

Abstract
An expanding body of preclinical evidence suggests EGCG, the major catechin found in green tea (Camellia sinensis), has the potential to impact a variety of human diseases. Apparently, EGCG functions as a powerful antioxidant, preventing oxidative damage in healthy cells, but also as an antiangiogenic and antitumor agent and as a modulator of tumor cell response to chemotherapy. Much of the cancer chemopreventive properties of green tea are mediated by EGCG that induces apoptosis and promotes cell growth arrest by altering the expression of cell cycle regulatory proteins, activating killer caspases, and suppressing oncogenic transcription factors and pluripotency maintain factors. In vitro studies have demonstrated that EGCG blocks carcinogenesis by affecting a wide array of signal transduction pathways including JAK/STAT, MAPK, PI3K/AKT, Wnt and Notch. EGCG stimulates telomere fragmentation through inhibiting telomerase activity. Various clinical studies have revealed that treatment by EGCG inhibits tumor incidence and multiplicity in different organ sites such as liver, stomach, skin, lung, mammary gland and colon. Recent work demonstrated that EGCG reduced DNMTs, proteases, and DHFR activities, which would affect transcription of TSGs and protein synthesis. EGCG has great potential in cancer prevention because of it’s safety, low cost and bioavailability. In this review, we discuss its cancer preventive properties and it’s mechanism of action at numerous points regulating cancer cell growth, survival, angiogenesis and metastasis. Therefore, non-toxic natural agent could be useful either alone or in combination with conventional therapeutics for the prevention of tumor progression and/or treatment of human malignancies.
Keywords: Chemoprevention, Cell cycle, Apoptosis, Cellular signaling cascades, EGCG
1. Introduction
Green tea (Camellia sinensis) is an extremely popular beverage worldwide, is next to water and its habitual consumption has long been associated with health benefits including chemo-preventive efficacy [1], because its chemistry compared with other teas is better known [1, 2]. A search of literature shows that there are >795 published studies showing the effects of green tea on cancer, mostly dealing with its chemopreventive effects [3]. It is generally agreed that much of cancer chemopreventive effects of green tea are mediated by its polyphenols known as catechins. The major catechins in green tea are EGCG, (-)-epicatechin-3-gallate, (-)-epigallocatechin, and (-)-epicatechin (Fig. 1). EGCG is the major catechin in green tea and accounts for 50% to 80% representing 200 to 300 mg/brewed cup of green tea [4]. EGCG has also demonstrated other beneficial effects in studies of diabetes, possesses antioxidant activity, Parkinson’s disease, Alzheimer’s disease, stroke, and obesity [4-6].
Fig. 1.
Structures of green tea catechins.
The cancer-preventive effects of EGCG are widely supported by results from epidemiological, cell culture, animal and clinical studies. EGCG is known antioxidant compound and it is proposed that this flavonoid suppresses the inflammatory processes that lead to transformation, hyperproliferation, and initiation of carcinogenesis [7]. Green tea polyphenols inhibit cell proliferation and exert a strong antiradical activity [7]. Polyphenols, specifically, EGCG, have been shown to increase antioxidant activity in a variety of mouse organs and thus, enhancing the overall chemo-preventative effect of antioxidants in those cells and tissues [5]. EGCG may enhance gap junctional communication between cells and thus protect cells from tumor development. The experimental studies suggest an effect of this polyphenol which may block the promotion of tumor growth by sealing receptors in the affected cells [8]. Another possible mechanism indicates that this compound may facilitate direct binding to certain carcinogens [8]. Its inhibitory influences may ultimately suppress the final steps of carcinogenesis as well, namely angiogenesis and metastasis [9]. Various animal studies have revealed that treatment with EGCG inhibits tumor incidence and multiplicity in different organ sites such as skin (UV radiation and chemically induced), lung, liver, breast, prostate, stomach, mammary gland and colon based on preclinical, observational, and clinical trial data [9]. In vitro cell culture studies show that EGCG potently induces apoptosis and promotes cell growth arrest, by altering the expression of cell cycle regulatory proteins, activating killer caspases, and suppressing NFκB activation [10]. Besides, it regulates and promotes IL-23 dependent DNA repair and stimulates cytotoxic T cells activities in a tumor microenvironment. It also blocks carcinogenesis by modulating the signal transduction pathways involved in cancer development [11]. This flavonoid was also shown to affect several biological pathways, including growth factor-mediated pathway, the mitogen activated protein (MAP) kinase-dependent pathway, and ubiquitin/proteasome degradation pathways [12]. Recently, tremendous progress has been made in elucidating the molecular mechanisms of cancer chemoprevention by EGCG [13]. The suppression of various tumor biomarkers including growth factor receptor tyrosine kinases, cytokine receptor kinases, PI3K, phosphatases, ras, raf, MAP kinase, IKK, PKA, PKB, PKC, c-jun, c-fos, c-myc, cdks, cyclins, and related transducing proteins by tea polyphenols have been studied in our laboratory and others (Fig. 2). The IKK activity in LPs-activated murine macrophages (RAW 264.7 cells) was found to be inhibited by EGCG [14].
Fig. 2.
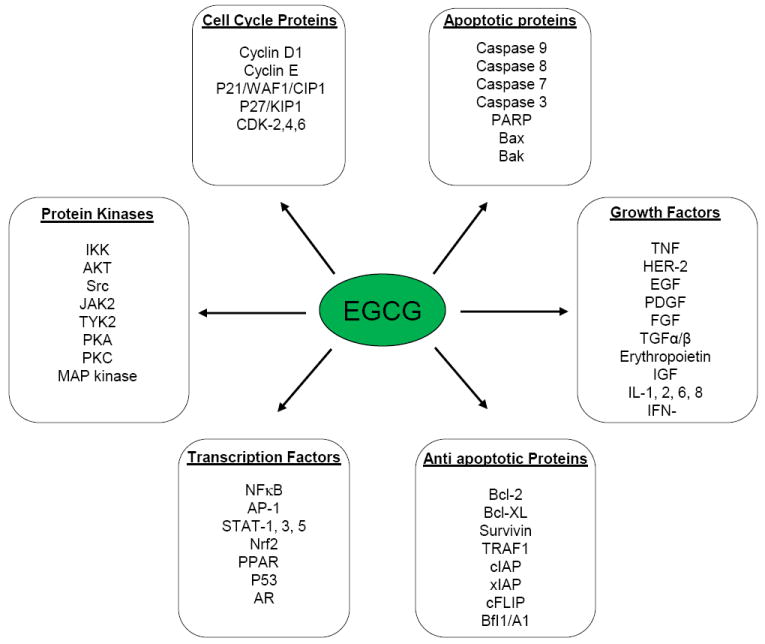
Mechanism of actions of EGCG.
Tumorigenesis is a multistep process that can be activated by any of various environmental carcinogens (such as cigarette smoke, industrial emissions, gasoline vapors), tumor promoters (such as phorbol esters and okadaic acid), and inflammatory agents (such as TNF-α and H2O2). These cancer promoting agents are known to modulate the transcription machinery factors (e.g., NFκB, AP-1, STAT3), anti-apoptotic proteins (e.g., Akt, Bcl-2, Bcl-XL), proapoptotic proteins (e.g., caspases, PARP), protein kinases (e.g., IKK, JNK, MAP kinase), cell cycle proteins (e.g., cyclins, cyclin-dependent kinases), cell adhesion molecules, cyclooxygenase (COX)-2, and growth factor signaling pathways [15]. Although several systematic reviews and meta-analyses have been published most are restricted to the effect of EGCG rich green tea consumption on specific cancer types or their risk factors. By contrast, a relatively small number of studies have shown that EGCG can inhibit certain biomedically important molecular targets such as DNMTs, HATs, and HDACs [16], antiapoptotic proteins [6], VEGFR signaling [17] and squalene epoxidase [18]. Recently, phase I and II clinical trials have been conducted to explore the chemopreventive effects of EGCG in humans [13]. A major challenge of cancer prevention is to integrate new molecular findings into clinical practice. This review summarizes recent research data focusing on EGCG induced cellular signal transduction events that seems to have implications in the inhibition of cell proliferation and transformation, induction of apoptosis of preneoplastic and neoplastic cells as well as inhibition of angiogenesis, tumor invasion, and metastasis. This justifies the need for a systematic review on this topic.
2. Cellular signaling in cancer
Cancer development (i.e. carcinogenesis) is generally recognized as a complex and multistep process in which distinct molecular and cellular modifications occur. In order to simplify understand the different possible options for chemoprevention and chemotherapy in cancer development and progression, three well defined stages have been described: (i) initiation is a rapid phase, comprises the exposure or uptake and interaction of cells, especially DNA, with a carcinogenic agent, and its distribution and transport to organs and tissues where metabolic activation and the covalent interaction with target cell DNA occur, leading to genotoxic damage, (ii) promotion is considered a relatively and reversible process in which actively proliferating abnormal cells persists, replicates and may originate a focus of preneoplastic cells, and (iii) progression stage is the final phase of neoplastic transformation, an uncontrolled growth of the cells (tumor) occurs, involves the gradual conversion of premalignant cells to neoplastic ones with an increase of invasiveness and metastasis potential, and new blood vessel formation (angiogenesis) [19]. The transcription factors, such as NFκB and AP-1, are transiently activated to regulate target gene expression in response to extracellular stimuli through specific intracellular signal transduction pathways [10]. Because advanced metastasized cancers are mostly incurable, an effort to prolong or block the process of carcinogenesis through chemoprevention has become an important and feasible strategy for cancer control and management.
Inhibition of oxidative damage constitutes the first line of defense system against carcinogenic insults and can be considered as most effective way for preventing cancer. It can be achieved by scavenging the reactive oxygen species (i.e. •OH and O2•-) or by inducing their detoxification through induction of phase-II conjugating enzymes (GST, glucuronidases and sulphotransferases) [5, 20]. Antioxidant enzymes (CAT, SOD, GPx and GR) are important components of the cellular stress response whereby a diverse array of electrophilic and oxidative toxicants can be removed from the cell before they are able to damage target cell DNA. In the tumor promotion step, mechanisms that stop or slow down cell division could be potentially beneficial (induction of cell cycle arrest, and apoptosis) in order to restore the lost balance between cell proliferation and apoptosis [21]. At the latest phase of carcinogenesis (progression), the interruption of angiogenesis or the prevention of malignant cells to escape from original location and invade other tissues could also be potentially useful. During all the stages of cancer development many key proteins related to cellular antioxidant defences (e.g. SOD, CAT, GST, GPx and GR), cellular proliferation and survival transduction pathways (e.g. AKT, PI3K, MAP kinases, and NFκB) are upregulated, and anti-apoptotic members of Bcl-2 family genes (e.g. Bax and Bak), and tumor suppressor genes (p53, BRAC1 and BRAC2) are downregulated [13].
Center to the cancer biology is disrupted intracellular signaling cascades, which transmit aberrant signals resulting in abnormal cellular functions. Consistent with this notion, targeting deregulated intracellular signaling cascades is considered to be a rational approach in achieving chemoprevention. Recent studies have shown that EGCG cancer chemopreventive agent exerts its effect by modulating one or more cell signaling pathways in a manner that interrupts the carcinogenic process [22]. Moreover, tea flavonoids display a vast array of cellular effects, they can affect all stages of cancer development by up- or down-regulating multiple key cellular proteins involved in diverse cellular signal transduction pathways: proliferation, differentiation, apoptosis, angiogenesis or metastasis, resulting in a potential beneficial effect [4, 23]. The potential chemopreventive effect of EGCG seems to be quite specific, and cancer cell lines appear to be more sensitive than normal cells, since EGCG has shown higher cytotoxicity in cancer cells than in their normal counterparts. The inhibition of lung tumor in A/J mice by EGCG was associated with decreased cell proliferation, induced apoptosis, and decreased angiogenesis as well as with lower levels of phospho-c-jun and phospho-ERK1/2 in lung adenomas and carcinomas [24].
3. Anticarcinogenic activity
Epigenetic alterations in particular, aberrant DNA methylation and acetylation of non-histone proteins associated with inappropriate gene silencing contribute significantly to the initiation and progression of human cancer. EGCG inhibits cancer-associated stages and exert an inhibitory effect on DNA methylation via blocking performance of DNMTs, strong free radical scavenging and antioxidant activities [25, 26]. The clinical studies suggest an effect of EGCG, which may block the promotion of tumor growth by blocking receptors in the affected cells. Another possible mechanism indicates that EGCG may facilitate direct binding to certain cancer developing carcinogens [25]. It has also been suggested that EGCG inhibits tumorigenesis in a variety of organs. Recently, EGCG inhibited lipopolysaccharide-induced nitric oxide production and inducible nitric oxide synthase gene expression in isolated peritoneal macrophages by decreasing the activation of NFκB [27]. EGCG inhibited PDGF-induced apoptosis and cell cycle regulating pathways of vascular smooth muscle cells, resulting in inhibition of tumor growth, metastasis, and angiogenesis in vivo [6, 28]. Moreover, green tea polyphenols and EGCG have been shown to induce apoptosis, antioxidant and detoxifying protein levels in human lymphoid leukaemia cells and human prostate cancer cells [29].
Procarcinogens such as N-nitrosodiethylamine and aflatoxins that are activated by cytochrome P450 enzymes are able to modify DNA and induce tumorigenesis [30]. Tea flavonoids can directly neutralise the procarcinogens by their strong antiradical activity, before cell membrane injury occur. EGCG exhibits the highest protection against DNA scissions, mutations, and in non-enzymatic interception of superoxide anions. Many animal studies indicate that EGCG can inhibit the growth of malignant cells and induce apoptosis even in cancerous cell lines resistant to CD95-mediated apoptosis. Some results suggest that EGCG induce apoptosis due to their pro-oxidant effect. In a study where EGCG has been tested on oral cancer cell lines along with curcumin, EGCG blocked cell division in G1, whereas curcumin blocked cell division in S/G2M. EGCG has antiproliferative activities on tumor cells through the blockage of growth factor binding to the receptor and the suppression of mitogenic signal transduction [31]. Volatiles in tea have been found to be moderately cytotoxic against human carcinoma cells, with β-ionone and nerolidol exhibiting the strongest activity [32]. EGCG blocks urokinase, an enzyme which is essential for cancer growth and metastasis formation, by interfering with the enzyme’s ability to recognize its substrates [33]. EGCG can also kill specifically transformed cells by adenovirus [13]. Tea catechin EGCG inhibits DNA synthesis of rat hepatoma cells, leukemia cells and lung carcinoma cells [15]. EGCG, in a transgenic mice model for skin cancer, has exhibited a preventive effect and/or improvement of the situation [34]. Sazuka et al. [35] reported that the adhesion of lung carcinoma cells to fibronectin, a plasma protein, can be inhibited by EGCG, hindering cancer progression.
It has been known that immune cells play an important role in host defence against tumor development and progression. Their plasma membranes are rich in polyunsaturated fatty acids, and are thus susceptible to oxidation by ROS [36]. Besides their antioxidant activity, flavonoids exhibit a modulating effect on cells responding to a stimulus or antigen-activated cells, like mast cells, lymphocytes, macrophages, platelets, hepatocytes, and smooth muscle [15]. The TNF-α is a cytokine induced by tumor promoters that stimulates the production of cell adhesion molecules and the inflammatory process response [23]. EGCG inhibits NFκB and expression of TNF-α, reduces cancer promotion [28]. The activation of immune B cells involved in antibody production induces the phosphorylation of tyrosine residues of proteins implicated in cancer cell proliferation. EGCG selectively inhibits the tyrosine phosphorylation in the intracellular transduction pathway and the spheroid and colony formation in vascular smooth muscle cells [37]. Antioxidant molecules, flavonoids inhibit the expression of the multi-drug resistance gene and modulate topoisomerase activity associated with tumor growth. It is evident that tea flavonoid EGCG exhibits many protective activities and different metabolic pathways are involved [15]. It acts as strong ROS scavenger and antioxidant, selectively inhibit specific enzyme cancer developing activities such as DNMTs, and repair DNA aberrations [25]. Further research is needed for a better understanding of effect of EGCG at the cellular and molecular levels of the complex processes of cancer development and inhibition. Then it will be possible to explain how EGCG promotes cancer prevention and inhibition.
4. Chemopreventive effects
Chemoprevention of cancer through the use of naturally occurring nontoxic dietary agents recently has received an increasing interest, and dietary polyphenols have become not only important potential chemopreventive, but also therapeutic, natural agents. Chemoprevention, by definition, is a means of the therapy of precancerous lesions, which are called preinvasive neoplasia, dysplasia, or intraepithelial neoplasia, depending on the organ system. One misconception about chemoprevention is the thinking for complete prevention of cancer, an unachievable goal. Since the mechanism of cancer development is carcinogenesis, we believe that our key aim should be to prevent carcinogenesis process, which in turn will lead to lower cancer burden. We, therefore, define chemoprevention as slowing the process of carcinogenesis, a goal that can be met [1]. Polyphenols have been demonstrated to act on multiple key elements in intracellular signal transduction pathways related to cell proliferation, differentiation, apoptosis, inflammation, angiogenesis and metastasis; however, these molecular mechanisms of action are not completely characterized and many features remain to be elucidated [15]. Among all the polyphenolic compounds, green tea polyphenols have especially received considerable attention because of demonstrated chemoprevention efficacy against a variety of human malignances. The cancer chemopreventive effects of EGCG may be the result of decreased cell transformation and proliferation or increased cell cycle arrest and apoptosis [28]. In vitro, EGCG has been shown to cause growth inhibition and apoptosis in a number of human cancer cell lines including leukemia, melanoma, breast cancer, lung, and colon [4, 15]. These effects have been extensively studied in vitro to try to elucidate the potential mechanism(s) of action of EGCG. Moreover, these chemopreventive effects have been observed in vivo in certain animal models, no clear and fully understandable mechanism(s) has been reported for EGCG.
EGCG has been shown to possess chemopreventive effects against broad spectrum of carcinogens by inhibiting N-methylbenzylnitrosamine-induced esophagus, azoxymethane- and N-mehylnitrosourea-induced colon, diethylnitrosamine-induced liver, 7,12-dimethylbenz(a)anthraceneinduced mammary, N-methyl-N’-nitro-N-nitrosoguanidine-induced glandular stomach, N-ethyl-N’-nitro-N-nitrosoguanidine-induced duodenum, 4-(methylnitrosamimo)-1-(3-pyridyl)-1-butanone-induced pulmonary, diethylnitrosamine- and benzo(a)pyrene-induced lung and forestomach, N-nitrosobis(2-oxopropyl)amine-induced pancreatic and UV-induced skin carcinogenesis in the animal model [9]. Currently, the molecular evidence to prove chemopreventive efficacy by animal studies is at its initial stage. However, an ever-growing number of studies are demonstrating that EGCG can prevent carcinogenesis.
Recently, it has been demonstrated that EGCG might lead to the modulation of histone as well as non-histone proteins in diverse cell signaling pathways and require the integration of different signals for the final preventive or therapeutic effects [38]. A prospective cohort study with 8,552 subjects from Saitama Prefecture in Japan has revealed that green tea has a potentially preventive effect against cancers in all organs including stomach, lung, colorectum and liver [39]. Most of the in vitro and in vivo studies conducted in the recent years have suggested that EGCG is the most acceptable and promising superb drug that could inhibit or delay various types of cancers by to interfering at the initiation, promotion and progression of cancer and reduce the cancer incidence and mortality due to cancers.
5. Inhibition of tumorigenesis and possible mechanism: molecular targets
Many mechanisms have been proposed for the biological activities of EGCG. This includes antioxidant activities, cell cycle arrest, induction of apoptosis, induction or inhibition of drug metabolism enzymes, modulation of cell signaling, inhibition of DNA methylation, effect on miRNA expression, DHFR, proteases, and telomerase (Fig. 2). With the availability of many reagents for signal transduction research, EGCG has been found to affect different signal transduction pathways, such as the inhibition of many protein kinases; suppression of the activation of transcription factors (e.g. AP-1 and NFκB) and blocking growth receptor mediated pathways. However, it is not clear which of these mechanisms occur in vivo and are relevant to the cancer-preventive activities of tea.
5.1. Antioxidant potential
Tea catechins are characterized by the di- or tri-hydroxyl groups on the B-ring and the meta-5,7-dihydroxyl groups on the A ring (Fig. 1). The antioxidant activities of EGCG are due to the presence of phenolic groups that are sensitive to oxidation and can generate quinone. The antioxidative activity is further increased by the presence of the trihydroxyl structure in the D ring in EGCG [9, 36]. EGCG is powerful radical scavengers; protected neurons from the oxidative damage induced by a commonly used pro-oxidant such as tert-butylhydroperoxide [40]. Murakami et al. [41] reported that EGCG can reduced the cytotoxicity evoked by H2O2 and increased the levels of the enzymes related to the oxidative stress, resulting in an enhanced cellular GSH content in a HepG2 (Table 1). In a clinical investigation, when HUVEC were incubated with EGCG or in pro-oxidant conditions, its restored cell viability and inhibited apoptosis, showing that phenolic compound differ in their antiapoptotic efficacy [42]. Moreover, EGCG from green tea induced H2O2 formation in human lung adenocarcinoma (H661) and in Ha-ras gene-transformed human bronchial (21BES) cells, but exogenously added catalase (CAT) prevented EGCG-induced cell apoptosis, which suggested that H2O2 is involved in the apoptotic process provoked by EGCG [43].
Table 1.
Molecular targets of EGCG for prevention and therapy of cancer
| S. No. | Molecular mechanisms | Molecular targets | References | |
|---|---|---|---|---|
| 1. | Induction of cell cycle arrest | ↓ cyclin D; ↓ cyclin E; ↓ CDK1; ↓ CDK2; ↓ CDK4; ↓ CDK6; ↓ PCNA; ↑ 16; ↑ p18; ↑ p21; ↑ p27; ↑ pRb; ↑ p53; ↑ mdm2 | [23, 51, 54, 64] | |
| 2. | Antioxidant (s) | ↓ H2O2-induced apoptosis; ↑ H2O2 production; ↑ ROS; ↑ GSH; ↓ Nrf-2-mediated HO-1 activation | [9, 36, 41-47, 49] | |
| Phase I and II enzyme(s) | ↓ CYP1A1 ↓ CYP2E | [60-62] | ||
| 3. | Induction of apoptosis | ↑ ROS; ↑ caspase-3; ↑ caspase-8; ↑ caspase-9; ↑ cytochrome c; ↑ Smac/DIABLO; ↓↑ Bax; Z Bak; ↓ cleaved PPAR; ↓↑ Bcl-2; ↓ Bcl-xL; ↓ Bid; ↓ c-myc; = c-IAP1; ↓ c-IAP2; ↓ Mcl-1; ↓ survivin; ↓ XIAP | [4, 15, 23, 50-54, 57, 59] | |
| 4. | Inhibition of proliferation and inflammation | ↓ PI3K; ↓↑ AKT; ↓↑ ERK; ↓ p90RSK; ↓ FKHR; ↓ PDGF; ↓ PDGFRb; ↓ EGFR; ↓↑ c-fos; ↓ egr-1; ↓ AP-1; ↓ NF-kB; ↓ IKK; ↓ COX-2; ↑ JNK; ↑ Ras; ↑ MEKK1; ↑ MEK3; ↓↑ p38; ↑ IjB; ↑ AMPK; ↑ PGE2; ↑ TNF-α | [10, 15, 21, 67, 69-71, 77] | |
| 5. | Angiogenesis | ↓ HIF-1α; ↓ VEGF; ↓ VEGFR1; ↓VEGFR2 ↓ ErbB2; ↓ErbB3 ↑FOXO | [6, 28, 72, 75, 89-91] | |
| 6. | Metastasis | ↓ MMP-2; ↓ MMP-9; ↓ FAK; proMMP-2; ↓ MRLC; ↓ vimentin; ↓ laminin; ↓ integrina2b1; ↓ uPA; ↓ HuR; ↑ proMMP-7; ↑ TIMP-2; ↑ MT1-MMP | [33, 34, 95-97] | |
| 7. | Epigenetic modifier(s) | ↓ DNMTs; ↓ HAT ↓ acetylation of H3/H4 | [25, 26, 38, 82, 83] | |
| 8. | Validate gene target(s) | RARb, MGMT, MLH1, CDKN2A, RECK, TERT, RXRa, CDX2, GSTP1, WIF1 | [26, 46, 83] | |
| 9. | Proteasomal activity | ↓20S/26S proteasome complex | [79, 80] | |
| 10. | Inhibition of cancer(s) | In vitro model: Esophageal; oral; prostate; breast; urinary; lung; colon; leukemia; lymphoma | In vivo model: skin, prostrate, colon and uterine cancer; human gastric, pancreatic and oral cancers | [6, 15, 26, 38, 80, 83] |
Treatment of 24 month-old rats with EGCG (100 mg/kg, i.g.) decreased 50% hepatic levels of lipid peroxides and 39% protein carbonyls formation [44]. EGCG treatment also increased the levels of antioxidant and antioxidant enzymes of hepatic compared to control one. These effects were not observed in young rats, suggesting that EGCG offered no improvement in antioxidant status in the absence of pre-existing oxidative stress [45]. This may explain why other studies have failed to observe an effect of tea phenolics treatment. Treatment of C57bl/6J mice with this catechin has been reported to increase gene expression of c-GST, glutamate cysteine ligase, and hemeoxygenase-1 in an Nrf2-ARE-dependent manner [46]. Similar results have been reported in the tumor cells of colon cancer xenograft-bearing nude mice treated with dietary EGCG [47]. These effects have been re-capitulated in vitro [48]. Increased expression of hemeoxygenase-1 and SOD was recorded when human mammary epithelial cells were treated with EGCG [36]. This effect was reduced by siRNA-mediated disruption of Nrf2, suggesting a role for this pathway in the EGCG-mediated induction of these endogenous antioxidant systems. EGCG induced apoptosis in 21BES cells. EGCG-mediated apoptosis was reduced by approximately 50% by inclusion of exogenous catalase. This result suggested that EGCG can induce apoptosis by an ROS dependent mechanism [36]. EGCG-mediated ROS production may underlie its ability to induce endogenous antioxidants, at least in vitro. Treatment of Hepa1c1c7 human hepatoma cells with EGCG resulted in dose-dependent increases in NADPH:quinone reductase-1 and glutathione genes expression through the EpRE. LC–MS analysis of the cell culture medium revealed the presence of EGCG-20-glutathione further supporting this conclusion [49].
5.2. Induction of apoptosis and cell cycle arrest
Apoptosis or programmed cell death has long been described as a key strategy for the elimination of neoplastic cells [6]. Apoptosis is a highly ordered protective mechanism by which unwanted or damaged cells are eliminated before malignancy manifests. It is essential for normal development, turnover, and replacement of cells in the living system. Cell apoptosis is characterized by typical morphological and biochemical hallmarks including chromatin condensation, membrane blebbing, cell shrinkage, nuclear DNA fragmentation and the formation of apoptotic bodies [6]. The cell subsequently breaks up into membrane-enclosed fragments, termed apoptosis bodies, which are rapidly recognized and engulfed by neighboring cells or macrophages [19]. Markedable, biochemical modifications occurs within the apoptosis bodies by phagocytosis. To occur cell shrinkage, cytoskeleton and cytoplasmic membranes modifications are required. This results in the loss of cell-cell contact, untethering of the plasma membrane and rapid blebbing or zeiosis. On the molecular basis, apoptosis can be induced by two major pathways: (i) at the plasma membrane upon ligation of the death receptor (extrinsic pathway) and (ii) at the mitochondria (intrinsic pathway). Trigger of death receptors of the TNF receptor superfamily such as TRAIL or CD95 (APO-1/Fas) receptors results in activation of the initiator caspase-8, which can propagate the apoptosis signal by direct cleavage of downstream effector caspases (i.e. caspase-3) [19].
Apoptosis induction by EGCG is more prominent in many cancer cells without affecting normal cells because NFκB is activated in the cancer cells (Table 1). Although EGCG has been shown to affect a number of factors associated with cell cycle progression, the direct inhibition of cyclin-dependent kinases is considered as the primary event [4]. EGCG-induced apoptosis in tumor cells may be mediated through NFκB inactivation [15]. In vitro and in vivo studies demonstrated that the inactivation of NFκB by EGCG was associated with enhancement of phosphorylation-dependent degradation of IkBα, subsequent increases in nuclear translocation of p65 protein and inhibition of IKK activity [50]. The induction of different negative regulators of the cell cycle may be the consequence of this inhibition. EGCG also induces the expression of p21 and p27 while decreasing the expression of cyclin D1 and the phosphorylation of retinoblastoma. However, EGCG inhibited LPs-induced phosphorylation of IKK, but failed to affect NFκB luciferase reporter gene activation in human colon cancer (HT-29) cells, suggesting that EGCG modulation of NFκB transcriptional activity is not necessarily dependent on IkBα degradation and subsequent release of NFκB proteins (Table 1;[51]).
EGCG down-regulates the NFκB inducing kinase expression in human lung cancer cell PC-9 [52]. Activation of NFκB promotes transcriptional up-regulation of Bcl-2 and Bcl-XL. Negative regulation of NFκB by EGCG decreases the expression of the proapoptotic protein Bcl-2 [52]. In human prostate carcinoma LNCaP cells, treatment with EGCG induced apoptosis and was associated with stabilization of p53 and also with a down-regulation of NFκB activity, resulting in a decreased expression of the anti-apoptotic protein Bcl-2 [53]. EGCG (70% lethal dose) at the suprapharmacological concentration of 10 μg/ml increased the proportion of HNSCC cells in the G1 phase, decreased cyclin D1 protein expression and increased the levels of p21WAF1/CIP1 and p27KIP1 proteins [54].
Many recent studies demonstrated that EGCG trigger cell growth arrest pathways at G1 stage of cell cycle through regulation of cyclin D1, cdk4, cdk6, p21/WAF1/CIP1 and p27/KIP1, and induced apoptosis through generation of ROS and caspase-3 and caspase-9 activation [23]. EGCG inhibited expressions of Bcl-2 and Bcl-XL and induced expressions of Bax, Bak, Bcl-XS and PUMA. Furthermore, ras, raf-1 activities and ERK1/2 were down regulated, whereas the activities of MEKK1, JNK1/2 and p38 MAP kinases were upregulated [23]. Inhibition of craf-1 or ERK enhanced EGCG-induced apoptosis, whereas inhibition of JNK or p38 MAP kinase inhibited EGCG-induced apoptosis. EGCG promoted the activation of p90 ribosomal protein S6 kinase, and induced the activation of c-jun. Xenograft and TRAMP models have shown that green tea or EGCG can decrease the tumorigenic potential of prostate cancer [23]. Treatment of MCF7 breast cancer cells with EGCG (30 μM) inhibited cell cycle arrest in G0/G1 phase. In prostate cancer cells, EGCG (10–80 μM) increased the expression of p16, p18, p21, p27 and p53, which are associated with negative regulation of cell cycle progression (Table 1). Overall, these findings suggest that green tea and its constituents induce growth arrest and apoptosis through multiple mechanisms and can be used for chemoprevention to target cancer cells. EGCG induces apoptosis by activating capase-3/7 and inhibiting the expression of Bcl-2, survivin and XIAP in prostate stem cancer cells. Furthermore, EGCG inhibits epithelial-mesenchymal transition by inhibiting the expression of vimentin, slug, snail and nuclear β-catenin, and the activity of LEF-1/TCF responsive reporter, and also retards CSC’s migration and invasion, suggesting the blockade of signaling involved in early metastasis [55]. Interestingly, quercetin synergizes with EGCG in inducing apoptosis, and blocking CSC’s migration and invasion. These data suggest that EGCG either alone or in combination with quercetin can eliminate CSC’s-characteristics.
Caspases, a ubiquitous family of cysteine proteases play key roles both as upstream initiators and downstream effectors in apoptosis. This cascade leads to proteolytic cleavage of a variety of cytoplasmic and nuclear proteins, thereby favoring the prevalence of proapoptotic activities on antiapoptotic activities. EGCG abrogated the expression of anti-apoptotic Bcl-2 and Bcl-XL proteins and enhanced the levels of proapoptotic Bax proteins followed by caspase-3 activation [56]. Moreover, EGCG–induced nuclear condensation, and poly(ADP)ribose polymerase cleavage. Treatment of human colorectal carcinoma HT-29 cells with EGCG resulted in nuclear condensation, DNA fragmentation, caspase activation, disruption of mitochondrial membrane potential and cytochrome c release, which all appeared to be mediated by the JNKs pathway [57]. In addition, EGCG also invokes Bax oligomerization and depolarization of mitochondrial membranes to facilitate cytochrome c release into cytosol. Treatment of colon cancer cells with EGCG decreased cell proliferation index (based on Ki-67 expression), increased apoptotic index (cleaved caspase-3), decreased nuclear β-catenin levels, and decreased phospho-Akt levels [58]. EGCG also reduced the protein expression of cyclin D1, cyclin E, CDK2, CDK4, and CDK6. EGCG also inhibited the activity of CDK2 and CDK4, and caused Rb hypophosphorylation [54]. Head and neck squamous cell carcinoma cells were found to be more sensitive to the effects of EGCG; EGCG induced G0/G1 phase cell cycle arrest at concentrations lower than 20 μM [54]. The results suggest that EGCG exerts cancer preventive potential by inhibiting cell proliferation, promoting apoptosis, and modulating β-catenin and Akt signaling. The direct binding of tea polyphenols to the BH3 pocket of antiapoptotic Bcl-2 family proteins is observed by using nuclear magnetic resonance spectroscopy, suggesting a mechanism for EGCG to inhibit the antiapoptotic function of Bcl-2 proteins. The BH3 domain was recognized as one of the binding sites of tea polyphenols [59]. In HepG2 cells, EGCG has been shown to stimulate apoptosis and inhibit cell cycle progression at G1 [23]. These effects were accompanied by increased expression of p53 and p21/WAF1 proteins and proapoptotic Fas and Bax proteins.
5.3. Effect on phase I and II enzymes
One of the most important mechanisms of chemoprevention by cruciferous plants extracts appears to be induction of phase II enzymes and inhibition of phase I enzymes. Procarcinogenic metabolism can be altered by EGCG by inhibiting phase-I drug-metabolizing enzymes CYPs, increasing the activity or modulating the gene expression of phase II conjugating-enzymes which can regulate several biological events such as acetylation, methylation, glucuronidation, sulphation and conjugation (Table 1). In vitro studies have demonstrated that EGCG possesses a strong inhibitory effect on CYP1A1 at the transcriptional level [60]. Phenolic compounds can induce phase II conjugating enzymes, they can be considered as potential candidates for preventing tumor development [60]. EGCG increased the activity of the phase II detoxifying enzymes GST and NQO in mouse liver, breast and prostate cancer cells. Similarly, drinks rich in tea phenols increased QR activity in the Hepa1c1c7 cells and in general, EGCG prevented glutathione depletion and ROS formation after an oxidative injury caused by different carcinogens which are formed reactive metabolites such as prooxidants [61]. Consequently, tea also showed in vitro a protective effect on rat hepatic extracts, which was associated with a reduced level of CYP2E and an enhanced activity of phase II detoxifying enzyme UDPGT, suggesting that EGCG can regulate phase I and phase II drug metabolizing enzymes, although GST activity was unaffected [60]. An ARE has been found in the promoters of antioxidant proteins (SOD, Gpx, and CAT) and several drug-metabolizing enzymes (GST, and GR). Several signaling pathways have been involved in the activation of the ARE that binds transcription factor Nrf-2. In a study, it has been observed that in human hepatoma HepG2 cells, an EGCG rich green tea polyphenol extract induced the transcriptional potency of phase II detoxifying enzymes through ARE and decreased in Nrf-2-ARE binding in lung adenocarcinoma A549 cells [62].
5.4. Modulation of intracellular signaling cascades
5.4.1. Inhibition of NFκB
NFκB is a family of closely related protein dimers or oxidative stress–sensitive transcription factor that bind to a common sequence motif in DNA called the kB site [15]. The identification of the p50 subunit of NFκB as a member of the REL family of viruses provided the first evidence that NFκB is linked to cancer. NFκB is sequestered in the cytoplasm in an inactive form through interaction with IkB. NFκB is activated by free radicals, inflammatory stimuli, cytokines, carcinogens, tumor promoters, endotoxins, γ-radiation, UV light, and X-rays. Upon activation, it is translocated to the nucleus, where it induces the expression of more than 200 genes that have been shown to suppress apoptosis and induce cellular transformation, proliferation, invasion, metastasis, chemo-resistance, radio-resistance, innate immunity and inflammation.
Recent results also totally confirmed its pivotal roles in suppressing apoptosis in cancer cells [15]. Phosphorylation and activation of IkB kinase is controlled by an NFκB-inducing kinase and there is crosstalk between activation of the MAP kinase/ERK pathway, and the NFκB - inducing kinase/InB kinase/ NFκB pathway. In the cytosol, NFκB is inactive when bound to I-kB (Fig. 2). The phosphorylation of I-kB by IKKs leads to proteasome dependent degradation of I-kB, resulting NFκB free. NFκB can activate the expression of a set of NFκB responsive genes when translocate into the nucleus. EGCG has been shown to inhibit the activation of NFκB in H891 head and neck cancer cells and MDA-MB-231 breast cancer cells [63]. Treatment of EGCG dose- and time-dependently increased I-kB level, and inhibited NFκB nuclear translocation in A431 epidermoid carcinoma cells [64]. In normal human epidermal keratinocytes, UVB irradiation-induced NFκB activation was associated with increased I-kB phosphorylation and degradation and EGCG was shown to inhibit NFκB activation and nuclear translocation [15]. Although, ROS have been suggested to be involved in the activation of the NFκB signaling system, and that its suppression by EGCG is due to its strong antioxidant and free radical quenching activities [65]. It has been shown that the galloyl and phenolic groups at the 3’ position on EGCG are responsible for its strong anti-inflammatory properties [27]. EGCG has been shown to inhibit NFκB activity in human colon cancer cells [15]. EGCG suppresses the TNF-induced activation of IKK that leads to the inhibition of TNF-dependent phosphorylation and degradation of Ik-Ba and translocation of the p65 subunit. Based on its ability to inhibit other kinases, emodin may act directly on the IKK complex to block phosphorylation of IkBα. Yang et al. [13] found that EGCG suppresses NFκB activation by inhibiting IKK activity. Some act by suppressing Ik-Bα degradation and p65 translocation or NFκB –DNA binding activity. Treatment with EGCG in a dose- and time-dependent manner was found to inhibit UVB-mediated activation of NFκB in normal human epidermal keratinocytes [66]. Gupta et al. [10] identified NFκB/p65 component of the NFκB complex as a target for specific cleavage by caspases during EGCG-mediated apoptosis. Thus, one of the probable mechanisms by EGCG exercise their anti-tumor property is through the suppression of the NFκB signaling pathway.
5.4.2. Activation of FOXO transcription factors
FOXO transcription factors including FOXO1, FOXO3a, and FOXO4, exert critical biological functions in response to genotoxic stress. In mammals four FOXOs proteins are known [67]. FOXOs induce cell cycle arrest, repair damaged DNA, or initiate apoptosis by modulating genes that control these processes. The importance of FOXO factors ascribes them under multiple levels of regulation including phosphorylation, acetylation/deacetylation, ubiquitination and protein-protein interactions [67]. This function of FOXO is essential for the regulation of cancer cells and CSCs and progenitor cell pool in the hematopoietic system [67]. Recently, we have demonstrated that EGCG, resveratrol, sulforaphane inhibited cell proliferation and colony formation, and induced apoptosis through caspase-3 activation in pancreatic cancer cells [58, 68]. The inhibition of PI3K/AKT and MEK/ERK pathways activated FOXO transcription factors. EGCG inhibited phosphorylation of AKT and ERK, and activated FOXO transcription factors, leading to cell cycle arrest and apoptosis and further enhanced FOXO activity and apoptosis [21].
5.4.3. Inhibition of AP-1 and MAP kinases
AP-1 was originally identified by its binding to a DNA sequence in the SV40 enhancer. AP-1 transcription factor is a protein dimer composed of homo- or heterodimers members of the basic region leucine zipper protein superfamily, specifically, the Jun, Fos, and activating transcription factor proteins. Many stimuli, most notably serum, growth factors, and oncoproteins, are potent inducers of AP-1 activity; it is also induced by TNF and IL-1, as well as by a variety of environmental stresses, such as UV radiation. High AP-1 activity or activation of AP-1 has also been shown to be involved in the tumor promotion and progression of various types of cancers, such as lung, breast, and skin cancer. AP-1 has been implicated in regulation of genes involved in apoptosis and proliferation and may promote cell proliferation by activating the cyclin D1 gene, and repressing TSGs, such as p53, p21cip1/waf1 and p16 [15]. Green tea polyphenol EGCG was shown to inhibit 12-O-tetradecanoylphorbol-13-acetate and epidermal growth factor-induced transformation of mouse epidermal cell line JB6 (Table 1; [69]). This finding correlates with the inhibition of AP-1 DNA binding and transcriptional activity. The inhibition of AP-1 activity by EGCG was associated with inhibition of JNK activation but not ERK activation. Interestingly, in another study where EGCG blocked the UVB-induced c-Fos activation in a human keratinocyte cell line HaCaT, inhibition of p38 activation was suggested as the major mechanism underlying the effects of EGCG [69].
MAP kinases have been known to involve in variety of key physiologic processes, including cell apoptosis, differentiation, and death. In mammalian cells, three major types of MAP kinases are presented; p38 MAP kinases, ERK, and c-JNK [15]. Activated MAP kinases such as ERK, JNK, and p38 can activate ELK and c-Jun. The second messenger, phosphatidyl inositol-3,4,5-triphosphate synthesized by activate PI3K, which is necessary for phosphorylation of Akt, then Akt directly phosphorylates the proapoptotic protein Bad, thus enhancing the antiapoptotic function of Bcl-xL. In an animal study, it was shown that EGCG (5-20 μmol/L) in concentration dependent manner inhibited the MAP kinase pathway in JB6 mouse epidermal cell line [69]. In NHEK cells, treatment with EGCG inhibited UVB-induced H2O2 production concomitant with block of UVB-induced phosphorylation of ERK1/2, JNK, and p38 proteins (Table 1; [70]).
Recently, EGCG has been shown to inhibit MAP kinase pathway and activator protein-1 (AP-1) activity in human colon cancer cells. In a study, orally feeding of EGCG inhibits PI3K pathway in TRAMP model system [71]. The confirm involvement of MAP kinase pathways in the regulation of AP-1 activity by EGCG has been investigated. Treatment of EGCG has been shown to inhibit c-Jun, ERK1/2 phosphorylation and the phosphorylation of ELK1 and MEK1/2 in Ha-ras-transformed human bronchial cells [70]. In contrast to these reports, markedly increase AP-1 factor-associated responses through a MAP kinase signaling mechanism in normal human keratinocytes has been measured by EGCG, suggesting that the signaling mechanism of EGCG action could be markedly different in different cell types [20]. The major pathways that lie downstream of the membrane-associated RTK are also regulated cell cycle and apoptosis. In this cascade, Ras interacts with and activates Raf-1, which in turn phosphorylates and activates MAP/ERK kinase 1/2 (MEK1/2) then phosphorylates ERK1/2. The JNK1/2/3 and p38α/β/γ pathways are parallel MAP kinase cascades in mammalian cells [69]. Because the deregulation of the MAP kinase pathway is frequently seen in a variety of cancers, modulation of MAP kinases by EGCG may provide novel strategies for the prevention or treatment of human cancer.
5.4.4. Inhibition of epidermal growth factor receptor (EGFR)–mediated signal transduction pathway
The EGFR (ErbB-1; HER1 in humans) is a cell surface glycoprotein receptor with an extracellular ligand-binding domain, a single transmembrane region, and an intracellular domain that exhibits intrinsic tyrosine kinase activity. The EGFR is a member of the ErbB family of receptors, a subfamily of four closely related receptor tyrosine kinases: EGFR (ErbB-1), HER2/neu (ErbB-2), HER3 (ErbB-3) and HER4 (ErbB-4). Mutations that lead to EGFR overexpression (known as upregulation) or overexpression have been associated with a number of cancers, including lung cancer, anal cancers and glioblastoma multiforme. A blockade of EGFR may lead the cancer cells to enter apoptosis [12]. Therefore, inhibition of EGFR abrogates the invasive potential of the cancer cells. Overexpression of HER-2/neu, an oncogene in the EGFR tyrosine kinase superfamily, is observed in breast, prostate, ovarian and lung cancers and it is recognized as a target for cancer therapy [12]. Downstream events, the phosphorylation of ERK, STAT3, and Akt were subsequently blocked by the treatment with EGCG (Table 1; [71]). However, the cells were pre-incubated with EGCG before the addition of TGF-α, resulting the inhibition of EGFR-dependent STAT3 activation subsequently retarded VEGF synthesis in the cancer cells. The inhibitory effect of NFκB by EGCG also contributed to the overall VEGF down-regulation. Treatment with EGCG inhibited the constitutive ligand-mediated activation of the EGFR in both YCU-H891 head and neck squamous cell carcinoma and MDA-MB-231 breast carcinoma cell lines, indicating that it has the potential to break the autocrine loops that are established in several advanced cancers [72]. The results suggested that blocking the EGFR signaling by EGCG would potentially inhibit both cancer cell proliferation and angiogenesis.
Recently, Ahmad observed that EGCG suppresses the expression of IL-6 and IL-8 in vitro [73]. EGCG has been shown to suppress the production of VEGF in swine granulosa cells and breast carcinoma cells [73]. EGCG inhibits EGFR signaling pathway, most likely through the direct inhibition of ERK1/2 and Akt kinases [58]. EGCG blocks PDGF-induced proliferation and migration of rat pancreatic stellate cells [6]. EGCG binds to a specific metastasis associated 67 kDa laminin receptor that is expressed on a variety of tumor cells [74]. Cells were treated with all trans-retinoic acid that the anticancer action of EGCG is mediated by laminin receptor and it allows EGCG to bind to the cell surface using a subtraction cloning strategy involving cDNA libraries. The cell-laminin interaction via receptor 67LR is a key step in several signaling pathways and kinase-phosphatase cascades. Authors found an association between 67LR and the integrin α6 subunit, which is a part of laminin-binding integrins α6β4 and α6β1. When EGCG incorporated in plasma membrane directly interacts with Platelet-derived Growth Factor-BB (PDGF-BB), thereby preventing specific receptor binding leading to the inhibitory effects of EGCG on PDGF-induced cell signaling and mitogenesis. Moreover, Givant-Horwitz and associates [75] reported that in human colon cancer cells, EGCG inhibits growth and activation of EGFR and human EGFR-2 signaling pathways. Based on these data, it was suggested that may be existence of EGCG receptor. This suggestion awaits follow-up and confirmation.
5.4.5. Inhibition of overexpression of cyclooxygenase-2 (COX-2)
COX is prostaglandin H synthase, the key regulatory enzyme for prostaglandin synthesis is transcribed from two distinct genes (COX-1 & COX-2). COX-1 is constitutively expressed in many tissues, but the expression of COX-2 is regulated by a variety of factors, such as mitogens, tumor promoters, cytokines, and growth factors. Inappropriate COX-2 activity has been observed in practically every premalignant and malignant condition involving the colon, liver, pancreas, breast, lung, bladder, skin, stomach, head and neck, and esophagus [15]. Several transcription factors including AP-1, and NFκB can stimulate COX-2 transcription [15]. EGCG inhibits mitogen-stimulated COX-2 expression in androgen-sensitive LNCaP and androgen-insensitive PC-3 human prostate carcinoma cells [76]. It has been shown that EGCG reduces the activity of COX-2 following interleukin-1A stimulation of human chondrocytes [77]. EGCG exhibited COX inhibition in LPs-induced macrophages (Table 1). Pretreatment with green tea extract enriched with EGCG inhibited COX-2 expression induced by TPA in mouse skin. EGCG can down-regulate COX-2 in TPA-stimulated human mammary cells (MCF-10A) in culture [78]. Recent clinical research findings strongly suggest that development of chemopreventive compounds, which can inhibit COX-2 expression preferably without affecting COX-1, is a high priority for cancer research.
5.4.6. Inhibition of proteasome activity
The proteasome is a massive multicatalytic protease complex that is responsible for degrading most of the damage or misfold proteins. The proteasomal degradation pathway is essential for many cellular processes, including cell proliferation, down-regulation of cell death, development of drug resistance in human tumor cells, regulation of gene expression and responses to oxidative stress, suggesting the use of proteasome inhibitors as potential novel anticancer drugs. In vitro and in vivo studies shown that EGCG inhibits the chymotrypsin-like but not trypsin-like activity of the proteasome. Inhibition of the chymotrypsin-like activity of the proteasome has been associated with induction of tumor cell apoptosis. The catalytic activities of the 20S/26S proteasome complex were inhibited by EGCG, resulting in intracellular accumulation of IkBα and subsequent inhibition of NFκB activation (Table 1; [79]). This finding suggests that the proteasome is a cancer related molecular target of EGCG and that inhibition of the proteasome activity by this phytomolecule may contribute to its cancer preventive potential. Application of EGCG resulted in G0/G1 cell cycle arrest and accumulation of p27 and IkBα in LNCaP prostate cancer cells with both of which are targets for proteasomes [80]. The difference of effective concentrations in cell-free systems (IC50 0.09–0.2 mM) and in cell lines (IC50 1–10 mM) suggests that EGCG may bind nonspecifically to proteins or other macromolecules in the cells, and therefore lower the effective concentration of EGCG at the active site of the protease and stability and cellular uptake of EGCG may also be an important factor.
5.5. Inhibition of epigenetic modifications
Epigenetics refers to heritable changes that are not encoded in the DNA sequence itself, but play an important role in the control of gene expression. In mammals, epigenetic mechanisms include changes in DNA methylation, histone modifications and non-coding RNAs. The tail and globular domains of eukaryotic nucleosomal histones can by modified by these epigenetic events and alter the gene expression [81]. Among them, DNA methylation of cytosines at CpG dinucleotides is perhaps the most extensively studied epigenetic modification in mammals with profound functional implications for wide range of cellular processes [81]. Although epigenetic changes are heritable in somatic cells, these modifications are also potentially reversible, which makes them attractive and promising avenues for tailoring cancer preventive and therapeutic strategies.
DNA methylation is a covalent biochemical modification, resulting in the addition of a methyl group to the 5th carbon position in the pyramidine ring of cytosine located in the CpG dinucleotides. Hypermethylation often limits the accessibility of transcription factors to promoters, promote the methyl-CpG binding domain (MBD) binding, which results in recruitment of additional silencing-associated proteins, and ultimately, gene silencing[81]. An estimated 60% of mammalian gene promoters contain CpG islands, many of these genes belongs to the house keeping category that are usually unmethylated and transcriptionally activate [81]. It has been well known that CpG islands in certain genes, especially tumor suppressor genes, often become aberrantly hypermethylated during the development of cancer (Fig. 4a & b). Patterns of DNA methylation are established by the coordinated action of DNMTs and associated factors, such as the polycomb proteins, in the presence of S-adenosylmethionine (SAM) that serves as a methyl donor for methyl group. The mammalian DNMT family include for active members, DNMT1, DNMT3A, DNMT3B, and DNMT3L. Demethylation of SAM results in the formation of S-adenosylhomocysteine (SAH), and evidences from clinical studies demonstrated that SAH is a potent inhibitor of DNMT [25].
Fig, 4. Different DNA methylation patterns, and histone modifications between normal and tumor cells.
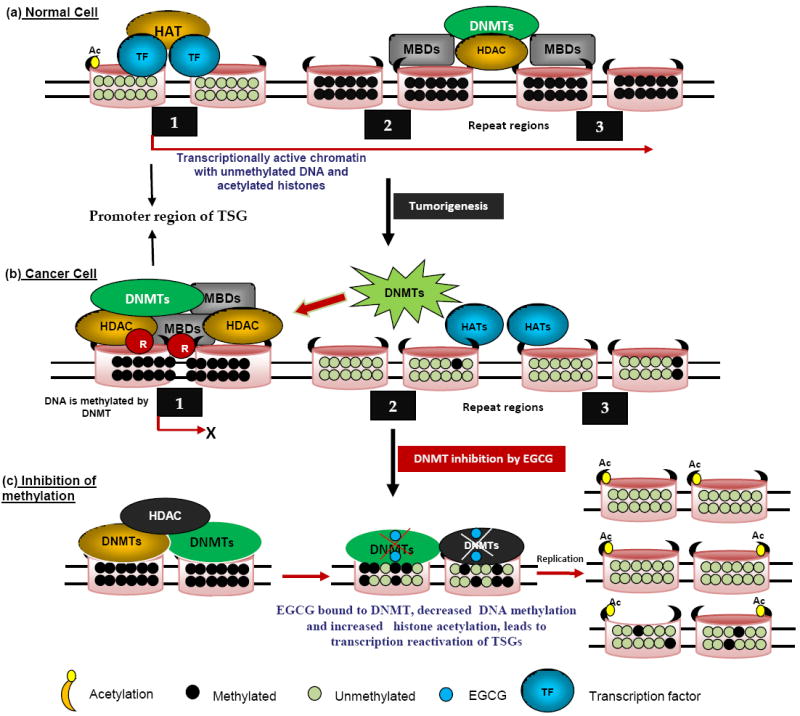
Molecular modeling of the interaction between EGCG and DNMT. In normal cells (a), genes are generally unmethylated and packaged with acetylated histone proteins associated with HAT as well as basal transcription factor machinery. These epigenetic elements constitute an ‘open’ chromatin structure which favors transcription. In cancer cells, the same genes may become hypermethylated (b), and the methylated CpG sites are recognized by the methyl-binding proteins (MBDs), which are coupled with repressor (R) and histone deacetyltransferase (HDAC) proteins to remove the acetyl group from the histones, generating a tightly closed chromatin status to shut down gene expression. (c) DNMT activity is blocked by EGCG through forming hydrogen bonds with amino acids (Pro, Glu, Cys, Ser, and Arg) in the catalytic pocket of DNMT. Newly synthesized DNA strands are hemi-methylated after the first round of DNA replication and become progressively more demethylated after several rounds of replication due to the dilution effect. Using EGCG as a DNMT inhibitor, the silenced epigenetic modifications could be switched to an active status.
EGCG can form hydrogen bonds with different residues in the catalytic pocket of DNMT, thus acting as a direct inhibitor of DNMT1 (Table1; Fig. 4c). The inhibition of DNMT may prevent the methylation of the newly synthesized DNA strand, resulting in the reversal of the hypermethylation and the re-expression of the silenced genes [25]. Finally, it has been shown that EGCG is also an efficient blocker of DHFR. EGCG acts through interaction with folic acid metabolism in cells, causing the inhibition of nucleic acid (DNA & RNA) synthesis and altering pattern of DNA methylation. EGCG was shown to affect various biologic pathways and inhibits DNMTs activity in human cancer cell lines (Fig. 4c). EGCG binds to DNMT1 and blocks the enzyme’s active site. EGCG generates oxidizing agent H2O2 in a substantial amount, which is oxidized to DNMTs and other proteins might contribute to its inhibition of DNA methylation as well [25, 82]. The same action, however, may also cause oxidative damage and substantially increase its cytotoxicity. EGCG caused a concentration and time-dependent reversal of hypermethylation of TSGs such as p16, RAR, MGMT, and MLH1 genes in human esophageal cancer cells [26, 83]. Treatment of human colon cancers and prostate cancer cells with EGCG reactivated some methylation-silenced genes. Since then, several groups found similar in vitro results. Partial demethylation of hypermethylated RARb by EGCG was demonstrated in breast cancer cells such as MCF-7 and MDA-MB-231 [38]. EGCG partially reversed the hypermethylation status of the RECK gene and significantly enhanced the expression level of RECK mRNA in oral cancer cells. Repetitive elements such as p16, RARb, MAGE-A1, MAGE-B2 and Alu in T24, HT29, and PC3 cell lines for their DNA methylation levels and their mRNA expression levels using several demethylating agents [84]. Recently, in vivo study showed that topical treatment of EGCG in hydrophilic cream inhibits UVB induced global DNA hypomethylation pattern in chronically UVB-exposed mice. Treatment of MCF-7 breast cancer cells with EGCG resulted in activation of several genes and decrease in hTERT promoter methylation [38].
5.6. Inhibition of dihydrofolate reductase (DHFR) and telomerase
DHFR, a methyl group shuttle required for the de novo synthesis of purines, thymidylic acid, and certain amino acids. DHFR as a novel modulator of β-catenin and GSK3 signaling and raise several implications for clinical and can use for inflammatory disease and cancer. Recently, it was reported that EGCG acts as a potential inhibitor of DHFR [85]. It exhibited kinetic characteristics of a slow tight binding inhibitor of 7,8-dihydrofolate reduction with bovine liver DHFR (Ki 0.11 mM). The very low Ki value observed for bovine liver DHFR might be due to the fact that very low levels of the enzyme were used in the assay, and the inhibition was due to the strong binding activity of EGCG to the enzyme (a slow tight binding inhibitor, as reported), not necessarily by binding to the active site (Table1; [86]). But EGCG acted as a classic reversible competitive inhibitor with chicken liver DHFR with a much larger Ki (10.3 mM). Folate depletion increased the sensitivity of these cell lines to the antifolate activities of EGCG. Telomerase is important for maintaining the telomere nuclear protein endcaps of the chromosome and it has been shown to be overexpressed in many human cancers. Long-term treatment with EGCG (5–10 mM) was reported to inhibit telomerase and induce cell senescence [87]. At high nanomolar to low micromolar concentrations of EGCG inhibited telomerase activity in cell-free systems at neutral pH. The authors were concluded that EGCG decomposes by auto-oxidation to form a galloyl radical, which can covalently modify telomerase activity [88]. In an animal study, treatment of mice bearing telomerase-positive colon cancer xenografts (HCT-L2) with 1.2 mg of EGCG per day for 80 d resulted in a 50% inhibition in tumor size [87]. Whereas mice bearing telomerase-negative tumors of the same parent cell line (HCT-S2R) were unresponsive to EGCG treatment. This interesting line of observation should be extended into other cell lines.
5.7. Inhibition of angiogenesis
Angiogenesis is a complex event which requires endothelial cell sprouting, lumen formation, tubulogenesis and is regulated by the coordinated action of different transcription factors [6]. Their interaction leads to endothelial cell differentiation and acquisition of arterial, venous and lymphatic properties. The transcription factors regulate the correct organization of the vascular system, controlling excessive endothelial growth and inducing apoptosis. FOXO transcription factors play a crucial role in the regulation of tissue homeostasis in organs such as the pancreas and the ovaries and complex diseases such as diabetes and cancer. We have recently shown that EGCG inhibits angiogenesis by enhancing FOXO transcriptional activity [28]. Inhibition of AKT and MEK kinases synergistically induced FOXO transcriptional activity, which was further enhanced in the presence of EGCG. Phosphorylation deficient mutants of FOXO induced FOXO transcriptional activity, inhibited HUVEC cell migration and capillary tube formation. Inhibition of FOXO phosphorylation also enhanced antiangiogenic effects of EGCG through transcriptional activation of FOXO [28]. Authors concluded that the activation of FOXO transcription factors through inhibition of these two pathways may have physiological significance in management of diabetic retinopathy, rheumatoid arthritis, psoriasis, cardiovascular diseases and cancer. Recently, Bartholome et al. [89] reported that EGCG at 1 μM stimulated FOXO transcription factor nuclear accumulation and DNA binding activity. EGCG decreases ET-1 expression and secretion from endothelial cells, in part, via Akt- and AMPK-stimulated FOXO1 regulation of the ET-1 promoter. These findings may be relevant to beneficial cardiovascular actions of green tea [90]. Moreover, EGCG exerts its insulin mimetic effects at least in part by phosphorylation of the FOXOs through a mechanism that is similar but not identical to insulin and IGF-1 induced FOXO phosphorylation [91].
5.8. Possible modulation of miRNA expression
Small molecules known as MicroRNAs are a contemporary class of noncoding endogenous RNA molecules, generating great excitement in the clinical and scientific communities. miRNAs have been recently discovered as key regulators of gene expression by controlling the translation of a specific type of RNA called messenger RNA which relays the genetic instructions for making proteins [92]. Previous research has indicated that miRNAs are express in a tissue-specific manner and control a wide spectrum of biological processes including cell proliferation, apoptosis and differentiation. Although miRNA are vital to normal cell physiology, aberrant expression of these small non-coding RNAs has been linked to carcinogenesis [92]. The recent discovery that miRNA expression is frequently dysregulated in cancer has uncovered an entirely new repertoire of molecular factors upstream of gene expression, which warrants extensive investigation to further elucidate their precise role in malignancy. Although there is still limited evidence, recent reports have identified that dietary agents can also alter gene expression by targeting various oncogenic or tumor suppressive miRNAs. One of the interesting features of miRNAs is that similar to regular genes, their own expression can be regulated by other DNA methylation. The influence of miRNA on the epigenetic machinery and the reciprocal epigenetic regulation of miRNA expression suggest that its aberrant expression during carcinogenesis has an important implication for global regulation of cancer. Additionally, interaction among various components of the epigenetic machinery re-emphasizes the integrated nature of epigenetic mechanisms involved in the maintenance of global gene expression patterns in mammals. Treatment of human hepatocellular carcinoma HepG2 cells with EGCG has been recently found to inhibit the expression of oncogenic miRNA and induce level of tumor suppressive miRNAs (Fig. 5). Treatment of HepG2 with EGCG resulted in the expressions of 61 miRNAs. miR-16, one of the miRNAs up-regulated by EGCG, is known to regulate the anti-apoptotic protein Bcl-2, and interestingly EGCG treatment induced apoptosis and downregulated Bcl-2 in HepG2 cells [93]. Transfection with anti-miR-16 inhibitor suppressed miR-16 expression and counteracted the EGCG effects on Bcl-2 down-regulation and also induction of apoptosis in cells.
Fig. 5. Impact of EGCG on microRNA (miRNA) expression.
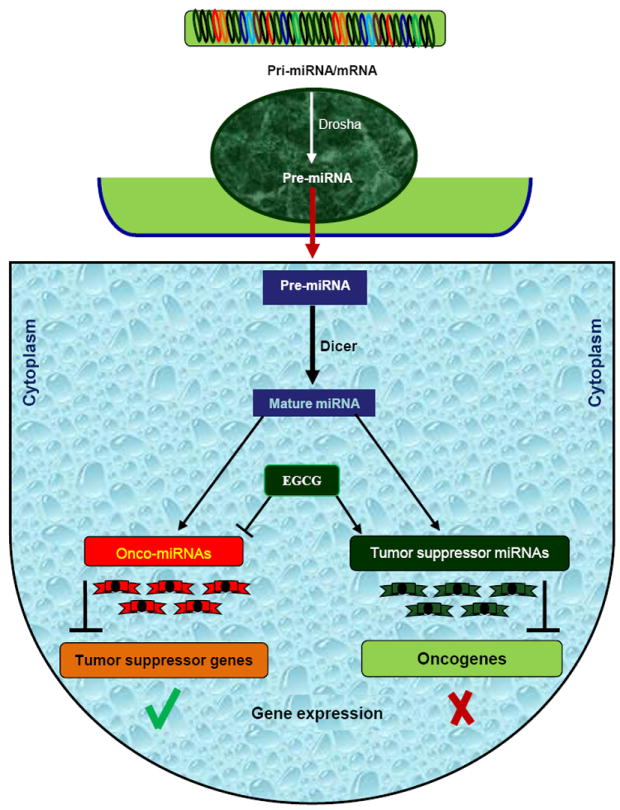
miRNA are transcribed in the nucleus into pri-miRNA (primary miRNA) which is further cleaved by Drosha into precursor miRNA (pre-miRNA). Pre-miRNA is exported from nucleus to the cytoplasm and further processed by Dicer into miRNA duplex. Single strand of miRNA duplex (also called mature miRNA) leads this complex to mRNA cleavage or translation repression, which is dependent on miRNA:mRNA complementarity. Dependent on various factors, miRNA can have either an oncogenic role (called oncomiRNAs) if the target mRNA is a tumor suppressor gene, or a tumor suppressive role (tumor-suppressor miRNAs) if the target molecule is an oncogene. EGCG can impact on expression level of miRNAs and participate in gene expression regulation.
5.9. Miscellaneous mechanisms
The uPA is a trypsin-like protease that converts the zymogen plasminogen into active plasmin. It is highly expressed in human cancer and has the ability to prevent apoptosis, stimulate angiogenesis, mitogenesis, cell migration, and to modulate cell adhesion [94]. uPA, one of the hydrolases implicated in the degradation of the extracellular matrix and tumor invasion, is directly inhibited by EGCG [33]. Inhibition of uPA can decrease tumor size or even cause complete remission of cancers in mice. EGCG-induced suppression of uPA promoter activity as well as expression appears to be mediated by blocking ERK and p38 MAP kinase, but not JNK and AKT. Jankun et al. [33] showed that EGCG binds to urokinase, blocking His57 and Ser195 of the uPA catalytic triad, and extending toward Arg35 from a positively charged loop of uPA. Thus, it was suggested that the cancer prevention action of EGCG is mediated by inhibition of uPA. EGCG was found to be a potent inhibitor of uPA expression in human fibrosarcoma HT 1080 cells. In addition, EGCG inhibited uPA promoter activity and also destabilized uPA mRNA [95]. EGCG was also shown to affect MMPs directly and indirectly. EGCG also inhibited the activation of MMPs by MT1-MMP [96]. EGCG increased the expression of the tissue inhibitor of MMPs (TIMP1 and 2) at even lower concentrations (~1 μM), which provides an additional mechanism to suppress the activity of MMPs. The activities of secreted MMP2 and MMP9 were inhibited by EGCG with IC50 values of 8–13 μM [96]. These activities may contribute to the reported inhibition of metastasis and invasion following treatment of tumor-bearing mice with EGCG. Most recently, in several human colon carcinoma cell lines, EGCG was found to inhibit the activity of topoisomerase I at 3–17 μM concentration. By comparison, EGCG at concentrations up to 550 μM did not inhibit topoisomerase II activity [97]. This is an interesting mechanism of action for EGCG given the relatively low concentrations necessary for inhibition and the correlation of topoisomerase I inhibition with phenomena, such as DNA damage, cell cycle arrest, and induction of apoptosis [97].
6. Clinical trails
Limited data are currently available from EGCG chemoprevention trials. EGCG offers several potential clinical advantages compared to other traditional cancer drugs. In contrast, EGCG is globally available as tea, is inexpensive to isolate, and can be administered orally [98]. While, traditional cancer drugs are often destroy some healthy cells along with cancerous cells [99]. EGCG appears to target biochemical and genetic functions unique to cancer cells [53]. Some of the anticarcinogenic agents currently in use have toxic adverse effects, but data from clinical trials reported to date suggest that EGCG has a very acceptable safety profile [53]. These benefits support further development of EGCG as a potentially useful anticarcinogenic agent. A prospective cohort study with over 8,000 individuals revealed that the daily consumption of green tea resulted in delayed cancer onset and a follow-up study of breast cancer patients found that stages I and II breast cancer patients experienced a lower recurrence rate and longer disease free period [100]. Moreover, EGCG delivered in the form of capsule (200 mg p.o.) for 12 weeks has been reported to be effective in the patients with human papilloma virus–infected cervical lesions [37]. The positive results observed in phase II and phase III clinical trials along with exciting preclinical results indicate that ways and means to take EGCG “from bench to real-life situations” are on the horizon.
7. Summery, conclusion and future prospects
Naturally occurring antioxidant substances such as polyphenols and flavonoids that are derived from herbs, fruits, vegetable and medicinal plants provides a new insight in prevention and therapy of cancer. The mechanisms of action of several dietary chemopreventive agents have gained considerable attention in cancer research. There is extensive research going on in elucidating the molecular mechanisms of cancer chemoprevention by green tea catchine EGCG. Although there are several studies supporting the preventive potential of EGCG against cancer, a proper understanding of the mechanisms by which EGCG reduces the risk is necessary to establish its efficacy for the population where it could be most useful. Despite the demonstration of cancer prevention by green tea and their polyphenolic compounds in many animal studies, epidemiological studies have yielded mixed results concerning the effectiveness of EGCG as a superb medicine for prevention and therapy of cancer in humans. Several mechanisms to explain the chemopreventive potentials of EGCG have been presented, among which its effect to target specific cell signaling pathways have received considerable attention for regulating cellular proliferation and apoptosis. The diversified effects of EGCG may explain its broad pharmacologic activities in modulating cellular signaling pathways in cells. EGCG, in addition to other mechanisms, at human achievable dose, is known to activate cell death signals and induce apoptosis in precancerous or cancer cells, resulting in the inhibition of tumor development and/or progression. Importantly, these anti-proliferative and proapoptotic effects of EGCG have been shown to be selective for cancer cells, as normal cells were not affected by its treatment. In cancer cells, EGCG also causes inhibition of the activity of specific receptor tyrosine kinases and related downstream pathways of signal transduction. This review summarizes recent research data focusing on EGCG induced cellular signal transduction events that seems to have implications in the inhibition of cell proliferation and transformation, induction of apoptosis of preneoplastic and neoplastic cells as well as inhibition of angiogenesis, tumor invasion, and metastasis.
Most modern medicines currently available for treating cancers are very expensive, toxic, and less effective in treating the disease. Thus, one must investigate further in detail the EGCG derived from green tea, described traditionally, for the prevention and treatment of cancer and other diseases. The understanding of the cell signaling pathways and the molecular events leading to carcinogenesis will provide more insight into the identification and development of potent chemopreventive/chemotherapeutic agents that specifically target these pathways. Future studies from in vitro systems should be integrated with studies in vivo, especially in ongoing clinical trials, to evaluate the applicability of these mechanisms in cancer prevention in humans. To fully elucidate the molecular mechanisms of action of EGCG with existing therapy in future studies, more in-depth in vitro and in vivo experiments are needed.
Fig. 3. Possible modulation of NF-kB pathway by EGCG.
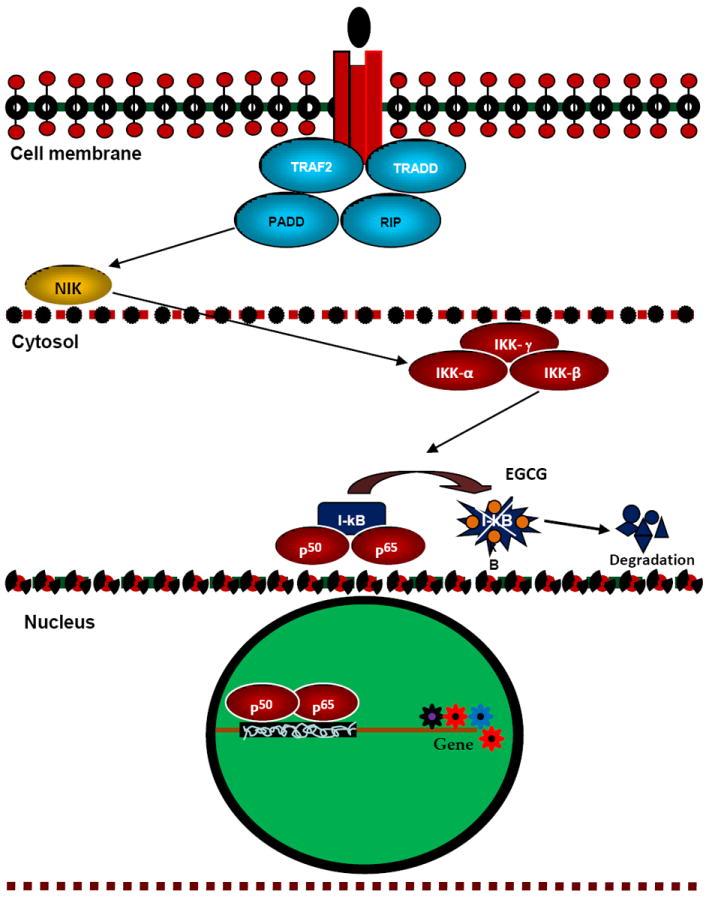
In the cytosol as a result of the binding of p50 and p65 to I-kB, NF-kB becomes inactive. When I-kB is phosphorylated by IKKs and degraded in a proteasome-dependent pathway, p50 and p65 are set free and are translocated into the nucleus to activate a specific set of genes. In vitro and in vivo this pathway has been shown to be inhibited by EGCG both, possibly by inhibiting IKK-catalyzed phosphorylation of I-kB.
Acknowledgments
We thank our lab members for critical reading of the manuscript. This work was supported in part by the grants from the National Institutes of Health (R01CA125262, RO1CA114469 and RO1CA125262-02S1) and Kansas Bioscience Authority.
Abbreviations
- AMPK
adenosine monophosphate-activated protein kinase
- AP1
activator protein 1
- AREs
antioxidant responsive elements
- Bak
Bcl-2 antagonist killer
- Bax
Bcl-2 associated x protein
- Bcl-2
B-cell lymphoma-2
- CAT
catalase
- CDKN2A
cyclin dependent kinase 2A
- Cdks
cyclin-dependent kinases
- c-IAP1
cellular inhibitor of apoptosis protein1
- COX-2
cyclooxygenase-2
- CpG
cytosine-phosphate-guanine
- CSCs
cancer stem cells
- CYP
cytochrome P450
- DHFR
dihydrofolate reductase
- DIABLO
direct inhibitor of apoptosis-binding protein with low pI
- DNMTs
DNA methyltransferases
- EGCG
(-)-epigallocatechin-3-gallate
- EGFR
epidermal growth factor receptor
- EpRE
electrophile-responsive element
- ERK
extracellular signal-regulated kinase
- FAK
focal adhesion kinase
- FKHR
forkhead homolog of rhabdosarcoma
- GFRs
growth factor receptors
- GPx
glutathione peroxidase
- GR
glutathione reductase
- GST
glutathione S-transferase
- H2O2
hydrogen peroxide
- HATs
histone acetyl transferases
- HDACs
histone deacetylases
- HER
human epidermal receptor
- HIF
hypoxia inducible factor
- hTERT
human telomerase reverse transcriptase
- HUVEC
human vascular endothelial cell
- IKK
I kappa B kinase
- IL-1
interleukin 1
- JNK
Jun NH2-terminal kinase
- LPs
Lipopolysaccharides
- MAP
mitogen-activated protein
- MBD
methyl-CpG binding domain
- Mcl-1
myeloid cell leukemia 1
- mdm2
mouse double minute 2
- MEKK1
mitogen-activated protein/ERK kinase 1
- MLH1
MutL homologue 1
- MMPs
matrix metalloproteinases
- MRLC
myosin regulatory light chain
- MT1-MMP
membrane Type 1-matrix metalloproteinase
- NFκB
nuclear factor kappaB
- NQO
NADPH quinone oxidoreductase
- Nrf
NF-E2 p45-related factor
- O2•-
superoxide anion radical
- •OH
hydroxyl radical
- p90RSK
90 kDa ribosomal S6 kinase
- PCNA
proliferating cell nuclear antigen
- PDGF
platelet-derived growth factor
- PGE2
prostaglandins E2
- PI3K
phosphatidylinositol- 3-kinase
- PKA
protein kinase A
- PKB
protein kinase B
- PKC
protein kinase C
- PPAR
peroxisome proliferator-activated receptor
- pRb
retinoblastoma protein
- PUMA
p53 upregulated modulator of apoptosis
- RAR
retinoic acid receptor
- RECK
reversion-inducing cysteine-rich protein with Kazal motifs
- ROS
reactive oxygen species
- RTK
receptor tyrosine kinase
- RXRα
retinoid X receptor alpha
- SAM
S-adenosyl-methionine
- siRNA
small-interfering RNA
- Smac
second mitochondria-derived activator of caspase
- SOD
superoxide dismutase
- STAT
signal transducers and activators of transcription
- TERT
telomerase reverse transcriptase
- TIMP
tissue inhibitor of metalloproteinase
- TRAIL
tumor necrosis factor-related apoptosis-inducing ligand
- TRAMP
transgenic adenocarcinoma of the mouse prostate
- TSGs
tumor suppressor genes
- uPA
urokinase plasminogen activator
- VEGF
vascular endothelial growth factor
- VEGFR-2
vascular endothelial growth factor receptor-2
- XIAP
X-linked inhibitor of apoptosis protein
- α-TNF
alpha-tumor necrosis factor
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Siddiqui IA, Asim M, Hafeez BB, Adhami VM, Tarapore RS, Mukhtar H. Green tea polyphenol EGCG blunts androgen receptor function in prostate cancer. The FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2010 doi: 10.1096/fj.10-167924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yang CS, Wang X, Lu G, Picinich SC. Cancer prevention by tea: animal studies, molecular mechanisms and human relevance. Nature reviews Cancer. 2009;9:429–39. doi: 10.1038/nrc2641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bettuzzi S, Brausi M, Rizzi F, Castagnetti G, Peracchia G, Corti A. Chemoprevention of human prostate cancer by oral administration of green tea catechins in volunteers with high-grade prostate intraepithelial neoplasia: a preliminary report from a one-year proof-of-principle study. Cancer research. 2006;66:1234–40. doi: 10.1158/0008-5472.CAN-05-1145. [DOI] [PubMed] [Google Scholar]
- 4.Khan N, Afaq F, Saleem M, Ahmad N, Mukhtar H. Targeting multiple signaling pathways by green tea polyphenol (-)-epigallocatechin-3-gallate. Cancer research. 2006;66:2500–5. doi: 10.1158/0008-5472.CAN-05-3636. [DOI] [PubMed] [Google Scholar]
- 5.Higdon JV, Frei B. Tea catechins and polyphenols: health effects, metabolism, and antioxidant functions. Critical reviews in food science and nutrition. 2003;43:89–143. doi: 10.1080/10408690390826464. [DOI] [PubMed] [Google Scholar]
- 6.Shankar S, Ganapathy S, Hingorani SR, Srivastava RK. EGCG inhibits growth, invasion, angiogenesis and metastasis of pancreatic cancer. Frontiers in bioscience : a journal and virtual library. 2008;13:440–52. doi: 10.2741/2691. [DOI] [PubMed] [Google Scholar]
- 7.Thawonsuwan J, Kiron V, Satoh S, Panigrahi A, Verlhac V. Epigallocatechin-3-gallate (EGCG) affects the antioxidant and immune defense of the rainbow trout, Oncorhynchus mykiss. Fish physiology and biochemistry. 2010;36:687–97. doi: 10.1007/s10695-009-9344-4. [DOI] [PubMed] [Google Scholar]
- 8.Sigler K, Ruch RJ. Enhancement of gap junctional intercellular communication in tumor promoter-treated cells by components of green tea. Cancer letters. 1993;69:15–9. doi: 10.1016/0304-3835(93)90026-6. [DOI] [PubMed] [Google Scholar]
- 9.Mukhtar H, Ahmad N. Tea polyphenols: prevention of cancer and optimizing health. The American journal of clinical nutrition. 2000;71:1698S–702S. doi: 10.1093/ajcn/71.6.1698S. discussion 703S-4S. [DOI] [PubMed] [Google Scholar]
- 10.Gupta S, Hastak K, Afaq F, Ahmad N, Mukhtar H. Essential role of caspases in epigallocatechin-3-gallate-mediated inhibition of nuclear factor kappa B and induction of apoptosis. Oncogene. 2004;23:2507–22. doi: 10.1038/sj.onc.1207353. [DOI] [PubMed] [Google Scholar]
- 11.Ahmed S, Wang N, Lalonde M, Goldberg VM, Haqqi TM. Green tea polyphenol epigallocatechin-3-gallate (EGCG) differentially inhibits interleukin-1 beta-induced expression of matrix metalloproteinase-1 and -13 in human chondrocytes. J Pharmacol Exp Ther. 2004;308:767–73. doi: 10.1124/jpet.103.059220. [DOI] [PubMed] [Google Scholar]
- 12.Khan N, Afaq F, Saleem M, Ahmad N, Mukhtar H. Targeting multiple signaling pathways by green tea polyphenol (-)-epigallocatechin-3-gallate. Cancer Res. 2006;66:2500–5. doi: 10.1158/0008-5472.CAN-05-3636. [DOI] [PubMed] [Google Scholar]
- 13.Yang CS, Lambert JD, Ju J, Lu G, Sang S. Tea and cancer prevention: molecular mechanisms and human relevance. Toxicology and applied pharmacology. 2007;224:265–73. doi: 10.1016/j.taap.2006.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Singh M, Tyagi S, Bhui K, Prasad S, Shukla Y. Regulation of cell growth through cell cycle arrest and apoptosis in HPV 16 positive human cervical cancer cells by tea polyphenols. Investigational new drugs. 2010;28:216–24. doi: 10.1007/s10637-009-9240-x. [DOI] [PubMed] [Google Scholar]
- 15.Aggarwal BB, Shishodia S. Molecular targets of dietary agents for prevention and therapy of cancer. Biochemical pharmacology. 2006;71:1397–421. doi: 10.1016/j.bcp.2006.02.009. [DOI] [PubMed] [Google Scholar]
- 16.Lee AH, Fraser ML, Binns CW. Possible role for green tea in ovarian cancer prevention. Future oncology. 2005;1:771–7. doi: 10.2217/14796694.1.6.771. [DOI] [PubMed] [Google Scholar]
- 17.Lamy S, Gingras D, Beliveau R. Green tea catechins inhibit vascular endothelial growth factor receptor phosphorylation. Cancer research. 2002;62:381–5. [PubMed] [Google Scholar]
- 18.Abe I, Seki T, Umehara K, Miyase T, Noguchi H, Sakakibara J, et al. Green tea polyphenols: novel and potent inhibitors of squalene epoxidase. Biochem Biophys Res Commun. 2000;268:767–71. doi: 10.1006/bbrc.2000.2217. [DOI] [PubMed] [Google Scholar]
- 19.Srivastava RK, Kurzrock R, Shankar S. MS-275 sensitizes TRAIL-resistant breast cancer cells, inhibits angiogenesis and metastasis, and reverses epithelial-mesenchymal transition in vivo. Molecular cancer therapeutics. 2010;9:3254–66. doi: 10.1158/1535-7163.MCT-10-0582. [DOI] [PubMed] [Google Scholar]
- 20.Mukhtar H, Ahmad N. Green tea in chemoprevention of cancer. Toxicological sciences : an official journal of the Society of Toxicology. 1999;52:111–7. doi: 10.1093/toxsci/52.2.111. [DOI] [PubMed] [Google Scholar]
- 21.Shankar S, Chen Q, Srivastava RK. Inhibition of PI3K/AKT and MEK/ERK pathways act synergistically to enhance antiangiogenic effects of EGCG through activation of FOXO transcription factor. J Mol Signal. 2008;3:7. doi: 10.1186/1750-2187-3-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shankar S, Suthakar G, Srivastava RK. Epigallocatechin-3-gallate inhibits cell cycle and induces apoptosis in pancreatic cancer. Front Biosci. 2007;12:5039–51. doi: 10.2741/2446. [DOI] [PubMed] [Google Scholar]
- 23.Shankar S, Suthakar G, Srivastava RK. Epigallocatechin-3-gallate inhibits cell cycle and induces apoptosis in pancreatic cancer. Frontiers in bioscience : a journal and virtual library. 2007;12:5039–51. doi: 10.2741/2446. [DOI] [PubMed] [Google Scholar]
- 24.Milligan SA, Burke P, Coleman DT, Bigelow RL, Steffan JJ, Carroll JL, et al. The green tea polyphenol EGCG potentiates the antiproliferative activity of c-Met and epidermal growth factor receptor inhibitors in non-small cell lung cancer cells. Clinical cancer research : an official journal of the American Association for Cancer Research. 2009;15:4885–94. doi: 10.1158/1078-0432.CCR-09-0109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fang MZ, Wang Y, Ai N, Hou Z, Sun Y, Lu H, et al. Tea polyphenol (-)-epigallocatechin-3-gallate inhibits DNA methyltransferase and reactivates methylation-silenced genes in cancer cell lines. Cancer research. 2003;63:7563–70. [PubMed] [Google Scholar]
- 26.Fang JY, Tsai TH, Lin YY, Wong WW, Wang MN, Huang JF. Transdermal delivery of tea catechins and theophylline enhanced by terpenes: a mechanistic study. Biological & pharmaceutical bulletin. 2007;30:343–9. doi: 10.1248/bpb.30.343. [DOI] [PubMed] [Google Scholar]
- 27.Lin YL, Lin JK. (-)-Epigallocatechin-3-gallate blocks the induction of nitric oxide synthase by down-regulating lipopolysaccharide-induced activity of transcription factor nuclear factor-kappaB. Molecular pharmacology. 1997;52:465–72. [PubMed] [Google Scholar]
- 28.Shankar S, Chen Q, Srivastava RK. Inhibition of PI3K/AKT and MEK/ERK pathways act synergistically to enhance antiangiogenic effects of EGCG through activation of FOXO transcription factor. Journal of molecular signaling. 2008;3:7. doi: 10.1186/1750-2187-3-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hibasami H, Achiwa Y, Fujikawa T, Komiya T. Induction of programmed cell death (apoptosis) in human lymphoid leukemia cells by catechin compounds. Anticancer Res. 1996;16:1943–6. [PubMed] [Google Scholar]
- 30.Singh BN, Singh BR, Sarma BK, Singh HB. Potential chemoprevention of N-nitrosodiethylamine-induced hepatocarcinogenesis by polyphenolics from Acacia nilotica bark. Chem Biol Interact. 2009;181:20–8. doi: 10.1016/j.cbi.2009.05.007. [DOI] [PubMed] [Google Scholar]
- 31.Khafif A, Schantz SP, Chou TC, Edelstein D, Sacks PG. Quantitation of chemopreventive synergism between (-)-epigallocatechin-3-gallate and curcumin in normal, premalignant and malignant human oral epithelial cells. Carcinogenesis. 1998;19:419–24. doi: 10.1093/carcin/19.3.419. [DOI] [PubMed] [Google Scholar]
- 32.Babich H, Zuckerbraun HL, Weinerman SM. In vitro cytotoxicity of (-)-catechin gallate, a minor polyphenol in green tea. Toxicology letters. 2007;171:171–80. doi: 10.1016/j.toxlet.2007.05.125. [DOI] [PubMed] [Google Scholar]
- 33.Jankun J, Selman SH, Swiercz R, Skrzypczak-Jankun E. Why drinking green tea could prevent cancer. Nature. 1997;387:561. doi: 10.1038/42381. [DOI] [PubMed] [Google Scholar]
- 34.Meeran SM, Mantena SK, Elmets CA, Katiyar SK. (-)-Epigallocatechin-3-gallate prevents photocarcinogenesis in mice through interleukin-12-dependent DNA repair. Cancer research. 2006;66:5512–20. doi: 10.1158/0008-5472.CAN-06-0218. [DOI] [PubMed] [Google Scholar]
- 35.Sazuka M, Itoi T, Suzuki Y, Odani S, Koide T, Isemura M. Evidence for the interaction between (-)-epigallocatechin gallate and human plasma proteins fibronectin, fibrinogen, and histidine-rich glycoprotein. Bioscience, biotechnology, and biochemistry. 1996;60:1317–9. doi: 10.1271/bbb.60.1317. [DOI] [PubMed] [Google Scholar]
- 36.Lambert JD, Elias RJ. The antioxidant and pro-oxidant activities of green tea polyphenols: a role in cancer prevention. Archives of biochemistry and biophysics. 2010;501:65–72. doi: 10.1016/j.abb.2010.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ahn WS, Yoo J, Huh SW, Kim CK, Lee JM, Namkoong SE, et al. Protective effects of green tea extracts (polyphenon E and EGCG) on human cervical lesions. Eur J Cancer Prev. 2003;12:383–90. doi: 10.1097/00008469-200310000-00007. [DOI] [PubMed] [Google Scholar]
- 38.Nandakumar V, Vaid M, Katiyar SK. (-)-Epigallocatechin-3-gallate reactivates silenced tumor suppressor genes, Cip1/p21 and p16INK4a, by reducing DNA methylation and increasing histones acetylation in human skin cancer cells. Carcinogenesis. 2011 doi: 10.1093/carcin/bgq285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Imai K, Suga K, Nakachi K. Cancer-preventive effects of drinking green tea among a Japanese population. Preventive medicine. 1997;26:769–75. doi: 10.1006/pmed.1997.0242. [DOI] [PubMed] [Google Scholar]
- 40.Sonee M, Sum T, Wang C, Mukherjee SK. The soy isoflavone, genistein, protects human cortical neuronal cells from oxidative stress. Neurotoxicology. 2004;25:885–91. doi: 10.1016/j.neuro.2003.11.001. [DOI] [PubMed] [Google Scholar]
- 41.Murakami C, Hirakawa Y, Inui H, Nakano Y, Yoshida H. Effect of tea catechins on cellular lipid peroxidation and cytotoxicity in HepG2 cells. Bioscience, biotechnology, and biochemistry. 2002;66:1559–62. doi: 10.1271/bbb.66.1559. [DOI] [PubMed] [Google Scholar]
- 42.Choi JH, Rhee IK, Park KY, Kim JK, Rhee SJ. Action of green tea catechin on bone metabolic disorder in chronic cadmium-poisoned rats. Life sciences. 2003;73:1479–89. doi: 10.1016/s0024-3205(03)00433-8. [DOI] [PubMed] [Google Scholar]
- 43.Yang TT, Koo MW. Inhibitory effect of Chinese green tea on endothelial cell-induced LDL oxidation. Atherosclerosis. 2000;148:67–73. doi: 10.1016/s0021-9150(99)00239-7. [DOI] [PubMed] [Google Scholar]
- 44.Kumaran G, Clamp AR, Jayson GC. Angiogenesis as a therapeutic target in cancer. Clinical medicine. 2008;8:455–8. doi: 10.7861/clinmedicine.8-4-455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Srividhya R, Jyothilakshmi V, Arulmathi K, Senthilkumaran V, Kalaiselvi P. Attenuation of senescence-induced oxidative exacerbations in aged rat brain by (-)-epigallocatechin-3-gallate. International journal of developmental neuroscience : the official journal of the International Society for Developmental Neuroscience. 2008;26:217–23. doi: 10.1016/j.ijdevneu.2007.12.003. [DOI] [PubMed] [Google Scholar]
- 46.Rietveld A, Wiseman S. Antioxidant effects of tea: evidence from human clinical trials. J Nutr. 2003;133:3285S–92S. doi: 10.1093/jn/133.10.3285S. [DOI] [PubMed] [Google Scholar]
- 47.Liu TT, Liang NS, Li Y, Yang F, Lu Y, Meng ZQ, et al. Effects of long-term tea polyphenols consumption on hepatic microsomal drug-metabolizing enzymes and liver function in Wistar rats. World journal of gastroenterology : WJG. 2003;9:2742–4. doi: 10.3748/wjg.v9.i12.2742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yuan JM, Gao YT, Yang CS, Yu MC. Urinary biomarkers of tea polyphenols and risk of colorectal cancer in the Shanghai Cohort Study. International journal of cancer Journal international du cancer. 2007;120:1344–50. doi: 10.1002/ijc.22460. [DOI] [PubMed] [Google Scholar]
- 49.Muzolf-Panek M, Gliszczynska-Swiglo A, de Haan L, Aarts JM, Szymusiak H, Vervoort JM, et al. Role of catechin quinones in the induction of EpRE-mediated gene expression. Chemical research in toxicology. 2008;21:2352–60. doi: 10.1021/tx8001498. [DOI] [PubMed] [Google Scholar]
- 50.Ahmad N, Gupta S, Mukhtar H. Green tea polyphenol epigallocatechin-3-gallate differentially modulates nuclear factor kappaB in cancer cells versus normal cells. Archives of biochemistry and biophysics. 2000;376:338–46. doi: 10.1006/abbi.2000.1742. [DOI] [PubMed] [Google Scholar]
- 51.Kim HS, Kim MH, Jeong M, Hwang YS, Lim SH, Shin BA, et al. EGCG blocks tumor promoter-induced MMP-9 expression via suppression of MAPK and AP-1 activation in human gastric AGS cells. Anticancer research. 2004;24:747–53. [PubMed] [Google Scholar]
- 52.Fujiki H, Suganuma M, Okabe S, Sueoka E, Sueoka N, Fujimoto N, et al. Cancer prevention with green tea and monitoring by a new biomarker, hnRNP B1. Mutat Res. 2001;480-481:299–304. doi: 10.1016/s0027-5107(01)00189-0. [DOI] [PubMed] [Google Scholar]
- 53.Hastak K, Gupta S, Ahmad N, Agarwal MK, Agarwal ML, Mukhtar H. Role of p53 and NF-kappaB in epigallocatechin-3-gallate-induced apoptosis of LNCaP cells. Oncogene. 2003;22:4851–9. doi: 10.1038/sj.onc.1206708. [DOI] [PubMed] [Google Scholar]
- 54.Masuda M, Suzui M, Weinstein IB. Effects of epigallocatechin-3-gallate on growth, epidermal growth factor receptor signaling pathways, gene expression, and chemosensitivity in human head and neck squamous cell carcinoma cell lines. Clinical cancer research : an official journal of the American Association for Cancer Research. 2001;7:4220–9. [PubMed] [Google Scholar]
- 55.Tang GQ, Yan TQ, Guo W, Ren TT, Peng CL, Zhao H, et al. (-)-Epigallocatechin-3-gallate induces apoptosis and suppresses proliferation by inhibiting the human Indian Hedgehog pathway in human chondrosarcoma cells. J Cancer Res Clin Oncol. 2010;136:1179–85. doi: 10.1007/s00432-010-0765-3. [DOI] [PubMed] [Google Scholar]
- 56.Smith DM, Wang Z, Kazi A, Li LH, Chan TH, Dou QP. Synthetic analogs of green tea polyphenols as proteasome inhibitors. Molecular medicine. 2002;8:382–92. [PMC free article] [PubMed] [Google Scholar]
- 57.Chan HY, Wang H, Tsang DS, Chen ZY, Leung LK. Screening of chemopreventive tea polyphenols against PAH genotoxicity in breast cancer cells by a XRE-luciferase reporter construct. Nutrition and cancer. 2003;46:93–100. doi: 10.1207/S15327914NC4601_12. [DOI] [PubMed] [Google Scholar]
- 58.Roy SK, Srivastava RK, Shankar S. Inhibition of PI3K/AKT and MAPK/ERK pathways causes activation of FOXO transcription factor, leading to cell cycle arrest and apoptosis in pancreatic cancer. Journal of molecular signaling. 2010;5:10. doi: 10.1186/1750-2187-5-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lambert JD, Lee MJ, Diamond L, Ju J, Hong J, Bose M, et al. Dose-dependent levels of epigallocatechin-3-gallate in human colon cancer cells and mouse plasma and tissues. Drug Metab Dispos. 2006;34:8–11. doi: 10.1124/dmd.104.003434. [DOI] [PubMed] [Google Scholar]
- 60.Maliakal PP, Coville PF, Wanwimolruk S. Tea consumption modulates hepatic drug metabolizing enzymes in Wistar rats. The Journal of pharmacy and pharmacology. 2001;53:569–77. doi: 10.1211/0022357011775695. [DOI] [PubMed] [Google Scholar]
- 61.Chandra S, De Mejia Gonzalez E. Polyphenolic compounds, antioxidant capacity, and quinone reductase activity of an aqueous extract of Ardisia compressa in comparison to mate (Ilex paraguariensis) and green (Camellia sinensis) teas. J Agric Food Chem. 2004;52:3583–9. doi: 10.1021/jf0352632. [DOI] [PubMed] [Google Scholar]
- 62.Syed DN, Afaq F, Kweon MH, Hadi N, Bhatia N, Spiegelman VS, et al. Green tea polyphenol EGCG suppresses cigarette smoke condensate-induced NF-kappaB activation in normal human bronchial epithelial cells. Oncogene. 2007;26:673–82. doi: 10.1038/sj.onc.1209829. [DOI] [PubMed] [Google Scholar]
- 63.Masuda M, Suzui M, Lim JT, Deguchi A, Soh JW, Weinstein IB. Epigallocatechin-3-gallate decreases VEGF production in head and neck and breast carcinoma cells by inhibiting EGFR-related pathways of signal transduction. Journal of experimental therapeutics & oncology. 2002;2:350–9. doi: 10.1046/j.1359-4117.2002.01062.x. [DOI] [PubMed] [Google Scholar]
- 64.Gupta S, Hussain T, Mukhtar H. Molecular pathway for (-)-epigallocatechin-3-gallate-induced cell cycle arrest and apoptosis of human prostate carcinoma cells. Archives of biochemistry and biophysics. 2003;410:177–85. doi: 10.1016/s0003-9861(02)00668-9. [DOI] [PubMed] [Google Scholar]
- 65.Lee KM, Yeo M, Choue JS, Jin JH, Park SJ, Cheong JY, et al. Protective mechanism of epigallocatechin-3-gallate against Helicobacter pylori-induced gastric epithelial cytotoxicity via the blockage of TLR-4 signaling. Helicobacter. 2004;9:632–42. doi: 10.1111/j.1083-4389.2004.00281.x. [DOI] [PubMed] [Google Scholar]
- 66.Afaq F, Adhami VM, Ahmad N, Mukhtar H. Inhibition of ultraviolet B-mediated activation of nuclear factor kappaB in normal human epidermal keratinocytes by green tea Constituent (-)-epigallocatechin-3-gallate. Oncogene. 2003;22:1035–44. doi: 10.1038/sj.onc.1206206. [DOI] [PubMed] [Google Scholar]
- 67.Srivastava RK, Unterman TG, Shankar S. FOXO transcription factors and VEGF neutralizing antibody enhance antiangiogenic effects of resveratrol. Molecular and cellular biochemistry. 2010;337:201–12. doi: 10.1007/s11010-009-0300-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chen Q, Ganapathy S, Singh KP, Shankar S, Srivastava RK. Resveratrol induces growth arrest and apoptosis through activation of FOXO transcription factors in prostate cancer cells. PloS one. 2010;5:e15288. doi: 10.1371/journal.pone.0015288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Dong Z, Ma W, Huang C, Yang CS. Inhibition of tumor promoter-induced activator protein 1 activation and cell transformation by tea polyphenols, (-)-epigallocatechin gallate, and theaflavins. Cancer research. 1997;57:4414–9. [PubMed] [Google Scholar]
- 70.Afaq F, Ahmad N, Mukhtar H. Suppression of UVB-induced phosphorylation of mitogen-activated protein kinases and nuclear factor kappa B by green tea polyphenol in SKH-1 hairless mice. Oncogene. 2003;22:9254–64. doi: 10.1038/sj.onc.1207035. [DOI] [PubMed] [Google Scholar]
- 71.Adhami VM, Siddiqui IA, Ahmad N, Gupta S, Mukhtar H. Oral consumption of green tea polyphenols inhibits insulin-like growth factor-I-induced signaling in an autochthonous mouse model of prostate cancer. Cancer research. 2004;64:8715–22. doi: 10.1158/0008-5472.CAN-04-2840. [DOI] [PubMed] [Google Scholar]
- 72.Masuda M, Suzui M, Lim JT, Weinstein IB. Epigallocatechin-3-gallate inhibits activation of HER-2/neu and downstream signaling pathways in human head and neck and breast carcinoma cells. Clinical cancer research : an official journal of the American Association for Cancer Research. 2003;9:3486–91. [PubMed] [Google Scholar]
- 73.Ahmed S. Green tea polyphenol epigallocatechin 3-gallate in arthritis: progress and promise. Arthritis research & therapy. 2010;12:208. doi: 10.1186/ar2982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Tachibana H, Fujimura Y, Yamada K. Tea polyphenol epigallocatechin-3-gallate associates with plasma membrane lipid rafts: lipid rafts mediate anti-allergic action of the catechin. BioFactors. 2004;21:383–5. doi: 10.1002/biof.552210174. [DOI] [PubMed] [Google Scholar]
- 75.Givant-Horwitz V, Davidson B, Reich R. Laminin-induced signaling in tumor cells: the role of the M(r) 67,000 laminin receptor. Cancer research. 2004;64:3572–9. doi: 10.1158/0008-5472.CAN-03-3424. [DOI] [PubMed] [Google Scholar]
- 76.Hussain T, Gupta S, Adhami VM, Mukhtar H. Green tea constituent epigallocatechin-3-gallate selectively inhibits COX-2 without affecting COX-1 expression in human prostate carcinoma cells. International journal of cancer Journal international du cancer. 2005;113:660–9. doi: 10.1002/ijc.20629. [DOI] [PubMed] [Google Scholar]
- 77.Ahmad N, Adhami VM, Gupta S, Cheng P, Mukhtar H. Role of the retinoblastoma (pRb)- E2F/DP pathway in cancer chemopreventive effects of green tea polyphenol epigallocatechin-3-gallate. Archives of biochemistry and biophysics. 2002;398:125–31. doi: 10.1006/abbi.2001.2704. [DOI] [PubMed] [Google Scholar]
- 78.Kundu JK, Na HK, Chun KS, Kim YK, Lee SJ, Lee SS, et al. Inhibition of phorbol ester-induced COX-2 expression by epigallocatechin gallate in mouse skin and cultured human mammary epithelial cells. J Nutr. 2003;133:3805S–10S. doi: 10.1093/jn/133.11.3805S. [DOI] [PubMed] [Google Scholar]
- 79.Khan N, Adhami VM, Mukhtar H. Apoptosis by dietary agents for prevention and treatment of prostate cancer. Endocrine-related cancer. 2010;17:R39–52. doi: 10.1677/ERC-09-0262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Nam S, Smith DM, Dou QP. Ester bond-containing tea polyphenols potently inhibit proteasome activity in vitro and in vivo. The Journal of biological chemistry. 2001;276:13322–30. doi: 10.1074/jbc.M004209200. [DOI] [PubMed] [Google Scholar]
- 81.Singh BN, Zhang G, Hwa YL, Li J, Dowdy SC, Jiang SW. Nonhistone protein acetylation as cancer therapy targets. Expert review of anticancer therapy. 2010;10:935–54. doi: 10.1586/era.10.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lee WJ, Shim JY, Zhu BT. Mechanisms for the inhibition of DNA methyltransferases by tea catechins and bioflavonoids. Molecular pharmacology. 2005;68:1018–30. doi: 10.1124/mol.104.008367. [DOI] [PubMed] [Google Scholar]
- 83.Kato K, Long NK, Makita H, Toida M, Yamashita T, Hatakeyama D, et al. Effects of green tea polyphenol on methylation status of RECK gene and cancer cell invasion in oral squamous cell carcinoma cells. British journal of cancer. 2008;99:647–54. doi: 10.1038/sj.bjc.6604521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Volate SR, Muga SJ, Issa AY, Nitcheva D, Smith T, Wargovich MJ. Epigenetic modulation of the retinoid X receptor alpha by green tea in the azoxymethane-Apc Min/+ mouse model of intestinal cancer. Molecular carcinogenesis. 2009;48:920–33. doi: 10.1002/mc.20542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Lee JH, Shim JS, Lee JS, Kim JK, Yang IS, Chung MS, et al. Inhibition of pathogenic bacterial adhesion by acidic polysaccharide from green tea (Camellia sinensis) J Agric Food Chem. 2006;54:8717–23. doi: 10.1021/jf061603i. [DOI] [PubMed] [Google Scholar]
- 86.Navarro-Martinez MD, Navarro-Peran E, Cabezas-Herrera J, Ruiz-Gomez J, Garcia-Canovas F, Rodriguez-Lopez JN. Antifolate activity of epigallocatechin gallate against Stenotrophomonas maltophilia. Antimicrobial agents and chemotherapy. 2005;49:2914–20. doi: 10.1128/AAC.49.7.2914-2920.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Naasani I, Seimiya H, Tsuruo T. Telomerase inhibition, telomere shortening, and senescence of cancer cells by tea catechins. Biochem Biophys Res Commun. 1998;249:391–6. doi: 10.1006/bbrc.1998.9075. [DOI] [PubMed] [Google Scholar]
- 88.Ju J, Hong J, Zhou JN, Pan Z, Bose M, Liao J, et al. Inhibition of intestinal tumorigenesis in Apcmin/+ mice by (-)-epigallocatechin-3-gallate, the major catechin in green tea. Cancer research. 2005;65:10623–31. doi: 10.1158/0008-5472.CAN-05-1949. [DOI] [PubMed] [Google Scholar]
- 89.Bartholome A, Kampkotter A, Tanner S, Sies H, Klotz LO. Epigallocatechin gallate- induced modulation of FoxO signaling in mammalian cells and C. elegans: FoxO stimulation is masked via PI3K/Akt activation by hydrogen peroxide formed in cell culture. Arch Biochem Biophys. 2010;501:58–64. doi: 10.1016/j.abb.2010.05.024. [DOI] [PubMed] [Google Scholar]
- 90.Reiter CE, Kim JA, Quon MJ. Green tea polyphenol epigallocatechin gallate reduces endothelin-1 expression and secretion in vascular endothelial cells: roles for AMP-activated protein kinase, Akt, and FOXO1. Endocrinology. 2010;151:103–14. doi: 10.1210/en.2009-0997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Anton S, Melville L, Rena G. Epigallocatechin gallate (EGCG) mimics insulin action on the transcription factor FOXO1a and elicits cellular responses in the presence and absence of insulin. Cell Signal. 2007;19:378–83. doi: 10.1016/j.cellsig.2006.07.008. [DOI] [PubMed] [Google Scholar]
- 92.Croce CM, Calin GA. miRNAs, cancer, and stem cell division. Cell. 2005;122:6–7. doi: 10.1016/j.cell.2005.06.036. [DOI] [PubMed] [Google Scholar]
- 93.Tsang WP, Kwok TT. Epigallocatechin gallate up-regulation of miR-16 and induction of apoptosis in human cancer cells. The Journal of nutritional biochemistry. 2010;21:140–6. doi: 10.1016/j.jnutbio.2008.12.003. [DOI] [PubMed] [Google Scholar]
- 94.Barber CG, Dickinson RP, Fish PV. Selective urokinase-type plasminogen activator (uPA) inhibitors. Part 3: 1-isoquinolinylguanidines. Bioorganic & medicinal chemistry letters. 2004;14:3227–30. doi: 10.1016/j.bmcl.2004.03.094. [DOI] [PubMed] [Google Scholar]
- 95.Kim HJ, Yum KS, Sung JH, Rhie DJ, Kim MJ, Min DS, et al. Epigallocatechin-3-gallate increases intracellular [Ca2+] in U87 cells mainly by influx of extracellular Ca2+ and partly by release of intracellular stores. Naunyn-Schmiedeberg’s archives of pharmacology. 2004;369:260–7. doi: 10.1007/s00210-003-0852-y. [DOI] [PubMed] [Google Scholar]
- 96.Oku N, Matsukawa M, Yamakawa S, Asai T, Yahara S, Hashimoto F, et al. Inhibitory effect of green tea polyphenols on membrane-type 1 matrix metalloproteinase, MT1-MMP. Biological & pharmaceutical bulletin. 2003;26:1235–8. doi: 10.1248/bpb.26.1235. [DOI] [PubMed] [Google Scholar]
- 97.Berger SJ, Gupta S, Belfi CA, Gosky DM, Mukhtar H. Green tea constituent (--)-epigallocatechin-3-gallate inhibits topoisomerase I activity in human colon carcinoma cells. Biochem Biophys Res Commun. 2001;288:101–5. doi: 10.1006/bbrc.2001.5736. [DOI] [PubMed] [Google Scholar]
- 98.Sartippour MR, Shao ZM, Heber D, Beatty P, Zhang L, Liu C, et al. Green tea inhibits vascular endothelial growth factor (VEGF) induction in human breast cancer cells. J Nutr. 2002;132:2307–11. doi: 10.1093/jn/132.8.2307. [DOI] [PubMed] [Google Scholar]
- 99.Ahn WS, Huh SW, Bae SM, Lee IP, Lee JM, Namkoong SE, et al. A major constituent of green tea, EGCG, inhibits the growth of a human cervical cancer cell line, CaSki cells, through apoptosis, G(1) arrest, and regulation of gene expression. DNA Cell Biol. 2003;22:217–24. doi: 10.1089/104454903321655846. [DOI] [PubMed] [Google Scholar]
- 100.Fujiki H, Suganuma M, Okabe S, Sueoka E, Suga K, Imai K, et al. Mechanistic findings of green tea as cancer preventive for humans. Proc Soc Exp Biol Med. 1999;220:225–8. doi: 10.1046/j.1525-1373.1999.d01-38.x. [DOI] [PubMed] [Google Scholar]