

Abstract
Background
Diabetic patients have a significantly worse prognosis after an acute myocardial infarction (AMI) than their counterparts. Previous studies have shown that the number of circulating endothelial progenitor cells (EPCs) significantly increase early after an AMI in normoglycemic patients. However, it is well known that type 2 diabetes mellitus (DM) is associated with impaired function and reduced circulating EPCs levels. Nonetheless, few studies have analyzed EPCs response of diabetics to an AMI and the EPC response of pre-diabetic patients has not been reported yet. Therefore, we hypothesized that in the acute phase of an AMI, diabetic and pre-diabetics have lower circulating EPCs levels than patients with normal glucose metabolism. We also evaluated the possible capacity of chronic antidiabetic treatment in the recovery of EPCs response to an AMI in diabetics.
Methods
One-hundred AMI patients were prospectively enrolled in the study. Using the high-performance flow cytometer FACSCanto II, circulating EPCs (CD45dimCD34+KDR+ and CD45dimCD133+KDR+ cells) were quantified, within the first 24 hours of admission. In addition, as an indirect functional parameter, we also analyzed the fraction of EPCs coexpressing the homing marker CXCR4.
Results
We found that in the acute phase of an AMI, diabetic patients presented significantly lower levels of circulating CD45dimCD34+KDR+ and CD45dimCD133+KDR+ EPCs by comparison with nondiabetics, with a parallel decrease in the subpopulations CXCR4+ (p < 0.001). Indeed, this study suggests that the impaired response of EPCs to an AMI is an early event in the natural history of DM, being present even in pre-diabetes. Our results, also demonstrated that numbers of all EPCs populations were inversely correlated with HbA1c (r = -0.432, p < 0.001 for CD45dimCD34+KDR+ cells). Finally, this study suggests that previous chronic insulin therapy (but not oral antidiabetic drugs) attenuate the deficient response of diabetic EPCs to an AMI.
Conclusion
This study indicates that there is a progressive decrease in EPCs levels, from pre-diabetes to DM, in AMI patients. Moreover, glycemic control seems to be determinant for circulating EPCs levels presented in the acute phase of an AMI and chronic insulin therapy may probably attenuate the deficit in EPCs pool seen in diabetics.
Keywords: Endothelial progenitor cells, Diabetes, Pre-diabetes, Insulin, Oral antidiabetic drugs, Acute myocardial infarction, Homing
Background
It is well recognized that patients with type 2 diabetes mellitus (DM) have accelerated atherosclerosis, increased risk of developing coronary artery disease (CAD) and worse prognosis after an acute myocardial infarction (AMI) [1].
Endothelial progenitor cells (EPCs), a subpopulation of adult stem cells, have emerged as critical to endothelial repair and vascular homeostasis. Although the mechanisms whereby EPCs protect the cardiovascular system are still not fully understood, it has been extensively demonstrated that these bone marrow-derived cells contribute to endothelial repair and postnatal neovascularization [2,3]. EPCs can differentiate into mature endothelial cells and be incorporated into new vessels or act by a paracrine manner, through the secretion of pro-angiogenic growth factors that enhance vascularization mediated by resident endothelial cells and/or promote angiogenesis [2-5].
The number of EPCs in peripheral circulation is generally low, and in normal physiological conditions, these endothelial precursor cells are very rare in blood, but they are mobilized from the bone marrow to the peripheral circulation in response to tissue injury, such as myocardial ischemia [6]. In fact, tissue ischemia is considered the strongest stimulus for EPCs mobilization and it has been shown that their numbers significantly increase in patients with an AMI [7,8]. However, it is well established that diabetic patients present impaired function and reduced numbers of circulating EPCs, reflecting a poor endogenous regenerative capacity that may contribute to the development of vascular complications and to the dismal prognosis associated with this prevalent disease [9-12]. Therefore, it is likely that, in the clinical context of myocardial infarction, diabetic patients also have lower levels of circulating EPCs, but regrettably the data addressing the dynamics of EPCs mobilization in diabetic patients with AMI are scarce. Furthermore, little is known about potential EPCs impairment in pre-diabetic states and no studies are available on the kinetics of EPCs mobilization in pre-diabetic patients with AMI. This is of great importance, since multiple studies have demonstrated that individuals with pre-diabetes are also at increased risk for cardiovascular events [13]. On the other hand, some drugs commonly prescribed in diabetic patients, like statins, angiotensin II receptor blockers (ARBs) and angiotensin-converting-enzyme (ACE)-inhibitors, have been shown to increase the number of EPCs in peripheral blood of patients with stable CAD [14]. However, we have no data available regarding the impact of previous antidiabetic treatment on EPC response to an AMI, in diabetic patients.
In this study, we tested the hypothesis that diabetes and pre-diabetes states were associated with reduced circulating EPCs levels in the acute phase of a myocardial infarction (MI) by comparison with patients with normal glucose metabolism. We also examined the impact of previous antidiabetic treatment on the dynamics of EPCs mobilization in diabetic patients following an AMI.
Methods
Study population and selection
A prospective cohort of 686 consecutive patients hospitalized in a single Coronary Care Unit (CCU) due to myocardial infarction, from 5 January 2009 to 23 September 2011, were screened on admission for inclusion. Screening included an interview, clinical examination, ECG and laboratory assessment. Patients were excluded if they were >80 years old, showed clinical or biochemical evidence of concomitant inflammatory disease, known auto-immune or malignant diseases, severe peripheral arterial occlusive disease, deep vein thrombosis or pulmonary embolism, atrial fibrillation, recent trauma or surgery (<1 month), recent major bleeding requiring blood transfusion (<6 months), renal insufficiency (creatinine > 2.0 mg/dl), anemia (hemoglobin < 8.5 g/dl) or thrombocytopenia (<100 000/L), previous coronary bypass surgery, myocardial infarction within the preceding 2 months, cardiogenic shock, severe valvular disease or congenital heart disease, co-morbidities associated with a life expectancy less than 2 years. A regular use of nonsteroidal anti-inflammatory drugs or anticoagulants, patients with pacemakers, implantable cardioverter defibrillators or resynchronization devices, and excessive alcohol consumption or illicit drugs abuse that may influence EPC kinetics were also exclusion criteria. A total of 100 patients were prospectively included (65% with ST segment elevation myocardial infarction – STEMI and 35% with non-ST segment elevation myocardial infarction - NSTEMI) (Figure 1).
Figure 1.
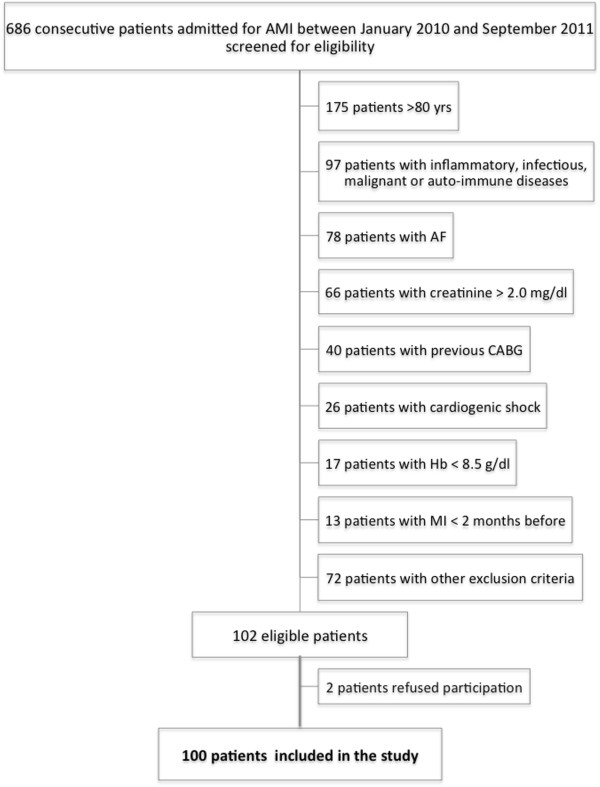
Flow diagram of patient recruitment. AF, atrial fibrillation; AMI, acute myocardial infarction; CABG, coronary artery bypass graft; Hb, hemoglobin; MI, myocardial infarction; yrs, years old.
All patients received the standard therapy for the acute phase of MI that included acetylsalicylic acid (ASA), clopidogrel and low-molecular-weight heparin, according to usual hospital practice.
Baseline demographic data, cardiovascular risk factors and previous medications were recorded in all patients. Smoking status was recorded as ever-smoker (past or current) or non-smoker.
Blood samples were collected to assess chemistry (including fasting plasma glucose (FPG) and glycosylated hemoglobin (HbA1C)), total cholesterol, low-density lipoprotein-cholesterol (LDL-C), high-density lipoprotein-cholesterol (HDL-C), triglycerides, high sensitivity C-reactive protein (hs-CRP), creatinine, and hematological parameters in all patients according to standard hospital practice.
The study was approved by the local ethics committee (Approval Number: HUC-23-08). All patients gave written informed consent and research was conducted according to the principles expressed in the Declaration of Helsinki.
Classification of glucose metabolism status
DM and glucose metabolism disorders were defined according to the American Diabetes Association (ADA) criteria [15,16]. All patients without previously known diabetes underwent an Oral Glucose Tolerance Test (OGTT) on day 4 or 5 of hospitalization. Therefore, patients were classified as having diabetes if they have a FPG ≥126 mg/dl, a 2-h glucose ≥200 mg/dl on OGTT, a A1c ≥ 6.5% or a random plasma glucose ≥200 mg/dl in a patient with classic symptoms of hyperglycemia or hyperglycemic crisis. For patients without diabetes, prediabetes was defined as FPG levels of 100–125 mg/dl (impaired fasting glucose – IFG), 2-hour OGTT glucose level of 140–199 mg/dl (impaired glucose tolerance - IGT) or HbA1c values of 5.7%-6.4%. Patients were classified as having a normal glucose metabolism (NGM) if they have FPG < 100 mg/dl, 2-hour OGTT glucose level < 140 mg/dl and HbA1c <5.7%.
Quantification of circulating EPCs by flow cytometry
For the identification and quantification of EPCs, we have used a standardized protocol - the modified International Society for Hematotherapy and Graft Engineering (ISHAGE) sequential gating strategy - proposed by Schmidt-Lucke et al. [17]. Briefly, within the first 24 h of CCU admission, 1 ml of whole blood was collected from a forearm vein into EDTA tubes, transported into the cytometry laboratory and processed within 1 to 2 hours of collection. Hence, 150 μl of whole blood were incubated with the following combination of anti-human monoclonal antibodies: 10 μl of anti-CD133 conjugated with allophycocyanin (APC) (Miltenyi Biotec), 5 μl of anti-CD45 conjugated with APC-H7 (Becton Dickinson), 10 μl of anti-KDR (also known as type 2 vascular endothelial growth factor receptor - VEGF-R2) conjugated with phycoerythrin (PE) (Sigma), 10 μl of anti-CD34 conjugated with fluorescein isothiocyanate (FITC) (Becton Dickinson) and 10 μl of anti-CD184 (also known as CXCR4) conjugated with PE-Cyanine 5 (PE-Cy5) (BD Pharmingen) for 30 min at 4°C, in the dark. Red blood cell lysis was performed using FACS Lysing Solution (BDBiosciences) diluted 1:10 (vol/vol) in distilled water and washed with phosphate-buffered-saline (PBS) before flow cytometry acquisition. Data acquisition was performed with a high-performance flow cytometer, a BDBioscience FACSCanto II, which can analyze with high resolution up to eight different fluorescent markers from a large number of events and we used the flow cytometry software Infinicyt 1.5 (Cytognos) for the analysis. According to the used standardized protocol, human circulating EPCs were identified by a minimal antigenic profile that includes at least one marker of stemness/immaturity (CD34 and/or CD133), plus at least one marker of endothelial commitment (KDR). CD45 staining was also performed to exclude leucocytes, as it has been previously demonstrated that only the fraction of CD45dim cells harbours the “true” circulating EPCs [18]. CXCR4, the receptor for stromal cell–derived factor-1 (SDF-1), is a cell surface antigen expressed in EPCs, which plays a key role in their transendothelial migration and homing to sites of vascular injury [19]. Therefore, by analyzing the subpopulation of progenitors coexpressing CXCR4, we could study a functional parameter of EPCs. As isotype controls are known to mask rare cell populations, none were used in this analysis, and baseline fluorescence was determined using unstained cells [20]. Because EPCs are extremely rare events in peripheral blood, additional strategies were applied in order to increase the sensitivity of the method and the accuracy of our work. These included: automatic compensation for minimizing fluorescence spillover, exclusion of dead cells, and use of specific high quality mononuclear antibodies. The total number of acquired events was increased to at least 1 million per sample, which is generally not needed for most other applications of flow cytometry. Circulating EPCs were measured in triplicate from the same patients, revealing a very close correlation (r = 0.87, p < 0.0001). The same trained operator, who was blind to the clinical status of the patients, performed all the cytometric analysis throughout the study.
Four different populations of EPCs were quantified: 1) CD45dimCD133+KDR+ cells; 2) CD45dimCD34+KDR+ cells; 3) CD45dimCD34+CD133+KDR+ triple positive cells; and 4) the subpopulation of CD45dimCD34+KDR+CXCR4+ EPCs.
Patients follow up for cardiovascular events
All patients were followed up for 24 months after discharge. The following cardiovascular events were recorded: cardiovascular death; nonfatal stroke or transient ischemic attack; re-infarction; unstable angina and re-hospitalization for unstable angina or heart failure. We also analyzed the combined endpoint of cardiovascular death, re-hospitalization for ACS and unplanned PCI – Major Adverse Cardiac Events (MACE). Cardiovascular death was defined as death due to a MI or stroke or documented sudden cardiac death. For patients experiencing more than one acute event, only the first event was considered in the analysis.
Statistical analysis
Statistical analyses were performed using SPSS software version 20.
Based on previous data, we estimated a 40% reduction in circulating EPCs of diabetics by comparison with nondiabetic patients. Therefore, a minimum sample size of 18 patients in each group would provide 90% power to detect difference in circulating EPCs between diabetic and nondiabetic patients, using a two-sided hypothesis test with a significance level (alpha) of 0.05.
Continuous variables were tested for normal distribution by Kolmogorov–Smirnov test and expressed as mean ± standard deviation or median ± interquartile range for parametric and nonparametric data, respectively. Categorical data are expressed as counts and percentages.
For comparison of continuous data unpaired Student t-tests or ANOVA tests were used when variables were normally distributed and nonparametric Mann–Whitney test or Kruskal-Wallis test for variables without a normal distribution. Categorical variables were compared with the chi-square test or with Fisher exact test as appropriate. The relationship between variables was calculated using Pearson’s or Spearman’s correlation coefficient, whichever appropriate. Multivariate linear regression analysis was used to assess the relationship between circulating EPCs levels and HbA1c, after adjustment for confounding variables. Kaplan-Meier survival analyses were performed to evaluate time-dependent outcomes. Differences between pairs of survival curves were tested by the log-rank test. For all analyses, a 2-sided value of P < 0.05 was considered statistically significant.
Results
Characteristics of the study population
There were 38 patients with DM, 13% of them with newly diagnosed DM. Overall, diabetics had similar age and cardiovascular risk factors as nondiabetic patients, except for hypertension that was significantly more frequent in diabetics (Table 1). Additionally, they tended to have more frequently previously known CAD and were more often medicated with ASA, ACE-inhibitors and ARB as well as oral hypoglycemic agents and insulin before admission than nondiabetics. As expected, diabetics had significantly higher levels of admission glycemia, fasting glycemia and HbA1c and also presented higher total cholesterol and LDL-cholesterol than nondiabetics.
Table 1.
Comparison of clinical characteristics between diabetic and nondiabetic patients
| Non-diabetics (N = 62) | Type 2 diabetics (N = 38) | p value | |
|---|---|---|---|
| Age (years)* |
59.8 ± 10.3 |
61.5 ± 11.0 |
0.300 |
| Male gender (%) |
90.3 |
89.5 |
0.891 |
| BMI (Kg/m2)* |
27.9 ± 4.4 |
29.2 ± 6.9 |
0.251 |
| Previous CAD (%) |
14.5 |
31.6 |
0.075 |
| Previous MI (%) |
11.3 |
18.4 |
0.319 |
| Type of MI | |||
| STEMI vs NSTEMI (%) |
66.1/33.9 |
63.2/36.8 |
0.762 |
| Cardiovascular risk factors | |||
| Hypertension (%) |
56.5 |
84.2 |
0.004 |
| Smoking habits (%) |
61.3 |
47.4 |
0.215 |
| Family history (%) |
37.1 |
28.9 |
0.492 |
| Hyperlipidemia (%) |
71.1 |
82.3 |
0.189 |
| Physical inactivity (%) |
56.5 |
60.5 |
0.689 |
| Previous cardiovascular or antidiabetic drugs | |||
| Statins (%) |
29.0 |
31.6 |
0.825 |
| ASA (%) |
19.4 |
42.1 |
0.021 |
| ACEI (%) |
12.9 |
36.8 |
0.007 |
| ARB (%) |
12.9 |
31.6 |
0.038 |
| Beta-blockers (%) |
9.7 |
21.1 |
0.141 |
| Insulin (%) |
0.0 |
26.3 |
<0.001 |
| Oral hypoglycemic (%) |
0.0 |
65.8 |
<0.001 |
| Baseline laboratory | |||
| Admission Troponin I (μg/L)§ |
0.7 ± 5.8 |
1.5 ± 3.6 |
0.798 |
| Peak Troponin I (μg/L)§ |
55.4 ± 71.6 |
56.7 ± 64.7 |
0.793 |
| HbA1C (%)§ |
5.6 ± 0.5 |
7.1 ± 2.2 |
<0.001 |
| Admission glycemia (mg/dl)§ |
109.0 ± 31.0 |
206.5 ± 110.8 |
<0.001 |
| First fasting glycemia (mg/dl)§ |
103.0 ± 24.5 |
156.0 ± 52.5 |
<0.001 |
| Total cholesterol (mg/dl)* |
178.5 ± 59.0 |
211.7 ± 54.9 |
0.007 |
| LDL cholesterol (mg/dl)* |
113.2 ± 39.6 |
145.3 ± 44.3 |
<0.001 |
| HDL cholesterol (mg/dl)§ |
40.2 ± 9.5 |
38.7 ± 12.9 |
0.164 |
| Triglycerides (mg/dl)§ |
138.5 ± 109.5 |
148.0 ± 88.5 |
0.801 |
| Uric acid (mg/dl)* |
5.6 ± 1.3 |
6.2 ± 1.4 |
0.096 |
| Baseline creatinine (mg/dl)§ |
0.8 ± 0.3 |
0.9 ± 0.4 |
0.123 |
| Baseline hemoglobin (g/dl)* |
14.8 ± 1.4 |
14.4 ± 1.2 |
0.200 |
| Admission hs-CRP (mg/dl)* |
0.9 ± 1.3 |
1.0 ± 1.4 |
0.872 |
| LVEF (%)* |
52.6 ± 9.6 |
50.0 ± 11.8 |
0.104 |
| Hospital length of stay§ | 5.4 ± 2.6 | 5.9 ± 3.0 | 0.424 |
ACEI, Angiotensin-Converting Enzyme Inhibitors; ARB, Angiotensin II receptor blockers; ASA, acetylsalicylic acid; CAD, coronary artery disease; hs-CRP, high sensitivity C-reactive protein; LVEF, left ventricular ejection fraction; MI, myocardial infarction; STEMI, ST elevation myocardial infarction; NSTEMI, non-ST elevation myocardial infarction.
*mean ± SD.
§median ± interquartile range.
There were no significant differences in MI presentation (STEMI versus NSTEMI), left ventricular function or renal function between groups (Table 1).
There were no significant differences in the extent of coronary atherosclerosis, number of stents deployed or other cath lab parameters between diabetics and nondiabetics (Table 2).
Table 2.
Comparison of catheterization lab data between diabetics and nondiabetics
| Non-diabetics (N = 62) | Type 2 diabetics (N = 38) | p value | |
|---|---|---|---|
| Catheterization during hospitalization (%) |
94.7 |
96.8 |
0.614 |
| Normal coronaries (%) |
5.0 |
0.0 |
0.289 |
| 1-vessel disease (%) |
41.7 |
41.7 |
1.000 |
| 2-vessel disease (%) |
26.7 |
30.6 |
0.682 |
| 3-vessel disease (%) |
26.7 |
27.8 |
0.906 |
| Left main disease (%) |
6.7 |
5.7 |
0.854 |
| LAD disease (%) |
69.5 |
80.6 |
0.235 |
| PCI before EPCs evaluation (%) |
72.6 |
68.4 |
0.656 |
| Complete revascularization before EPCs evaluation (%) |
44.9 |
45.3 |
0.912 |
| Number of stents deployed before EPCs evaluation | 1.6 ± 1.1 | 1.8 ± 1.2 | 0.649 |
LAD; left anterior descending, PCI; percutaneous coronary intervention.
Reduction of circulating EPCs in diabetic patients
Circulating EPCs levels were expressed for one million cytometric events (Figure 2). Diabetic patients had circulating numbers of CD45dimCD34+KDR+ cells reduced by 63% when compared with nondiabetics, with a parallel decrease in the subpopulation CXCR4+ (Table 3, Figure 3). There was also a significant reduction in the more immature population of CD45dimCD34+CD133+KDR+ EPCs to around half the levels of nondiabetics, and numbers of its precursors CD45dimCD133+KDR+ in peripheral circulation were also significantly decreased. The subpopulation coexpressing the homing marker CXCR4 (CD45dimCD133+KDR+CXCR4+) was also significantly reduced in diabetics (Table 3).
Figure 2.
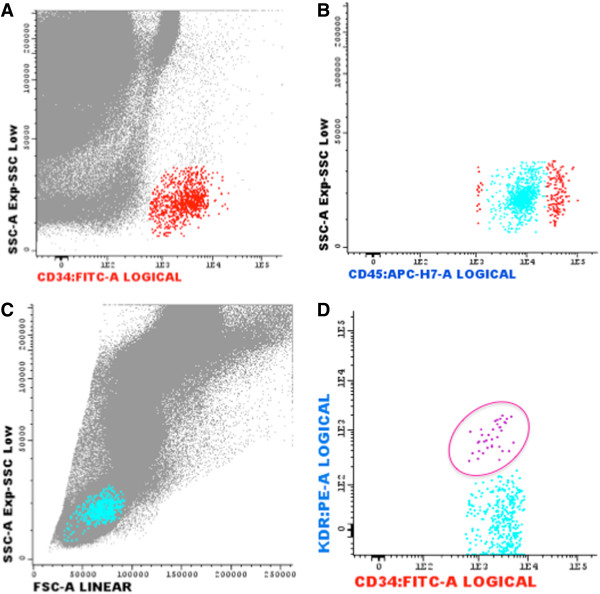
Representative flow cytometry quantification of CD45low/CD34+/KDR+ EPCs in a a nondiabetic patient with STEMI. (A) The cluster of CD34+ cells (red points) was analyzed using FITC-labeled antibodies against CD34 vs. SSC. B) CD45dim subset analysis was conducted using APC-H7-labeled antibodies against CD45 vs SSC. At this step, events forming a cluster of characteristic low SSC and low CD45 fluorescence (SSClowCD45dim cells) expressed CD34+CD45dim cells (light blue events). C) The events fulfilling previous criteria were then displayed on a forward light scatter (FSC) vs. SSC dot plot to confirm that the selected cells fall into the lymphocyte region. D) Finally, CD45dimCD34+KDR+ endothelial progenitor cells (pink cells) are deducted in the dot plot of PE-labeled antibodies against KDR vs CD34.
Table 3.
Comparison of circulating EPCs levels between diabetics and nondiabetics
| Non-diabetics (n = 62) | Type 2 diabetics (n = 38) | p value | |
|---|---|---|---|
| Time from PCI to blood sampling (hours) |
13.8 ± 14.7 |
11.6 ± 11.7 |
0.649 |
| CD34+ cells/106 WBC |
228.8 ± 136.7 |
197.0 ± 115.2 |
0.098 |
| CD133+/106 WBC |
54.4 ± 35.7 |
36.0 ± 18.0 |
0.020 |
| CD45dimCD34+KDR+ cells/106 WBC |
6.2 ± 3.0 |
2.3 ± 0.9 |
<0.001 |
| CD45dimCD34+KDR+CXCR4+ cells/106 WBC |
1.8 ± 1.1 |
0.8 ± 0.7 |
<0.001 |
| CD45dimCD34+CD133+KDR+ cells/106 WBC |
2.1 ± 1.1 |
1.0 ± 0.8 |
<0.001 |
| CD133+KDR+/106 WBC |
4.6 ± 2.9 |
3.1 ± 1.6 |
<0.001 |
| CD133+KDR+CXCR4+/106 WBC | 3.5 ± 1.9 | 2.0 ± 1.2 | <0.001 |
PCI, percutaneous coronary intervention; WBC, white blood cells.
Figure 3.
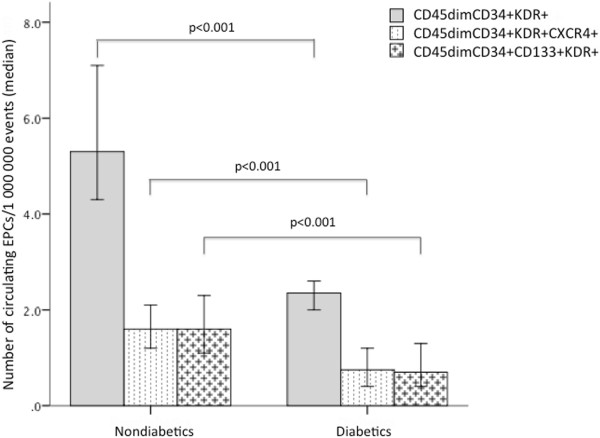
Comparison of circulating EPCs levels between diabetics and nondiabetics. Bars represent median and error bars interquartile range of circulating EPCs numbers quantified by flow cytometry. Mann Whitney U test was used for the comparison between diabetic and nondiabetic EPCs levels.
Circulating EPCs levels across the different disorders of glucose metabolism
Upon OGTT, 24 of the nondiabetic patients had pre-diabetes (29.2% with impaired fasting glucose - IFG, 58.3% with impaired glucose tolerance – IGT and 12.5% with both disorders of glucose metabolism).
Circulating CD45dimCD34+KDR+ EPCs decreased as a continuum from NGM to DM, as there was a reduction of approximately 40% in patients with pre-diabetes as compared with NGM patients (p = 0.018) and there was an additional reduction of these EPCs of about 40% (p = 0.042) when diabetics were compared with patients with pre-diabetes (Table 4). Nonetheless, the population of more immature progenitor cells (CD45dimCD133+KDR+) and the subpopulations coexpressing the CXCR4 marker (CD45dimCD34+KDR+CXCR4+ and CD45dimCD133+KDR+CXCR4+) were not significantly reduced in pre-diabetic patients by comparison with NGM patients (5.4 ± 2.4 vs 3.9 ± 2.8, p = 0.314; 1.8 ± 0.9 vs 1.3 ± 1.2, p = 0.175; and 3.5 ± 2.1 vs 3.2 ± 1.3, p = 0.290, respectively), whereas a significant reduction was apparent from pre-diabetic to diabetic patients on these cells levels (p = 0.022; p = 0.045 and p = 0.015, respectively) (Table 4).
Table 4.
Comparison of circulating EPCs levels between the different glucose metabolism status
| NGM (n = 38) | Pre-diabetes (n = 24) | Diabetes (n = 38) | p value | |
|---|---|---|---|---|
| CD34+ cells/106 WBC |
417.3 ± 266.9 |
225.4 ± 97.5 |
176.5 ± 148.8 |
0.006 |
| CD133+/106 WBC |
41.5 ± 23.7 |
34.1 ± 21.2 |
34.4 ± 19.2 |
0.101 |
| CD45dimCD34+KDR+ cells/106 WBC |
7.0 ± 3.5 |
4.3 ± 2.7 |
2.4 ± 1.2 |
<0.001 |
| CD45dimCD34+KDR+CXCR4+ cells/106 WBC |
1.8 ± 0.9 |
1.3 ± 1.2 |
0.8 ± 0.7 |
0.002 |
| CD45dimCD34+CD133+KDR+ cells/106 WBC |
1.7 ± 1.0 |
1.3 ± 1.1 |
0.7 ± 0.6 |
0.001 |
| CD133+KDR+/106 WBC |
5.4 ± 2.4 |
3.9 ± 2.8 |
3.0 ± 1.9 |
0.002 |
| CD133+KDR+CXCR4+/106 WBC | 3.5 ± 2.1 | 3.2 ± 1.3 | 2.0 ± 1.4 | 0.002 |
NGM, normal glucose metabolism; WBC, white blood cells.
Circulating EPCs numbers according to previous antidiabetic treatment
Regarding the antidiabetic strategy before admission, there were 53% of diabetic patients on oral hypoglycemic drugs, 26% insulin-treated diabetics, and 21% of patients who were not taking any antidiabetic drug (because they were on diet-only therapy or new onset DM was diagnosed during hospitalization). As expected, diabetes duration was significantly longer in insulin-treated patients (13.5 ± 9.8 years versus 6.8 ± 5.0 in patients on oral hypoglycemic drugs versus 1.7 ± 1.2 in diabetics not receiving any antidiabetic drug, p = 0.001). Insulin-treated DM (ITDM) patients and diabetics not previously treated with antidiabetic drugs presented a worse glycemic control as compared with patients on oral hypoglycemic drugs (Figure 4).Numbers of CD45dimCD34+KDR+ EPCs were significantly reduced in diabetic patients previously treated with oral antidiabetic drugs and in diabetics not taking any hypoglycemic drug when compared with nondiabetic patients (Figure 5, A). However, despite the worse glycemic control of diabetics on chronic insulin, their CD45dimCD34+KDR+ EPCs levels were not significantly reduced compared to that of nondiabetic patients (p = 0.160) (Figure 5-A). Regarding the subpopulation of CD45dimCD34+KDR+ cells also expressing the homing marker CXCR4+, all diabetes treatment categories presented significantly decreased circulating levels by comparison with nondiabetic patients (Figure 5-B). Circulating CD45dimCD133+KDR+ cell levels showed a progressive decline from nondiabetics, untreated DM, DM on oral hypoglycemic drugs and finally, ITDM, with patients receiving insulin and patients on oral hypoglycemic drugs presenting significantly lower levels as compared with nondiabetics (p = 0.002 and p = 0.004, respectively) (Figure 5-C). Circulating levels of the CD45dimCD133+KDR+CXCR4+ subpopulation were also significantly lower in all diabetic treatment categories than in nondiabetic patients (Figure 5-D).
Figure 4.
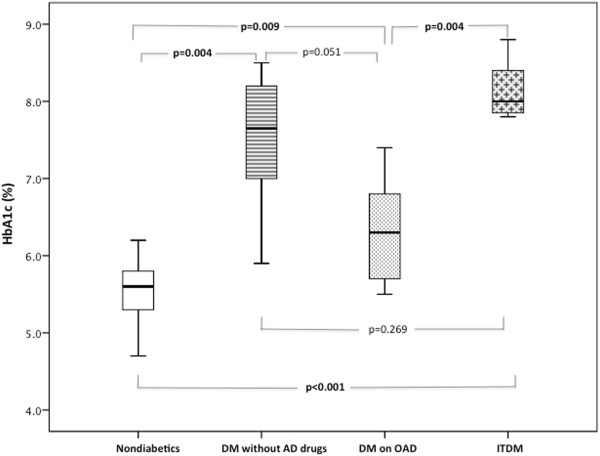
Glycosylated hemoglobin levels according to chronic antidiabetic treatment. Box plots represent the interquartile range of values, the horizontal lines show the median, and whiskers represent the maximum and minimum values. In order to find between which antiadiabetic treatment categories there were significant differences, we compared two by two groups using the Mann Whitney U test.
Figure 5.
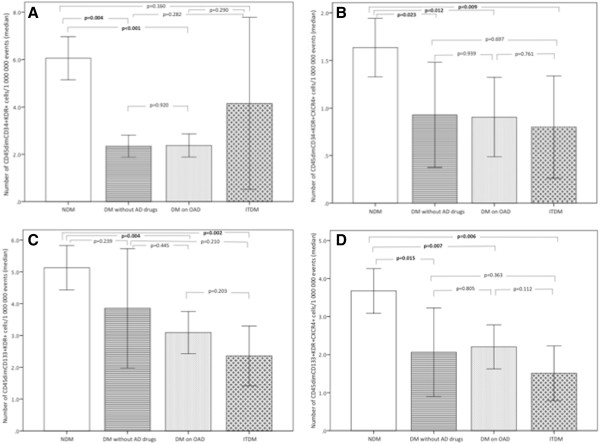
Comparison of circulating EPCs levels between nondiabetic patients and diabetics under different antidiabetic treatments. (A) Circulating numbers of CD45dimCD34+KDR+ cells; (B) Circulating numbers of CD45dimCD34+KDR+CXCR4+ cells; (C) Circulating numbers of CD45dimCD133+KDR+ cells and (D) Circulating numbers of CD45dimCD133+KDR+CXCR4+ cells. Bars represent median and error bars interquartile range of circulating EPCs numbers quantified by flow cytometry. We compared two by two groups using the Mann Whitney U test, to evaluate between which antiadiabetic treatment category there were significant differences.
Impact of glycemic control on EPCs levels
There were significant negative correlations between levels of circulating CD45dimCD34+KDR+ (Figure 6, A), CD45dimCD133+KDR+ progenitors (Figure 6, C), their CXCR4+ subpopulations (Figure 6, B and D) and HbA1c. CD45dimCD34+KDR+EPCs and their subpopulation CD45dimCD34+KDR+CXCR4+ were also inversely correlated with fasting glycemia (r = - 0.371, p < 0.001 and r = - 0.213, p = 0.046, respectively). Nonetheless, EPCs levels were not correlated with DM duration. Levels of circulating CD45dimCD34+KDR+ and CD45dimCD133+KDR+ progenitors were also negatively correlated with age (r = - 0.285, p = 0.007 and r = - 0.343, p = 0.001, respectively).
Figure 6.
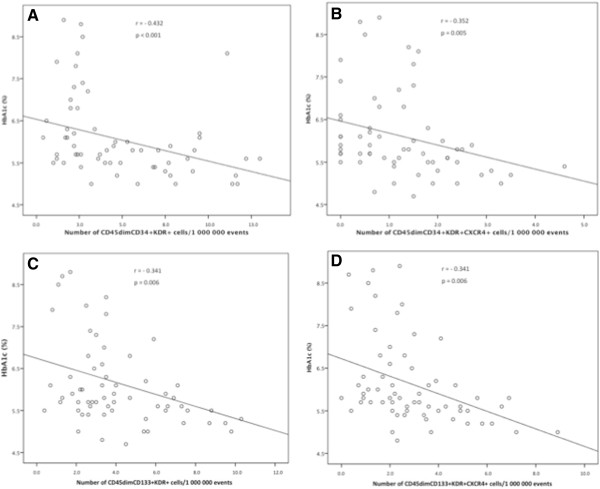
Relationship between levels of circulating EPCs and HbA1c. Scatter plots showing significant negative relationship relationship (Pearson correlation) between HbA1c and circulating levels of: A) CD45dimCD34+KDR+ cells; B) CD45dimCD34+KDR+CXCR4+ cells; C) CD133+KDR+ cells; D) CD133+KDR+CXCR4+ cells.
Remarkably, correlations with HbA1c remain significant even after adjustment for age, gender, hypertension, LDL-cholesterol, family history of CAD, smoking habits and physical inactivity (Table 5).
Table 5.
Multivariate regression analysis assessing the correlation between HbA1c and circulating progenitor cells levels, after adjustment for other cardiovascular risk factors than diabetes
| Variable |
CD45dimCD34+KDR+ levels |
CD45dimCD34+KDR+CXCR4+ levels |
CD133+KDR+ levels |
CD133+KDR+CXCR4+ levels |
||||
|---|---|---|---|---|---|---|---|---|
| Standard coefficient (ß) | p | Standard coefficient (ß) | p | Standard coefficient (ß) | p | Standard coefficient (ß) | p | |
| HbA1c |
-0.308 |
0.019 |
-0.260 |
0.031 |
-0.342 |
0.009 |
-0.416 |
0.001 |
| Age |
-0.188 |
0.217 |
-0.107 |
0.482 |
-0.254 |
0.090 |
-0.052 |
0.720 |
| Gender |
0.208 |
0.067 |
0.207 |
0.119 |
0.044 |
0.740 |
0.158 |
0.233 |
| Hypertension |
-0.071 |
0.597 |
-0.075 |
0.579 |
0.025 |
0.855 |
-0.015 |
0.905 |
| LDL-cholesterol |
-0.057 |
0.683 |
-0.047 |
0.727 |
-0.176 |
0.202 |
-0.148 |
0.267 |
| Family history of CAD |
-0.129 |
0.361 |
-0.170 |
0.202 |
0.081 |
0.560 |
0.078 |
0.563 |
| Smoking habits |
0.003 |
0.985 |
-0.204 |
0.188 |
-0.086 |
0.563 |
-0.133 |
0.369 |
| Physical inactivity |
-0.203 |
0.139 |
-0.080 |
0.556 |
-0.057 |
0.657 |
-0.167 |
0.180 |
| Adjusted R2 |
0.264 |
… |
0.256 |
… |
0.246 |
… |
0.247 |
… |
| Significance (ANOVA) | … | 0.032 | … | 0.040 | … | 0.046 | … | 0.033 |
CAD, coronary artery disease; HbA1c, hemoglobin A1c; LDL, low density lipoprotein.
Prognostic impact of EPCs
Clinical outcomes during the 24 months follow-up period are represented in Table 6.
Table 6.
Comparison of clinical outcomes after AMI between diabetics and nondiabetics
| Nondiabetics (N = 62) | Diabetics (N = 38) | Odds ratio | P value | |
|---|---|---|---|---|
| Cardiovascular mortality (%) |
1.6 |
7.9 |
5.2 |
0.120 |
| Stroke or TIA (%) |
0 |
5.4 |
- |
0.064 |
| Re-infarction (%) |
3.3 |
0 |
- |
0.266 |
| Unstable Angina (%) |
3.3 |
18.9 |
6.9 |
0.009 |
| Re-hospitalization for UA or HF (%) |
8.1 |
29.7 |
4.8 |
0.005 |
| MACE (%) | 9.8 | 31.6 | 4.2 | 0.006 |
AMI, acute myocardial infarction; HF, heart failure; MACE, Major Adverse Cardiac Events; TIA, transient ischemic accident; UA, unstable angina.
There were no significant differences in re-infarction, nonfatal stroke/transient ischemic attack or cardiovascular mortality rates between groups. However, the occurrence of unstable angina, the composite endpoints MACE and re-hospitalization for unstable angina or heart failure were significantly higher in diabetics, with the following odds ratios 6.89 (95% CI, 1.35-35.19), 4.23 (95% CI 1.43-12.53) and 4.82 (95% CI 1.52-15.30), respectively.Regarding baseline circulating EPCs levels, patients with unstable angina, unplanned PCI or MACE during follow-up presented significantly lower levels of CD45dimCD34+KDR+ and CD45dimCD133+KDR+ EPCs. Levels of the CD45dimCD133+KDR+CXCR4+ EPCs subpopulation were also significantly reduced, at baseline, in patients who underwent unstable angina or MACE during the 2-year follow-up period (Figure 7). Additionally, the Kaplan–Meier survival curves for freedom from MACE according to EPCs levels showed a significantly lower event-free survival rate in patients with lower EPCs levels in the early phases of AMI (log-rank test, p = 0.023 for CD45dimCD34+KDR+ EPCs and log-rank test, p = 0.004 for CD45dimCD133+KDR+ cells) (Figure 8).
Figure 7.
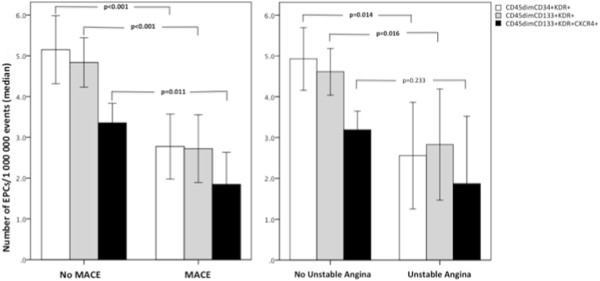
Comparison of baseline EPCs levels between patients with or without cardiovascular events during the 2-year follow-up period. Bars represent median and error bars interquartile range of circulating EPCs numbers, quantified by flow cytometry within the first 24 h of admission. We compared EPCs levels between patients with versus without MACE (left panel) and between patients with versus without unstable angina (right panel), using the Mann Whitney U test.
Figure 8.
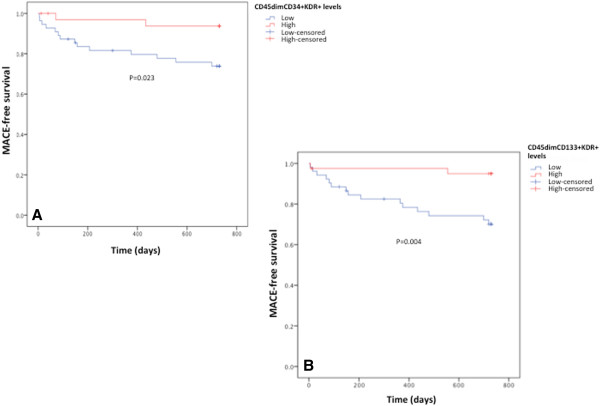
Kaplan-Meier MACE-free survival curves following an AMI, according to circulating EPCs levels presented in the acute phase. A) Comparison of MACE-free survival curves between patients with levels of CD45dimCD34+KDR+ cells above (red line) or below (blue line) the median. B) Comparison of MACE-free survival curves between patients with levels of CD133+KDR+ cells above (red line) or below (blue line) the median level. The p value was determined by log-rank test.
Discussion
There were four major findings in the present study. First, we confirmed that, in the acute phase of a MI, diabetic patients present dramatically reduced levels of circulating EPCs by comparison with nondiabetics. Second, this study showed for the first time that even pre-diabetes reduces EPCs response to an AMI, since EPCs levels were significantly reduced in pre-diabetics and further reduced in diabetics as compared with patients with NGM. Third, previous chronic insulin therapy (but not oral antidiabetic drugs) seems to attenuate the deficit in circulating EPCs seen in diabetic patients with an AMI. Finally, we have demonstrated that the degree of glycemic control is an important determinant of circulating EPCs numbers in the setting of an AMI.
An AMI is a recognized pathological stimulus for EPCs mobilization. In fact, patients with AMI present significantly increased numbers of circulating EPCs as compared with control subjects or with patients with stable angina [8]. It has been shown that circulating EPCs increase immediately after the onset of an AMI, with a subsequent peak at day 5 and a rapid decline thereafter, normalizing within 2 months [21,22]. Circulating EPCs constitute a key endogenous repair mechanism to counteract ongoing endothelial cell injury, replace dysfunctional endothelium, and enhance tissue repair after ischemic vascular injury [23]. Of note, depletion of circulating EPCs pool and impaired migratory activity of these progenitor cells have been shown to be predictive of future adverse cardiovascular events [24,25]. In accordance with these previous studies, our work showed that freedom from MACE following an AMI was significantly poorer in patients with lower baseline EPCs levels.
It has been extensively demonstrated that patients with DM have a profound reduction of EPCs levels in peripheral blood, which has been correlated with the high cardiovascular morbidity and mortality associated with diabetes [10,26]. Additionally, reduced EPCs numbers have been independently associated with impaired myocardial function in diabetic patients [27]. Fadini et al. have demonstrated in diabetic animals, a deficient EPCs mobilization and impaired compensatory angiogenesis after hindlimb ischaemia-reperfusion injury [28]. However, in the clinical setting of AMI, and despite the important vascular protective role of EPCs, to date, only three clinical studies have studied the dynamics of EPCs mobilization in diabetic patients [21,22,29]. In those studies, circulating EPCs levels were decreased in diabetics [21,22] (or hyperglycemic patients, in the Marfella et al. study) [29] compared with non-diabetic patients immediately after the onset of AMI (day 1). Moreover, it has been demonstrated that the peak level of circulating EPCs was delayed in diabetic patients compared with that of nondiabetic patients (from day 5 in nondiabetic patients to day 7 in diabetic patients) [21,22]. Consistent with these previous studies, the present work confirmed that circulating EPCs levels were strikingly reduced in the early phases of an AMI in diabetic patients as compared with nondiabetic patients. Of note, this importantly reduction in EPCs levels seen in diabetic patients does not seem justified by differences in myocardial ischemia or different coronary revascularization procedures between groups, as values of troponin I (a highly specific marker of myocardial injury) and coronary revascularization were similar in diabetic and nondiabetic patients.
It has become evident that circulating EPCs numbers were inversely correlated to the severity of CAD [30,31]. However, in the present study the huge difference in EPCs levels between AMI diabetics and nondiabetics cannot be explained by differences in CAD severity, since there were no significant differences in the extension of coronary stenosis between both groups.
A large body of evidence links classical cardiovascular risk factors, such as hypertension, with reduction in circulating EPCs [32]. In this study population, diabetics presented a significantly higher prevalence of hypertension that could exacerbate the difference in EPCs levels as compared with nondiabetics. However, diabetics were also more frequently treated with drugs that recognizably increase circulating EPCs numbers, such as ACE-inhibitors and ARBs, what would counterbalance the possible reduction on EPCs numbers due to the higher prevalence of hypertension [14].
EPCs can be identified on the basis of the expression of surface markers, by flow cytometry, a method considered the gold standard for the quantification of these cells in peripheral blood [33]. Of note, there are no unique or specific surface antigen that can be used to identify circulating EPCs. Therefore, FACS protocols must use the combination of various membrane markers for EPCs quantification. In the present work, we used a standardized polychromatic FACS protocol based upon the detection of CD34 (an adhesion molecule expressed mainly on haematopoietic stem cells) [34], CD133/AC133 (a surface marker expressed in an immature subset of EPCs, which share more characteristics of stem/progenitor cells) [35], KDR/VEGF-R2 (a typical endothelial marker) [36], CXCR4/CD184 (a homing marker) [19] and CD45dim (critical to exclude myeloid cells and because it has been previously demonstrated that only the fraction of CD45dim cells harbors the “true” circulating EPCs) [17,18]. Importantly, there are no studies in the literature that have attempted to quantify, at the same time, both CD45dimCD34+KDR+EPCs and the more immature population of CD45dimCD133+KDR+ progenitors in patients with AMI. Thus, until now there has been no data available on the relation between these 2 populations in diabetics with an AMI, which would be important to elucidate the mechanisms underlying their impaired response. In this study, we showed for the first time that, not only CD45dimCD34+KDR+ but also 2the more immature precursors CD45dimCD34+CD133+KDR+ and CD45dimCD133+KDR+ were significantly reduced in diabetic AMI patients by comparison with nondiabetics. Based on these results, it is tempting to speculate that EPCs reduction in diabetes was due, at least in part, to impaired bone marrow mobilization. Because, if the reduction in EPCs levels was motivated by a decrease in survival alone it would be expected to have reduced levels of CD45dimCD34+KDR+ but increased, or at least normal, levels of the more immature population of CD45dimCD133+KDR+ cells, due to positive feedback stimulation of bone marrow recruitment. What we verified here was that the reduction in the more mature EPCs population was not accompanied by the expected up regulation of the more immature ones. In fact, despite the reduction in CD45dimCD34+KDR+ levels, CD45dimCD133+KDR+ and CD45dimCD34+CD133+KDR+ precursors were also reduced, pointing to impairment in recruitment mechanisms.
Besides the reduction in EPCs counts, we found that the fraction of EPCs coexpressing the homing receptor CXCR4 were also significantly reduced in diabetic AMI patients what may represent an impaired homing capacity of these cells to sites of vascular damage. In fact, CXCR4, the only known receptor for SDF-1, has been reported to play an important role in EPCs homing [19]. Moreover, CXCR4/SDF-1 interaction influences proliferation and mobilization of EPCs from the bone marrow [37]. Since functional study of EPCs, in large populations, with in vitro assays is prohibitively expensive and time consuming, the analysis by flow cytometry of EPCs coexpressing CXCR4 may provide a promising alternative parameter to assess EPCs function. This is the first study to show a reduction in numbers of EPCs coexpressing CXCR4 in diabetic patients with AMI compared with AMI nondiabetics. It is probable that this down regulation in CXCR4+ cells denotes a homing impairment, which in addition to the markedly reduction in circulating EPCs levels may contribute to the worsened outcome post-AMI observed in diabetics.
Pre-diabetes is a general term that refers to an intermediate stage between NGM and overt DM, including IFG and IGT. These disorders of glucose metabolism confer an increased risk for developing both DM and cardiovascular events [13,15,38]. In the present study we have found that CD45dimCD34+KDR+ EPCs were significantly lower in pre-diabetic patients and further reduced in those with DM, as compared with individuals with NGM, suggesting that the reduction in the more mature EPCs population follows the continuum of DM development. These findings suggest that circulating EPCs reduction is an early event in the natural history of DM, what is in accordance to a previous work of Fadini et al. [39]. That study has shown, in individuals from a metabolic outpatient clinic, that circulating CD34+KDR+ cells present a progressive decline from NGM, to prediabetics and diabetic patients and that both fasting and post-challenge glucose were inversely related to circulating CD34+KDR+ EPCs levels [39]. Our work further extends these findings by the quantification of more immature EPCs populations and the study of homing function by the analysis of CXCR4+ subpopulations. Interestingly, we verified that CD45dimCD133+KDR+ EPCs and both subpopulations of CXCR4+ EPCs (CD45dimCD34+KDR+CXCR4+ and CD45dimCD133+KDR+CXCR4+ cells) were not significantly reduced in pre-diabetic AMI patients, compared to patients with NGM. One possible explanation for this divergent influence on different EPCs populations is that pre-diabetes reduces EPCs survival (with subsequent reduction CD45dimCD34+KDR+ EPCs levels) but, does not impair neither bone marrow recruitment of EPCs (leading to no differences in levels of CD45dimCD133+KDR+ EPCs) nor homing processes (explaining the normal proportion of EPCs coexpressing CXCR4).
Previous in vitro and several animal studies have demonstrated that insulin therapy has a protective role over EPCs function [40-42]. More recently, Marfella et al. have demonstrated, in hyperglycemic patients with AMI, that EPCs levels increased after insulin infusion for intensive glycemic control [29]. Regarding oral antidiabetic drugs, several clinical studies have shown that PPAR-γ agonists, such as rosiglitazone and pioglitazone and also DPP-4 inhibitor sitagliptin increase EPCs levels and improve their function in diabetic patients [43-45]. However, little is known about the molecular mechanisms that regulate the beneficial effects of all these antidiabetic drugs over EPCs.
Importantly, evidence demonstrates that the degree of hyperglycemic control in diabetic patients is closely related to circulating EPCs levels [46,47]. However, despite the obvious interest to know the impact of chronic antidiabetic therapy on EPCs response of diabetic patients to an AMI, until now there have been no studies in the literature addressing this subject. Therefore, in the present work we have studied this issue and verified that, despite the longer DM duration and the worse glycemic control, insulin treated patients presented levels of CD45dimCD34+KDR+ EPCs that tended to approach that of nondiabetics. Conversely, CD45dimCD133+KDR+ EPCs and subpopulations coexpressing the CXCR4 receptor were not ameliorated by chronic insulin therapy, presenting the lowest levels in patients previously under insulin. Regarding oral antidiabetic drugs we were surprised to find no beneficial effect on EPCs levels, since these results differ from some published studies [43-45]. Notably, in accordance with the literature our results further demonstrated that levels of both CD45dimCD34+KDR+ and CD45dimCD133+KDR+ EPCs and even their subpopulations coexpressing the CXCR4 surface marker were inversely correlated with HbA1c, underscoring the importance of the glycemic control for EPCs response to an AMI. Taken together, these results suggest that insulin, but not oral antidiabetic drugs, may increase survival of circulating EPCs (denoted by the trend to the normalization of CD45dimCD34+KDR+ levels). So, it is tempting to speculate that the favorable clinical outcomes associated with glycemic control during AMI may be partly dependent on stimulation of EPCs-mediated neovascularization in the ischemic myocardium. However, even chronic insulin treatment seemed unable to correct the characteristic dysfunction of diabetics EPCs (here illustrated by the decrease in CD45dimCD133+KDR+ EPCs, which may represent an impairment in mobilization from bone marrow, and reduction in CXCR4+ subpopulations, denoting a possible homing dysfunction). Yet, since patients under insulin therapy had the highest HBA1c levels, it is still unknown if with a better glycemic control chronic insulin therapy could reverse EPCs dysfunction of diabetic patients and completely normalize their response to an AMI. Altogether, our results suggest that chronic hyperglycemia and not diabetes per se, is the responsible for impaired EPCs response of diabetic patients to myocardial ischemia.
Limitations
The limitations of our study should be acknowledged: 1) the widespread interlaboratory variations in FACS methodology used to quantify circulating EPCs is still a problem. In this study we used a standardized protocol, which has demonstrated a high accuracy in the detection of different EPCs subpopulations with angiogenic properties and enable us to study the differentiation and commitment of these cells, from early precursors to more mature circulating EPCs [17,48]. However, we recognize that further standardization of EPCs definitions and FACS protocols would be important to better compare results between different groups; 2) the long list of exclusion criteria limited the enrollment of higher number of AMI patients in this study, resulting in a relatively small number of patients in each antidiabetic treatment group. Therefore, the data regarding the comparison of EPCs levels between the different antidiabetic treatment categories should be interpreted with caution because of the risk of error type II and further studies to explore how insulin therapy may interact and affect diabetic EPC numbers and function in patients with AMI, are obviously warranted; 3) since investigation of the molecular mechanisms regulating circulating EPCs levels in AMI diabetic patients was not under the scope of this study, the signaling pathways underlying the observed reduction in EPCs levels during the early phases of AMI in diabetic as compared to nondiabetic patients are unknown”.
Conclusions
In summary, our data demonstrates that there is a progressive decrease in EPCs response to an AMI, according to the glycemic continuum, from NGM to pre-diabetes and finally DM, and that the exhaustion of the EPCs pool is influenced by the degree of glycemic control. Furthermore, it seems conceivable to use therapeutic interventions, such as insulin, to try to reverse the impaired response to an AMI of diabetics and possibly improve the dismal prognosis of these patients.
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
NA designed the study, contributed to clinical data acquisition and has written the first draft of the manuscript. RF contributed to data interpretation and critically revised the manuscript. AS helped to draft the manuscript and performed the statistical analysis. FS participated in the acquisition of clinical data. AL participated in interpretation of the data and helped to draft the manuscript. TC carried out the flow cytometry analysis and participated in interpretation of the data. AP contributed to the refinement of the research protocol, to the data analysis and interpretation, and to the development of the manuscript. GMP and LAP contributed to obtaining funding, and critically revised the manuscript. LG participated in the study design, contributed to obtaining funding and critically revised the manuscript. CFR conceived the study, participated in the data interpretation, oversaw the development of the manuscript and supervised the project. All authors read and approved the final manuscript.
Contributor Information
Natália António, Email: natalia.antonio@gmail.com.
Rosa Fernandes, Email: rcfernandes@fmed.uc.pt.
Ana Soares, Email: soares.amr@gmail.com.
Francisco Soares, Email: francisco.campos.soares@gmail.com.
Ana Lopes, Email: ana.lopes.231@gmail.com.
Tiago Carvalheiro, Email: tiago.carvalheiro@gmail.com.
Artur Paiva, Email: artur.paiva@ipst.min-saude.pt.
Guilherme Mariano Pêgo, Email: marianopego@huc.min-saude.pt.
Luís A Providência, Email: laprovid@gmail.com.
Lino Gonçalves, Email: lgoncalv@ci.uc.pt.
Carlos Fontes Ribeiro, Email: fontes.ribeiro@gmail.com.
Acknowledgments
This work was supported by unrestricted research grants from “Pfizer, Inc”, Portuguese Society of Cardiology, Foundation for Science and Technology (PEst-C/SAU/UI3282/2011 and COMPETE) and Faculty of Medicine of the University of Coimbra (Programa de Estímulo à Investigação).
References
- Harjai KJ, Stone GW, Boura J, Mattos L, Chandra H, Cox D, Grines L, O’Neill W, Grines C. Comparison of outcomes of diabetic and nondiabetic patients undergoing primary angioplasty for acute myocardial infarction. Am J Cardiol. 2003;91(9):1041–1045. doi: 10.1016/s0002-9149(03)00145-0. [DOI] [PubMed] [Google Scholar]
- Asahara T, Murohara T, Sullivan A, Silver M, van der Zee R, Li T, Witzenbichler B, Schatteman G, Isner JM. Isolation of putative progenitor endothelial cells for angiogenesis. Science. 1997;275(5302):964–967. doi: 10.1126/science.275.5302.964. [DOI] [PubMed] [Google Scholar]
- Zampetaki A, Kirton JP, Xu Q. Vascular repair by endothelial progenitor cells. Cardiovasc Res. 2008;78(3):413–421. doi: 10.1093/cvr/cvn081. [DOI] [PubMed] [Google Scholar]
- Hristov M, Erl W, Weber PC. Endothelial progenitor cells: mobilization, differentiation, and homing. Arterioscler Thromb Vasc Biol. 2003;23(7):1185–1189. doi: 10.1161/01.ATV.0000073832.49290.B5. [DOI] [PubMed] [Google Scholar]
- Urbich C, Aicher A, Heeschen C, Dernbach E, Hofmann WK, Zeiher AM, Dimmeler S. Soluble factors released by endothelial progenitor cells promote migration of endothelial cells and cardiac resident progenitor cells. J Mol Cell Cardiol. 2005;39(5):733–742. doi: 10.1016/j.yjmcc.2005.07.003. [DOI] [PubMed] [Google Scholar]
- Hill JM, Zalos G, Halcox JP, Schenke WH, Waclawiw MA, Quyyumi AA, Finkel T. Circulating endothelial progenitor cells, vascular function, and cardiovascular risk. N Engl J Med. 2003;348(7):593–600. doi: 10.1056/NEJMoa022287. [DOI] [PubMed] [Google Scholar]
- Shintani S, Murohara T, Ikeda H, Ueno T, Honma T, Katoh A, Sasaki K, Shimada T, Oike Y, Imaizumi T. Mobilization of endothelial progenitor cells in patients with acute myocardial infarction. Circulation. 2001;103(23):2776–2779. doi: 10.1161/hc2301.092122. [DOI] [PubMed] [Google Scholar]
- Massa M, Rosti V, Ferrario M, Campanelli R, Ramajoli I, Rosso R, De Ferrari GM, Ferlini M, Goffredo L, Bertoletti A, Klersy C, Pecci A, Moratti R, Tavazzi L. Increased circulating hematopoietic and endothelial progenitor cells in the early phase of acute myocardial infarction. Blood. 2005;105(1):199–206. doi: 10.1182/blood-2004-05-1831. [DOI] [PubMed] [Google Scholar]
- Hamed S, Brenner B, Aharon A, Daoud D, Roguin A. Nitric oxide and superoxide dismutase modulate endothelial progenitor cell function in type 2 diabetes mellitus. Cardiovasc Diabetol. 2009;8:56. doi: 10.1186/1475-2840-8-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fadini GP, Miorin M, Facco M, Bonamico S, Baesso I, Grego F, Menegolo M, de Kreutzenberg SV, Tiengo A, Agostini C, Avogaro A. Circulating endothelial progenitor cells are reduced in peripheral vascular complications of type 2 diabetes mellitus. J Am Coll Cardiol. 2005;45(9):1449–1457. doi: 10.1016/j.jacc.2004.11.067. [DOI] [PubMed] [Google Scholar]
- Tepper OM, Galiano RD, Capla JM, Kalka C, Gagne PJ, Jacobowitz GR, Levine JP, Gurtner GC. Human endothelial progenitor cells from type II diabetics exhibit impaired proliferation, adhesion, and incorporation into vascular structures. Circulation. 2002;106(22):2781–2786. doi: 10.1161/01.cir.0000039526.42991.93. [DOI] [PubMed] [Google Scholar]
- Li H, Zhang X, Guan X, Cui X, Wang Y, Chu H, Cheng M. Advanced glycation end products impair the migration, adhesion and secretion potentials of late endothelial progenitor cells. Cardiovasc Diabetol. 2012;11:46. doi: 10.1186/1475-2840-11-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeFronzo RA, Abdul-Ghani M. Assessment and treatment of cardiovascular risk in prediabetes: impaired glucose tolerance and impaired fasting glucose. Am J Cardiol. 2011;108(3 Suppl):3B–24B. doi: 10.1016/j.amjcard.2011.03.013. [DOI] [PubMed] [Google Scholar]
- Antonio N, Fernandes R, Rodriguez-Losada N, Jimenez-Navarro MF, Paiva A, de Teresa GE, Goncalves L, Ribeiro CF, Providencia LA. Stimulation of endothelial progenitor cells: a new putative effect of several cardiovascular drugs. Eur J Clin Pharmacol. 2010;66(3):219–230. doi: 10.1007/s00228-009-0764-y. [DOI] [PubMed] [Google Scholar]
- Ryden L, Grant PJ, Anker SD, Berne C, Cosentino F, Danchin N, Deaton C, Escaned J, Hammes HP, Huikuri H, Marre M, Marx N, Mellbin L, Ostergren J, Patrono C, Seferovic P, Uva MS, Taskinen MR, Tendera M, Tuomilehto J, Valensi P, Zamorano JL. ESC Guidelines on diabetes, pre-diabetes, and cardiovascular diseases developed in collaboration with the EASD: the Task Force on diabetes, pre-diabetes, and cardiovascular diseases of the European Society of Cardiology (ESC) and developed in collaboration with the European Association for the Study of Diabetes (EASD) Eur Heart J. 2013;34(39):3035–3087. doi: 10.1093/eurheartj/eht108. [DOI] [PubMed] [Google Scholar]
- American Diabetes Association. Diagnosis and classification of diabetes mellitus. Diabetes Care. 2012;35(Suppl 1):S64–S71. doi: 10.2337/dc12-s064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt-Lucke C, Fichtlscherer S, Aicher A, Tschope C, Schultheiss HP, Zeiher AM, Dimmeler S. Quantification of circulating endothelial progenitor cells using the modified ISHAGE protocol. PLoS One. 2010;5(11):e13790. doi: 10.1371/journal.pone.0013790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingram DA, Caplice NM, Yoder MC. Unresolved questions, changing definitions, and novel paradigms for defining endothelial progenitor cells. Blood. 2005;106(5):1525–1531. doi: 10.1182/blood-2005-04-1509. [DOI] [PubMed] [Google Scholar]
- Walter DH, Haendeler J, Reinhold J, Rochwalsky U, Seeger F, Honold J, Hoffmann J, Urbich C, Lehmann R, Arenzana-Seisdesdos F, Aicher A, Heeschen C, Fichtlscherer S, Zeiher AM, Dimmeler S. Impaired CXCR4 signaling contributes to the reduced neovascularization capacity of endothelial progenitor cells from patients with coronary artery disease. Circ Res. 2005;97(11):1142–1151. doi: 10.1161/01.RES.0000193596.94936.2c. [DOI] [PubMed] [Google Scholar]
- Sutherland DR, Anderson L, Keeney M, Nayar R, Chin-Yee I. The ISHAGE guidelines for CD34+ cell determination by flow cytometry. International Society of Hematotherapy and Graft Engineering. J Hematother. 1996;5(3):213–226. doi: 10.1089/scd.1.1996.5.213. [DOI] [PubMed] [Google Scholar]
- Sun JY, Zhai L, Li QL, Ye JX, Kang LN, Xie J, Xu B. Effects of ACE inhibition on endothelial progenitor cell mobilization and prognosis after acute myocardial infarction in type 2 diabetic patients. Clinics (Sao Paulo) 2013;68(5):665–673. doi: 10.6061/clinics/2013(05)14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling L, Shen Y, Wang K, Jiang C, Fang C, Ferro A, Kang L, Xu B. Worse clinical outcomes in acute myocardial infarction patients with type 2 diabetes mellitus: relevance to impaired endothelial progenitor cells mobilization. PLoS One. 2012;7(11):e50739. doi: 10.1371/journal.pone.0050739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeh ET, Zhang S, Wu HD, Korbling M, Willerson JT, Estrov Z. Transdifferentiation of human peripheral blood CD34+-enriched cell population into cardiomyocytes, endothelial cells, and smooth muscle cells in vivo. Circulation. 2003;108(17):2070–2073. doi: 10.1161/01.CIR.0000099501.52718.70. [DOI] [PubMed] [Google Scholar]
- Werner N, Kosiol S, Schiegl T, Ahlers P, Walenta K, Link A, Bohm M, Nickenig G. Circulating endothelial progenitor cells and cardiovascular outcomes. N Engl J Med. 2005;353(10):999–1007. doi: 10.1056/NEJMoa043814. [DOI] [PubMed] [Google Scholar]
- Fortunato O, Spinetti G, Specchia C, Cangiano E, Valgimigli M, Madeddu P. Migratory activity of circulating progenitor cells and serum SDF-1alpha predict adverse events in patients with myocardial infarction. Circ Res. 2013;100(2):192–200. doi: 10.1093/cvr/cvt153. [DOI] [PubMed] [Google Scholar]
- Rosenson RS, Reasner CA. Therapeutic approaches in the prevention of cardiovascular disease in metabolic syndrome and in patients with type 2 diabetes. Curr Opin Cardiol. 2004;19(5):480–487. doi: 10.1097/01.hco.0000133111.66486.c6. [DOI] [PubMed] [Google Scholar]
- Zhao CT, Wang M, Siu CW, Hou YL, Wang T, Tse HF, Yiu KH. Myocardial dysfunction in patients with type 2 diabetes mellitus: role of endothelial progenitor cells and oxidative stress. Cardiovasc Diabetol. 2012;11:147. doi: 10.1186/1475-2840-11-147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fadini GP, Sartore S, Schiavon M, Albiero M, Baesso I, Cabrelle A, Agostini C, Avogaro A. Diabetes impairs progenitor cell mobilisation after hindlimb ischaemia-reperfusion injury in rats. Diabetologia. 2006;49(12):3075–3084. doi: 10.1007/s00125-006-0401-6. [DOI] [PubMed] [Google Scholar]
- Marfella R, Rizzo MR, Siniscalchi M, Paolisso P, Barbieri M, Sardu C, Savinelli A, Angelico N, Del Gaudio S, Esposito N, Rambaldi PF, D'Onofrio N, Mansi L, Mauro C, Paolisso G, Balestrieri ML. Peri-procedural tight glycemic control during early percutaneous coronary intervention up-regulates endothelial progenitor cell level and differentiation during acute ST-elevation myocardial infarction: effects on myocardial salvage. Int J Cardiol. 2013;168(4):3954–3962. doi: 10.1016/j.ijcard.2013.06.053. [DOI] [PubMed] [Google Scholar]
- Chen MC, Chen CJ, Yang CH, Liu WH, Fang CY, Hsieh YK, Chang HW. Relationship of the percentage of circulating endothelial progenitor cell to the severity of coronary artery disease. Heart Vessels. 2008;23(1):47–52. doi: 10.1007/s00380-007-1006-9. [DOI] [PubMed] [Google Scholar]
- Bozdag-Turan I, Turan RG, Turan CH, Ludovicy S, Akin I, Kische S, Arsoy NS, Schneider H, Ortak J, Rehders T, Hermann T, Paranskaya L, Kohlschein P, Bastian M, Ulus AT, Sahin K, Ince H, Nienaber CA. Relation between the frequency of CD34(+) bone marrow derived circulating progenitor cells and the number of diseased coronary arteries in patients with myocardial ischemia and diabetes. Cardiovasc Diabetol. 2011;10:107. doi: 10.1186/1475-2840-10-107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasa M, Fichtlscherer S, Aicher A, Adler K, Urbich C, Martin H, Zeiher AM, Dimmeler S. Number and migratory activity of circulating endothelial progenitor cells inversely correlate with risk factors for coronary artery disease. Circ Res. 2001;89(1):E1–E7. doi: 10.1161/hh1301.093953. [DOI] [PubMed] [Google Scholar]
- Khan SS, Solomon MA, McCoy JP Jr. Detection of circulating endothelial cells and endothelial progenitor cells by flow cytometry. Cytometry B Clin Cytom. 2005;64(1):1–8. doi: 10.1002/cyto.b.20040. [DOI] [PubMed] [Google Scholar]
- Yoder MC. Human endothelial progenitor cells. Cold Spring Harb Perspect Med. 2012;2(7):a006692. doi: 10.1101/cshperspect.a006692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Handgretinger R, Gordon PR, Leimig T, Chen X, Buhring HJ, Niethammer D, Kuci S. Biology and plasticity of CD133+ hematopoietic stem cells. Ann N Y Acad Sci. 2003;996:141–151. doi: 10.1111/j.1749-6632.2003.tb03242.x. [DOI] [PubMed] [Google Scholar]
- Peichev M, Naiyer AJ, Pereira D, Zhu Z, Lane WJ, Williams M, Oz MC, Hicklin DJ, Witte L, Moore MA, Rafii S. Expression of VEGFR-2 and AC133 by circulating human CD34(+) cells identifies a population of functional endothelial precursors. Blood. 2000;95(3):952–958. [PubMed] [Google Scholar]
- Yamaguchi J, Kusano KF, Masuo O, Kawamoto A, Silver M, Murasawa S, Bosch-Marce M, Masuda H, Losordo DW, Isner JM, Asahara T. Stromal cell-derived factor-1 effects on ex vivo expanded endothelial progenitor cell recruitment for ischemic neovascularization. Circulation. 2003;107(9):1322–1328. doi: 10.1161/01.cir.0000055313.77510.22. [DOI] [PubMed] [Google Scholar]
- Barr EL, Zimmet PZ, Welborn TA, Jolley D, Magliano DJ, Dunstan DW, Cameron AJ, Dwyer T, Taylor HR, Tonkin AM, Wong TY, McNeil J, Shaw JE. Risk of cardiovascular and all-cause mortality in individuals with diabetes mellitus, impaired fasting glucose, and impaired glucose tolerance: the Australian Diabetes, Obesity, and Lifestyle Study (AusDiab) Circulation. 2007;116(2):151–157. doi: 10.1161/CIRCULATIONAHA.106.685628. [DOI] [PubMed] [Google Scholar]
- Fadini GP, Pucci L, Vanacore R, Baesso I, Penno G, Balbarini A, Di Stefano R, Miccoli R, de Kreutzenberg S, Coracina A, Tiengo A, Agostini C, Del Prato S, Avogaro A. Glucose tolerance is negatively associated with circulating progenitor cell levels. Diabetologia. 2007;50(10):2156–2163. doi: 10.1007/s00125-007-0732-y. [DOI] [PubMed] [Google Scholar]
- Dong L, Kang L, Ding L, Chen Q, Bai J, Gu R, Li L, Xu B. Insulin modulates ischemia-induced endothelial progenitor cell mobilization and neovascularization in diabetic mice. Microvasc Res. 2011;82(3):227–236. doi: 10.1016/j.mvr.2011.09.006. [DOI] [PubMed] [Google Scholar]
- Zhao L, Cao F, Yin T, Sun D, Cheng K, Zhang J, Wang H. Moderate dose insulin promotes function of endothelial progenitor cells. Cell Biol Int. 2011;35(3):215–220. doi: 10.1042/CBI20100205. [DOI] [PubMed] [Google Scholar]
- Humpert PM, Djuric Z, Zeuge U, Oikonomou D, Seregin Y, Laine K, Eckstein V, Nawroth PP, Bierhaus A. Insulin stimulates the clonogenic potential of angiogenic endothelial progenitor cells by IGF-1 receptor-dependent signaling. Mol Med. 2008;14(5–6):301–308. doi: 10.2119/2007-00052.Humpert. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang CH, Ting MK, Verma S, Kuo LT, Yang NI, Hsieh IC, Wang SY, Hung A, Cherng WJ. Pioglitazone increases the numbers and improves the functional capacity of endothelial progenitor cells in patients with diabetes mellitus. Am Heart J. 2006;152(6):1051. doi: 10.1016/j.ahj.2006.07.029. e1051-1058. [DOI] [PubMed] [Google Scholar]
- Fadini GP, Boscaro E, Albiero M, Menegazzo L, Frison V, de Kreutzenberg S, Agostini C, Tiengo A, Avogaro A. The oral dipeptidyl peptidase-4 inhibitor sitagliptin increases circulating endothelial progenitor cells in patients with type 2 diabetes: possible role of stromal-derived factor-1alpha. Diabetes Care. 2010;33(7):1607–1609. doi: 10.2337/dc10-0187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pistrosch F, Herbrig K, Oelschlaegel U, Richter S, Passauer J, Fischer S, Gross P. PPARgamma-agonist rosiglitazone increases number and migratory activity of cultured endothelial progenitor cells. Atherosclerosis. 2005;183(1):163–167. doi: 10.1016/j.atherosclerosis.2005.03.039. [DOI] [PubMed] [Google Scholar]
- Yue WS, Lau KK, Siu CW, Wang M, Yan GH, Yiu KH, Tse HF. Impact of glycemic control on circulating endothelial progenitor cells and arterial stiffness in patients with type 2 diabetes mellitus. Cardiovasc Diabetol. 2011;10:113. doi: 10.1186/1475-2840-10-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Churdchomjan W, Kheolamai P, Manochantr S, Tapanadechopone P, Tantrawatpan C, U-pratya Y, Issaragrisil S. Comparison of endothelial progenitor cell function in type 2 diabetes with good and poor glycemic control. BMC Endocr Disord. 2010;10:5. doi: 10.1186/1472-6823-10-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estes ML, Mund JA, Mead LE, Prater DN, Cai S, Wang H, Pollok KE, Murphy MP, An CS, Srour EF, Ingram DA Jr, Case J. Application of polychromatic flow cytometry to identify novel subsets of circulating cells with angiogenic potential. Cytometry A. 2010;77(9):831–839. doi: 10.1002/cyto.a.20921. [DOI] [PMC free article] [PubMed] [Google Scholar]