogenetic old-brain area, is located in the vicinity of strategically important areas such as the hypothalamus and mamillar bodies and has been implicated in affect regulation. Interestingly, clinical information revealed that patient II:2 had demonstrated aggressive behavior and disturbed impulse control. Photos of PK31 II:2 (G and H) and II:1 (I and J) are shown.
For Austrian family LFKK1, a single large homozygosity-by-descent (HBD) region of about 4.8 Mb on chromosome 17q25.1–q25.3 was identified (Fig. 2); this area contained 107 genes. No other HBD stretch present in all affected family members and not in unaffected members could be identified. One possible candidate gene within the HBD region on 17q was TSEN54, which encodes one subunit from the tRNA splicing endonuclease complex.21 Mutation in this gene causes pontocerebellar hypoplasia type 2A (OMIM #277470) and type 4 (OMIM #225753)-neurodegenerative autosomal recessive disorders with chorea, dystonia or spasticity (9,10). However, Sanger sequencing of all exons of TSEN54 identified no mutation. After whole-exome sequencing for LFKK1 individual II:5, selection for homozygous exonic mutations in the 17q HBD region and exclusion of entries of the dbSNP131 database, only two genetic changes remained: A synonymous single nucleotide variant (SNV) (chr17:74392666G>A; NM_022066.3:c.2352C>T; p.Ala784Ala) in the UBE2O (ubiquitin-conjugating enzyme E2O) gene and a 5 bp frameshift deletion (chr17:74729256_74729260delAAGAT; NM_001206983.1: c.281_285delAAGAT; p. (Gln94Hisfs*6) resulting in a predicted 98 amino acids METTL23 protein) in exon 3 of the C17orf95 (METTL23, methyltransferase-like protein 23) gene (Fig. 2). As the synonymous SNV is a known SNP (rs145605062; dbSNP build 138; MAF = 0.58%), it is not predicted to affect splicing (analyzed through BDGP: www.fruitfly.org), and therefore seems unlikely to be disease related. In contrast, small indels have frequently been associated with severe, sporadic nonsyndromic ID (11), and therefore we focused on investigating METTL23 further. In addition to this, all known genes associated with NS-ARID were screened for compound heterozygosity in our exome data (12). The homozygous METTL23 variant was confirmed by Sanger sequencing in all four affected family members, whereas the unaffected sister and the parents were all heterozygous. For controls, we analyzed 200 unrelated, unaffected Austrian individuals for variants in METTL23 coding exons (exons 2–5) by denaturing high-performance liquid chromatography (DHPLC). In three samples, we identified the known SNV rs138247613 (c.G496A, p.D166N) in exon 5 with a comparable frequency of 2% for the heterozygous G/A genotype in the European population. However, we detected no other variants in this cohort.
Figure 2.

Ideogram of chromosome 17 is shown. Localization, indicated by red rectangles and base pair positions (referring to hg19) defining the homozygous regions of family FLKK1 and PK31 are given. The exon/intron structure of METTL23 is shown: blue blocks represent coding regions and gray blocks represent untranslated regions, introns are shown as dark lines. Black vertical lines indicate the position of the mutations for LFKK1 and PK31. Electropherograms show the homozygous familial mutations in METTL23 for LFKK1 and PK31 compared with WT sequences. Haplotypes and simplified pedigrees of LFKK1 and PK31 are shown. The disease haplotype is indicated by black bars. All alleles are recoded. Mutation status (mut) of all tested individuals is indicated by 1 for WT and 2 for mutated.
In Pakistani family PK31, a 5.1 Mb HBD region was observed on 17q25.1–q25.3, flanked by SNPs rs513643 and rs4313838 (chr17:72 599 418–77 698 582; hg19) (Fig. 2). Because of the overlap of HBD with the Austrian family and the similar phenotypes, we Sanger sequenced the METTL23 coding exons in family PK31. We identified a nonsense mutation in exon 4, which segregated with the disease (chr17:74729449C>T; NM_001080510.3: c.397C>T; p.(Gln133*) (Fig. 2). We screened 300 healthy Pakistani individuals by restriction fragment length polymorphism-PCR with the enzyme MseI, but did not find this allele. Neither this variant, nor the 5 bp deletion were present in the Exome Variant Server, NHLBI GO Exome Sequencing Project, Seattle, WA, USA (http://eversusgs.washington.edu/EVS/) (accessed Dec 2013) or in 1000 Genomes (http://browser.1000genomes.org/index.html).
Marker segregation for the two families was confirmed using microsatellites markers as well as the METTL23 variants (Fig. 2), and linkage analysis using SimWalk2 (version 2.91) (13) under a recessive model of inheritance gave a maximum LOD score of 2.66.
There are seven different METTL23 mRNA-transcripts reported (UCSC Genome Browser), six coding and one non-coding. Transcript variants 1, 2 and 3 encode the protein isoform 1 (190 amino acids) and the transcripts 4, 5 and 6 for the shorter protein isoform 2 (123 amino acid). The mRNAs have different length because of differing sizes of the non-coding first exon or because of alternative splicing of exon 2. In order to get a global overview of the expression of these mRNAs, we analyzed the 20 tissue samples from the Human Total RNA Panel (Clontech Takara, Madison, WI) by quantitative real-time PCR. We found that METTL23 is expressed at low levels but ubiquitously in all tissues analyzed, in comparison with the beta-2-microglobulin (B2M) and HPRT1-mRNA-amplification-levels. Both long- and short-isoform-encoding transcripts of METTL23 are expressed at very similar levels in all tissues (see Supplementary Material).
Quantitative RT-PCR using mRNA from lymphoblasts from PK31 family members showed similar levels of mRNA for the two main isoforms of METTL23 NM_001206983.1 (isoform 1) and NM_001206986.1 (isoform 2) in the affected siblings (II:1 and II:2) and a gender-matched unaffected individual, as well as in the tissues from the RNA panel (lymphocyte, brain and testis). Thus, nonsense-mediated mRNA decay does not appear to be involved in the mechanism of pathogenicity (see Supplementary Material).
To confirm that the nonsense mutation in METTL23 from patient PK31 II:1 definitively results in a truncated METTL23 protein (position 1–132 amino acids, lacking the catalytic domain) a polyclonal antiserum (raised in our lab, recognizing full-length METTL23 protein expressed in Escherichia coli, Fig. 3A) was used. Immunoprecipitation of METTL23 from lymphoblastoid cells and subsequent detection with a sequence-specific (C17orf95) antibody [cross-reacting with the more C-terminal portion (position 137–166 amino acids) of intact METTL23] revealed a strong immunoreactive signal of 21.5 kDa in controls but not in the patient (Fig. 3C). These observations confirm that the PK31 nonsense mutation does result in the loss of C-terminal METTL23 protein sequence.
Figure 3.
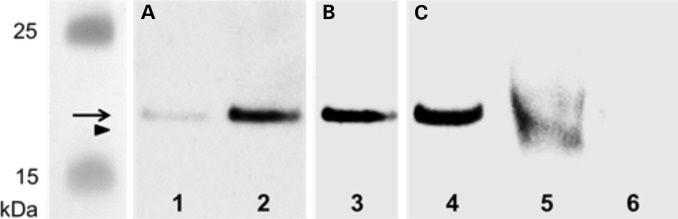
Western blot for METTL23 protein: Purified METTL23-His8-tag protein (expressed in E. coli) was loaded in lane 1 (50 ng) and 2–4 (100 ng). METTL23 proteins were immunoprecipitated from lymphoblastoid cells from healthy control (lane 5) and METTL23-mutant patient PK31 II:1 (lane 6) using polyclonal anti-METTL23. Immunoreactive bands were visualized using (A) rabbit polyclonal anti-METTL23-His8-tag antiserum, (B) rabbit polyclonal anti-His-tag (C-terminal) antibody and (C) sequence-specific anti-METTL23 (C17orf95) antibody as primary antibodies. Arrow (METTL23-His8-tag protein, 22.6 kDa); arrowhead (immunoprecipitated METTL23 protein, 21.5 kDa).
Based on sequence comparison, METTL23 appears to be a putative methyltransferase. Accordingly, we modeled the 3D structure of human METTL23. The final model has optimal confidence scores along the entire sequence length (190 residues, i.e. isoform 1) and is shown in Figure 4A. Based on this homology model, possible ligands and their binding sites in METTL23 were predicted using the server 3DLigandSite (14). In total, 22 similar structures were identified in the PDB, which all contain either S-adenosyl-methionine (SAM) or S-adenosyl-homocysteine (SAH) as a cofactor. Twenty-five residues of METTL23 were predicted to build up the SAM/SAH-binding site (colored green in Fig. 4A), ∼50% of which were completely conserved among this set of SAM/SAH-binding proteins. According to this structural bioinformatics analysis, it is very likely that METTL23 isoform 1 adopts a 3D fold typical for methyltransferases and contains a binding site for the methylation cofactor SAM. Thus, we conclude that METTL23 is indeed functioning as a methyltransferase. Both truncation variants, on the other hand, are predicted to lack a significant portion of the core fold (central β-sheet) of the protein (Fig. 4B). Therefore, it is very likely that these variants are not properly folded and thus do not show methyltransferase activity.
Figure 4.
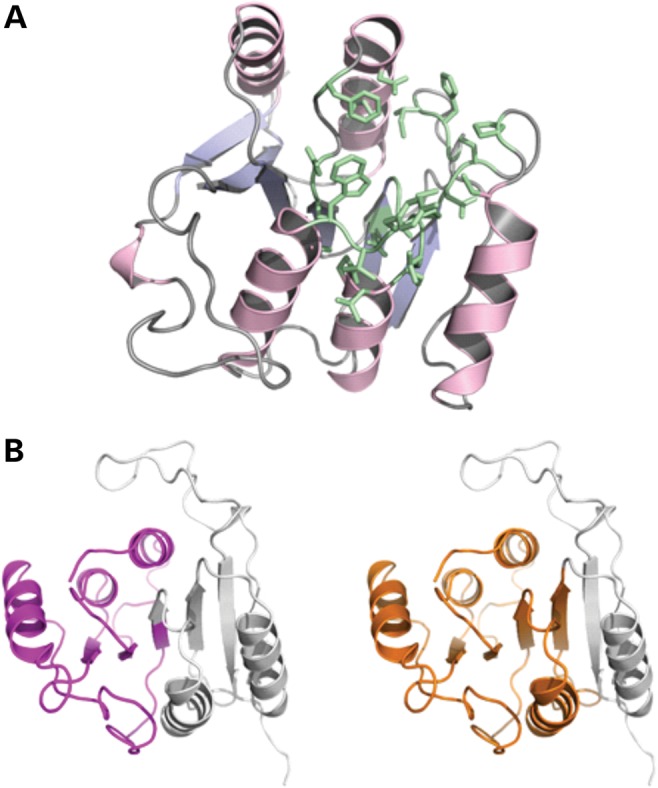
(A) Homology model of human METTL23 generated using the Phyre2 server (23). Amino acid residues predicted to build an SAM/SAH-binding site are shown in green. (B) Cartoon representations of the protein model with purple and orange colored portions representing the extent of the truncation variants (left: 1–98; right: 1–132). The parts missing in these variants are shown in gray.
To obtain more information on the catalytic properties of METTL23, we heterologously expressed the gene in E. coli BL21 host cells. The protein was produced in good yield but was insoluble and formed inclusion bodies (Fig. 5A). We then generated the protein in the presence of chaperones (e.g. GroEL) yielding soluble METTL23. Interestingly, affinity purification of his8-tagged METTL23 and size exclusion chromatography showed that the protein co-eluted with the chaperone (SDS–PAGE: fig. 5B). Attempts to release METTL23 from the chaperone, for example by addition of ATP, were unsuccessful indicating that METTL23 is tightly bound. This finding is in accordance with the suspected association of METTL23 with the endoplasmic reticulum (ER) membrane (as shown in Fig. 6). On the other hand, Cloutier and coworkers (15) recently reported that several human methyltransferases interact with molecular chaperones and regulate their activity by methylation of a conserved lysine residue.
Figure 5.

Expression and purification of METTL23 in E. coli in the absence and presence of chaperone coexpression. (A)The expression of METTL23 in the absence of chaperone coexpression in E. coli. METTL23 was found solely in inclusion bodies (left lane), whereas the supernatant was devoid of METTL23 (right lane). (B) The purification of METTL23 coexpressed with GroEL-GroES by Ni-NTA affinity and size exclusion chromatography. The middle lane was supernatant of METTL23 coexpressed with GroEL-GroES chaperones after cell lysis. The right lane is a fraction of METTL23 after Ni-NTA affinity and size exclusion chromatography.
Figure 6.

Subcellular localization of METTL23-GFP fusion proteins for WT isoform 1, WT isoform 2 and the corresponding 5 bp-deletion mutant proteins are shown in CHO cells after transient transfection. The WT of isoform 1 is predominantly located in the ER and to a lower extent in the nucleus. The corresponding mutant protein shows a similar distribution but additionally forms highly fluorescent protein aggregates. The WT protein of isoform 2 appears to be located in comparable concentrations in the nucleus and in the cytoplasm, whereas the corresponding mutant protein located in the cytoplasm forms numerous concentric aggregates. Green fluorescent signal could also be observed in the nucleus.
The 5 bp deletion in exon 3 is predicted to lead to a 98 amino acids-protein isoform 1 and 31 amino acids-protein isoform 2, whereas the nonsense mutation in exon 4 is predicted to lead to 132 and 65 amino acids-long gene products, for isoform 1 and 2, respectively. Wild-type-isoform 1 appears to be localized at or in the ER-membrane, whereas isoform 2 is mainly localized in the nucleoplasm in all analyzed cells (Fig. 6). These findings support the predicted transmembrane domain in isoform 1 but not isoform 2. However, this overexpression of the mutant protein leads to the formation of protein aggregates in the cytoplasm in all cells, for both isoform 1 and isoform 2 (Fig. 6).
DISCUSSION
The findings presented here add support for METTL23 as an important gene for ARID. Interestingly, a homozygous 4-bp deletion (c.169_172delCACT) in the METTL23 gene was very recently described in a consanguineous family of Arab origin (16). In contrast to our two families, Reiff et al. noted some dysmorphic features in their affected family members. Clinical findings common to all affected individuals included flat occiput, large eyes, depressed nasal bridge, short upturned nose, long philtrum, thin lips and incomplete syndactyly. The cognitive impairment was classified as ‘severe, with autistic symptoms’ in one individual and as ‘moderate’ in two other affected family members. Altogether, this suggests that truncating mutations can lead to nonsyndromic ARID as well as ARID with dysmorphic or syndromic features. The cognitive impairment also seems to be milder in families LFKK1 and PK31, as the affected individuals of both families have a relatively high degree of comprehension, including long sentences, and have at least very simple reading, writing and math abilities. There was no evidence of autistic traits. As the three mutations found thus far in METTL23 are different and present with different severity of ID, a genotype/phenotype correlation seems probable. Importantly, the mutation reported by Reiff et al. (16) is located in an exon that is only coding in isoform 1 (and is in a 5′ untranslated region in isoform 2), whereas the mutations in LFKK1 and PK31 are truncating for both isoforms. This perhaps represents something of a paradox, as the symptoms associated with the mutation that is exclusive to isoform 1 are severer.
Localization studies suggest that wild-type (WT) METTL23 isoform 1 is likely to function in the ER, whereas isoform 2 has a nuclear localization. Expression of the mutant (truncated) protein appeared to result in cytoplasmic protein aggregates. It is likely that these aggregates are a secondary effect related to the hydrophobicity and overexpression of the protein, rather than being the primary effect of the mutation. Protein aggregation and inclusion bodies are frequently associated with neurodegeneration; however, there is no evidence of a gain-of-function associated with the two METTL23 mutations identified as a pathogenic mechanism in these two families. It is most likely that the neurodevelopmental phenotype in our two families is a result of a deficit in the enzymatic function of the methyltransferase.
Methyltransferases catalyze the transfer of a methyl group to diverse substrates, including nucleic acids (DNA and RNA), proteins and lipids. They have been grouped into superfamilies according to their structural properties. Today, 208 proteins in the human proteome, representing ∼0.9% of human gene products, are already characterized as putative methyltransferases, and ∼30% of these have already been linked to diseases (17). By modifying their targets, they essentially influence multiple cellular regulatory mechanisms. Via epigenetic effects, methyltransferases are involved in tissue differentiation and proliferation, and have therefore been associated with a variety of diseases including cancer and neuropsychiatric disorders (18–20).
Many genes encoding demethylases and methyltransferases have now been implicated in syndromic or nonsyndromic forms of ID. These include histone demethylases, histone methyltransferases, tRNA and rRNA methyltransferases (4–8). Epigenetic events influenced by methylases and demethylases have been shown to have a substantial involvement in development and function of the central nervous system, and mutations in the underlying genes may result in cognitive impairment and intellectual disabilities. In the case of METTL23, it appears likely that abnormal methylation may also result in disrupted neuronal development via other (non-epigenetic) pathways. The METTL group of genes is likely to contain many more ID-related genes yet to be discovered.
A newly uncovered group of distantly related lysine methyltransferases preferentially interact with molecular chaperones to regulate their activity (15). Our data suggest tight binding of METTL23 to a chaperone protein, GroEL, which is highly homologous to human heat shock protein HSP60 (known to be mutated in hereditary spastic paraplegia; MIM 605280). Thus, studies of the interaction of METTL23 with various chaperones may eventually lead to the discovery of the cognate substrate(s) of the enzyme. Currently, we are investigating this putative function of METTL23 and developing heterologous expression systems that will yield soluble protein for further characterization of its enzymatic properties.
In summary, we present two different families with METTL23 mutations, one of them located in Europe, i.e. Austria, the other in Pakistan, which extends the geographical distribution and prevalence of METTL23 as a gene affecting cognitive function. In fact, our study suggests that the prevalence of mutations in this gene in individuals with ID may be relatively high. Our study adds to the known phenotypic consequences of mutations in METTL23. In contrast to the study by Reiff and coworkers (16), our mutations resulted in a nonsyndromic and mild form of ID. We also describe MRI results, which revealed some minor abnormalities in affected individuals. It remains to be determined whether METTL23 isoform 2, as predicted, has a physiological role. Since the mutation reported in Reiff and coworkers (16) only disrupts isoform 1, yet the resulting phenotype is more severe, we conclude that the function of isoform 2 may be restricted. Further functional characterization of the different METTL23 protein isoforms is recommended.
MATERIALS AND METHODS
Family ascertainment
We ascertained an Austrian ID family (LFKK1) with five children, through the Human Genetics Department of the Landes-Frauen und Kinderklinik, Linz. In four siblings—two male and two female—mild, nonsyndromic ID was diagnosed, whereas one sister was unaffected. Consanguinity was not known, however both parents originated from the same village from a mountainous region, which has been geographically isolated throughout previous centuries. We performed a detailed clinical genetic examination of the four affected and the non-affected siblings, and blood was drawn for genetic investigations. Photographs of the individuals were assessed for dysmorphic features by several experienced clinical geneticists and pediatricians, and medical records were studied. The family from Pakistan (PK31) in which the parents were first cousins was recruited through the Lahore Institute of Research and Development, as part of a study researching ID among consanguineous families. The family is located within Lahore, in Punjab Province, and presented with two girls affected with mild ID, and a third female sibling who is unaffected.
The study was approved by the respective ethics committees, and conducted according to the Declaration of Helsinki, and written informed consent were obtained from all included members from the Austrian and Pakistani families. Blood was drawn from family members and DNA extracted by standard procedures.
Single nucleotide polymorphism microarray analysis
For the Austrian family (LLKK1), DNAs from individuals II:1–II:5 were genotyped using Affymetrix 6.0 SNP microarrays, and data were analyzed using Chromosome Analysis Suite (ChAS) version 1.2. For the Pakistani family, PK31, both affected individuals (II:1, II:2) and unaffected sibling (II:3) were genotyped using Affymetrix 500K NspI microarrays (Affymetrix; Santa Clara, CA, USA). Homozygosity analysis was performed using dCHIP (http://biosun1.harvard.edu/complab/dchip; 21) and HomozygosityMapper (http://www.homozygositymapper.org; 22).
Whole-exome sequencing
Whole-exome sequencing of one ID affected family member (II:5) from family LFKK1 was performed by Macrogen Korea Inc. on the Illumina HiSeq1000 platform using Illumina TruSeq Exome Enrichment. Analysis was performed using the Macrogen exome sequencing standard analysis pipeline, and variants were filtered for homozygous exonic and splice site variants within the 17q HBD region, and excluding known SNPs in the dbSNP131 database. Sequence validation was performed by Sanger sequencing using standard procedures.
Protein 3D modeling
We used the Phyre2 server (23) for modeling the 3D structure of METTL23. The software chose Protein Data Bank (PDB) entries 4LG1, 4LEC and 3BZB as templates. These proteins exhibit 19–26% sequence identity to METTL23 and are annotated as human methyltransferase-like protein 21A (4LEC), and 21D (4LG1) as well as an uncharacterized protein CMQ451C from Cyanidioschyzonmerolae (3BZB). We then used the 3DLigandSite server (14) to predict potential ligands and their binding sites in METTL23.
EBV transformation and culture of lymphoblasts
Patient and control blood samples collected in acid-citrate-dextrose BD Vacutainer® blood tubes (Becton, Dickinson and Company) were diluted (1:1) with Roswell Park Memorial Institute medium, and white blood cells separated using ACCUSPIN tubes (Sigma), and transformed with Epstein-Barr virus.
RNA extraction, cDNA synthesis and quantitative RT-PCR
RNA was extracted from lymphoblast cells using the Trizol method. cDNA was synthesized through reverse transcription of 1 µg of RNA using Superscript III™ Reverse Transcriptase (Invitrogen, Carlsbad, CA, USA) and random hexamers according to manufacturer's guidelines.
mRNA from Leukocytes was extracted from peripheral blood samples using the Chemagic Magnetic Separation Module I (Perkin Elmer, Baesweiler, Germany). cDNA was synthesized through reverse transcription using M-MLV Reverse Transcriptase (Promega, Madison, WI, USA) and random hexamers according to manufacturer's guidelines. Primers were designed to amplify the coding DNA sequence of the METTL23 gene, using the primer express software (Applied Biosystems Inc., Foster city, CA, USA) and their details are given in Supplementary Material, Table S1. The Primer-Probe-Sets for B2M (Life Technologies, Carlsbad, CA, USA) and HPRT1 (Life Technologies) were used as an internal reference for all the runs. For METTL23 isoform-specific qRT-PCR, primers and a FAM-dyed probe were designed. In addition, cDNATotal RNA from a multitissue panel (Clontech) was reverse transcribed into cDNA duplicates and used for qRT-PCR. PCR was performed in quadruplicates on the Roche LightCycler 1.0 System (Roche Diagnostics, Basel, Switzerland) using the LightCycler FastStart DNA Master HybProbe Assay (Roche) T. The Ct for all reactions was calculated automatically by the LightCycler® Analysis software version 3.5. (Roche Diagnostics). Due to the variability among the internal controls compared with the Ct values for METTL23 isoform 1 and METTL23 isoform 2 for the 20 analyzed tissue samples, a relative normalization analysis was not performed.
Immunoprecipitation of WT and mutant METTL23
Lymphoblastoid cells were homogenized in RIPA buffer (50 mmol/l Tris–HCl, pH 7.5, 300 mmol/l NaCl, 5 mmol/l EDTA, 50 mmol/l NaF, 0.1%, v/v, Triton X-100, 0.02%, w/v, NaN3, pH 7.4) containing a protease inhibitor cocktail tablet (Sigma–Aldrich). Cell lysates were centrifuged at 10 000 rpm (4°C, 10 min) to pellet debris and were equilibrated against immunoprecipitation buffer containing (in mmol/l): 50 Tris–HCl (pH 8.0), 10 MgCl2 and 150 NaCl. Lysates containing equal protein concentrations (500 µg) were mixed with 10 µl rabbit polyclonal anti-METTL23 antiserum (raised in the own lab against full-length METTL23 protein (BMWF-2013-10-22T11:53:23) containing a His8-tag, expressed in E. coli, see Supplementary Material part) for 2 h at 4°C. Immune complexes were precipitated by mixing 20 µl of protein A/G plus Agarose (Santa Cruz Biotechnology, Heidelberg, Germany) overnight (4°C with shaking). Immune complexes were pelleted by centrifugation at 10 000 rpm (4°C, 1 min). After washing three times with RIPA buffer, immune complexes were resuspended in 40 µl of 4× NuPAGE LDS sample buffer and heated (70°C, 10 min). Western blot analyses were performed as described below.
Western blot analysis
After protein estimation using the Lowry method, indicated concentrations of purified METTL23-His8-tag protein (22.6 kDa) were added to 10 µl of 4× NuPAGE LDS sample buffer containing 2 µl sample reducing agent (Invitrogen, Austria) and heated (70°C, 10 min). Purified METTL23-His8-tag protein and immunoprecipitates were separated by electrophoresis on 12% Bis–Tris gel and transferred to nitrocellulose membranes. Membranes were blocked with 5% (w/v) non-fat milk in TBST (Tris-buffered saline containing Tween 20) (25°C, 2 h) and incubated with either (i) rabbit polyclonal anti-METTL23 antiserum (1:2000 in 5%, w/v, BSA), (ii) rabbit polyclonal anti-His-tag (C-terminal) antibody (1:1000 in 5%, w/v, BSA, Relia Tech, Germany) and (iii) rabbit sequence-specific anti-METTL23 (C17orf95) antibody (1:300 in 5%, w/v, BSA, Abgent, Germany, raised against a synthetic peptide; position 137–166 amino acids) (4°C, overnight). Immunoreactive bands were visualized with either Clean-Blot IP Detection Kit [horseradish peroxidase (HRP)] (1:100 000 in 5%, w/v, non-fat milk in TBST, Thermo scientific, Austria) or HRP-conjugated goat anti-rabbit IgG (1:100 000 in 5%, w/v, non-fat milk in TBST) (25°C, 2 h) followed by Super Signal West Pico Chemiluminescent substrate (Thermo Scientific, Austria) and developed by Bio-Rad ChemiDoc MP Imaging System.
Expression and purification of METTL23
Full-length METTL23 including a C-terminal hexa-histidine tag was synthesized (Life Technology™) and inserted into the NdeI/XhoI restriction site of pMA-T. One shot® Top10 E. coli harboring the plasmid were cultured in 100 ml LB medium containing 100 µg/ml ampicillin to propagate the plasmid. Extraction and purification of the plasmid was performed according to the protocol of NucleoSpin® Plasmid (Macherey-Nagel, Darmstadt, Germany). The isolated plasmid was digested with NdeI and XhoI to release METTL23. The insert was then cloned into pET21a (mettl23-pET21a) and used for transformation of competent E. coli BL21 (DE3). Expression of the gene was induced by addition of 0.1 mm IPTG at an OD600 ∼1 at 20°C for 14 h. Coexpression of GroEL-GroES chaperones was induced by addition of 1 mg/ml of l-arabinose.
Purification of METTL23 was achieved by Ni-NTA affinity (5-ml Ni-Sepharose™ High Performance HisTrap™ HP column) and size exclusion chromatography (Superdex 200 prep grade column). For the latter step, the protein solution was applied to the column after equilibration with 50 mm Tris–HCl buffer, pH 8.0, containing 100 mm NaCl. Protein fractions were pooled according to the absorption at 280 nn. Aliquots of each fraction were analyzed by SDS–PAGE (staining with Coomassie Brilliant Blue R-250) to monitor the progress of purification.
METTL23 wild type and mutant protein localization
To study the cellular localization of METTL23 WT isoform 1, WT isoform 2 and the 5 bp-deletion mutant proteins, we fused the GFP-peptide to the C-terminal end using the GFP Fusion TOPO TA Expression Kit (Invitrogen). For transfection, we used 6.5 µg of DNA from WT and mutant constructs and added OptiMEM I Reduced Serum Medium (Life Technologies) and FuGENE (R) HD Transfection Reagent (Promega) to CHO-K cells. The transfection was performed in duplicates, and 26 h later analyzed using inverted fluorescence microscopy (TE2000, Nikon Corporation).
SUPPLEMENTARY MATERIAL
FUNDING
This research was supported by a grant from the Canadian Institutes of Health Research (#MOP-102758) and the Austrian Science Fund (FWF) DK-MCD W1226-B18. Funding to pay the Open Access publication charges for this article was provided by the Austrian Science Fund (FWF, DK-MCD W1226).
Supplementary Material
ACKNOWLEDGEMENTS
We thank the family members for their willing participation and cooperation with this study. We also want to thank Univ. Prof. Franz Fazekas (Department of Neurology, Medical University of Graz, Graz, Austria) for inspection and evaluation of the MRI Scans and Assoc. Prof. Roland Malli (Institute of Molecular Biology and Biochemistry, Center of Molecular Medicine, Medical University of Graz, Graz, Austria) for inspection and interpretation of the GFP-fluorescence images.
Conflict of Interest statement. None declared.
REFERENCES
- 1.Maulik P.K., Mascarenhas M.N., Mathers C.D., Dua T., Saxena S. Prevalence of intellectual disability: a meta-analysis of population-based studies. Res. Dev. Disabil. 2011;32:419–436. doi: 10.1016/j.ridd.2010.12.018. [DOI] [PubMed] [Google Scholar]
- 2.Kaufman L., Ayub M., Vincent J.B. The genetic basis of non-syndromic intellectual disability: a review. J. Neurodev. Disord. 2010;2:182–209. doi: 10.1007/s11689-010-9055-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Musante L., Ropers H.H. Genetics of recessive cognitive disorders. Trends Genet. 2014;30:32–39. doi: 10.1016/j.tig.2013.09.008. [DOI] [PubMed] [Google Scholar]
- 4.Khan M.A., Rafiq M.A., Noor A., Hussain S., Flores J.V., Rupp V., Vincent A.K., Malli R., Ali G., Khan F.S., et al. Mutation in NSUN2, which encodes an RNA methlytransferase, causes autosomal-recessive intellectual disability. Am. J. Hum. Genet. 2012;90:856–863. doi: 10.1016/j.ajhg.2012.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Abbasi-Moheb L., Mertel S., Gonsior M., Nouri-Vahid L., Kahrizi K., Cirak S., Wieczorek D., Motazacker M.M., Esmaeeli-Nieh S., Cremer K., et al. Mutations in NSUN2 cause autosomal-recessive intellectual disability. Am. J. Hum. Genet. 2012;90:847–855. doi: 10.1016/j.ajhg.2012.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kleefstra T., Brunner H.G., Amiel J., Oudakker A.R., Nillesen W.M., Magee A., Genevieve D., Cormier-Daire V., van Esch H., Fryns J.P., et al. Loss-of-function mutations in euchromatin histone methyl transferase 1 (EHMT1) cause the 9q34 subtelomeric deletion syndrome. Am. J. Hum. Genet. 2006;79:370–377. doi: 10.1086/505693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Freude K., Hoffmann K., Jensen L.-R., Delatycki M.B., des Portes V., Moser B., Hamel B., van Bokhoven H., Moraine C., Fryns J.-P., et al. Mutations in the FTSJ1 gene coding for a novel S-adenosylmethionine-binding protein cause nonsyndromic X-linked mental retardation. Am. J. Hum. Genet. 2004;75:305–309. doi: 10.1086/422507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Najmabadi H., Hu H., Garshasbi M., Zemojtel T., Abedini S.S., Chen W., Hosseini M., Behjati F., Haas S., Jamali P., et al. Deep sequencing reveals 50 novel genes for recessive cognitive disorders. Nature. 2011;478:57–63. doi: 10.1038/nature10423. [DOI] [PubMed] [Google Scholar]
- 9.Budde B.S., Namavar Y., Barth P.G., Poll-The B.T., Nürnberg G., Becker C., van Ruissen F., Weterman M.A., Fluiter K., te Beek E.T., et al. tRNA splicing endonuclease mutations cause pontocerebellar hypoplasia. Nat. Genet. 2008;40:1113–1118. doi: 10.1038/ng.204. [DOI] [PubMed] [Google Scholar]
- 10.Barth P.G. Pontocerebellar hypoplasias. An overview of a group of inherited neurodegenerative disorders with fetal onset. Brain Dev. 1993;15:411–422. doi: 10.1016/0387-7604(93)90080-r. [DOI] [PubMed] [Google Scholar]
- 11.Rauch A., Wieczorek D., Graf E., Wieland T., Endele S., Schwarzmayr T., Albrecht B., Bartholdi D., Beygo J., DiDonato N., et al. Range of genetic mutations associated with severe non-syndromic sporadic intellectual disability: an exome sequencing study. Lancet. 2012;380:1674–1682. doi: 10.1016/S0140-6736(12)61480-9. [DOI] [PubMed] [Google Scholar]
- 12.Jamra R.A., Wohlfart S., Zweier M., Uebe1 S., Priebe L., Ekici A., Giesebrecht S., Abboud A., Ayman M., Khateeb A., Fakher M., et al. Homozygosity mapping in 64 Syrian consanguineous families with non-specific ID reveals 11 novel loci and high heterogeneity. Eur. J. Hum. Genet. 2011;19:1161–1166. doi: 10.1038/ejhg.2011.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sobel E., Lange K. Descent graphs in pedigree analysis: applications to haplotyping, location scores, and marker sharing statistics. Am. J. Hum.Genet. 1996;58:1323–1337. [PMC free article] [PubMed] [Google Scholar]
- 14.Wass M.N., Kelley L.A., Sternberg M.J. 3DLigandSite: predicting ligand-binding sites using similar structures. Nucleic Acids Res. 2010;38:469–473. doi: 10.1093/nar/gkq406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cloutier P., Lavallée-Adam M., Faubert D., Blanchette M., Coulombe B. A newly uncovered group of distantly related lysine methyltransferases preferentially interact with molecular chaperones to regulate their activity. PLoS Genet. 2013;9:e1003210. doi: 10.1371/journal.pgen.1003210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Reiff R.E., Ali B.R., Baron B., Yu T.W., Ben-Salem S., Coulter M.E., Schubert C.R., Hill R.S., Akawi N.A., Al-Younes B., et al. METTL23, a transcriptional partner of GABPA, is essential for human cognition. Hum. Mol. Genet. 2014 doi: 10.1093/hmg/ddu054. [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Petrossian T.C., Clarke S.G. Uncovering the human methyltransferasome. Mol. Cell. Proteom. 2011;10 doi: 10.1074/mcp.M110.000976. M110.000976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Poleshko A., Einarson M.B., Shalginskikh N., Zhang R., Adams P.D., Skalka A.M., Katz R.A. Identification of a functional network of human epigenetic silencing factors. J. Biol. Chem. 2010;285:422–433. doi: 10.1074/jbc.M109.064667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Albert H., Helin K. Histone methyltransferases in cancer. Semin. Cell Dev. Biol. 2010;21:209–220. doi: 10.1016/j.semcdb.2009.10.007. [DOI] [PubMed] [Google Scholar]
- 20.Feng J., Fan G. The role of DNA methylation in the central nervous system and neuropsychiatric disorders. Int. Rev. Neurobiol. 2009;89:67–84. doi: 10.1016/S0074-7742(09)89004-1. [DOI] [PubMed] [Google Scholar]
- 21.Lin M., Wei L.J., Sellers W.R., Lieberfarb M., Wong W.H., Li C. dChipSNP: significance curve and clustering of SNP-array-based loss-of-heterozygosity data. Bioinformatics. 2004;20:1233–1240. doi: 10.1093/bioinformatics/bth069. [DOI] [PubMed] [Google Scholar]
- 22.Seelow D., Schuelke M., Hildebrandt F., Nürnberg P. HomozygosityMapper-an interactive approach to homozygosity mapping. Nucleic Acids Res. 2009;37:W593–W599. doi: 10.1093/nar/gkp369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kelley L.A., Sternberg M.J.E. Protein structure prediction on the web: a case study using the Phyre server. Nat. Protoc. 2009;4:363–371. doi: 10.1038/nprot.2009.2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.