

Abstract
Background
Hutchinson-Gilford progeria syndrome is an ultra-rare segmental premature aging disease resulting in early death from heart attack or stroke. There is no approved treatment, but starting in 2007, several recent single arm clinical trials have administered inhibitors of protein farnesylation aimed at reducing toxicity of the disease-producing protein progerin. No study has assessed whether treatments influence patient survival. The key elements necessary for this analysis are a robust natural history of survival and comparison with a sufficiently large patient population that has been treated for a sufficient time period with disease-targeting medications.
Methods and Results
We generated survival Kaplan-Meier survival analyses for the largest untreated Hutchinson-Gilford progeria syndrome cohort to date. Mean survival was 14.6 years. Comparing survival for treated versus age-and-gender-matched untreated cohorts, hazard ratio was 0.13 (95% CI 0.04-0.37; P<0.001) with median follow-up of 5.3 years from time of treatment initiation. There were 21/43 deaths in untreated versus 5/43 deaths among treated subjects. Treatment increased mean survival by 1.6 years.
Conclusions
This study provides a robust untreated disease survival profile, which can be utilized for comparisons now and in the future to assess changes in survival with treatments for HGPS. The current comparisons estimating increased survival with protein farnesylation inhibitors provide the first evidence of treatments influencing survival for this fatal disease.
Clinical Trial Registration Information
www.clinicaltrials.gov. Indentifiers: NCT00425607, NCT00879034 and NCT00916747.
Keywords: progeria, Hutchinson-Gilford progeria syndrome, lamin, farnesylation, FTI, aging, atherosclerosis
Background
Hutchinson-Gilford progeria syndrome (HGPS) is a sporadic autosomal dominant segmental premature aging disease with an incidence of 1 in 4 million1. Cardiovascular disease in HGPS is characterized by early and pervasive vascular stiffening, along with later-stage arterial occlusive disease2-4. These factors are major contributors to an accelerated form of premature atherosclerosis that culminates in early death from heart attack or, less often, stroke.
The genetic mutations causing HGPS are a series of silent point mutations in the LMNA gene that increase the use of an internal splice site5, 6 resulting in translation of the disease-causing abnormal lamin A protein, progerin. The normal LMNA gene encodes lamin A, a principal protein of the nuclear lamina, which is a complex molecular interface located between the inner membrane of the nuclear envelope and chromatin (reviewed in Broers et al7). The integrity of the lamina is central to many cellular functions, creating and maintaining structural integrity of the nuclear scaffold, DNA replication, RNA transcription, organization of the nucleus, nuclear pore assembly, chromatin function, cell cycling, and apoptosis.
Disease in HGPS is produced by a dominant negative mechanism; it is the effect of progerin, not the diminution of lamin A, which causes the disease phenotype8. Progerin is found in increased concentration in skin and the vascular wall of normal older compared to younger individuals, suggesting a role in normal aging2. Unlike lamin A, progerin lacks the proteolytic cleavage site required for removal of its post-translationally attached farnesyl moiety9. Progerin is postulated to remain associated with the inner nuclear membrane, unable to be released for degradation due to persistent farnesylation10-13.
The pathologic effects of progerin farnesylation form the central hypothesis underlying treatment protocols utilizing protein farnesylation inhibitors in HGPS. Preclinical studies administering farnesylation inhibitors have demonstrated positive effects on both in vitro14, 12, 15 and murine in vivo progeria disease models16-20. The preclinical data in support of farnesylation inhibitors was encouraging, but complicated. With treatment, HGPS fibroblasts displayed improved nuclear morphology, gene expression, cellular lifespan, and nuclear stiffness14, 12, 15, 21. However, HGPS fibroblasts also exhibited the potential for alternative prenylation 19, and lack of improved sensitivity to mechanical strain21 with FTI treatment. In vivo, several progeroid mouse models displayed improved phenotype22, 17, 19, 20, and in some cases extended lifespan22, 17, 19. However, some mouse models display bone or neurological morbidity without overt Cardiovascular (CV) morbidity, and cause of death is undetermined for any mouse model. Given the complicated preclinical results, extended survival in humans could not be assumed, and could only be tested with adequate human cohort numbers and treatment duration.
The first human clinical treatment trial for HGPS administered the protein farnesyltransferase inhibitor (FTI) lonafarnib for 2 years23. CV and neurovascular (NV) results demonstrated evidence for decreased vascular stiffness23, incidence of stroke, TIA and headache24. There was also evidence for skeletal and audiologic benefit23. Improvements occurred in some, but not all subjects, and some disease phenotypes were not improved with lonafarnib. Trial duration was inadequate to test influence on survival. The second and currently ongoing trial added two additional medications to lonafarnib, also aimed at inhibiting progerin farnesylation. The statin pravastatin inhibits HMG-CoA reductase and the bisphosphonate zoledronate inhibits farnesyl-pyrophosphate (PP) synthase19; each enzyme functions along the protein prenylation pathway (Fig. 1).
Figure 1.

Current HGPS treatment strategies aimed at preventing formation of progerin protein by inhibiting post-translational farnesylation of preprogerin. Enzymes facilitating each step are italicized. Dashed line indicates multiple steps in pathway not shown. Medications aimed at inhibiting protein farnesylation are circled. ICMT = isoprenylcysteine carboxyl methyltransferase
Along with their influences on protein prenylation, both pravastatin and zoledronate affect disease in non-HGPS subjects using mechanisms of action independent of the prenylation pathway. There exists both direct and indirect support for efficacy of these drugs specifically through inhibiting progerin prenylation in HGPS versus alternative mechanisms of action. In vitro, phenotypic improvements in progeroid mouse fibroblasts treated with pravastatin plus zoledronate are completely abolished when cells are allowed to specifically by-pass the need for HMG-CoA reductase and farnesyl-PP synthase19. In vivo, statins have been shown to exert beneficial cardiovascular effects through mechanisms distinct from their effect in lowering cholesterol and low-density-lipoproteins 25. Additional statin effects have been demonstrated in pathways of inflammation, immunomodulation and thrombosis. However, statin's usual target pathways do not appear as significant components in the HGPS population. Children with HGPS exhibit normal values for serum total cholesterol and LDL, serum high-sensitivity C-reactive protein 26, 27, arterial intima-media thickness27, 3 and demonstrate no overt evidence of endothelial dysfunction. Finally, although zoledronate exhibits its major effects by decreasing bone resorption and ultimately improving bone density28, and both bone density and skeletal morphology are affected in HGPS29; fracture rate is normal in HGPS30 and subjects do not die from bone disease. Thus, influence on HGPS lifespan in humans stemming from zoledronate would likely be due to effects outside of the skeletal system.
Assessing change in population survival due to treatment necessitates a robust analysis of the untreated HGPS comparison population. Two studies have estimated mean survival for this disease group at 13.431 and 12.61 years, based mainly on literature searches. Neither included subjects who were living or lost to follow-up (censored data), nor did they generate survival curves. We developed Kaplan-Meier curves and survival estimates for a large untreated HGPS cohort. To assess whether treatment has an influence on survival for children with HGPS, we provide comparisons between this untreated cohort and a treated cohort that received HGPS-specific treatments during clinical trials. A robust comparison required subject matching with regard to age, gender, dates, and other potential confounding factors.
Methods
Inclusion Criteria and Demographics
This project was approved by the Rhode Island Hospital Institutional Review Board. Some data were obtained through a Data Use Agreement between The Progeria Research Foundation, Rhode Island Hospital, and Brown University. The clinical trials were approved by the Boston Children's Hospital Committee on Clinical Investigation.
Study subjects were identified using The Progeria Research Foundation International Registry (www.progeriaresearch.org), published scientific and news articles, and publicly available databases. Minimum inclusion criteria were phenotype confirmation by study investigators, living age or age of death, inclusion history in progeria clinical trials NCT00425607 (lonafarnib monotherapy), NCT00879034 and NCT00916747 (lonafarnib, zoledronate, and pravastatin combination therapy), and treatment duration. Due to institutional restrictions, ten subjects with HGPS included in an open label clinical trial conducted in Marseilles (NCT00731016) were unavailable for inclusion in this study (N. Levy, personal communication).
Untreated subjects had not received clinical trial medications within any clinical treatment trial for HGPS. Treated subjects received trial medications for any length of time; treatment initiation and duration varied.
HGPS was defined by clinical phenotype, which is consistent and unique from non-progerin producing progeroid laminopathies. The main differential diagnosis for HGPS includes mandibulo-acral dysplasia and restrictive dermopathy, which are both due to alternative mutations in LMNA. These are both quite distinct in appearance, so there is little possible confusion with the classic HGPS 32. Where genotype was known, all positive cases by phenotype contained a progerin-producing mutation in the LMNA gene. While exclusion of non-progerin producing laminopathies is reliably accomplished using phenotype in the absence of genotype, there are cases where the splicing mutation yields very low levels of progerin and a clinically different phenotype which is not categorized as HGPS 33, 34. These subjects were not included in the analysis because they are considered non-HGPS by phenotype.
Statistics
Authors JM and RBD performed all statistical analyses. Demographic characteristics are presented with counts and percentages and compared between groups using Fisher's exact test. Untreated patient survival age was estimated by the Kaplan-Meier method. Untreated subjects living as of the start of data analysis, and subjects appearing in a published report living at the time of the report, but then lost to follow-up (Table S1), were censored at time of last known living age. Treatment trial subjects were included as part of the untreated cohort until age of treatment initiation.
Cox proportional hazards regression was used to compare treated and untreated (i.e., never treated) groups for survival. To control for potential confounding variables, gender- and age-matching was performed, and the untreated subject pool included only those born on or after 1991, the year on or after which all treated subjects were born. For every treated patient, all untreated subjects of the same gender who were alive at the age when the treated patient began treatment were identified; from this group of untreated subjects, one was randomly selected and used as the matched untreated patient in the analysis; once an untreated patient was matched to a treated patient, the untreated patient was no longer available for matching. Patient follow-up begins at time 0, where time 0 is set to the age of treatment initiation for the treated patient in the matched pair. Supportive analyses were performed where all subjects were followed from birth and placed at risk at the age of treatment initiation. Age, gender and continent of residency were included as covariates in these Cox models.
Treated and untreated subjects born on or after 1991 were compared using treatment (yes/no) as a time-dependent covariate, where all treated subjects were considered untreated until time of treatment initiation and where gender and continent of residence were included as covariates. For at least the first 2 years of age, all subjects are untreated, yielding 0 treated subjects during this time frame and 8 treated subjects through approximately ages 0-4. In other words, while all subjects (treated and untreated) are theoretically placed at risk for mortality at birth in the time-dependent analysis, in reality the treated subjects are only truly at risk at the age they began treatment, which was at least 2 years of age for all treated subjects, theoretically yielding a survival advantage in at least the first 2 years of life for the treated patients over the untreated patients. The survival advantage was not large, as only 1 untreated patient born after 1991 died before two years of age; nevertheless for this potential bias in favor of the treated group, we considered the time-dependent analysis as supportive.
Hazard ratios and their two-sided 95% confidence intervals for mortality in treated vs. untreated were calculated. Sensitivity analyses were conducted by removing or censoring subjects with various confounding variables listed in results section.
Estimated extension in mean survival with treated versus untreated subjects was calculated by comparing areas under the treated and untreated Kaplan-Meier curves for the matched sample set.
Statistical analysis was carried out by SAS Version 9.3 and STATA v12. P-values are two-sided and deemed significant at 0.05.
Results
Patient Characteristics
Overall, 161 untreated subjects and 43 treated subjects (100% of HGPS clinical trial subjects) were eligible for analysis. Subject sources are detailed in Supplementary Appendix. For matched analysis of untreated versus treated cohorts, gender (P=1.00), continent (P=0.39) and known mutation subgroups (P=0.16) were similar (Table 1).
Table 1. Patient Characteristics.
| Variable | All (N=204) N (%) |
Untreated (N=161) N (%) |
Treated* (N=43) N (%) |
Matched Untreated (N=43) N (%) |
|---|---|---|---|---|
| Females | 98 (48.0) | 72 (44.7) | 26 (60.5) | 26 (60.5) |
| Males | 106 (52.0) | 89 (55.3) | 17 (39.5) | 17 (39.5) |
| Born on or After 1986 | 136 (66.7) | 93 (57.8) | 43 (100.0) | 43 (100.0) |
| Born on or After 1991 | 118 (57.8) | 75 (46.6) | 43 (100.0) | 43 (100.0) |
| Known Genotype | 105 (51.5) | 62 (38.5) | 43 (100.0) | 24 (55.8) |
| Continent | ||||
| Africa | 10 (4.9) | 5 (3.1) | 5 (11.6) | 1 (2.3) |
| Asia | 37 (18.1) | 30 (18.6) | 7 (16.3) | 9 (20.9) |
| Australia | 2 (1.0) | 2 (1.2) | 0 (0.0) | 0 (0.0) |
| Europe | 45 (22.1) | 35 (21.7) | 10 (23.3) | 11 (25.6) |
| North America | 78 (38.2) | 63 (39.1) | 15 (34.9) | 12 (27.9) |
| South America | 32 (15.7) | 26 (16.2) | 6 (14.0) | 10 (23.3) |
| Mutation Subgroup** | ||||
| c.1824 C>T; p.G608G | 89 (84.8) | 50 (80.6) | 39 (90.7) | 18 (75) |
| c.1822 G>A, p.G608S | 5 (4.8) | 3 (4.9) | 2 (4.7) | 1 (4.2) |
| Intron 11, c.1968+1 G>C | 2 (1.9) | 2 (3.3) | 0 (0.0) | 1 (4.2) |
| Intron 11, c.1968+1 G>A | 5 (4.8) | 4 (6.6) | 1 (2.3) | 2 (8.3) |
| Intron 11, c.1968+2 T>A | 2 (1.9) | 2 (3.3) | 0 (0.0) | 1 (4.2) |
| Intron 11, c.1968+2 T>C | 1 (1.0) | 1 (1.6) | 0 (0.0) | 1 (4.2) |
| Intron 11, c 1968+5 G>C | 1 (1.0) | 0 (0.0) | 1 (2.3) | 0 (0.0) |
There were no significant differences when comparing treated versus matched untreated cohorts for sex, continent of origin, birth year or known mutation subgroups.
Percentages of known mutations
Cause of death was identified for 50 of the 102 deceased untreated subjects, and was attributed to CV failure (N=40; 80%), head injury or trauma (N=5; 10%), stroke (N=2; 4%), respiratory infection superimposed on CV disease (N=2; 4%), and complications from anesthesia during surgery (N=1; 2%). Similarly, cause of death in the 5 deceased trial participants was CV failure (N=3; 60%), head injury (N=1; 20%), and stroke (N=1; 20%).
Trial Medication Side Effects
Notable lonafarnib monotherapy-related side effects included mild diarrhea, fatigue, nausea, vomiting, anorexia, transiently elevated aspartate aminotransferase and alanine aminotransferase, and depressed serum hemoglobin, all of which generally improved with time and are detailed in Gordon et al.23. As the combination trial is ongoing, a detailed account of toxicities is not available. However, to date the most notable side effects include zoledronate-related post-infusion flu-like symptoms35, pravastatin-induced transient muscle discomfort and mildly elevated creatine phosphokinase36.
Survival Analysis for Untreated Group
Analysis of the full untreated cohort (N=204), including treatment trial participants censored at time of treatment initiation, provided a Kaplan-Meier survival curve for HGPS (Fig. 2A). Mean and median survival were 14.6 and 14.5 years, respectively.
Figure 2.



Kaplan-Meier-survival estimates for untreated and treated HGPS cohorts. The number of patients at risk are presented below the x-axis. Numbers in parentheses are number of deaths during that time interval. A. Untreated cohort. Treated subjects were included, but censored at age of treatment initiation. Mean and median survival were 14.6 and 14.5 years, respectively. B. Kaplan-Meier-survival Estimates Comparing Untreated (solid line) to Treated (dashed line) Cohorts Using Matched Analysis (age-adjusted P<0.001) where Time 0 on the x-axis (i.e., beginning of patient being at risk) is defined for each matched pair as the age of treatment initiation for the treated patient in the matched pair. C. Kaplan-Meier-survival Estimates Comparing Untreated (solid line) to Treated (dashed line) Cohorts Using Matched Analysis (unadjusted P<0.001) where Time 0 on the x-axis (i.e., beginning of patient follow-up) is defined as patient birth and subject becomes at risk at the age of treatment initiation for the treated patient in the matched pair.
Subgroup comparisons were conducted, with no significance found. (Table S2). These included male versus female and known versus unknown genotype. The possibility that general medical advances over time would improve survival for more recent subjects was addressed by comparing subjects born before 1986 to those born on or after 1986 (approximately 50% of subjects). The possibility that healthier subjects would be removed from the untreated cohort as they enrolled in treatment trials was addressed by censoring the entire patient cohort at the clinical trial initiation date, May 2007.
For use in future comparison studies by other investigators, data elements for all subjects are provided (Tables S1, S3, S4, S5).
Association Between Farnesylation Inhibitors and Survival
There were 5/43 (11.6%) deaths in the treated group and 21/43 (48.8%) deaths in the matched untreated group. Median follow-up from time of treatment initiation in both treatment groups (untreated subjects matched to treated subjects) is 5.3 years (quartiles of 3.3 – 5.5 years). Kaplan-Meier estimates demonstrated increased mortality for the untreated cohort over the treated cohort when follow-up begins at age of treatment initiation for the treated patient in the matched pair (age- and sex- adjusted p<0.001; Fig. 2B). Age-, sex- and continent-adjusted hazard ratio for mortality of treated subjects in the matched analysis was 0.15 and therefore positively associated with survival (95% CI 0.04-0.43). Kaplan-Meier estimates similarly demonstrated increased mortality for untreated when follow-up begins at birth with subjects placed at risk at the age of treatment initiation for the treated patient in the matched pair (P<0.001; Fig. 2C). Time-dependent analyses on patients born after 1991 yielded increased survival with P=0.017, and sex and continent-adjusted HR=0.28 (95% CI 0.10-0.79) (Fig. 3).
Figure 3.
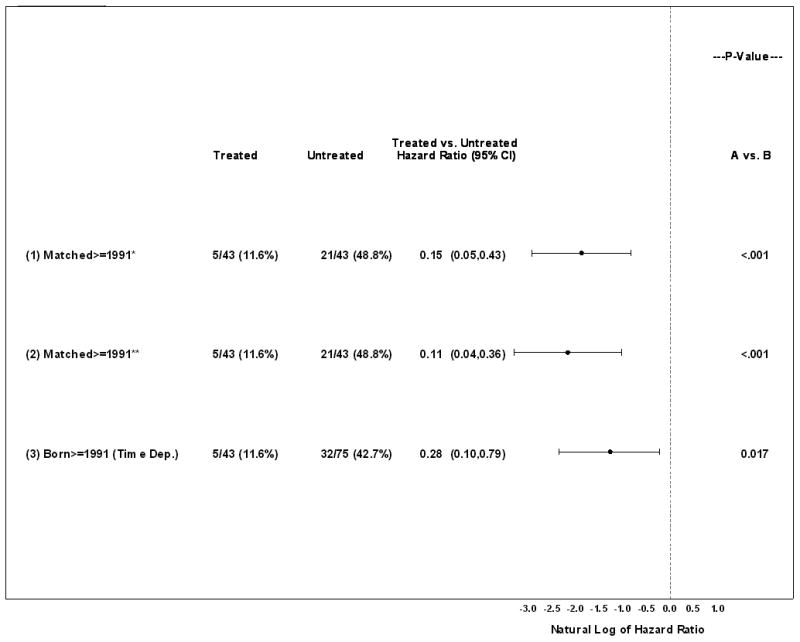
Hazard ratios (HRs) comparing untreated to treated cohorts using matched analyses and time-dependent analysis for patients born on or after 1991. HRs and P-values were generated from Cox proportional hazards regression and adjusted for sex and continent. *For each matched pair, follow-up begins at time 0 defined as the age of treatment initiation for the treated patient in the matched pair; HR and P-value further adjusted for age at risk. **For each matched pair, follow-up begins at birth and patient is placed at risk at the age of treatment initiation for the treated patient in the matched pair; HR and P-value further adjusted for age at risk.
During the first 6 years following treatment initiation for the treated patient in the matched pair, extension in mean survival with treatment was 1.6 years with a 95% CI of 0.8 to 2.4 years (P<0.001). There was a 33% increase in Kaplan-Meier AUC for treated versus untreated. To account for potential confounding variables within comparisons between untreated and treated cohorts, a sensitivity analysis which either excluded or censored specific subjects was conducted as follows: Two prospective subjects could not enroll due to health issues and were omitted from the untreated group. Five trial subjects taking recombinant growth hormone were omitted from the treated group. Clinical trial subjects did not generally receive clinical care at the trial hospital site, as clinical care was the responsibility of the subjects' home physicians. However, one trial patient received clinical care from the clinical trial group starting at age 18.4 years due to urgent clinical need while at the trial site. This subject returned home after care was completed, and subsequently passed away at home at age 20.3 years. To account for this trial site clinical care, sensitivity analysis was performed where this patient was censored at age 18.4. Other than these variables, there were no known differences between subjects who enrolled and those who did not enroll in the clinical trials. Results of this sensitivity analysis were similar to those described above.
Discussion
This study demonstrates that without treatment, HGPS survival distribution is stable and independent of gender or medical advances, since male compared with female, as well as pre-compared with post-1986 Kaplan-Meier curves were similar. This implies that the progerin-associated morbidity is the overriding factor in survival. The quality and quantity of data for the reference population is key to current and future success in assessing changes in survival. Given that the estimated prevalence of HGPS is currently 1 in 18 million37, this study has captured a significant portion of the population.
This study is the first to demonstrate a positive effect of any treatment on estimated survival in HGPS. Results were consistent across 8 different possible confounding variables (sex, continent of origin, mutation status, birth year, medical advances, growth hormone treatment, failing health, trial site clinical treatment and various analytic methods), strengthening our assertion that farnesylation inhibitors positively influenced patient survival. Because these children die from sequelae of a pervasive premature, progressive form of atherosclerosis, we speculate that extended survival is attributable to cardiovascular and possibly cerebrovascular benefit. This premise is supported by secondary outcomes showing evidence for decreased pulse wave velocity, carotid artery wall echodensity, and incidence of stroke, headache and seizures in subjects treated with lonafarnib monotherapy23, 24.
Because each treatment trial was sequential and of relatively short duration (2 years on lonafarnib monotherapy and 3.5 years on combination therapy), the analysis did not distinguish individual drug influences on longevity. As lonafarnib is the drug to which all subjects have been exposed, and for the longest period of time in most instances, we speculate that this drug is largely responsible the estimated life extension. This speculation takes into account the CV and NV systems being responsible for most deaths, and the improvements seen in some CV and NV properties with lonafarnib treatment. To evaluate further whether addition of zoledronate and pravastatin may be beneficial, neutral, or harmful to morbidity and mortality, it will be crucial to compare CV and other clinical changes with combination therapy to those of lonafarnib monotherapy, once the combination therapy trial is completed.
In the treated group 5/43 subjects died, compared with 21/43 in the untreated matched comparison group, both with median follow-up of 5.3 years. Treatment group inclusion was independent of duration of treatment, age, or stage of disease at treatment initiation. The hazard ratio of 0.13 indicates that given a specific point in time, HGPS subjects receiving farnesylation inhibitors demonstrated an 80% reduction in risk of death compared with untreated subjects. Interpretation of this effect is complicated by the longitudinal nature of the Kaplan-Meier curve and variable treatment times for different subjects. The estimated 1.6 years of extended survival may be conservative due to the fact that many subjects started treatment late in their disease course and may potentially benefit from earlier initiation of FTI therapy, and given that most subjects were still living at time of analysis due to the short follow-up time. This is a statistical estimate; it will take approximately 6 years until a true extension in mean survival can be determined from actual treated cohort age.
This study was limited by the use of an external untreated control group. For HGPS and other ultra-rare, fatal pediatric diseases with no known treatments, only single arm clinical trials have been conducted to date and are therefore the sole source of data to demonstrate safety and efficacy of any potential new treatment. We attempted to address this issue by using a matching statistical analysis and integrating potential confounding variables.
There are no previously established life-extending treatments for HGPS. Farnesylation inhibitors are clearly not curative, as many features of disease persist despite treatment23. However, evidence suggesting that survival may be improved by these medications offers a first step in recognizing that treatments aimed at further reducing progerin could thwart its fatal effects.
Supplementary Material
Acknowledgments
We are extremely grateful to the children with Progeria and their families for participation in this study. We thank Erin Bettencourt and Lynne MacKenzie for assistance with data collection at PRF; Alan Hubbard, PhD for initial project discussions; The Sunshine Foundation, Nicolas Levy, MD, Marjet Stamsnijder, and Raoul Hennekam, MD for patient communications; Boston team members Annette Corriea, O.T., Brian Fligor, Sc.D., Anita Giobbie-Hurder, M.S., Marilyn G. Liang M.D., Donna Neuberg, Sc.D., Christine Ploski, P.T., Nicolle Quinn, M.S., R.D., Amy Regen, D.M.D., Susan Riley, P.T., Andrew Sonis, D.M.D.; Merck members Paul Statkevich, PhD, David Harris, PhD.
Funding Sources: Natural History Study supported by The Progeria Research Foundation. Clinical Trials supported by The Progeria Research Foundation grants PRFCLIN2007-01, PRFCLIN2009-02, PRFCLIN2009-03, National Heart, Lung and Blood Institute grant 1RC2HL101631-0, National Institutes of Health grants to the Boston Children's Hospital General Clinical Research Center (MO1-RR02172), and Harvard Catalyst Clinical & Translational Science Center (UL1 RR025758-01).
Appendix
Additional Participating Progeria Clinical Trials Collaborative Investigators (alphabetical): W. Robert Bishop, PhD., Robert Cleveland, M.D., Marie Gerhard-Herman, M.D., Catherine M. Gordon, M.D., MSc., Susanna Y. Huh, M.D., M.P.H., David T. Miller, M.D., Ph.D., Marsha Moses, PhD., Ara Nazarian, Ph.D., Michele Silvera, MD, Leslie Smoot, M.D., Brian D. Snyder, M.D., Ph.D., Nicole J. Ullrich, M.D., Ph.D. From the Merck Research Labs, Kenilworth, NJ (R.B.); Departments of Radiology (R.C.), Gastroenterology and Nutrition (S.Y.H.), Dermatology (M.G.L.), Genetics and Laboratory Medicine (D.T. M.), Surgery (M.M.), Cardiology (L.S.), Orthopedics (B.D.S.), Physical Therapy (S.R.); and Neurology (N.J.U.), at Boston Children's Hospital and Harvard Medical School, Boston, MA 02115; Department of Cardiology ( M.G-H), Brigham & Women's Hospital, Boston, MA 02115; Department of Pediatrics (C.M.G.), Hasbro Children's Hospital and Warren Alpert Medical School of Brown University, Providence, RI ; Center for Advanced Orthopedic Studies, Department of Orthopedic Surgery (A.N.), Beth Israel Deaconess Medical Center, Harvard Medical School, Boston, MA
Footnotes
Conflict of Interest Disclosures: Author LBG is the parent of a child who participated in the study.
References
- 1.Hennekam RC. Hutchinson-gilford progeria syndrome: Review of the phenotype. Am J Med Genet A. 2006;140:2603–2624. doi: 10.1002/ajmg.a.31346. [DOI] [PubMed] [Google Scholar]
- 2.Olive M, Harten I, Mitchell R, Beers JK, Djabali K, Cao K, Erdos MR, Blair C, Funke B, Smoot L, Gerhard-Herman M, Machan JT, Kutys R, Virmani R, Collins FS, Wight TN, Nabel EG, Gordon LB. Cardiovascular pathology in hutchinson-gilford progeria: Correlation with the vascular pathology of aging. Arterioscler Thromb Vasc Biol. 2010;30:2301–2309. doi: 10.1161/ATVBAHA.110.209460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gerhard-Herman M, Smoot LB, Wake N, Kieran MW, Kleinman ME, Miller DT, Schwartzman A, Giobbie-Hurder A, Neuberg D, Gordon LB. Mechanisms of premature vascular aging in children with hutchinson-gilford progeria syndrome. Hypertension. 2012;59:92–97. doi: 10.1161/HYPERTENSIONAHA.111.180919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ullrich NJ, Kieran MW, Miller DT, Gordon L, Cho YJ, Silvera VM, Giobbie-Hurder A, Neuberg D, Kleinman M. Neurologic features of hutchinson-gilford progeria syndrome after lonafarnib treatment. Neurology. 2013;81:427–430. doi: 10.1212/WNL.0b013e31829d85c0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.De Sandre-Giovannoli A, Bernard R, Cau P, Navarro C, Amiel J, Boccaccio I, Lyonnet S, Stewart CL, Munnich A, Le Merrer M, Levy N. Lamin a truncation in hutchinson-gilford progeria. Science. 2003;300:2055. doi: 10.1126/science.1084125. [DOI] [PubMed] [Google Scholar]
- 6.Eriksson M, Brown WT, Gordon LB, Glynn MW, Singer J, Scott L, Erdos MR, Robbins CM, Moses TY, Berglund P, Dutra A, Pak E, Durkin S, Csoka AB, Boehnke M, Glover TW, Collins FS. Recurrent de novo point mutations in lamin a cause hutchinson-gilford progeria syndrome. Nature. 2003;423:293–298. doi: 10.1038/nature01629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Broers JL, Ramaekers FC, Bonne G, Yaou RB, Hutchison CJ. Nuclear lamins: Laminopathies and their role in premature ageing. Physiol Rev. 2006;86:967–1008. doi: 10.1152/physrev.00047.2005. [DOI] [PubMed] [Google Scholar]
- 8.Fong LG, Ng JK, Lammerding J, Vickers TA, Meta M, Cote N, Gavino B, Qiao X, Chang SY, Young SR, Yang SH, Stewart CL, Lee RT, Bennett CF, Bergo MO, Young SG. Prelamin a and lamin a appear to be dispensable in the nuclear lamina. J Clin Invest. 2006;116:743–752. doi: 10.1172/JCI27125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Eriksson M, Brown WT, Gordon L, Glynn MW, Singer J, Scott L, Erdos MR, Robbins CM, Moses TY, Berglund P, Dutra A, Pak E, Durkin S, Csoka AB, Boehnke M, Glover TW, Collins FS. Recurrent de novo point mutations in lamin a cause hutchinson-gilford progeria syndrome. Nature. 2003;423:293–297. doi: 10.1038/nature01629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Goldman RD, Shumaker DK, Erdos MR, Eriksson M, Goldman AE, Gordon LB, Gruenbaum Y, Khuon S, Mendez M, Varga R, Collins FS. Accumulation of mutant lamin a causes progressive changes in nuclear architecture in hutchinson-gilford progeria syndrome. Proc Natl Acad Sci U S A. 2004;101:8963–8968. doi: 10.1073/pnas.0402943101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Scaffidi P, Misteli T. Reversal of the cellular phenotype in the premature aging disease hutchinson-gilford progeria syndrome. Nat Med. 2005;11:440–445. doi: 10.1038/nm1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Capell BC, Erdos MR, Madigan JP, Fiordalisi JJ, Varga R, Conneely KN, Gordon LB, Der CJ, Cox AD, Collins FS. Inhibiting farnesylation of progerin prevents the characteristic nuclear blebbing of hutchinson-gilford progeria syndrome. Proc Natl Acad Sci U S A. 2005;102:12879–12884. doi: 10.1073/pnas.0506001102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Scaffidi P, Misteli T. Lamin a–dependent nuclear defects in human aging. Science. 2006;312:1059–1063. doi: 10.1126/science.1127168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Glynn MW, Glover TW. Incomplete processing of mutant lamin a in hutchinson-gilford progeria leads to nuclear abnormalities, which are reversed by farnesyltransferase inhibition. Hum Mol Genet. 2005;14:2959–2969. doi: 10.1093/hmg/ddi326. [DOI] [PubMed] [Google Scholar]
- 15.Toth JI, Yang SH, Qiao X, Beigneux AP, Gelb MH, Moulson CL, Miner JH, Young SG, Fong LG. Blocking protein farnesyltransferase improves nuclear shape in fibroblasts from humans with progeroid syndromes. Proc Natl Acad Sci U S A. 2005;102:12873–12878. doi: 10.1073/pnas.0505767102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yang SH, Meta M, Qiao X, Frost D, Bauch J, Coffinier C, Majumdar S, Bergo MO, Young SG, Fong LG. A farnesyltransferase inhibitor improves disease phenotypes in mice with a hutchinson-gilford progeria syndrome mutation. J Clin Invest. 2006;116:2115–2121. doi: 10.1172/JCI28968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fong LG, Frost D, Meta M, Qiao X, Yang SH, Coffinier C, Young SG. A protein farnesyltransferase inhibitor ameliorates disease in a mouse model of progeria. Science. 2006;311:1621–1623. doi: 10.1126/science.1124875. [DOI] [PubMed] [Google Scholar]
- 18.Yang SH, Qiao X, Fong LG, Young SG. Treatment with a farnesyltransferase inhibitor improves survival in mice with a hutchinson-gilford progeria syndrome mutation. Biochim Biophys Acta. 2008;1781:36–39. doi: 10.1016/j.bbalip.2007.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Varela I, Pereira S, Ugalde AP, Navarro CL, Suarez MF, Cau P, Cadinanos J, Osorio FG, Foray N, Cobo J, de Carlos F, Levy N, Freije JM, Lopez-Otin C. Combined treatment with statins and aminobisphosphonates extends longevity in a mouse model of human premature aging. Nat Med. 2008;14:767–772. doi: 10.1038/nm1786. [DOI] [PubMed] [Google Scholar]
- 20.Capell BC, Olive M, Erdos MR, Cao K, Faddah DA, Tavarez UL, Conneely KN, Qu X, San H, Ganesh SK, Chen X, Avallone H, Kolodgie FD, Virmani R, Nabel EG, Collins FS. A farnesyltransferase inhibitor prevents both the onset and late progression of cardiovascular disease in a progeria mouse model. Proc Natl Acad Sci U S A. 2008;105:15902–15907. doi: 10.1073/pnas.0807840105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Verstraeten V, Ji J, Cummings K, Lee R, Lammerding J. Hutchinson-gilford progeria cells: Effects of farnesyltransferase inhibitors. Aging cell. 2008;7:383–393. doi: 10.1111/j.1474-9726.2008.00382.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yang SH, Bergo MO, Toth JI, Qiao X, Hu Y, Sandoval S, Meta M, Bendale P, Gelb MH, Young SG, Fong LG. Blocking protein farnesyltransferase improves nuclear blebbing in mouse fibroblasts with a targeted hutchinson-gilford progeria syndrome mutation. Proc Natl Acad Sci U S A. 2005;102:10291–10296. doi: 10.1073/pnas.0504641102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gordon LB, Kleinman ME, Miller DT, Neuberg DS, Giobbie-Hurder A, Gerhard-Herman M, Smoot LB, Gordon CM, Cleveland R, Snyder BD, Fligor B, Bishop WR, Statkevich P, Regen A, Sonis A, Riley S, Ploski C, Correia A, Quinn N, Ullrich NJ, Nazarian A, Liang MG, Huh SY, Schwartzman A, Kieran MW. Clinical trial of a farnesyltransferase inhibitor in children with hutchinson-gilford progeria syndrome. Proc Natl Acad Sci U S A. 2012;109:16666–16671. doi: 10.1073/pnas.1202529109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ullrich NJ, Kieran MW, Miller DT, Gordon LB, Cho YJ, Silvera VM, Giobbie-Hurder A, Neuberg D, Kleinman ME. Neurologic features of hutchinson-gilford progeria syndrome after lonafarnib treatment. Neurology. 2013;81:427–430. doi: 10.1212/WNL.0b013e31829d85c0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Halcox JP, Deanfield JE. Beyond the laboratory: Clinical implications for statin pleiotropy. Circulation. 2004;109:II42–48. doi: 10.1161/01.CIR.0000129500.29229.92. [DOI] [PubMed] [Google Scholar]
- 26.Gordon LB, Harten IA, Patti ME, Lichtenstein AH. Reduced adiponectin and hdl cholesterol without elevated c-reactive protein: Clues to the biology of premature atherosclerosis in hutchinson-gilford progeria syndrome. J Pediatr. 2005;146:336–341. doi: 10.1016/j.jpeds.2004.10.064. [DOI] [PubMed] [Google Scholar]
- 27.Merideth MA, Gordon L, Clauss S, Sachdev V, Smith A, Perry M, Brewer C, Zalewski C, Kim H, Solomon B, Brooks B, Gerber L, Turner M, Domingo DL, Hart TC, Graf J, Reynolds J, Gropman A, Yanovski J, Gerhard-Herman M, Collins FS, Nabel EG, Cannon R, Gahl WA, Introne WJ. Phenotype and course of hutchinson-gilford progeria syndrome. New Engl J Med. 2008;358:592–604. doi: 10.1056/NEJMoa0706898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rakel A, Boucher A, Ste-Marie LG. Role of zoledronic acid in the prevention and treatment of osteoporosis. Clin Interv Aging. 2011;6:89–99. doi: 10.2147/CIA.S7282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gordon CM, Gordon LB, Snyder BD, Nazarian A, Quinn N, Huh S, Giobbie-Hurder A, Neuberg D, Cleveland R, Kleinman M, Miller DT, Kieran MW. Hutchinson-gilford progeria is a skeletal dysplasia. J Bone Miner Res. 2011;26:1670–1679. doi: 10.1002/jbmr.392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gordon LB, McCarten KM, Giobbie-Hurder A, Machan JT, Campbell SE, Berns SD, Kieran MW. Disease progression in hutchinson-gilford progeria syndrome: Impact on growth and development. Pediatrics. 2007;120:824–833. doi: 10.1542/peds.2007-1357. [DOI] [PubMed] [Google Scholar]
- 31.DeBusk FL. The hutchinson-gilford progeria syndrome. Report of 4 cases and review of the literature. J Pediatr. 1972;80:697–724. doi: 10.1016/s0022-3476(72)80229-4. [DOI] [PubMed] [Google Scholar]
- 32.Hennekam R, K ID, A JE. Gorlin's syndromes of the head and neck. 5th. Oxford University Press; 2010. p. 447. [Google Scholar]
- 33.Shalev SA, De Sandre-Giovannoli A, Shani AA, Levy N. An association of hutchinson-gilford progeria and malignancy. Am J Med Genet A. 2007;143A:1821–1826. doi: 10.1002/ajmg.a.31803. [DOI] [PubMed] [Google Scholar]
- 34.Hisama FM, Lessel D, Leistritz D, Friedrich K, McBride KL, Pastore MT, Gottesman GS, Saha B, Martin GM, Kubisch C, Oshima J. Coronary artery disease in a werner syndrome-like form of progeria characterized by low levels of progerin, a splice variant of lamin a. Am J Med Genet A. 2011;155A:3002–3006. doi: 10.1002/ajmg.a.34336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Munns CF, Rajab MH, Hong J, Briody J, Hogler W, McQuade M, Little DG, Cowell CT. Acute phase response and mineral status following low dose intravenous zoledronic acid in children. Bone. 2007;41:366–370. doi: 10.1016/j.bone.2007.05.002. [DOI] [PubMed] [Google Scholar]
- 36.Pfeffer MA, Keech A, Sacks FM, Cobbe SM, Tonkin A, Byington RP, Davis BR, Friedman CP, Braunwald E. Safety and tolerability of pravastatin in long-term clinical trials: Prospective pravastatin pooling (ppp) project. Circulation. 2002;105:2341–2346. doi: 10.1161/01.cir.0000017634.00171.24. [DOI] [PubMed] [Google Scholar]
- 37.Gordon LB. PRF by the numbers. [Accessed on April 2, 2014];2013 HGPS Information Resource (website). http://www.progeriaresearch.org/prf-by-the-numbers.html.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.