

Abstract

We have demonstrated a multistep 2-dimensional paper network immunoassay based on controlled rehydration of patterned, dried reagents. Previous work has shown that signal enhancement improves the limit of detection in 2-dimensional paper network assays, but until now, reagents have only been included as wet or dried in separate conjugate pads placed at the upstream end of the assay device. Wet reagents are not ideal for point-of-care because they must be refrigerated and typically limit automation and require more user steps. Conjugate pads allow drying but do not offer any control of the reagent distribution upon rehydration and can be a source of error when pads do not contact the assay membrane uniformly. Furthermore, each reagent is dried on a separate pad, increasing the fabrication complexity when implementing multistep assays that require several different reagents. Conversely, our novel method allows for consistent, controlled rehydration from patterned reagent storage depots directly within the paper membrane. In this assay demonstration, four separate reagents were patterned in different regions of the assay device: a gold-antibody conjugate used for antigen detection and three different signal enhancement components that must not be mixed until immediately before use. To show the viability of patterning and drying reagents directly onto a paper device for dry reagent storage and subsequent controlled release, we tested this device with the malaria antigen Plasmodium falciparum histidine-rich protein 2 (PfHRP2) as an example of target analyte. In this demonstration, the signal enhancement step increases the visible signal by roughly 3-fold and decreases the analytical limit of detection by 2.75-fold.
Lateral flow tests (LFTs) have been widely accepted for a variety of applications, ranging from home pregnancy tests to rapid diagnostic tests for infectious diseases in low-resource settings. A major advantage of these devices over microfluidic point-of-care diagnostics (such as the Cepheid GeneXpert) is that the fluid flow in LFTs is driven entirely by capillary pressure, eliminating the need for any external pumps or vacuum sources. Furthermore, LFTs have widespread appeal because they satisfy many of the ASSURED criteria that were developed to describe ideal characteristics of point-of-care tests: Affordable, Sensitive, Specific, User-friendly, Rapid and Robust, Equipment-free, and Delivered to those in need.1 LFTs are low-cost, rapid, easy to use, and require little to no instrumentation to interpret results; however, most are still severely lacking in sensitivity.2
There has been a recent push in the point-of-care diagnostics community to develop paper-based diagnostic tests that incorporate sophisticated functions into otherwise simple devices, stretching their capabilities while maintaining the core benefits of LFTs. These advancements include 2D and 3D paper devices that are capable of tasks such as multiplexing,3−5 sample processing,6 and signal enhancement,7,8 sophisticated functions that have previously been reserved for laboratory-based or traditional microfluidic tests. Conversely, traditional LFTs are only capable of performing a single step and cannot incorporate enhancement steps or any sample processing.
One key characteristic of most of these newer paper-based devices that remains consistent with early LFTs is the inclusion of dried reagents in the assay device. There are several benefits to including all of the essential reagents dried in situ within a device. First, the number and complexity of user steps are reduced: the user does not need to identify, measure, and add assay reagents to the device, which is particularly important when incorporating more sophisticated processes in a point-of-care test. Second, dried reagents are more resistant to damage at ambient temperatures, particularly when stabilizing additives, such as sucrose and trehalose,9,10 are added. Third, reagents dried in situ can facilitate device automation because device geometry and reagent location within the device can be designed to automate multistep processes. Together, these advantages improve the robustness, affordability, and ease-of-use of the device while decreasing the equipment needs. These four traits are crucial for designing devices appropriate for point-of-care diagnosis in low-resource settings.11,12
Traditionally, conjugate pads have been used to store dry reagents in both lateral flow8,13 and conventional microfluidic devices.9 However, there are two significant disadvantages to using these separate pads: they offer limited control over the release of rehydrated reagents, and they require additional materials and components that add to manufacturing costs. The fabrication costs of including conjugate pads is particularly substantial when used in two-dimensional paper diagnostics because, unlike LFTs, 2D devices cannot be fabricated by slicing dozens of strips from a single sheet (Figure 1A). Instead, conjugate pads must be added in a “pick-and-place” process (Figure 1B).
Figure 1.

Fabrication schematics of different types of tests. (A) Slicing lateral flow tests apart is relatively simple and straightforward. However, (B) the “pick and place” process of laying down several pads containing a variety of dry reagents, which is required when conjugate pads are used in 2DPN devices, is inefficient and error-prone. Printing reagents directly onto the assay membrane, as described here, is a first step toward roll-to-roll fabrication (C), which is more high-throughput than pick-and-place methods.
Methods for dry reagent storage, other than conjugate pads, in microfluidic devices have achieved such storage directly within the channels of these devices, for later controlled release. Some examples of these techniques include cavities in channel walls to control the reconstitution of dried proteins14 and “reagent integrators” to store and subsequently release predetermined dilutions of reagents into microfluidic devices.15 Inkjet printing has been used in a variety of cases to deposit reagents or channel barriers onto paper microfluidic assays,16−19 and our previous work developed methods for controlled release of reagents stored directly on a paper microfluidic device20 that are analogous to some of the controlled release mechanisms used in traditional microfluidic devices. Those novel methods for printing reagents on porous substrates enabled controlled spatial and temporal concentration gradients of reagents rehydrating during capillary flow within a porous device. Furthermore, patterning reagents into arrays of individual spots allows the storage of incompatible reagents, such as the multicomponent gold enhancement system that we used here for signal amplification, which loses functionality if the components are mixed prior to drying. Our previous work showed that the multiple components of this gold enhancement system can be reconstituted and recombined when stored dry within arrays patterned directly onto nitrocellulose membranes.20 Here, we have expanded upon those methods to present an application of controlled reagent rehydration in the implementation of a signal-enhanced immunoassay for the malaria antigen Plasmodium falciparum histidine-rich protein 2 (PfHRP2).
Though this is just a proof-of-concept assay example, malaria is an interesting target for an inexpensive highly sensitive point-of-care diagnostic test, because while current malaria rapid diagnostics have improved dramatically, these tests are insufficient for accurate diagnosis in settings where malaria control efforts have dramatically reduced the malaria rates in many endemic areas.21−23 A key part of the malaria control effort requires rapid and extremely sensitive diagnostic tests to detect infections early, begin treatment, and implement further mosquito-control measures in the area in which the infection was acquired.21 Another example of a disease antigen used in currently poorly performing rapid tests is the influenza nucleoprotein (NP), which is used to distinguish between influenza A and influenza B infection. Current influenza rapid tests for NP generally exhibit mediocre (10–70%) sensitivity24−26 and would dramatically benefit from the increased sophistication enabled by 2DPN devices.
Previous work from our group has shown high performance two-dimensional paper-based assays,8,27 but the work presented here is particularly significant because all of the necessary reagents were patterned, dried, and stored on a single porous membrane and rehydrated with the addition of sample and buffer upon the initiation of the assay. This method is a significant simplification of conventional methods that require storage in separate pads and subsequent placement in the device and is a first step toward roll-to-roll fabrication of 2DPN devices (Figure 1C). Here, the proof-of-concept device was tested with mock samples of antigen spiked into fetal bovine serum. The signal enhancement step increases the visible signal by roughly 3-fold and increases the analytical limit of detection by 2.75-fold.
Experimental Section
Device Construction and Patterning
All porous devices were fabricated using untreated backed nitrocellulose membranes with a nominal pore diameter of 8 μm (Millipore Hi Flow Plus 135, Millipore, Billerica, MA). Glass fiber pads (Ahlstrom, Helsinki, Finland) were used as fluid sources, and cellulose pads were used as downstream wicks to drive capillary flow throughout the duration of the assay. All of these materials were cut using a CO2 laser (Universal Laser Systems, Scottsdale, AZ), using a previously described cutting protocol.28 Nitrocellulose membranes were cut into 3-inlet networks (as shown in Figure 2). After cutting, 14 μL of blocking solution was applied to the first inlet to minimize nonspecific protein adsorption during storage. This blocking solution consisted of 0.125% poly(vinylpyrrolidone) + 0.125% bovine serum albumin + 2.5% sucrose + 7.5 mM sodium azide + 0.1% Tween-20 in water. After blocking, membranes were placed in a desiccated oven at 37 °C for 2 h and then transferred to a desiccator for storage. An antigen-capture line (0.375 μL, 1 mg/mL mouse monoclonal anti-PfHRP2 IgM, Immunology Consultants Lab) and a process control (0.375 μL, 0.5 mg/mL ImmunoPure Antibody goat anti-mouse IgG, Thermo Scientific) were immobilized at the downstream end of the common channel of the device via printing with a piezoelectric spotter (SciFLEXARRAYER S3, Scienion AG) (Figure 1). The nonspecific adsorption of proteins to nitrocellulose in this way has long been used in the fabrication of LFTs.13
Figure 2.
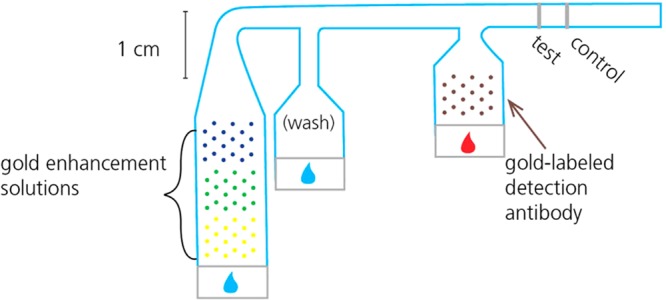
Schematic of patterned 2D paper network PfHRP2 assay indicating locations of patterned reagents. Mock sample is indicated by the red drop; buffer is indicated by the blue drops.
The detection antibody (Immunogold conjugate mouse monoclonal anti-PfHRP2, BBInternational) and gold enhancement solutions (GoldEnhance LM, Nanoprobes, Yaphank, NY) were also printed onto the porous device using the piezoelectric printer, but these reagents were deposited in patterns to prevent nonspecific immobilization and facilitate controlled rehydration (see Figure 1 for a schematic of pattern and locations). To achieve this controlled rehydration, the detection antibody was mixed with 5% sucrose, 5% trehalose, and 1% BSA prior to printing it onto the nitrocellulose surface. In our previous work, we demonstrated that these additives improve the uniformity of rehydration of proteins after storage.20 Then, a total of 2 μL of the detection antibody was patterned on the first inlet in a single array of 29 spots, spaced at 1 mm from each other to prevent disruption of wet-out flow. Each of the three components of the gold enhancement reagent was patterned in a separate 2 μL, 29-spot array onto the third inlet. These three reagent arrays were sequenced in the order that the three solutions were required to mix: “enhancer” solution first must mix with “activator” and then that combined solution must mix with the “initiator”. These components are proprietary so their exact composition is unknown to us; however, the basic process is as follows: the “enhancer” solution contains a gold salt that is chemically reduced to gold atoms by the combination of the “activator” and “initiator” in the reconstituted solution, in the presence of the gold conjugate. These gold atoms are deposited onto the surface of the gold conjugate nanoparticle, and this changes the light absorption of the conjugates captured at the detection region. The third inlet of the device was not preblocked, and no protein was added to the gold enhancement solutions because it was found to inhibit the enhancement activity of those reagents. After printing, strips were wrapped in foil to protect them from light, then dried in a desiccator, and stored there until use (2–5 days).
Assay Demonstration
In initial experiments, nitrocellulose devices were affixed to a PMMA substrate using double-sided tape (Scotch, 3M, St. Paul, MN), and an untreated glass fiber pad (Ahlstrom, Helsinki, Finland), cut using the CO2 laser cutter, was placed at the upstream end of each inlet as a fluid application zone (Figure 2). A 30 μL “mock sample” consisting of known concentrations of stock PfHRP2 antigen spiked into fetal bovine serum (FBS, Certified, One Shot, US Origin, Gibco, 16000-077, Invitrogen, Carlsbad, CA) was applied to the first inlet, while 40 and 100 μL of phosphate buffered saline (PBS) were applied to the second and third inlets, respectively. The mock sample rehydrated the gold conjugate antibody in the first inlet, and PBS rehydrated and combined the gold enhancement reagents in the third inlet. Rehydrated reagents were then delivered sequentially to the detection region of the assay. Time-lapse uncompressed AVI videos of assay experiments were acquired using HandyAvi software (AZcendant, Tempe, AZ) on a web camera (Logitech, Fremont, CA) at 1 frame per 30 s for 1 h (Figure 3).
Figure 3.

Time series of schematics and images illustrating gold signal development and enhancement. (A) Device prior to fluid addition. Anti-PfHRP2 IgM was immobilized as a capture line, while goat anti-mouse IgG was used as a control line. Thirty μL of a mock sample consisting of PfHRP2 spiked at 100 ng/mL into fetal bovine serum was applied to the right-most inlet, where Anti-PfHRP2 IgG-gold conjugate was patterned for rehydration. Immediately afterward, 40 and 100 μL of PBS were applied to the middle and left inlets, respectively. (B) The initial gold signal appears 15 min after fluid addition. At the test line, the capture IgM, PfHRP2 antigen, and gold conjugate form a “sandwich”, while the control line consists of goat anti-mouse IgG binding to the mouse IgGs of the gold conjugate. (C) Finally, the three gold enhancement solutions patterned on the left-most leg recombine upon rehydration and deposit more gold ions onto the surface of the gold nanoparticles, shifting the absorbance and turning the lines a dark black color. By 60 min, the gold enhancement has increased the signal to 3.2-fold (s.d. 0.2) of the unenhanced intensity.
To quantify the baseline analytical limit of detection of the PfHRP2 assay, mock samples were generated by diluting PfHRP2 antigen (ImmunoDx, Woburn, MA) in FBS to 0, 5, 10, 25, and 50 ng/mL on the same day that the assays were performed. All assays for limit of detection calculation were performed on the same day. In these experiments, assay device enclosures were cut from adhesive-coated Melinex sheets (Fralock, San Carlos, CA) using the CO2 laser cutter. (See Fu et al. for a full description of the development of analogous enclosures.8) In these cases, strips were scanned to uncompressed image files with a flatbed scanner (Epson Perfection V700 Photo, Epson, Long Beach, CA) with gamma set to 1. Devices were imaged at 15 and 60 min. Four replicates were performed at each concentration (resulting in 20 devices overall), but two devices were designated as enhancement failures because the control line did not fully enhance, and these are not included in the graphical representation of results or LOD analysis (Figure 5).
Figure 5.

Unenhanced (blue) and enhanced (red) assay results for varying concentrations of PfHRP2 antigen. Four replicates of each concentration were performed, and assay devices were imaged at 15 min to quantify unenhanced signal and then again at 60 min to quantify the enhanced signal. The average signal for each concentration of antigen is plotted here (n = 4, error bars = s.d.* n = 2 for the enhanced 10 ng/mL data due to two tests failing to enhance fully.) Dashed lines indicate the limit of detection signal intensity for both enhanced (red) and unenhanced (blue) assays, and the short vertical lines intersecting those dashed lines indicate the PfHRP2 concentration corresponding to that signal (at 3.6 and 9.9 ng/mL, respectively). The dotted lines indicate the limit of blank for both enhanced (red) and unenhanced (blue) assays.
Assay Analysis
Uncompressed AVI files of preliminary experiments were analyzed with ImageJ to determine the signal development over time (Figure 4). The reported intensity values were determined as follows: average grayscale intensity of the test line was quantified for each frame, and background intensity was subtracted and then normalized by the background to account for lighting variations. The enhancement ratio was determined by quantifying the fully enhanced assay signal at 60 min and dividing it by the unenhanced signal at 15 min. To quantify signal for varying concentrations of PfHRP2 (Figure 5), scanned images were quantified using Matlab (MathWorks, Natick, MA), using a script that autodetected the location and intensity of the test lines based on the location of the control line. The grayscale intensity of the test line was measured, and then, the background value was subtracted. The limit of detection (LOD) of the assay was calculated as follows: LOD = (LOB + 1.645σt)/m, where LOB = limit of blank (to be explained below), σt = the standard deviation of the lowest antigen test, and m is the slope of the signal response curve between zero and the lowest antigen test. The limit of blank is defined as LOB = averageblank + σb, where σb = standard deviation of the blank (no antigen control).29
Figure 4.
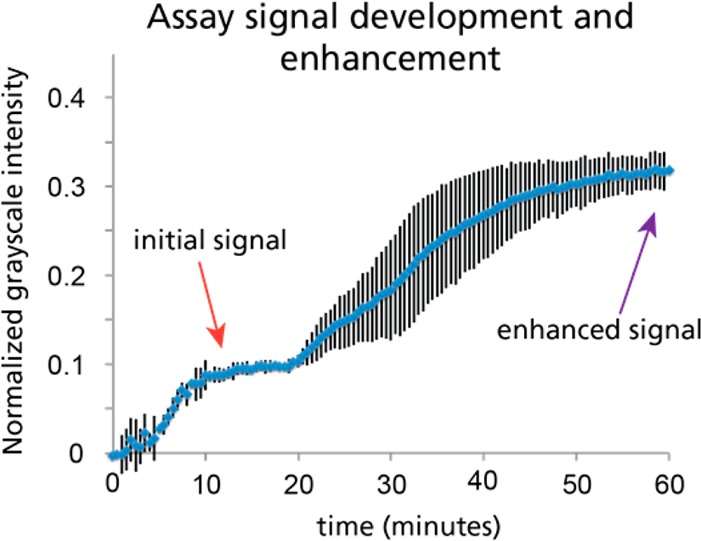
Assay signal vs time. Time-lapse uncompressed AVI files were acquired (1 frame per 30 s) and were analyzed using ImageJ (n = 4, error bars = s.d.). The large error bars between 25 and 50 min are due to variability in the time at which enhancement began, rather than the rate of enhancement. Over 3-fold enhancement is achieved through the use of the gold enhancement system.
Results and Discussion
Figure 3 shows a schematic of the different assay steps performed in this device and an image of the device at each time point taken from videos of the entire assay performed with mock samples at 100 ng/mL antigen spiked into fetal bovine serum. Videos were analyzed to observe the assay signal development over time. After 60 min, the rehydration and midflow combination of printed gold enhancement reagents produced a 3.2-fold signal enhancement (n = 4, s.d. 0.2), relative to the initial signal observed at 15 min (Figure 4). Just 2 μL of gold-antibody conjugate and 2 μL of each gold enhancement reagent were used. These volumes are significantly lower than those required in previous demonstrations of signal enhancement in paper-based assays, where at least 9 μL of each reagent was used,7,8 resulting in at least 4.5 times less reagent cost for each device. Though the enhancement ratio achieved with this particular gold enhancement reagent is modest, this is a clear demonstration of the viability of patterning and drying reagents for dry reagent storage in paper-based assays.
To quantify the enhanced and unenhanced analytical limit of detection of this device design, mock samples at several different antigen concentrations (0, 5, 10, 25, and 50 ng/mL) were tested using the patterned reagent device. Clear visible signal was observed in all enhanced assay containing nonzero PfHRP2 levels. The unenhanced signal generated by the 10 ng/mL assay was detectable by the eye; however, the signal from the 5 ng/mL test was not visible. For quantitative analysis, the pre- and post-enhanced assay signal was determined from scanned images using the Matlab script mentioned above. This quantification confirmed reproducible signal at all concentrations for the enhanced assay and as low as 10 ng/mL for the unenhanced assay. Both pre- and post-enhancement images had very low background signal in the negative controls, though the post-enhancement negatives did exhibit slightly darker and noisier negatives, which led to a slightly higher “limit of blank” for the enhanced assay (Figure 5). The intermediate wash step is used for two purposes: first, to rinse over the detection region to reduce false positive signal and, second, to prevent the gold conjugate from contacting the reconstituted gold enhancement system, which would also lead to higher false positives.
The wash and enhancement steps lead to an increase in signal observed in the enhanced assay that is significantly more than the increase in the limit of blank, however, which yields a 2.75-fold improvement in the overall limit of detection in the enhanced case. The quantified limit of detection of the unenhanced assay was 9.9 ng/mL, whereas the enhanced assay resulted in a limit of detection of 3.6 ng/mL (Figure 5).
Though this is just an initial proof-of-concept demonstration of the viability of patterning reagents for dry storage and subsequent rehydration, it is useful to compare these values with published limits of detection for a variety of malaria diagnostics (Table 1) to benchmark whether this sensitivity is even within a relevant clinical range. Comparing to other limits of detection published in the literature, the analytical limit of detection for our assay is much lower than detection by microscopy and lower than observed LODs for commercially available RDTs and is comparable to the upper end of reported ELISA detection LOD (see references included in table). Even at this early stage of development, this technique of patterning and later rehydrating reagents from dry storage within the membrane achieves a limit of detection within the clinical range, and the addition of a signal enhancement step significantly improves the limit of detection achieved by this device.
Table 1. Reported Limits of Detection for Various Malaria Diagnostics.
| method | reported limit of detection | converted limit of detection (parasites/μL ⇔ ng/mL)a | source |
|---|---|---|---|
| microscopy | 20–50 parasites/μL | 18–44 ng/mL | Moody et al.31 and Guerin et al.32 |
| ELISA | 0.1–4 ng/mL | roughly 0.1–4.5 parasites/μL | Butterworth et al.,33 Dondorp et al.,34 Kifude et al.35 |
| in-lab RDTs (FirstSign–ParaView; SD BIOLINE; Carestart; ICT Malaria Combo Cassette) | 6.94–27.75 ng/mL | 8–30 parasites/μLa | Marquart et al.36 |
| RDTs in the field | varies widely; ∼100 parasites/μL | ∼90 ng/mL | Moody et al.,31 Hendriksen et al.37 |
Calculated on the basis of the model developed by Marquart et al. correlating PfHRP2 concentration to parasite levels, using their maximum circulating approximation: 6.94 ng/mL ∼ 7.8 parasites/μL.36
The current device could benefit from several process improvements, however, to lower the limit of detection even further. The second leg of the device (Figure 2) is a wash step that prevents the gold conjugate from coming into direct contact with the gold enhancement solution and rinses the detection region to reduce nonspecific binding of conjugate. A more rigorous wash step may be able to further reduce this nonspecifically bound gold conjugate. Additionally, an improved assay quantification algorithm could be designed to differentiate between stray marks and true signal (for example, one negative test had a dark smudge at the upper edge, which was included in the quantitation method of the current algorithm, but was clearly not spanning the width of the strip). Another feature of this device that needs some improvement is the reproducibility of enhancement: out of the 20 devices that were tested, 2 failed to enhance fully. Using the detection algorithm and the measured limit of detection, both of these tests would be correctly designated as positive results, though if the level of antigen were quantified, it would be underestimated due to the incomplete enhancement. We hypothesize that the incomplete enhancement is due to a defect in timing that caused the enhancement and wash fluid streams to pass over the detection line simultaneously, with the enhancement stream at the top edge of the strip and the wash stream at the lower edge of the strip. Improvements in device actuation, such as the implementation of inlet capillaries as shown by Dharmaraja et al.,30 could mitigate this problem.
In the devices described here, all reagents were patterned onto the paper membranes and allowed to dry at room temperature for between 2 and 5 days before use in the assay. This demonstration of reagent drying within the porous nitrocellulose matrix, successful rehydration, and subsequent viability in the sample assay is a valuable first step toward incorporating these techniques in clinically relevant assays. These methods are very well suited for roll-to-roll manufacturing techniques, which are much more efficient for mass-scale device fabrication than the pick-and-place methods that would be required to fabricate an analogous 2DPN with many different dried reagent pads (see comparison in Figure 1.) This is a proof-of-concept study to demonstrate the feasibility of patterned reagents in the context of paper-based assays; however, even after improving the details of device operation described above, there are several important next steps needed before this assay device could be used in a clinical setting: (1) future studies designed to determine longer-term shelf stability and resistance to damage at extreme temperatures, (2) clinical sample validation, using blinded samples both positive and negative for P. falciparum malaria at a wide range of parasitemia levels, and (3) manufacturing optimization to implement these fabrication methods within a roll-to-roll process. Another area of future work that is currently being actively pursued in the laboratory is implementing similar methods in other assay systems. The gold enhancement chemistry is effective for any gold-conjugate-based detection, so for another immunoassay, the only part of the assay that needs to be replaced is the specific detection antibodies both at the capture region and those conjugated to the gold nanoparticle for detection.
In this paper, we have shown a proof-of-concept demonstration of the potential of a novel system for incorporating dry reagents into paper-based diagnostic devices. Dry reagents are an essential component of rapid diagnostics for point-of-care use, primarily because wet reagents require refrigeration and limit automation. Previously, dry reagents in paper-based devices had only been included via conjugate pads. Here, a novel method was described to pattern reagents directly onto a paper substrate for dry-down and subsequent rehydration in the context of a multistep immunoassay. There are many advantages to patterning reagents directly onto porous substrates rather than incorporating several individual dried reagent pads. First, patterned reagents provide improved control over dry reagent rehydration, allowing customized rehydration profiles.20 Second, smaller volumes of reagents can be used, reducing the material costs of an assay. Third, applying reagents directly to the assay substrate reduces the number of pieces required in an assay and, thus, the possible sources of malfunction or error.
Conclusions
Here, we have presented a simple and effective demonstration of the feasibility and utility of patterning multiple reagents sequentially on a porous device; it is just one example of the potential applications enabled by printing reagents directly on porous devices for controlled rehydration and use in assays. Generally, two-dimensional paper network assays offer a significant improvement over traditional lateral flow rapid tests by incorporating additional assays steps to dramatically improve assay sensitivity: our methods to pattern reagents for storage and rehydration take these advancements a step further by reducing the amount of reagents required to achieve the same level of performance, while also simplifying device manufacturing. Though there are many more steps required before this demonstration is suitable for commercial-scale manufacturing and clinical use, fabrication considerations are absolutely essential for engineers who are seeking to develop technologies that will have the ability to make a real impact on point-of-care diagnostic medicine; technologies that cannot be manufactured efficiently will never achieve the low cost and robustness that are required to make them deliverable to the patients and health centers that most desperately need them.
Acknowledgments
This material is based upon work supported by the National Science Foundation Graduate Research Fellowship for G.E.F. under Grant No. DGE – 0718124, as well as NIH 1 R01 AI 096184-01. Additional funding was provided by the University of Washington Bioengineering Department. The authors would also like to thank Carly Holstein, Shefali Oza, and Elain Fu for valuable discussions and Dean Stevens for use of his Adobe Illustrator assay schematics (used in Figure 3).
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
References
- Peeling R. W.; Holmes K. K.; Mabey D.; Ronald A. Sex. Transm. Infect. 2006, 82, V1–V6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Posthuma-Trumpie G. A.; Korf J.; van Amerongen A. Anal. Bioanal. Chem. 2009, 393, 569–582. [DOI] [PubMed] [Google Scholar]
- Martinez A. W.; Phillips S. T.; Butte M. J.; Whitesides G. M. Angew. Chem., Int. Ed. 2007, 46, 1318–1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez A. W.; Phillips S. T.; Whitesides G. M.; Carrilho E. Anal. Chem. 2010, 82, 3–10. [DOI] [PubMed] [Google Scholar]
- Martinez A. W.; Phillips S. T.; Nie Z.; Cheng C.-M.; Carrilho E.; Wiley B. J.; Whitesides G. M. Lab Chip 2010, 10, 2499–2504. [DOI] [PubMed] [Google Scholar]
- Govindarajan A. V.; Ramachandran S.; Vigil G. D.; Yager P.; Boehringer K. F. Lab Chip 2012, 12, 174–181. [DOI] [PubMed] [Google Scholar]
- Fu E.; Liang T.; Houghtaling J.; Ramachandran S.; Ramsey S. A.; Lutz B.; Yager P. Anal. Chem. 2011, 83, 7941–7946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu E.; Liang T.; Spicar-Mihalic P.; Houghtaling J.; Ramachandran S.; Yager P. Anal. Chem. 2012, 84, 4574–4579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens D. Y.; Petri C. R.; Osborn J. L.; Spicar-Mihalic P.; McKenzie K. G.; Yager P. Lab Chip 2008, 8, 2038–2045. [DOI] [PubMed] [Google Scholar]
- Crowe J. H.; Carpenter J. F.; Crowe L. M. Annu. Rev. Physiol. 1998, 60, 73–103. [DOI] [PubMed] [Google Scholar]
- Mabey D.; Peeling R. W.; Ustianowski A.; Perkins M. D. Nat. Rev. Microbiol. 2004, 2, 231–240. [DOI] [PubMed] [Google Scholar]
- Yager P.; Domingo G. J.; Gerdes J. Annu. Rev. Biomed. Eng. 2008, 10, 107–144. [DOI] [PubMed] [Google Scholar]
- Wong R., Tse H.Lateral Flow Immunoassay; Humana Press: New York, 2009. [Google Scholar]
- Garcia E.; Kirkham J. R.; Hatch A. V.; Hawkins K. R.; Yager P. Lab Chip 2004, 4, 78–82. [DOI] [PubMed] [Google Scholar]
- Hitzbleck M.; Gervais L.; Delamarche E. Lab Chip 2011, 11, 2680–2685. [DOI] [PubMed] [Google Scholar]
- Abe K.; Kotera K.; Suzuki K.; Citterio D. Anal. Bioanal. Chem. 2010, 398, 885–893. [DOI] [PubMed] [Google Scholar]
- Komuro N.; Takaki S.; Suzuki K.; Citterio D. Anal. Bioanal. Chem. 2013, 405, 5785–5805. [DOI] [PubMed] [Google Scholar]
- Hossain S. M. Z.; Luckham R. E.; Smith A. M.; Lebert J. M.; Davies L. M.; Pelton R. H.; Filipe C. D. M.; Brennan J. D. Anal. Chem. 2009, 81, 5474–5483. [DOI] [PubMed] [Google Scholar]
- Wang J. Y.; Monton M. R. N.; Zhang X.; Filipe C. D. M.; Pelton R.; Brennan J. D. Lab Chip 2014, 14, 691–695. [DOI] [PubMed] [Google Scholar]
- Fridley G. E.; Le H. Q.; Fu E.; Yager P. Lab Chip 2012, 12, 4321–4327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- UNITAID. Malaria Diagnostic Technology Landscape Report; World Health Organization: Geneva, 2011. [Google Scholar]
- Shakely D.; Elfving K.; Aydin-Schmidt B.; Msellem M. I.; Morris U.; Omar R.; Xu W. P.; Petzold M.; Greenhouse B.; Baltzell K. A.; Ali A. S.; Bjorkman A.; Martensson A. Plos One 2013, 8, e72912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris I.; Sharrock W. W.; Bain L. M.; Gray K.-A.; Bobogare A.; Boaz L.; Lilley K.; Krause D.; Vallely A.; Johnson M.-L.; Gatton M. L.; Shanks G. D. Cheng Q.. Malaria J. 2010, 9; DOI: 10.1186/1475-2875-9-254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cazacu A. C.; Greer J.; Taherivand M.; Demmler G. J. J. Clin. Microbiol. 2003, 41, 2132–2134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uyeki T. M.; Prasad R.; Vukotich C.; Stebbins S.; Rinaldo C. R.; Ferng Y.-h.; Morse S. S.; Larson E. L.; Aiello A. E.; Davis B.; Monto A. S. Clin. Infect. Dis. 2009, 48, E89–E92. [DOI] [PubMed] [Google Scholar]
- Vasoo S.; Stevens J.; Singh K. Clin. Infect. Dis. 2009, 49, 1090–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu E.; Kauffman P.; Lutz B.; Yager P. Sens. Actuators, B: Chem. 2010, 149, 325–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spicar-Mihalic P.; Toley B.; Houghtaling J.; Liang T.; Yager P.; Fu E.. J. Micromech. Microeng., 2013, 23; DOI: 10.1088/0960-1317/23/6/067003. [DOI] [Google Scholar]
- Armbruster D. A.; Pry T. Clin. Biochem. Rev. 2008, 29Suppl 1S49–S52. [PMC free article] [PubMed] [Google Scholar]
- Dharmaraja S.; Lafleur L.; Byrnes S.; Kauffman P.; Buser J.; Toley B.; Fu E.; Yager P. Lutz B.. Programming paper networks for point of care diagnostics. Proceedings of SPIE, 8615, Microfluidics, BioMEMS, and Medical Microsystems XI, 86150X, 2013; DOI: 10.1117/12.2006138. [DOI] [Google Scholar]
- Moody A. Clin. Microbiol. Rev. 2002, 15, 66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerin P. J.; Olliaro P.; Nosten F.; Druilhe P.; Laxminarayan R.; Binka F.; Kilama W. L.; Ford N.; White N. J. Lancet Infect. Dis. 2002, 2, 564–573. [DOI] [PubMed] [Google Scholar]
- Butterworth A. S.; Robertson A. J.; Ho M.-F.; Gatton M. L.; McCarthy J. S.; Trenholme K. R. Malaria J. 2011, 10, 95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dondorp A. M.; Desakorn V.; Pongtavornpinyo W.; Sahassananda D.; Silamut K.; Chotivanich K.; Newton P. N.; Pitisuttithum P.; Smithyman A. M.; White N. J.; Day N. P. J. PloS Med. 2005, 2, 788–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kifude C. M.; Rajasekariah H. G.; Sullivan D. J. Jr.; Stewart V. A.; Angov E.; Martin S. K.; Diggs C. L.; Waitumbi J. N. Clin. Vaccine Immunol. 2008, 15, 1012–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marquart L.; Butterworth A.; McCarthy J. S.; Gatton M. L. Malaria J. 2012, 11, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendriksen I. C. E.; Mtove G.; Pedro A. J.; Gomes E.; Silamut K.; Lee S. J.; Mwambuli A.; Gesase S.; Reyburn H.; Day N. P. J.; White N. J.; von Seidlein L.; Dondorp A. M. Clin. Infect. Dis. 2011, 52, 1100–1107. [DOI] [PMC free article] [PubMed] [Google Scholar]