

Abstract
Osteogenesis imperfecta (OI) is a heritable disorder of connective tissue characterized by bone fragility and low bone mass. Recently, our group and others reported that WNT1 recessive mutations cause OI, whereas WNT1 heterozygous mutations cause early onset osteoporosis. These findings support the hypothesis that WNT1 is an important WNT ligand regulating bone formation and bone homeostasis. While these studies provided strong human genetic and in vitro functional data, an in vivo animal model to study the mechanism of WNT1 function in bone is lacking. Here, we show that Swaying (Wnt1sw/sw) mice previously reported to carry a spontaneous mutation in Wnt1 share major features of OI including propensity to fractures and severe osteopenia. In addition, biomechanical and biochemical analyses showed that Wnt1sw/sw mice exhibit reduced bone strength with altered levels of mineral and collagen in the bone matrix that is also distinct from the type I collagen-related form of OI. Further histomorphometric analyses and gene expression studies demonstrate that the bone phenotype is associated with defects in osteoblast activity and function. Our study thus provides in vivo evidence that WNT1 mutations contribute to bone fragility in OI patients and demonstrates that the Wnt1sw/sw mouse is a murine model of OI caused by WNT1 mutations.
INTRODUCTION
Osteogenesis imperfecta (OI; MIM #166200, #166210, #259420, #166220, #610967, #610968, #610682, #610915) is a genetic disorder characterized by bone fragility coupled with low bone mass (1,2). The majority of OI cases are dominantly inherited and caused by mutations in type I collagen encoded by COL1A1 and COL1A2 (3–6). These mutations result in either qualitative or quantitative defects of type I procollagen. In addition to the dominant forms of OI, recessively inherited OI has been reported to be caused by mutations in several genes, such as CRTAP, P3H1, CYPB, FKBP10 and HSP47, which are involved in collagen processing (7–13). Although the majority of OI genes are related to type I collagen, recent studies have identified a new set of OI genes which are not directly involved in collagen synthesis or processing. For example, mutations in Serpin Peptidase Inhibitor Clade F (SERPINF1), which encodes pigment epithelium-derived factor, cause OI type VI characterized primarily by an osteomalacia phenotype, and mutations in WNT1 cause recessively inherited OI with mild-to-severe phenotypes (14–20). These findings suggest that there is a broad spectrum of pathogenic mechanisms of OI. Therefore, it will be necessary to develop additional genetic tools to gain a mechanistic understanding of OI and to test genotype-specific treatment.
Wnt signaling is a well-known major regulator of bone homeostasis (21). Both human and mouse genetic studies of LRP5/6 co-receptors and β-catenin have provided evidence that canonical Wnt/β-catenin signaling is essential for skeletal development and postnatal bone formation (22–33). However, the identity of the key WNT ligands that regulate human bone homeostasis has remained unknown until now. Recently, several studies have demonstrated that mutations in the WNT1 gene cause recessively inherited OI as well as a dominant form of early-onset osteoporosis in humans (17–20). These surprising findings raised the possibility that WNT1 is one of the major WNT ligands regulating bone homeostasis. Despite the human genetic evidence and in vitro functional studies, there are no in vivo animal model data supporting that WNT1 mutations cause bone fragility.
The Swaying mouse (Wnt1sw/sw) was first described in 1967 and carries a mutant allele of Wnt1 (34,35). The mutation is a single nucleotide deletion (G565) in the third exon of Wnt1. The deletion causes a frame shift and results in a premature termination 30 bp downstream from the point mutation. Although homozygous Wnt1 null mice are embryonic lethal, the Wnt1sw/sw mice can survive postnatally thereby offering an opportunity to study the postnatal skeletal phenotype (35–37). The Wnt1sw/sw mice show a severe cerebellar defect that is also observed in some OI patients with the WNT1 mutation (19,35). The genetic and phenotypic evidence strongly suggest that the Wnt1sw/sw mice could constitute a murine model of OI. However, the skeletal phenotype of the Wnt1sw/sw mice had not been characterized.
In this study, we show that the Wnt1sw/sw mice have spontaneous fractures and severe osteopenia, which are hallmarks of OI. Our histomorphometric analysis revealed that the bone phenotype is caused by a decrease in osteoblast activity. Further biomechanical analysis showed that Wnt1sw/sw mouse bones exhibited reduced bone strength in comparison with wild-type mice. Finally, Raman spectroscopic data suggest that swaying mice showed altered mineral and collagen properties in the bone matrix. Our studies provide strong in vivo evidence that WNT1 mutations can cause bone fragility and the swaying mouse (Wnt1sw/sw) is an appropriate mouse model for OI caused by WNT1 mutations in humans. Moreover, the material properties of WNT1 mutant bone are distinct from collagen-related OI bone.
RESULTS
The Wnt1sw/sw mice show spontaneous tibial fractures and severe osteopenia
To gain insight into the pathogenic mechanism of OI caused by WNT1 mutations, we characterized Wnt1sw/sw mice carrying a spontaneous mutation in the Wnt1 gene. Heterozygous Wnt1sw/+ mice were intercrossed to generate homozygous Wnt1sw/sw mice. Among a total 117 mice, 20.5% of mice were homozygous Wnt1sw/sw mice, and 29% of the Wnt1sw/swmice showed postnatal lethality before 1 month of age (Table 1). All Wnt1sw/sw mice exhibited an ataxia phenotype that has been previously reported. Wnt1sw/sw mice also showed bidirectional rotation behavior evidenced by pivoting with hindlimbs. All surviving Wnt1sw/sw and wild-type mice were used for phenotypic analysis at 6 weeks of age.
Table 1.
Analysis of genotype, postnatal lethality and fracture rate of wild-type (+/+), heterozygous Wnt1sw/+ (sw/+) and homozygous Wnt1sw/sw (sw/sw) mice
| Genotype | Number of mice | % of mice | Postnatal lethality (%) | Fracture rate (%) |
|---|---|---|---|---|
| +/+ | 24 | 20.5 | 0 | 0 |
| sw/+ | 69 | 59 | 0 | 0 |
| sw/sw | 24 | 20.5 | 29 | 65 |
| Total | 117 | 100% |
The Wnt1sw/sw mice showed growth retardation throughout postnatal development resulting in a reduction in body weight (Fig. 1A). Consistent with growth retardation, whole body radiography showed that the overall skeleton of the Wnt1sw/sw mice was slightly smaller than that of their wild-type controls (Fig. 1B). Besides growth retardation, spontaneous tibial fractures were observed in the whole body X-rays of the Wnt1sw/sw mice (Fig. 1B, white arrow). The area of fracture was easily detected by callus formation during dissection of the Wnt1sw/sw mice. The callus was subsequently confirmed by radiography of tibia and H&E stained sections (Fig. 1C–E). All fractures were located in tibia, and the frequency of fracture was 65% (Table 1). Thirty-six percent of fractures were unilateral and 64% were bilateral. Overall, the Wnt1sw/sw mice showed growth retardation and spontaneous fractures, which are major characteristics of OI.
Figure 1.

The Wnt1sw/sw mice show growth retardation and tibial fractures. (A) Growth curve of wild-type (+/+), heterozygous Wnt1sw/+ (sw/+) and homozygous Wnt1sw/sw (sw/sw) male mice (P < 0.05 for all time points. Means ±SDs are shown). The weight was measured every 10 days from P10 to P40. (B) Whole body radiography of wild-type and Wnt1sw/sw mice. Arrows indicate tibial fractures. Spontaneous fracture in tibia evidenced by callus formation (C), X-ray (D) and H&E stained section (E).
In addition to tibial fractures, severe osteopenia was observed by radiographic analysis in the Wnt1sw/sw mice, as indicated by increased lucency of the trabecular and cortical bones (Fig. 1B and D). Histological analysis of long bones confirmed a dramatic decrease of trabecular bone and demonstrated thinner cortical bone in the Wnt1sw/sw mice (Fig. 2A). However, the growth plate of the Wnt1sw/sw mice was normal. To quantify the low bone mass phenotype, we analyzed long bones using a microcomputed tomography (μCT) system (Fig. 2B–D). 3D reconstruction images generated from μCT verified the decreased trabecular bone and thinner cortical bone in the Wnt1sw/sw mice (Fig. 2B–C). Quantification of femurs revealed a 12-fold decrease in trabecular bone volume (BV/TV) compared with wild-type controls (Fig. 2D). The lower bone mass was also associated with a decreased trabecular number (Tb.N) and thickness (Tb.Th) and increased trabecular separation (Tb.Sp). The thickness of cortical bone (Cort.Th) in the Wnt1sw/sw mice was half that of the wild-type bones. Quantification of vertebrae also showed a low bone mass phenotype, but the phenotype was milder than that in the femur (Supplementary Material, Fig. S1). The BV/TV of vertebrae in the Wnt1sw/sw mice was 3-fold less than that of wild-type mice. Both male and female Wnt1sw/sw mice presented with the low bone mass phenotype (Fig. 2; Supplementary Material, Fig. S1). In summary, the Wnt1sw/sw mice exhibited severe osteopenia which is also characteristic of human OI patients.
Figure 2.

The Wnt1sw/sw mice exhibit severe osteopenia. (A) H&E stained longitudinal sections of femurs of wild-type (+/+) and Wnt1sw/sw (sw/sw) mice at 6 weeks of age. μCT reconstruction image of trabecular bone (B) and cortical bone (C) at 6 weeks of age. (D) μCT analysis shows decreased bone volume per total volume (BV/TV), decreased trabecular number (Tb.N) and trabecular thickness (Tb. Th), increased trabecular separation (Tb. Sp), and decreased cortical bone thickness (Cort.Th) when compared with wild type (male mice, n = 6 per group, ***P < 0.001).
The bones in the Wnt1sw/sw mice exhibit reduced bone strength
The frequency of fractures prompted us to test the biomechanical properties of Wnt1sw/sw bones. We performed three-point bending experiments using femurs of 6-week-old mice. Wnt1sw/sw mice displayed a dramatic decrease in maximum load, stiffness and energy to failure, which indicate that the bones in the Wnt1sw/sw mice are weaker and have decreased resistance to bending (Table 2). However, these parameters do not account for geometric factors such as the length and diameter of bone. Because the Wnt1sw/sw mice exhibited growth retardation, they have shorter and thinner bones when compared with wild type. Thus, it is possible that the changes in the geometric parameters were the major contributors to the overall weakness in the Wnt1sw/sw mouse bone. To eliminate the effect of the geometric factors on bone strength, we determined the ultimate strength and elastic modulus. Consistent with other parameters, the ultimate strength and elastic modulus were significantly decreased in the Wnt1sw/sw mouse bone (Table 2), indicating that the bones in the Wnt1sw/sw mice have substantially weaker intrinsic properties compared with those in wild-type mice.
Table 2.
Biomechanical testing of femurs by three-point bending (6-week-old mice, n = 6 for wild type, n = 4 for Wnt1sw/sw)
| Maximum load (N) | Stiffness (N/mm) | Energy to failure (N/mm) | Ultimate strength (MPa) | Elastic modulus (N/mm2) | Postyield displacement (mm) | Postyield strain (non-dimensional) | |
|---|---|---|---|---|---|---|---|
| +/+ (SD) | 13.55 (1.92) | 92.43 (13.15) | 12.33 (1.69) | 100.58 (5.91) | 3416.34 (398.94) | 1.14 (0.13) | 0.23 (0.03) |
| sw/sw (SD) | 3.60 (0.85) | 14.36 (5.69) | 3.41 (0.62) | 73.61 (13.44) | 1935.19 (637.99) | 1.45 (0.09) | 0.22 (0.01) |
| P-value | <0.001 | <0.001 | <0.001 | 0.004 | 0.002 | 0.034 | 0.51 |
OI caused by mutations in collagen and collagen processing genes is mainly characterized by brittle bones. To assess the brittleness of the Wnt1sw/sw mice, we first measured postyield displacement, which does not account for geometric factors. Interestingly, the postyield displacement was significantly increased in the Wnt1sw/sw mouse bone compared with wild-type mouse bone, which indicates that the Wnt1sw/sw mice bones are in fact less brittle (Table 2). To account for geometric factors, we calculated the postyield strain. Both the Wnt1sw/sw and wild-type mouse bones showed similar levels of postyield strain (Table 2). These data indicate that geometric factors are more important than the basic material properties for determining brittleness in the Wnt1sw/sw mice model. Overall, the bones in Wnt1sw/sw mice are weak, but not brittle, in contrast to the material properties of classical collagen-related forms of OI.
Wnt1sw/sw bones have less collagen and mineral
Following biomechanical testing, we evaluated the bone mineral and matrix composition of the Wnt1sw/sw mice by Raman spectroscopy. Bones in Wnt1sw/sw mice showed a decreased ratio of proline to amide I (Fig. 3A), which indicates a lower quantity of collagen in the bone matrix of Wnt1sw/sw mice. Relative mineral content was calculated by the ratio of phosphate to amide I is also reduced in the Wnt1sw/sw mice (Fig. 3B). Finally, the Wnt1sw/sw mice exhibited a reduction in collagen mineralization, which was evident from the decreased ratio of phosphate to proline (Fig. 3C). Overall, Raman spectroscopy analysis demonstrated that Wnt1sw/sw mouse bones have decreased mineral and collagen composition in their bone matrix.
Figure 3.

Wnt1sw/sw mice have altered biochemical properties in the bone matrix. Biochemical properties of cortical bone as measured by Raman spectroscopy in wild-type (+/+) and Wnt1sw/sw (sw/sw) mice. The relative quantity of collagen to matrix was measured by the ratio of proline to amide 1 (A). The relative amount of mineral to matrix was measured by the ratio of phosphate to amide (B). The relative amount of mineral to collagen was measured by the ratio of phosphate to proline (C). n = 5 each group, and *P < 0.05; ***P < 0.001.
Osteoblast activity is decreased in the Wnt1sw/sw mice
To understand the cellular mechanism leading to the fractures and osteopenic phenotypes in the Wnt1sw/sw mice, we performed bone histomorphometric analysis of vertebral sections. In this analysis, osteoclast number (N.Oc/BS) and surfaces (Oc.S/BS) were not significantly changed in the Wnt1sw/sw mice when compared with wild-type littermate controls (Fig. 4A). Additionally, osteoblast numbers (N.Ob/BS) were also not changed in the Wnt1sw/sw mice (Fig. 4B). These data prompted us to perform dynamic histomorphometric analysis using calcein and alizarin red double labeling. Interestingly, the bone formation rate and mineral apposition rate were decreased in the Wnt1sw/sw mice (Fig. 4B). These data strongly suggest that the fractures and osteopenia phenotype in the Wnt1sw/sw mice are caused by decreased osteoblast activity and function.
Figure 4.

Osteoblast activity is decreased in the Wnt1sw/sw mouse bone. (A and B) Bone histomorphometry of male wild-type (+/+) and Wnt1sw/sw (sw/sw) mice at 6 weeks of age (n = 5 each, **P < 0.01). (A) Osteoclast number (N.Oc/Bs) and surface (Oc.S/BS) normalized by bone surface. (B) Osteoblast number (N.Ob/BS) normalized by bone surface, mineral apposition rate (MAR) and bone formation rate (BFR/BS) per bone surface. (C) Relative expression of osteoblast and osteoclast markers in long bone of 6-week-old wild-type versus Wnt1sw/sw male mice (n = 3 each, *P < 0.05).
To confirm the defects in osteoblast activity in the Wnt1sw/sw mice, we examined osteoblast differentiation in vivo (Fig. 4C). We isolated RNA directly from the long bones and assessed osteoblast differentiation markers by real-time PCR. Despite no difference in the Runx2 expression levels as one marker of differentiation, the levels of other osteoblast markers such as Col1a1, Alkaline phosphatase (Alp) and Osteocalcin (Ocal) were significantly reduced in the Wnt1sw/sw mouse bone. As expected, the osteoclast markers, such as Ctsk and TRAP, were unchanged. Consistent with the bone histomorphometric data, the gene expression study supports that osteoblast function was decreased in the Wnt1sw/sw mice.
DISCUSSION
In the current study, we characterized Wnt1sw/sw mice in order to understand the pathogenic mechanism of OI caused by WNT1 mutations. Wnt1sw/sw mice exhibited spontaneous fractures and severe osteopenia, which are major features of OI. Subsequent biomechanical and biochemical analysis revealed that Wnt1sw/sw mice have weaker bone properties with less collagen and mineral in the bone matrix. Finally, histomorphometric analyses and gene expression studies provided evidence that the bone phenotype could be caused by defects in osteoblast activity and function, which could be one of the possible pathogenic mechanisms of OI caused by WNT1 mutations. This study also supports that the Wnt1sw/sw mice could be used as a murine model of OI caused by WNT1 mutations.
Previous human genetic and in vitro functional studies showed that mutations in WNT1 cause OI characterized by increased bone fragility. The current work characterizes the first in vivo mouse model for this type of OI. Homozygous Wnt1sw/sw mice share the major features of OI including propensity for spontaneous fractures and severe osteopenia. Interestingly, the genomic location of the Swaying mutation is exactly the same as the previously reported human WNT1 mutation. The reported human mutation is a single nucleotide substitution resulting in a nonsense mutation (c.565G>T, p.Glu189*), and the mouse swaying mutation is a single nucleotide deletion that results in a stop codon 30 bp downstream of the mutation site (c.565delG, p.Glu189Argfs*10) (18,35). Therefore, our study provides a strong in vivo model of a reported human mutation causing OI.
Because the Wnt1sw/sw mice carry the spontaneous mutation throughout their entire body, we could not determine any tissue or cell-type-specific function of Wnt1. The Wnt1sw/sw mice show cerebellar defects resulting in abnormal behavioral phenotypes. The neuronal or behavior phenotype could also contribute to the bone fragility phenotype given data supporting central control of bone formation (38). Bone is also a multicellular organ including osteoblasts, osteoclasts, chondrocytes and bone marrow cells. We still do not know which cell type is the major source of Wnt1 in bone, although our previous lineage tracing studies suggested that osteoblasts, osteocytes and B cells would be good candidates (19). Tissue-specific Wnt1 loss- and gain-of-function mouse models will be useful in addressing the question about the tissue and cell specificity of Wnt1 function.
Tibial fractures are an interesting phenotype of Wnt1sw/sw mice. There are several mouse models of OI, and only some exhibit multiple bone fractures (39). Interestingly, the Wnt1sw/sw mice presented with fractures in the tibia. We found the severe osteopenia phenotype in both the forelimbs and hindlimbs, but we never observed any callus formation or fractures in the forelimbs. One possible explanation is that these fractures are caused by the combination of both bone and behavior phenotypes. In our biomechanical study, the Wnt1sw/sw mice exhibited very weak bone properties. Wnt1sw/sw mice always walk by swaying and often pivot on one side of the hindlimb. This aberrant movement could afford greater mechanical loading on the weakened tibia, which would result in fractures.
Because of their antiresorptive function, bisphosphonates (BP) are widely used to treat OI patients. Several studies have shown that BP treatment increases bone mineral density (BMD) in OI patients (40–42). Despite increased BMD, BP treatment has variable effects on fracture frequency. Recent studies have also shown that long-term treatment with BP may have adverse effects by impairing bone modeling (43), which raises concerns with respect to BP treatment of adolescent OI patients. Therefore, new therapeutic reagents that enhance anabolic activity of osteoblasts will be required. Recently, the anti-sclerostin antibody, which is an agonist of Wnt signaling (bone anabolic signaling), was developed and is under phase III clinical trials (NCT02016716) (44–47). Anti-sclerostin antibody treatment could be beneficial to OI patients. However, this treatment may not improve the bone fragility in OI patients with WNT1 mutations because the WNT1 ligand is upstream of sclerostin. This means that anabolic WNT signaling in bone which can be inhibited by sclerostin maybe suboptimal in OI patients with WNT1 mutations, although response may also be determined by contribution of other WNT ligands. The Wnt1sw/sw mice will be a useful mouse model to test the therapeutic potential of the anti-sclerostin antibody for OI patient with WNT1 mutations.
MATERIALS AND METHODS
Animal
The swaying (Wnt1sw/sw) mice were purchased from the Jackson Laboratory on a mixed C57BL/6 background. The swaying mutation is a single nucleotide deletion that results in a new BSL1 restriction site; therefore, we performed genotyping by PCR with enzyme restriction. Specifically, two primers (Sway-F: 5′ GGTCACACATTCCGTGGC 3′, Sway-R: 5′ TTGGTTCCGGACACACCG 3′) were designed to amplify a 246 base amplicon of the Wnt1 genomic DNA sequence that includes the swaying mutation. This 246 base amplicon was subsequently purified with a PCR purification kit and digested with BSL1. The digested PCR products were separated by 2% agarose gel electrophoresis and different band pattern were observed between wild type (1 band with 246 base), Wnt1sw/+ (3 band with 246, 164 and 82 base) and Wnt1sw/sw (2 band with 164 and 82 base) genotypes.
Skeletal analyses and bone histomorphometry
To analyze bone phenotypes, femurs and vertebra were isolated from the wild-type and Wnt1sw/sw mice at 6 weeks of age. The bones were fixed in 10% buffered formalin for 24 h for subsequent analysis. Radiography of bones was performed with an Xpert 80 system (Kubtec), and microcomputed tomography (μCT) scans were conducted with μCT-40 system (Scanco Medical). Lumber vertebrae were used to generate plastic sections for bone histomorphometry. Specifically, lumber vertebrae were embedded in methyl methacrylate, and embedded samples were sectioned using tungsten carbide blades. The stained plastic sections were used for evaluation of bone formation parameters with Bioquant Osteo software (Bioquant). Tartrate-resistant acid phosphatase staining was used to quantify osteoclast number and surfaces, and trichrome staining was used to quantify osteoblast numbers. Calcein and Alizarin Red were injected into mice with a 5-day interval following the first injection of Calcein to assess dynamic histomorphometry. The 4th lumbar vertebrae were used for μCT and bone histomorphometry.
Three-point bending
Femurs were tested in three-point bending using a span of ∼6 mm using a Bose ElectroForce3200 device (Bose, Eden Prairie, MN, USA). All the femurs were tested wet at room temperature. They were preloaded to 1 N for 5 s and then were compressed to failure at a rate of 0.1 mm/s. Load and displacement data were captured at the rate of 40 Hz by using WinTest software (Bose). Following testing, all datafiles were analyzed using a MatLab program written to extract the pertinent data. Stress was calculated using the following formula:
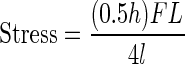 |
where F is the load applied on the femur in N, L is the span length in mm, h is the specimen diameter in mm, I is the cross-sectional moment of inertia in mm4. Both h and I were obtained by analyzing a midshaft micro-CT image. Strain was calculated using the following formula:
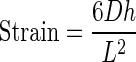 |
where D is the actuator displacement in mm, h is the diameter in mm and L is the span length in mm. To determine the yield point, a 0.2% offset from the maximum slope of the linear portion of the stress–strain curve was used. Stiffness and elastic modulus were defined from this maximum slope of the load–displacement and stress–strain curve, respectively. The elastic region was identified as the region from the completion of the preload to the yield point. Postyield region was identified as the region from the yield point until the failure point. Failure was defined as either fracture or the point when the specimen had achieved 20% strain after the maximum load, whichever came first. A trapezoidal numerical integration method was used to determine both energy (area under the load–displacement curve) and toughness (area under the stress–strain curve). Maximum load and ultimate strength were determined by finding the highest load and strength values recorded by WinTest, before the specimen failed.
Raman spectroscopy
Raman spectra were acquired from the cortex of intact tibiae using a confocal Raman microscope (Renishaw Invia, Gloucestershire, England, UK), as described previously (48,49). In brief, the tibiae were mounted on a microscope slide using polymer clay with the proximal metaphyseal surface leveled horizontally. About 30 mW of 785 nm laser light was focused onto the cortical surface of the tibiae through a Leica N PLAN L50X/0.50 objective. The scattered Raman signals were collected via the same objective and coupled to a spectrometer inside the Invia Raman system. Three spectra per tibia were collected from the midshaft. Each measurement consists of three accumulations of 10 s exposure and has a spectral resolution of ∼4 cm−1. Custom written Matlab scripts were used for baseline correction through polynomial fitting and for Raman peaks calculation. Spectral signatures of bone used in the analysis include phosphate v1 (960 cm−1), proline (856 cm−1) and amide I (1665 cm−1), among which peak heights of proline and amide I represent the abundance of collagen and organic matrix, respectively.
qRT-PCR analysis
For qRT-PCR analysis, total RNA from mouse femurs was extracted with Trizol at 6 weeks of age. In order to minimize contamination with other connective tissues such as muscle and tendon, we carefully dissected all the attached tissues from each femur. The proximal and distal ends of each femur were removed to minimize contamination by chondrocytes. In addition, bone marrow was removed by centrifugation. Superscript III First Strand RT-PCR kit (Invitrogen) was used for cDNA synthesis, and the qRT-PCR was executed on a LightCycler instrument (Roche). β2-Microglobulin was used as the internal control to normalize gene expression.
SUPPLEMENTARY MATERIAL
FUNDING
Research funding includes NIH grants PO1 HD22657 (B.H.L.), PO1 HD070394 (B.H.L.), K25 CA149194 (X.B.) and The Rolanette and Berdon Lawrence Bone Disease Program of Texas. K.S.J. was supported by NRSA fellowship F32 AR063616 (NIAMS/NIH). This work was also supported by the BCM Intellectual and Developmental Disabilities Research Center (HD024064) from the Eunice Kennedy Shriver National Institute Of Child Health & Human Development, and the BCM Advanced Technology Cores with funding from the NIH (AI036211, CA125123, and RR024574).
Supplementary Material
ACKNOWLEDGEMENTS
We thank Erica Homan, Elda Munivez and Brian Dawson for editing manuscript and technical support.
Conflict of Interest statement. None declared.
REFERENCES
- 1.Basel D., Steiner R.D. Osteogenesis imperfecta: recent findings shed new light on this once well-understood condition. Genet. Med. 2009;11:375–385. doi: 10.1097/GIM.0b013e3181a1ff7b. [DOI] [PubMed] [Google Scholar]
- 2.Rauch F., Glorieux F.H. Osteogenesis imperfecta. Lancet. 2004;363:1377–1385. doi: 10.1016/S0140-6736(04)16051-0. [DOI] [PubMed] [Google Scholar]
- 3.Barsh G.S., Byers P.H. Reduced secretion of structurally abnormal type I procollagen in a form of osteogenesis imperfecta. Proc. Natl. Acad. Sci. USA. 1981;78:5142–5146. doi: 10.1073/pnas.78.8.5142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chu M.L., Williams C.J., Pepe G., Hirsch J.L., Prockop D.J., Ramirez F. Internal deletion in a collagen gene in a perinatal lethal form of osteogenesis imperfecta. Nature. 1983;304:78–80. doi: 10.1038/304078a0. [DOI] [PubMed] [Google Scholar]
- 5.Williams C.J., Prockop D.J. Synthesis and processing of a type I procollagen containing shortened pro-alpha 1(I) chains by fibroblasts from a patient with osteogenesis imperfecta. J. Biol. Chem. 1983;258:5915–5921. [PubMed] [Google Scholar]
- 6.Byers P.H., Tsipouras P., Bonadio J.F., Starman B.J., Schwartz R.C. Perinatal lethal osteogenesis imperfecta (OI type II): a biochemically heterogeneous disorder usually due to new mutations in the genes for type I collagen. Am. J. Hum. Genet. 1988;42:237–248. [PMC free article] [PubMed] [Google Scholar]
- 7.Barnes A.M., Chang W., Morello R., Cabral W.A., Weis M., Eyre D.R., Leikin S., Makareeva E., Kuznetsova N., Uveges T.E., et al. Deficiency of cartilage-associated protein in recessive lethal osteogenesis imperfecta. N. Engl. J. Med. 2006;355:2757–2764. doi: 10.1056/NEJMoa063804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Morello R., Bertin T.K., Chen Y., Hicks J., Tonachini L., Monticone M., Castagnola P., Rauch F., Glorieux F.H., Vranka J., et al. CRTAP is required for prolyl 3- hydroxylation and mutations cause recessive osteogenesis imperfecta. Cell. 2006;127:291–304. doi: 10.1016/j.cell.2006.08.039. [DOI] [PubMed] [Google Scholar]
- 9.Cabral W.A., Chang W., Barnes A.M., Weis M., Scott M.A., Leikin S., Makareeva E., Kuznetsova N.V., Rosenbaum K.N., Tifft C.J., et al. Prolyl 3-hydroxylase 1 deficiency causes a recessive metabolic bone disorder resembling lethal/severe osteogenesis imperfecta. Nat. Genet. 2007;39:359–365. doi: 10.1038/ng1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Baldridge D., Schwarze U., Morello R., Lennington J., Bertin T.K., Pace J.M., Pepin M.G., Weis M., Eyre D.R., Walsh J., et al. CRTAP and LEPRE1 mutations in recessive osteogenesis imperfecta. Hum. Mutat. 2008;29:1435–1442. doi: 10.1002/humu.20799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Barnes A.M., Carter E.M., Cabral W.A., Weis M., Chang W., Makareeva E., Leikin S., Rotimi C.N., Eyre D.R., Raggio C.L., et al. Lack of cyclophilin B in osteogenesis imperfecta with normal collagen folding. N. Engl. J. Med. 2010;362:521–528. doi: 10.1056/NEJMoa0907705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Christiansen H.E., Schwarze U., Pyott S.M., AlSwaid A., Al Balwi M., Alrasheed S., Pepin M.G., Weis M.A., Eyre D.R., Byers P.H. Homozygosity for a missense mutation in SERPINH1, which encodes the collagen chaperone protein HSP47, results in severe recessive osteogenesis imperfecta. Am. J. Hum. Genet. 2010;86:389–398. doi: 10.1016/j.ajhg.2010.01.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kelley B.P., Malfait F., Bonafe L., Baldridge D., Homan E., Symoens S., Willaert A., Elcioglu N., Van Maldergem L., Verellen-Dumoulin C., et al. Mutations in FKBP10 cause recessive osteogenesis imperfecta and Bruck syndrome. J. Bone Miner. Res. 2011;26:666–672. doi: 10.1002/jbmr.250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Becker J., Semler O., Gilissen C., Li Y., Bolz H.J., Giunta C., Bergmann C., Rohrbach M., Koerber F., Zimmermann K., et al. Exome sequencing identifies truncating mutations in human SERPINF1 in autosomal-recessive osteogenesis imperfecta. Am. J. Hum. Genet. 2011;88:362–371. doi: 10.1016/j.ajhg.2011.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Homan E.P., Rauch F., Grafe I., Lietman C., Doll J.A., Dawson B., Bertin T., Napierala D., Morello R., Gibbs R., et al. Mutations in SERPINF1 cause osteogenesis imperfecta type VI. J. Bone Miner. Res. 2011;26:2798–2803. doi: 10.1002/jbmr.487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Venturi G., Gandini A., Monti E., Dalle Carbonare L., Corradi M., Vincenzi M., Valenti M.T., Valli M., Pelilli E., Boner A., et al. Lack of expression of SERPINF1, the gene coding for pigment epithelium-derived factor, causes progressively deforming osteogenesis imperfecta with normal type I collagen. J. Bone Miner. Res. 2012;27:723–728. doi: 10.1002/jbmr.1480. [DOI] [PubMed] [Google Scholar]
- 17.Fahiminiya S., Majewski J., Mort J., Moffatt P., Glorieux F.H., Rauch F. Mutations in WNT1 are a cause of osteogenesis imperfecta. J. Med. Genet. 2013;50:345–348. doi: 10.1136/jmedgenet-2013-101567. [DOI] [PubMed] [Google Scholar]
- 18.Keupp K., Beleggia F., Kayserili H., Barnes A.M., Steiner M., Semler O., Fischer B., Yigit G., Janda C.Y., Becker J., et al. Mutations in WNT1 cause different forms of bone fragility. Am. J. Hum. Genet. 2013;92:565–574. doi: 10.1016/j.ajhg.2013.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Laine C.M., Joeng K.S., Campeau P.M., Kiviranta R., Tarkkonen K., Grover M., Lu J.T., Pekkinen M., Wessman M., Heino T.J., et al. WNT1 mutations in early-onset osteoporosis and osteogenesis imperfecta. N. Engl. J. Med. 2013;368:1809–1816. doi: 10.1056/NEJMoa1215458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pyott S.M., Tran T.T., Leistritz D.F., Pepin M.G., Mendelsohn N.J., Temme R.T., Fernandez B.A., Elsayed S.M., Elsobky E., Verma I., et al. WNT1 mutations in families affected by moderately severe and progressive recessive osteogenesis imperfecta. Am. J. Hum. Genet. 2013;92:590–597. doi: 10.1016/j.ajhg.2013.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Baron R., Kneissel M. WNT signaling in bone homeostasis and disease: from human mutations to treatments. Nat. Med. 2013;19:179–192. doi: 10.1038/nm.3074. [DOI] [PubMed] [Google Scholar]
- 22.Babij P., Zhao W., Small C., Kharode Y., Yaworsky P.J., Bouxsein M.L., Reddy P.S., Bodine P.V., Robinson J.A., Bhat B., et al. High bone mass in mice expressing a mutant LRP5 gene. J. Bone Miner. Res. 2003;18:960–974. doi: 10.1359/jbmr.2003.18.6.960. [DOI] [PubMed] [Google Scholar]
- 23.Boyden L.M., Mao J., Belsky J., Mitzner L., Farhi A., Mitnick M.A., Wu D., Insogna K., Lifton R.P. High bone density due to a mutation in LDL-receptor-related protein 5. N. Engl. J. Med. 2002;346:1513–1521. doi: 10.1056/NEJMoa013444. [DOI] [PubMed] [Google Scholar]
- 24.Clement-Lacroix P., Ai M., Morvan F., Roman-Roman S., Vayssiere B., Belleville C., Estrera K., Warman M.L., Baron R., Rawadi G. Lrp5-independent activation of Wnt signaling by lithium chloride increases bone formation and bone mass in mice. Proc. Natl. Acad. Sci. USA. 2005;102:17406–17411. doi: 10.1073/pnas.0505259102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cui Y., Niziolek P.J., MacDonald B.T., Zylstra C.R., Alenina N., Robinson D.R., Zhong Z., Matthes S., Jacobsen C.M., Conlon R.A., et al. Lrp5 functions in bone to regulate bone mass. Nat. Med. 2011;17:684–691. doi: 10.1038/nm.2388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Glass D.A., 2nd, Bialek P., Ahn J.D., Starbuck M., Patel M.S., Clevers H., Taketo M.M., Long F., McMahon A.P., Lang R.A., et al. Canonical Wnt signaling in differentiated osteoblasts controls osteoclast differentiation. Dev. Cell. 2005;8:751–764. doi: 10.1016/j.devcel.2005.02.017. [DOI] [PubMed] [Google Scholar]
- 27.Gong Y., Slee R.B., Fukai N., Rawadi G., Roman-Roman S., Reginato A.M., Wang H., Cundy T., Glorieux F.H., Lev D., et al. LDL receptor-related protein 5 (LRP5) affects bone accrual and eye development. Cell. 2001;107:513–523. doi: 10.1016/s0092-8674(01)00571-2. [DOI] [PubMed] [Google Scholar]
- 28.Holmen S.L., Zylstra C.R., Mukherjee A., Sigler R.E., Faugere M.C., Bouxsein M.L., Deng L., Clemens T.L., Williams B.O. Essential role of beta-catenin in postnatal bone acquisition. J. Biol. Chem. 2005;280:21162–21168. doi: 10.1074/jbc.M501900200. [DOI] [PubMed] [Google Scholar]
- 29.Hu H., Hilton M.J., Tu X., Yu K., Ornitz D.M., Long F. Sequential roles of Hedgehog and Wnt signaling in osteoblast development. Development. 2005;132:49–60. doi: 10.1242/dev.01564. [DOI] [PubMed] [Google Scholar]
- 30.Joeng K.S., Schumacher C.A., Zylstra-Diegel C.R., Long F., Williams B.O. Lrp5 and Lrp6 redundantly control skeletal development in the mouse embryo. Dev. Biol. 2011;359:222–229. doi: 10.1016/j.ydbio.2011.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kato M., Patel M.S., Levasseur R., Lobov I., Chang B.H., Glass D.A., 2nd, Hartmann C., Li L., Hwang T.H., Brayton C.F., et al. Cbfa1-independent decrease in osteoblast proliferation, osteopenia, and persistent embryonic eye vascularization in mice deficient in Lrp5, a Wnt coreceptor. J. Cell. Biol. 2002;157:303–314. doi: 10.1083/jcb.200201089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Little R.D., Carulli J.P., Del Mastro R.G., Dupuis J., Osborne M., Folz C., Manning S.P., Swain P.M., Zhao S.C., Eustace B., et al. A mutation in the LDL receptor-related protein 5 gene results in the autosomal dominant high-bone-mass trait. Am. J. Hum. Genet. 2002;70:11–19. doi: 10.1086/338450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Riddle R.C., Diegel C.R., Leslie J.M., Van Koevering K.K., Faugere M.C., Clemens T.L., Williams B.O. Lrp5 and Lrp6 exert overlapping functions in osteoblasts during postnatal bone acquisition. PLoS ONE. 2013;8:e63323. doi: 10.1371/journal.pone.0063323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lane P.W. Swaying. Mouse News Lett. 1967;36:40. [Google Scholar]
- 35.Thomas K.R., Musci T.S., Neumann P.E., Capecchi M.R. Swaying is a mutant allele of the proto-oncogene Wnt-1. Cell. 1991;67:969–976. doi: 10.1016/0092-8674(91)90369-a. [DOI] [PubMed] [Google Scholar]
- 36.McMahon A.P., Bradley A. The Wnt-1 (int-1) proto-oncogene is required for development of a large region of the mouse brain. Cell. 1990;62:1073–1085. doi: 10.1016/0092-8674(90)90385-r. [DOI] [PubMed] [Google Scholar]
- 37.McMahon A.P., Joyner A.L., Bradley A., McMahon J.A. The midbrain-hindbrain phenotype of Wnt-1-/Wnt-1- mice results from stepwise deletion of engrailed-expressing cells by 9.5 days postcoitum. Cell. 1992;69:581–595. doi: 10.1016/0092-8674(92)90222-x. [DOI] [PubMed] [Google Scholar]
- 38.Yadav V.K., Oury F., Suda N., Liu Z.W., Gao X.B., Confavreux C., Klemenhagen K.C., Tanaka K.F., Gingrich J.A., Guo X.E., et al. A serotonin-dependent mechanism explains the leptin regulation of bone mass, appetite, and energy expenditure. Cell. 2009;138:976–989. doi: 10.1016/j.cell.2009.06.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Forlino A., Cabral W.A., Barnes A.M., Marini J.C. New perspectives on osteogenesis imperfecta. Nat. Rev. Endocrinol. 2011;7:540–557. doi: 10.1038/nrendo.2011.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sakkers R., Kok D., Engelbert R., van Dongen A., Jansen M., Pruijs H., Verbout A., Schweitzer D., Uiterwaal C. Skeletal effects and functional outcome with olpadronate in children with osteogenesis imperfecta: a 2-year randomised placebo-controlled study. Lancet. 2004;363:1427–1431. doi: 10.1016/S0140-6736(04)16101-1. [DOI] [PubMed] [Google Scholar]
- 41.Ward L.M., Rauch F., Whyte M.P., D'Astous J., Gates P.E., Grogan D., Lester E.L., McCall R.E., Pressly T.A., Sanders J.O., et al. Alendronate for the treatment of pediatric osteogenesis imperfecta: a randomized placebo-controlled study. J. Clin. Endocrinol. Metab. 2011;96:355–364. doi: 10.1210/jc.2010-0636. [DOI] [PubMed] [Google Scholar]
- 42.Barros E.R., Saraiva G.L., de Oliveira T.P., Lazaretti-Castro M. Safety and efficacy of a 1-year treatment with zoledronic acid compared with pamidronate in children with osteogenesis imperfecta. J. Pediatr. Endocrinol. Metab. 2012;25:485–491. doi: 10.1515/jpem-2012-0016. [DOI] [PubMed] [Google Scholar]
- 43.Saleh A., Hegde V.V., Potty A.G., Lane J.M. Bisphosphonate therapy and atypical fractures. Orthop. Clin. North. Am. 2013;44:137–151. doi: 10.1016/j.ocl.2013.01.001. [DOI] [PubMed] [Google Scholar]
- 44.Lin C., Jiang X., Dai Z., Guo X., Weng T., Wang J., Li Y., Feng G., Gao X., He L. Sclerostin mediates bone response to mechanical unloading through antagonizing Wnt/beta-catenin signaling. J. Bone Miner. Res. 2009;24:1651–1661. doi: 10.1359/jbmr.090411. [DOI] [PubMed] [Google Scholar]
- 45.Padhi D., Jang G., Stouch B., Fang L., Posvar E. Single-dose, placebo-controlled, randomized study of AMG 785, a sclerostin monoclonal antibody. J. Bone Miner. Res. 2011;26:19–26. doi: 10.1002/jbmr.173. [DOI] [PubMed] [Google Scholar]
- 46.Costa A.G., Bilezikian J.P. Sclerostin: therapeutic horizons based upon its actions. Curr. Osteoporos. Rep. 2012;10:64–72. doi: 10.1007/s11914-011-0089-5. [DOI] [PubMed] [Google Scholar]
- 47.Spatz J.M., Ellman R., Cloutier A.M., Louis L., van Vliet M., Suva L.J., Dwyer D., Stolina M., Ke H.Z., Bouxsein M.L. Sclerostin antibody inhibits skeletal deterioration due to reduced mechanical loading. J. Bone Miner. Res. 2013;28:865–874. doi: 10.1002/jbmr.1807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bi X., Patil C.A., Lynch C.C., Pharr G.M., Mahadevan-Jansen A., Nyman J.S. Raman and mechanical properties correlate at whole bone- and tissue-levels in a genetic mouse model. J. Biomech. 2011;44:297–303. doi: 10.1016/j.jbiomech.2010.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bi X., Sterling J.A., Merkel A.R., Perrien D.S., Nyman J.S., Mahadevan-Jansen A. Prostate cancer metastases alter bone mineral and matrix composition independent of effects on bone architecture in mice--a quantitative study using microCT and Raman spectroscopy. Bone. 2013;56:454–460. doi: 10.1016/j.bone.2013.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.