

Abstract
Pancreatic adenocarcinoma is highly resistant to conventional therapeutics and has been shown to evade apoptosis by deregulation of the X‐linked and cellular inhibitors of apoptosis proteins (XIAP and cIAP). Second mitochondria‐derived activator of caspases (Smac) induces and amplifies cell death by reversing the anti‐apoptotic activity of IAPs. Thus, Smac‐derived peptide analogues (peptidomimetics) have been developed and shown to represent promising cancer therapeutics. Sigma‐2 receptors are overexpressed in many proliferating tumor cells including pancreatic cancer. Selected ligands to this receptor are rapidly internalized by cancer cells. These characteristics have made the sigma‐2 receptor an attractive target for drug delivery because selective delivery to cancer cells has the potential to increase therapeutic efficacy while minimizing toxicity to normal tissues.
Here, we describe the initial characterization of SW IV‐134, a chemically linked drug conjugate between the sigma‐2 ligand SW43 and the Smac mimetic SW IV‐52 as a novel treatment option for pancreatic adenocarcinoma. The tumor killing characteristics of our dual‐domain therapeutic SW IV‐134 was far greater than either component in isolation or in an equimolar mix and suggests enhanced cellular delivery when chemically linked to the sigma‐2 ligand. One of the key findings was that SW IV‐134 retained target selectivity of the Smac cargo with the involvement of the NF‐κB/TNFα signaling pathway. Importantly, SW IV‐134 slowed tumor growth and improved survival in murine models of pancreatic cancer. Our data support further study of this novel therapeutic and this drug delivery strategy because it may eventually benefit patients with pancreatic cancer.
Keywords: Sigma‐2 receptor, Smac, IAPs, Pancreas cancer, Selective delivery
Highlights
We report a cancer‐selective drug delivery concept targeting the sigma‐2 receptor.
SW IV‐134 is a dual‐domain drug conjugate with delivery and effector component.
All features of the individual components are well retained in the drug conjugate.
SW IV‐134 represents a potent pancreatic cancer drug in vitro and in vivo.
This delivery concept can be expanded to other sigma‐2 receptor expressing cancers.
Abbreviations
- IAP
inhibitor of apoptosis proteins
- XIAP
X-linked inhibitor of apoptosis proteins
- cIAP
cellular inhibitor of apoptosis proteins
- Smac
second mitochondria-derived activator of caspases
- TNFα
tumor necrosis factor alpha
- NF-κB
nuclear factor kappa-light-chain-enhancer of activated B cells
- AST
aspartate aminotransferase
- ALT
alanine aminotransferase
- BUN
blood urea nitrogen
- Cr
creatinine
- MUC1
mucin 1
- i.p
intra peritoneal
- PDAC
pancreatic ductal adenocarinoma
- NIK
NF-қ-B-inducing kinase
- Ab
antibody
1. Introduction
Pancreatic cancer is the fourth leading cause of cancer related deaths in the United States with a 5‐year survival of 6% (Siegel et al., 2012). Unfortunately this number has not significantly changed in decades. Even more depressing is the fact that about 75% of patients will not live a year from the date of their diagnosis (America cancer society, 2010). Since the majority of patients present with advanced disease and 80% of those resected develop metastases after resection, there is a desperate need for a novel strategy that will address the disease on a systemic level.
In this study we deliver a novel apoptotic molecule selectively to pancreatic cancer cells by targeting sigma‐2 receptors, which are overexpressed in pancreatic ductal andenocarcinoma (PDAC). Sigma receptors are present in a variety of tissues but their physiologic function has not yet been completely defined. These receptors were first classified as a subtype of opioid receptors (Martin et al., 1976) and in the central nervous system they are involved in the regulation of neurotransmitter release, modulation of neurotransmitter receptor function, learning and memory processes, and regulation of movement and posture (Su, 1993). They are expressed in peripheral tissues such as liver, kidney, and endocrine organs but their function in these tissues has been poorly understood (Ogawa et al., 2009).
At least two types of sigma receptors have been identified. Sigma‐1 receptors were first characterized and cloned early on (Quirion et al., 1992; Walker et al., 1990) however it was several years before the sigma‐2 receptor was characterized. Our group first reported Progesterone receptor membrane component 1 (PGRMC1) as the binding component of the sigma‐2 receptor complex (Xu et al., 2011). Sigma‐2 receptors are biomarkers for proliferation in solid tumors and expression is about 10 times higher in proliferative cells as compared to quiescence cells (Mach et al., 1997). As such, they have been shown to be useful clinically for PET/CT imaging with targeted ligands (Dehdashti et al., 2013).
We have previously demonstrated that sigma‐2 receptors are overexpressed in PDAC and have developed sigma‐2 ligands that have a very high affinity for pancreatic cancer cells (Kashiwagi et al., 2007). In addition, we have suggested that it might be feasible to deliver drugs selectively to pancreatic cancer cells by conjugating small molecules to these ligands for delivery (Spitzer et al., 2012). Cancer‐selective drug delivery is important pharmacologically because it improves the therapeutic window, thus improving drug concentrations at tumor sites while reducing drug concentrations at non‐tumor sites and minimizing potential off‐target toxicities. In addition to cancer selectivity, sigma‐2 ligands have demonstrated the ability to rapidly internalize once they bind to a cancer cell (Spitzer et al., 2012; Zeng et al., 2007). This internalization property is another potential advantage of this delivery platform because there are a multitude of potential intracellular targets which have thus far been difficult to exploit.
Second mitochondria‐derived activator of caspases (Smac) is a mitochondrial protein that is released into the cytosol to induce or amplify apoptosis by inhibiting the inhibitors of apoptosis (Du et al., 2000; Verhagen et al., 2000). These targets are the inhibitor of apoptosis proteins (IAP) which are upregulated in many different types of cancer (Deveraux and Reed, 1999; Wu et al., 2007). Endogenous Smac induces auto‐ubiquitination and degradation of cIAP1, which subsequently creates an autocrine feedback loop leading to NF‐κB activation and TNFα secretion, which then engages the extrinsic death pathway via caspase‐8 (Wu et al., 2007). Another function is to antagonize XIAP's inhibitory role on caspases. BIR‐3 domain of XIAP binds and blocks caspase‐9 (Oost et al., 2004), while its BIR‐2 domain binds caspase‐3/7 (Deveraux et al., 1997; Shiozaki et al., 2003). Smac competitively bind baculovirus IAP repeat domains BIR resulting in release of active caspases. These multifaceted activities make Smac an ideal candidate for cancer drug development. In fact, there are an increasing number of studies describing Smac‐derived peptides and peptidomimetics for cancer therapy (Li et al., 2004, 2008, 2008, 2008, 2006).
In this study, we describe the biologic activity of the small molecule therapeutic SW IV‐134 and its efficacy in PDAC. This new dual‐domain compound was synthesized by chemical linkage of the sigma‐2 ligand SW43 (Vangveravong et al., 2006) and the Smac mimetic SW IV‐52 (Oost et al., 2004; Sun et al., 2008a) (with specificity for the BIR‐3 domain of XIAP) in order to selectively deliver the drug to PDAC cells. We have recently shown efficacy of a sigma‐2/Smac conjugate SW III‐123 in ovarian cancer (Zeng et al., 2013) and now we have developed SW IV‐134 in an effort to improve efficacy and binding affinity of these compounds in PDAC. First, SW IV‐134 is a pure enantiomer of the L‐configuration, while SW III‐123 is a racemic mixture of both enantiomers. As well, the binding affinity of SW IV‐134 (Ki = 22.6 ± 1.8 nM) for the sigma‐2 receptor is nearly ten times higher compared to SW III‐123 (Ki = 189.9 ± 12.8 nM) (Zeng et al., 2013). This strategy represents a promising step in cancer treatment with the potential to increase cancer cell death while minimizing off‐target toxicities.
2. Materials and methods
2.1. Compounds
Chemical synthesis of the sigma‐2 ligand SW43 was performed as previously described (Vangveravong et al., 2006) and the Smac mimetic SW IV‐52 exactly matches the structure from previously published work (Oost et al., 2004) (Sun et al., 2008a). Detailed synthesis schematics are shown in the supplementary data (Fig. S1).
2.2. Cell lines
CFPAC‐1, BxPC‐3, AsPC‐1, PANC‐1 and MiaPaCa‐2 were obtained from American Type Culture Collection (ATCC, Manassas, VA). KCM cell line was isolated from a human MUC1 expressing pancreatic tumor of transgenic mouse. The KCM mouse model was generated on the C57BL/6 background by mating the P48‐Cre with the LSL‐KRASG12D mice and further mating to the MUC1.Tg (Besmer et al., 2011; Tinder et al., 2008). The KCM cell line was kindly provided by Dr. Pinku Mukherjee (University of North Carolina–Charlotte). CFPAC‐1 and PANC1 were cultured in Iscove's modified Dulbecco's medium with 4 mM l‐glutamine, 1.5 g/L Sodium bicarbonate, and 10% fetal bovine serum (FBS). MiaPaCa‐2 was cultured in Dulbecco's Modified Eagle's with 10% FBS and 2.5% horse serum. BxPC‐3, AsPC‐1 and KCM were cultured in RPMI‐ 1640 medium with 10% FBS. Antibiotics penicillin (100 μg/ml) and streptomycin (100 μg/ml) were added to the media and cells maintained in humidified incubator at 37 °C with 5% CO2.
2.3. Sigma receptor binding studies
SW IV‐134 binding affinity to both sigma‐1 and 2 receptors were tested for as previously described for SW43 compound (Vangveravong et al., 2006). Briefly, guinea pig brain for sigma‐1 binding assay and rat liver for sigma‐2 binding assay membrane homogenates (∼300 μg protein) were diluted with 50 mM Tris–HCl, pH 8.0 and incubated with either ∼5 nM [3H](+)‐pentazocine (34.9 Ci mmol−1 (Perkin–Elmer, Boston, MA) for sigma‐1 assay) or 1 nM [3H]RHM‐1 (80 Ci mmol−1(American Radiolabeled Chemicals Inc., St. Louis, MO) for sigma‐2 assay) in a total volume of 150 μl in 96‐well plates at 25 °C. The concentrations of SW IV‐134 ranged from 0.1 nM to 10 μM. After 60 min inscubation, the reactions were terminated by the addition of 150 μl of cold wash buffer (10 mM Tris–HCl, 150 mM NaCl, pH 7.4) using a 96‐channel transfer pipette (Fisher Scientific, Pittsburgh, PA, USA), and the samples harvested and filtered rapidly into a 96‐well fiberglass filter plate (Millipore, Billerica, MA, USA) that had been presoaked with 100 μl of 50 mM Tris–HCl, at pH 8.0 for 1 h. Each filter was washed three times with 200 μl of ice‐cold wash buffer, and then the bound radioactivity quantified using a Wallac 1450 MicroBeta liquid scintillation counter (Perkin Elmer, Boston, MA, USA). Nonspecific binding was determined from samples that contained 10 μM of cold haloperidol.
2.4. Evaluation of cytotoxicity in vitro
Cytotoxicity of the drugs was evaluated by Cell titer‐Glo, Luminescent cell viability assay (Promega, Madison, WI). Pancreatic cell lines were plated at a density of 2 × 104/well in white 96 well, clear bottom plates for 24 h prior to treatment. Drugs were dissolved in DMSO and serially diluted in culture medium to achieve final concentration with DMSO less than 1%. Cells were treated for 24 h and 100 μl of the Titer Glo reagent was added to each well. The content of the plate was mixed using an orbital shaker and incubated for 10 min at room temperature. Luminescence signal was measured using multi‐mode microplate reader (BioTek instruments, Winooski, VT). Different drug concentrations were assayed in triplicate.
2.5. In vitro caspase assays
Caspase‐3/7, 8 and 9 activities were measured in AsPC‐1 cells using Caspase‐Glo® Assay Systems according to the manufacturer's instructions (Promega). Briefly, the assays are based on caspase‐specific substrates, which are activated by cleavage, resulting in caspase‐specific luminescence signals. Cells were seeded at a density of 2 × 104 in white 96‐well, clear bottom plates for 24 h before treatment with 12 μM of compound. The contents were mixed using plate shaker for 30 s, then incubated at room temperature for 90 min. Luminescence signal was measured using multi‐mode microplate reader (BioTek). Assay was performed in triplicates, and caspase activity was plotted compared cells treated with DMSO as a control.
2.6. In vitro evaluation of apoptosis via flow cytometry
Phosphatidylserine (PS) translocation to the plasma membrane is an early event during apoptosis induction and can be detected by flow cytometry. AsPC‐1 cells were seeded in 6‐well plates at a density of 8 × 105/well for 24 h and treated with 12 μM of the compounds. After 24 h they were assayed using Annexin‐V FITC Kit (Biolegend, San Diego, CA). Propidium iodide was added to differentiate early apoptotic cells from necrotic and late stage apoptotic cells. Cells were prepared according to the manufacturer's instructions and analyzed with a FACSCalibur flow cytometer from (BD Biosciences, San Jose, CA).
2.7. Immunoblotting
PANC‐1 cells were plated in 6 wells plate at a density of 8 × 105/well. Cells were treated with DMSO as a control and SW IV‐134 at concentrations of 4, 8 and 12 μM for 1, 6 and 24 h. Cells were harvested and lysed in RIPA buffer containing protease inhibitor cocktail (Roche, Mannheim, Germany) and phosphatase inhibitor cocktail (Sigma chemical, St Louis, MO). Protein concentration was measured by BCA protein assay kit (Thermo Fisher Scientific, Rockford, IL). Samples containing equal amount of protein were run on NuPAGE Bis‐Tris 4–12% gradient gel and then transferred onto PVDF membranes (Life Technologies, Grand Island, NY). The membranes were incubated in blocking buffer (5% dry milk) for 1 h then in primary antibody at 4 °C overnight. Membranes were washed 3 times with TBS‐T and incubated in secondary antibody at room temperature for 1 h. SuperSignal West Dura Substrate (Thermo Fisher Scientific Rockford, IL) was used to detect secondary antibody. Primary rabbit antibodies for XIAP, cIAP‐1, NIK, NF‐κB p65, Phospho NF‐қB p65, and NF‐κB2 p100/p52s were purchased from Cell Signaling Technology (Danvers, MA). Primary actin goat antibody was purchased from Santa Cruz (Dallas, TX). Primary antibody dilutions were made according to the manufacturer's instructions. HRP‐linked goat anti‐rabbit secondary antibodies were purchased from Cell Signaling. HRP linked donkey anti‐goat secondary antibody was purchased from Santa Cruz (Dallas, TX). Secondary antibody dilutions were 1:2000 for goat anti‐rabbit, 1:4000 for donkey anti‐goat.
2.8. In vivo caspase‐3 assay
Caspase‐3 Assay Kit Fluorometric (Abcam, Cambridge, MA) was used to detect in vivo caspase‐3 activity. Female C57BL/6 mice (6 weeks old) were purchased from Harlan laboratories (Indianapolis, IN). Mice were injected in the right flank with 200 μL of single‐cell suspension of KCM (2.5 × 104 cells per mouse). Treatment was started when the mean tumor diameter was 5 mm. Mice were stratified into 2 groups (n = 3)/group. One group (mouse 4–6) received daily intra peritoneal injections with 750 nmoles of SW IV‐134 in 100 μL of vehicle (25% Cremophor and 75% H2O) and the other group (mouse 1–3) was injected with vehicle once a day for 3 days. This drug dose and the vehicle were used in all the in vivo experiments in the study. Tumors were harvested and single cell suspensions prepared using a tumor dissociation kit (Miltenyi Biotec, Auburn, CA). Cells from each tumor were suspended in 50 μl/well of lysis buffer in white 96 wells at a density of 1 × 106/well, each tumor was assayed in triplicate. The assay was performed according to manufacturer's instructions. The fluorescence signal was detected using a multi‐mode microplate reader (BioTek). The fold increase in caspase‐3 activity was determined by comparing the results with the maximum level of the control as a baseline.
2.9. In vivo assessment of apoptosis
Athymic female nude mice (6 weeks old) were purchased from Harlan laboratories. They were injected in the right flank with 200 μL single cell suspension of AsPC‐1 in RPMI medium (1 × 106 cells per mouse). Treatment was started when the tumor diameter was between 5 and 6 mm. Mice received daily intra peritoneal injections with SW IV‐134 and vehicle once a day for 3 days. Tumors were harvested and fixed with 10% Neutral Buffered Formalin. Immunohistochemistry staining for terminal deoxynucleotidyl transferase mediated (dUTP) nick end labeling (TUNEL) was performed by Washington University Digestive Diseases Research Core Center (DDRCC).
2.10. In vivo assessment of tumor growth and survival
Animal studies were performed according to the animal studies protocol approved by the Washington University Institutional Animal Care Facility. In vivo studies with mice were performed to compare the effect of SW IV‐134, SW43, SW IV‐52, a combination of SW43 with SW IV‐52, and vehicle. Toxicity evaluation was also performed. C57BL/6 mice (6 weeks old) were injected subcutaneously in the right flank with KCM cells described above. Treatments started when the mean tumor diameter was ∼7 mm. Mice received daily intra peritoneal injections with 750 nmoles in 100 μL/mouse of SW IV‐134 and vehicle for 10 days. Tumors were measured every other day with a digital caliper. Several mice from different treatment groups were sent to the Division of Comparative Medicine in our institution for pathologic evaluation. Blood was collected for complete blood count (CBC) and biochemical analysis (AST, ALT, BUN, total bilirubin, and Cr). Organs were examined grossly and histologically.
Another in vivo experiment was performed to evaluate the effect of the drug on tumor growth and survival. Athymic female nude mice (6 weeks old, Harlan Laboratories) were injected in the right flank with 200 μL single cell suspension of 1 × 106 AsPC‐1 cells in RPMI medium. Treatment started when the mean tumors diameter was ∼6 mm. Mice received daily intra peritoneal injections with 750 nmoles in 100 μL/mouse of SW IV‐134 and vehicle for 2 weeks. Tumors were measured every other day. Mice were euthanized when tumors reached a diameter of 2 cm or had ulcerated.
2.11. Statistics
Statistical analyses and data plotting were performed using GraphPad Prism software version 5 (San Diego, CA). Results were expressed as mean ± SEM of at least 3 biological replicates. IC50 values were calculated by curve fitting normalized viability versus drug concentration. One‐way ANOVA was used to analyze the differences in caspase assays, AnnexinV and IC50 values. Two‐way ANOVA was used to analyze the difference in tumor sizes and in vivo caspase‐3 assays. Unpaired two‐tailed t‐test was used to evaluate the difference in CBC, biochemistry analyses and the TNFα blocking study. The Kaplan–Meier survival curve was plotted and the difference between the groups was compared with a log‐rank test. A p‐value <0.05 was considered significant for all analyses.
3. Results
3.1. SW IV‐134 and SW43 have similar binding kinetics for the sigma‐2 receptor
The major goal of our current study was to expand the versatility of the sigma‐2 receptor for the selective delivery of anti‐apoptotic drug cargoes into the cancer cells. We therefore chemically linked our well‐established sigma‐2 ligand SW43 with the Smac peptidomimetic SW IV‐52, resulting in the dual‐domain drug SW IV‐134 (Figure 1A). The question that arises when combining two unrelated molecules is whether this linkage will change the active domains of the individual components of the conjugate. In order to address these initial concerns, we performed competitive binding assays with SW IV‐134 to determine the impact the Smac domain might have caused with regard to the affinity for the sigma‐2 receptor. These assays demonstrated that the affinity of SW IV‐134 for the sigma‐2 receptor was only slightly reduced compared to the parent compound, Ki = 22.6 ± 1.8 nM [SW IV‐134] and Ki = 7.1 ± 1.3 nM [SW43] (Table 1). Of note, the affinity for the other family member of the sigma receptors (sigma‐1) was much more reduced for the drug conjugate and resulted in a more than 250‐fold higher sigma‐2 selectivity compared to only 19 for the parental compound SW43. The reason(s) for this affinity change are currently unknown but are the focus of future investigations.
Figure 1.
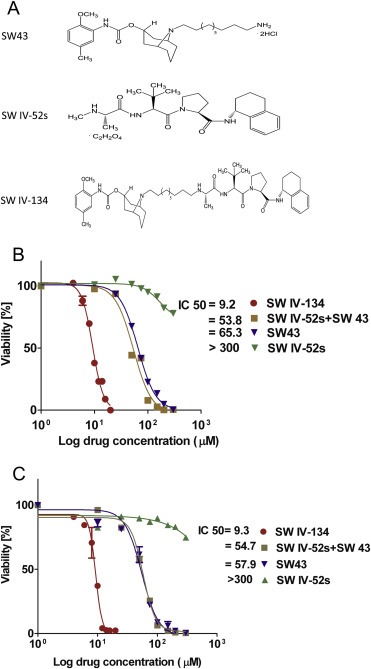
Compound structures and cell killing characteristics of SW IV‐134. (A) Chemical structures of the drugs used in the current study. IC50 of (B) KCM and (C) AsPC‐1. Cells were treated with different concentrations of SW IV‐134, SW43, SW IV‐52, and a combination of SW IV‐52 with SW43. Titer‐Glo viability assay was performed 24 h later. IC50 was expressed as mean ± SEM, n ≥ 3.
Table 1.
Binding affinities of SW IV‐134 and SW43 to the sigma‐1 and sigma‐2 receptors.
| Compound | Ki (nM)a | σ1/σ2 d | |
|---|---|---|---|
| σ1 b | σ2 c | ||
| SW 43 | 134.3 ± 11.9 | 7.1 ± 1.3 | 19 |
| SW IV‐134 | 5737 ± 476 | 22.6 ± 1.8 | 253 |
Abbreviations: σ1 for Sigma‐1 receptor. σ2 for Sigma‐2 receptor.
Mean ± SEM. Ki values were determined by at least three experiments.
Ki for inhibiting the binding of [3H](+)‐pentazocine to guinea pig brain homogenates.
Ki for inhibiting the binding of [3H](+)DTG to rat liver homogenates.
Ki for σ1 receptors/ Ki for σ2 receptors.
3.2. SW IV‐134 has potent killing activity against pancreas cancer cell lines
We next assessed the pharmacologic activity of our drugs on a panel of human and mouse pancreatic cancer cell lines in vitro. The cells were treated for 24 h with SW IV‐134, SW43, SW IV‐52, and an equimolar mixture of the individual compounds SW IV‐52 and SW43 to determine the killing profiles of the various drugs. As a general rule, the Smac mimetic alone had the least biologic activity for all cell lines with IC50 > 300 μM (Figure 1B, C and Table 2). SW43 treatment resulted in a robust killing pattern for all cell lines that was only slightly augmented in combination with SW IV‐52. In stark contrast, however, delivery of Smac via the sigma‐2 ligand SW43 (SW IV‐134) reduced the IC50 from 2‐fold (PANC‐1) to more than 8‐fold (MiaPaCa‐2) compared to the combination treatment SW43 + SW IV‐52 (Table 2). Cell death was induced by apoptosis, gauged macroscopically on membrane blebbing events (not shown) and the apoptotic marker (Annexin‐V positivity) (Figure 2A, B), which both correlated well with the viability data described above (Figure 1B, C).
Table 2.
IC50 (μM) of pancreatic cell lines treated with different compounds for 24 h.
| Drugs | Cell line | |||||
|---|---|---|---|---|---|---|
| AsPC‐1 | BxPC‐3 | KCM | MiaPaCa‐2 | PANC‐1 | CFPAC‐1 | |
| IC50 ± SEM | IC50 ± SEM | IC50 ± SEM | IC50 ± SEM | IC50 ± SEM | IC 50 ± SEM | |
| SW IV‐134 | 9.2 ± 0.4 | 6.8 ± 0.2 | 9.3 ± 0.2 | 7.8 ± 0.3 | 6.3 ± 0.1 | 7.4 ± 0.3 |
| SW 43 + SW IV‐52s | 54.8 ± 1.9 | 40.8 ± 1.2 | 53.8 ± 0.4 | 65.3 ± 0.9 | 13.4 ± 0.2 | 27.2 ± 2.3 |
| SW 43 | 56.8 ± 2.8 | 56.3 ± 0.8 | 65.3 ± 1.0 | 81.8 ± 1.3 | 21.4 ± 0 | 48.2 ± 1.7 |
| SW IV‐52s | >300 | >300 | >300 | >300 | >300 | >300 |
(Mean ± SEM), n ≥ 3. P < 0.05.
Figure 2.
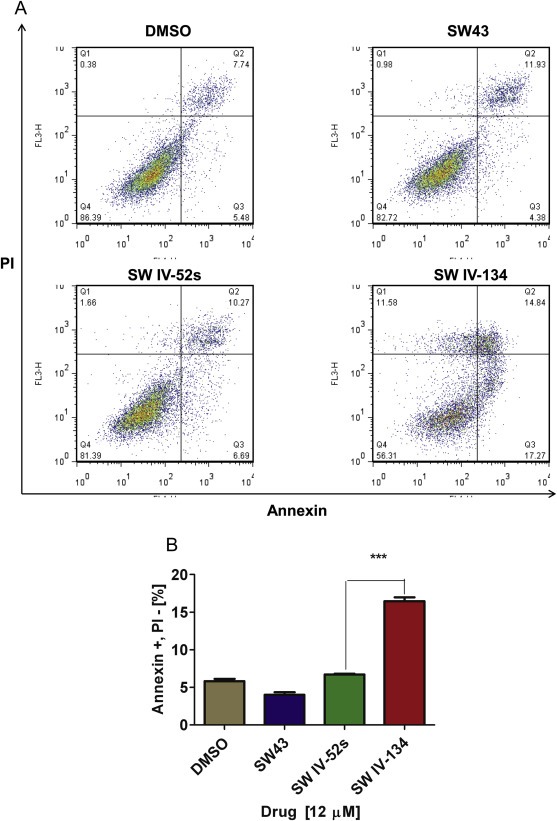
SW IV‐134 induces target cell apoptosis. (A) Flow cytometry dot plot of AsPC‐1 cells treated with different compounds for 24 h. There is significant shift of cells treated with SW IV‐134 from Annexin‐V/PI negative to Annexin‐V positive/PI negative quadrant (early apoptotic cells) compared to controls. (B) Flow cytometry of AsPC‐1 showing the percent of early apoptotic cells (Annexin‐V positive/PI negative), ***p < 0.0001.
These results were very encouraging and strongly suggest that the sigma‐2 ligand SW43 of the drug conjugate SW IV‐134 efficiently manages to deliver the Smac mimetic SW IV‐52 into the cancer cells which leads to enhanced cell death induction. This scenario is further supported by the notion that the combination treatment with the isolated compounds SW43 and SW IV‐52 was incapable to augment cancer cell killing at the same concentration.
3.3. SW IV‐134 activates intrinsic and extrinsic apoptotic pathways
Smac mimetics have been shown to competitively bind the BIR‐3 domain of XIAP, which results in the induction of caspase‐9 activity (intrinsic apoptotic pathway) (Oost et al., 2004). In addition to the activation of the intrinsic death pathway, the same peptidomimetics have been reported to also trigger the extrinsic apoptotic pathway via TNFα expression (Vince et al., 2007). In order to test the effects of SW IV‐134 on both death pathways, activation of caspase‐8 (extrinsic) and caspase‐9 (intrinsic) were assessed. To obtain a more complete picture, caspase‐3 was also included as both pathways converge at the level of this key executioner caspase. AsPC‐1 cells were treated with SW IV‐134, SW43, SW IV‐52, and a combination of SW IV‐52 and SW43. The drug conjugate SW IV‐134 was found to activate all three caspases more than SW IV‐52 and an equimolar combination of SW43 and SW IV‐52, while SW43 did not cleave the caspases at these concentrations, similar to the DMSO control (Figure 3). More specifically, SW IV‐134 induced a strong activation of caspase‐8 to nearly 5‐fold above baseline, while SW43, SW IV‐52 and the combination of the latter drugs were quite ineffective under these conditions (Figure 3A, p < 0.0001). These results are consistent with a robust involvement of the extrinsic death pathway by our dual‐domain compound.
Figure 3.
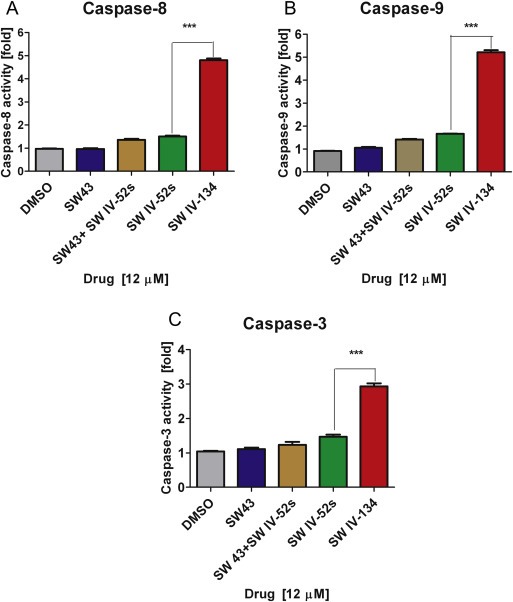
SW IV‐134 treatment leads to caspase activation. Caspase‐Glo® Assay of AsPC‐1 treated with 12 μM of different compounds for 24 h. Cells treated with SW IV‐134 had significant increase in caspase‐3, 8, and 9 ***p < 0.0001.
Regarding activation of the intrinsic death pathway, SW IV‐134 also increased the level of activated caspase‐9 more than 5‐fold, while SW43, SW IV‐52 and the SW43/SW IV‐52 combination were again rather ineffective (Figure 3B, p < 0.0001). These data suggest that the Smac domain of SW IV‐134 efficiently displaces XIAP from the inactive caspase‐9 precursor, subsequently leading to an increase in caspase‐9 activity.
Both death pathways converge at the level of active caspase‐3. Thus, it was not surprising to see an increase in cleaved caspase‐3 to nearly 3‐fold above baseline, while SW43, SW IV‐52 and a cotreatment with SW43 and SW IV‐52 only resulted in a marginally elevated level of the executioner caspase (Figure 3C, p < 0.0001). Overall, these results suggest that SW IV‐134 activates both intrinsic and extrinsic apoptosis pathways via the preserved structural features of the Smac mimetic as part of our dual‐domain drug conjugate.
3.4. SW IV‐134 induces NF‐κB‐mediated TNFα dependent extrinsic apoptotic pathway activation
Smac was previously reported to mediate degradation of cIAP‐1, which culminates in NF‐қB‐mediated TNFα production (Vince et al., 2007). This feedback loop amplifies the intrinsic death pathway and leads to an augmentation of apoptosis via the extrinsic TNF receptor route. We therefore speculated that our drug conjugate would induce the same amplification pathway. To evaluate this hypothesis, we treated PANC‐1 cells with SW IV‐134 and assessed the key modulators of this pathway by Western blot analysis.
We noticed that after exposure to our drug, the intracellular cIAP‐1 level quickly decreased, while XIAP was not affected (Figure 4A). This was not unexpected since antagonism of Smac is viewed as a displacement mechanism of XIAP from caspase‐9 and is not associated with XIAP degradation (Oost et al., 2004). The consequence of cIAP‐1 degradation leads to accumulation of NF‐қB‐inducing kinase (NIK), which, under steady‐state circumstances, is constitutively degraded by cIAP‐1. As a result, NIK readily accumulates within the target cells as shortly as 1 h post‐treatment. NIK activates IKKα which results in phosphorylation of the p100 NF‐κB subunit, leading to production of p52 of p52‐RelB dimers, that act as a transcription factor. Results showed an increase in NF‐κB p52 level with increasing time of SW IV‐134 treatment. Both increase in NIK and NF‐κB p52 levels indicate the activation of the non‐canonical NF‐қB pathway (Oeckinghaus and Ghosh, 2009). We also demonstrated an increase in activated phospho‐NF‐қB (p65), which indicates activation of the canonical NF‐қB pathway (Oeckinghaus and Ghosh, 2009) (Figure 4A).
Figure 4.
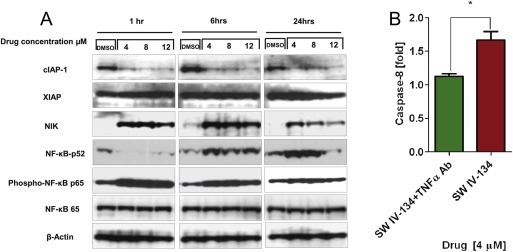
SW IV‐134 activates the NF‐қB pathway and TNFα dependent apoptosis. (A) Western blot of PANC‐1 cells treated with DMSO as control and SW IV‐134 in different time points and concentrations. (B) Caspase‐8 assay PANC‐1 cells treated with and without anti‐TNFα antibody for 1 h, followed by 24 h incubation with SW IV‐134. Activated caspase‐8 was monitored as a surrogate for TNFα induction, *p < 0.05.
A prominent transcriptional target of NF‐қB is TNFα (Vince et al., 2007). In order to evaluate the role of TNFα in activating the extrinsic death pathway, PANC‐1 cells were pre‐treated with or without 2 μg/ml of anti‐TNFα antibody from R&D Systems (Minneapolis, MN) to block the extrinsic TNF receptor pathway. Then cells were treated with 4 μM of SW IV‐134 for 24 h. Activation of caspase‐8 was used as a marker for pathway activation one day post‐treatment. In the absence of TNFα blocking antibody, a marked cleavage of caspase‐8 was noted, which was reduced to background levels in the presence of the antibody (Figure 4B). Together, our data support the notion that SW IV‐134 induces the extrinsic death pathway via the cIAP‐1/NIK/NF‐қB axis, which in turn leads to transcriptional and translational upregulation of TNFα, the key mediator of drug‐induced apoptosis. These data further suggest that both domain structures of SW IV‐134 are well preserved and accessible for the targeting (SW43) and execution of the effector function (SW IV‐52) of our dual‐domain cancer therapeutic.
3.5. SW IV‐134 induces apoptosis in vivo, reduces tumor growth and improves survival in preclinical mouse models of pancreatic cancer
The most stringent evaluation of a novel cancer therapeutic is its pharmacologic profile in a relevant animal model. In order to address in vivo applicability of SW IV‐134, we employed a syngeneic (C57BL/6) as well as a xenograft (nude) mouse model of pancreatic cancer. In both cases, female mice were injected subcutaneously into the right flanks with the respective tumor cell lines (KCM and AsPC‐1, respectively). Treatment was initiated at a mean tumor diameter of 6–7 mm, 2 weeks post‐implantation. The mice were then treated daily by i. p. injection with 750 nmoles in 100 μL of SW IV‐134, SW43, SW IV‐52 and an equimolar mix of SW43 and SW IV‐52 for the indicated time. Vehicle alone served as a negative control.
In the syngeneic cancer model (KCM in C57BL/6), only the dual‐domain therapeutic SW IV‐134 was capable of reducing the average tumor size (Figure 5A, p < 0.0001), while none of the other reagents alone or in combination resulted in a reduction in tumor growth with similar growth rates as the vehicle control (Figure 5A, p = 0.7). Of note, no gross abnormalities in mouse behavior (grooming) and drug‐related deaths were recorded, supported by unchanged serum labs (CBC, AST, ALT, BUN, total bilirubin, and Cr) Supplementary table S1 (A) and (B). Organ analyses (brain, heart, lungs, alimentary tract, kidneys, liver and pancreas), which did not reveal obvious signs of adverse drug effects, except a mild and transient peritonitis (data not shown).
Figure 5.
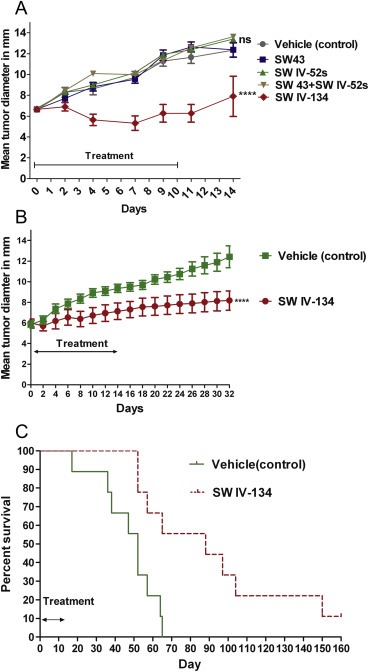
SW IV‐134 slows tumor growth and improves survival. (A) C57BL/6 (n = 10 mice/group) mice with right flank KCM subcutaneous tumors. Mice treated with daily i.p. injections with 750 nmoles in 100 μL of different compounds for 10 days. There was no significant difference between DMSO, WS 43, SW IV‐52s and combination of SW43 + SW IV52s p (ns) > 0.05. Tumor diameters of mice treated with SW IV‐134 were significantly smaller than other groups ****p < 0.0001. (B) Athymic nude mice (n = 9 mice/group) with right flank AsPC‐1 xenograft were treated with daily i.p. injections of SW IV‐134 and vehicle for 14 days. There was significant difference between tumor volumes of the 2 groups, ****p < 0.0001. (C) Kaplan–Meier survival curve of mice in (B). Median survival of mice treated with SW IV‐134 was 88 days compared to 52 days of the control, p = 0.003.
Similar results were obtained in a xenograft model using AsPC‐1 cells in nude mice. Due to the limited activity of the parental compounds from our previous investigation, we only assessed the efficiency of SW IV‐134 compared with a single control. After tumors were established, the mice were treated daily with either 750 nmoles in 100 μL SW IV‐134 or vehicle. In this model, the drug conjugate markedly reduced the growth rate of the established tumors while under treatment and resulted in a long‐lasting delay in tumor progression with mean tumor diameters significantly smaller in the SW IV‐134‐tretaed group (Figure 5B, p < 0.0001). This treatment benefit culminated in a significant life extension of 36 days (median survival SW IV‐134 vs. vehicle, 88 vs. 52 days, respectively; Figure 5C, p = 0.0001). Of note, one mouse in the treatment group was completely cured and remains tumor free for 11 months.
We sought to correlate tumor shrinkage in vivo with an apoptotic cell death mechanism and therefore assessed activation of the executioner caspase‐3 following SW IV‐134 treatments. In a first experiment, C57BL/6 mice with established KCM tumors received i.p. injections with 750 nmoles in 100 μL of SW IV‐134 daily for 3 days while a control group received daily vehicle injections. Tumors were harvested, lysed and assessed for caspase‐3 activity. Cleaved caspase‐3 levels in the tumors treated with SW IV‐134 was nearly 2‐fold higher than that of the control group (Figure 6A, p < 0.0001). An additional experiment was designed to verify the caspase‐3 data, this time by immunohistochemistry utilizing TUNEL stain. Here, nude mice with established AsPC‐1 tumors received i.p. injections with 750 nmoles in 100 μL of SW IV‐134 daily for 3 days while a control group received daily vehicle injections. Tumors were harvested and prepared for immunohistochemistry. Mice treated with SW IV‐134 demonstrated a widespread signal development in the tumor with close to undetectable levels of apoptosis in the control group (Figures 6B and C). Both sets of experiments are consistent with the potent activity of SW IV‐134 in vivo.
Figure 6.
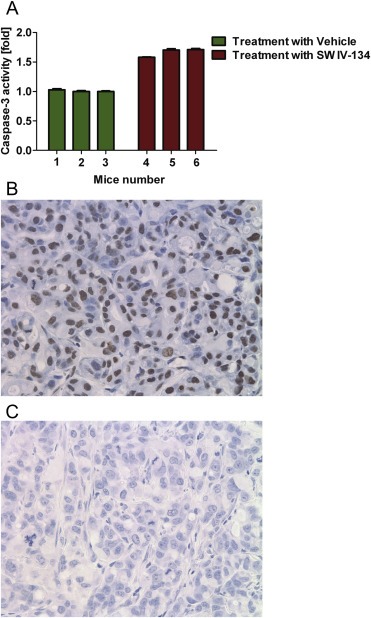
SW IV‐134 induces apoptosis in vivo. (A) C57BL/6 mice (n = 3 mice/group) with right flank KCM subcutaneous tumors received daily i.p. injections of vehicle (mouse 1–3) and 750 nmoles in 100 μL of SW IV‐134 (mouse 4–6) for 3 days. Caspase‐3 assay was performed on the tumors. Cleaved caspase‐3 level in the mice treated with SW IV‐134 was significantly higher than the control group, p < 0.0001(A). Athymic nude mice with right flank AsPC‐1 subcutaneous tumors were treated as mentioned above and tumors were evaluated for apoptosis induction via TUNEL staining. Tumors treated with the drug (B) had a remarkable number of TUNEL‐positive cells compared to the control (C), magnification = 40x.
4. Discussion
Currently available chemotherapeutics have limited efficacy in pancreatic cancer patients. There are many factors which contribute to the observed drug resistance. Unselective drug delivery and resistance to cell death through upregulation of prosurvival factors are most relevant to this discussion. Current chemotherapeutic drugs enter normal and cancer tissues with similar kinetics. As a result, dose limiting toxic effects on normal tissues often lead to changes in the chemotherapeutic plan including, dose reductions, switching to less effective drug combinations, and discontinuation of therapy. While normal cells are struggling to maintain homeostasis in this toxic environment, many pancreatic cancer cells have been shown to evade sensitivity to apoptosis by blocking caspase activation. They often do this by over‐expression of pro‐survival factors such as cIAP, XIAP, and survivin (Arlt et al., 2013). In this study we proposed to use sigma‐2 ligands as a way to deliver a drug selectively to cancers and decided to target this prosurvival advantage by introducing a Smac inhibitor as the delivery package.
The idea to use sigma‐2 ligands for drug delivery evolved from previous observations that several of these ligands localized to tumors and have been considered as potential imaging agents (Mach et al., 1997). We have previously shown that sigma‐2 receptors are overexpressed in human pancreatic adenocarcinoma (Kashiwagi et al., 2007). We have also demonstrated that sigma‐2 ligands can enter a pancreatic cancer cell and deliver additional drug cargos (Spitzer et al., 2012). These two findings provided the rationale for our efforts to develop sigma‐2 ligands as cancer selective drug delivery agents. We have also shown that selected sigma‐2 ligands at high doses have some ability to induce pancreatic cancer cell death. By choosing a small molecule that activates the proapoptotic pathway as our delivery package we hoped that we might find that sigma‐2 ligands ability to induce cell death might enhance this apoptotic effect in cancer cells. The mechanism by which sigma‐2 ligands, at high doses, induces cell death is not completely understood. We do know that sigma‐2 ligands induce apoptosis by caspase‐3 dependent and independent mechanisms (Kashiwagi et al., 2009; Ostenfeld et al., 2005; Wei et al., 2006). This was observed for the sigma‐2 ligands we utilized to develop SW IV‐134. For example we found that SW43 induced caspase‐3 activity in pancreatic cancers (Hornick et al., 2010). In addition we also found that SW43 induced cellular oxidative stress by increase lysosomal membrane permeabilization (Hornick et al., 2012).
Small molecules have been identified that sensitize pancreatic cancer to apoptosis including Smac mimetics (Dineen et al., 2010; Huang and Sinicrope, 2008). Smac was found to rapidly induce auto‐ubiquitination and degradation of cIAPs (Varfolomeev et al., 2007). This results in activation of NF‐κB pathway and TNFα production. TNFα mediates cytotoxicity by autocrine method through TNF‐R1 receptor signaling and caspase‐8 activation (Vince et al., 2007). The canonical NF‐κB pathway activation leads to IkBα ubiquitination and proteosomal degradation. This results in release and translocation of NF‐κB dimer (p50, 65) to the nucleus to induce transcription (Oeckinghaus and Ghosh, 2009). cIAP‐1 was reported to be associated with TRAF1 and TRAF2 through its N terminal BIR motif‐comprising domain (Rothe et al., 1995). cIAP‐1 has been shown to suppress caspase‐8 recruit to TNF‐R1 and cooperate with TRAF2 (Oeckinghaus and Ghosh, 2009). The noncanonical NF‐κB pathway depends on NIK which activates IKKα. This results in phosphorylation of the p100 NF‐κB subunit, leading to production of p52 of p52‐RelB dimmers that act as a transcriptional factor to induce apoptosis and cell death (Oeckinghaus and Ghosh, 2009).
We looked to see if these two pro‐apoptotic molecules would have additive or synergistic effects and were able to demonstrate a significant decrease in the IC50 of combination of SW43 and SW IV‐52s compared to these individual compounds. However the real benefits were observed only when we conjugated the sigma‐2 ligand SW43 to the Smac‐derived peptidomimetic (SW IV‐52) to create a tumor selective pro‐apoptotic anti‐cancer therapeutic, designated SW IV‐134. We believe that SW43 not only works as a carrier to selectively deliver SW IV‐52s, but both compounds work synergistically to induce cell death.
We tested several aspects of the dual domain therapeutic (SW IV‐134) with the goal of demonstrating that delivery of the drug was achieved and drug function was as expected. In competitive binding assays we found that binding affinity of SW IV‐134 to the sigma‐2 receptor was nearly unaltered compared with SW43. Smac frees up caspase‐9 in an active form (Oost et al., 2004). We therefore tested for these effects in our cell lines and animal models and found that SW IV‐134 increased caspase‐9 level 5.2 fold over the baseline cleavage. Subsequently we tested the efficacy of SW IV‐134 compared with the parent compounds. We found enhanced cell killing compared with either parent compound or the combination of the parent compounds suggesting that we were effectively delivering the Smac mimetic with the sigma‐2 ligand.
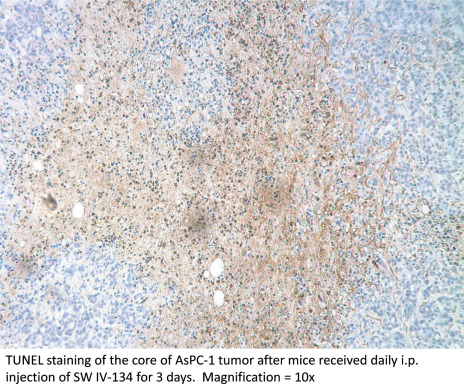
One challenge of chemotherapeutic delivery to pancreatic tumor is the poor blood supply. It has been reported that malignant pancreatic tumors have 60% decreased in blood flow compared to normal pancreatic tissue (Komar et al., 2009). In order to see if the selective delivery and high efficacy of SW IV‐134 can overcome this problem we tested our compound in murine models of pancreatic cancer. Immunohistochemical analysis of TUNEL staining showed that mice with tumor xenografts treated with SW IV‐134, demonstrated large areas of apoptosis throughout the tumors (supplementary data Fig. S2). SW IV‐134 showed promising results in both human (AsPC‐1) and mouse (KCM) pancreatic cell lines in vivo. A short course of the drug slowed tumor growth. We were able to measure significant differences between tumor size of the group treated with SW IV‐134 compared with controls (SW43, SW IV‐52 and SW43 + SW IV‐52). We observed no significant drug‐related toxicity with this therapy. Necropsy and laboratory evaluation demonstrated that SW IV‐134 had no major off target effects and mice tolerated the drug well without signs of weight loss. No mouse died as a result of treatment.
5. Conclusion
We advanced a novel tumor‐selective pro‐apoptotic anti‐cancer therapeutic (SW IV‐134) based on delivery of a Smac mimetic using a sigma‐2 ligand. This strategy was effective in treating pancreatic cancer in murine models and minimal toxicity was observed. While the model we utilized was pancreatic cancer the principles are likely applicable to additional cancer types. This dual‐domain (delivery‐therapy) concept represents an exciting opportunity for the development of additional small molecule therapeutics with dual functionality. Based on the observed mechanism of action we would expect that this drug will synergize with existing therapies (chemotherapy and/or radiotherapy). Taken together, we believe that this novel dual domain therapeutic has potential as a therapy for patients with pancreatic cancer and further investigation and investment is warranted.
Financial support
This work was funded by National Institute of Health R01 grant (US NIH 5R01CA16376402, #22‐004125‐52508) (W.G. Hawkins), the Mallinckrodt Institute of Radiology (R.H. Mach). Statistical support was provided by the Biostatistics Core, Siteman Comprehensive Cancer Center, and NCI Cancer Center Support Grant P30 CA091842. Salary support was provided for YH by the Washington University Surgical Oncology Training Grant (5T32CA00962124).
Disclosure
W.G. Hawkins and R.H. Mach have intellectual property rights and have patents related to the work. No additional conflicts or potential conflicts of interest were disclosed by the authors.
Grant support
This work was supported by National Institute of Health R01 grant (US NIH 5R01CA16376402, #22‐004125‐52508). Salary support was provided for YH by the Washington University Surgical Oncology Training Grant (5T32CA00962124). Statistical support was provided by the Biostatistics Core, Siteman Comprehensive Cancer Center, and NCI Cancer Center Support Grant P30 CA091842.
Supporting information
The following are the supplementary data related to this article:
Table S1 (A) SW IV‐134 does not induce changes in blood cytology (CBC) following treatment of tumor bearing C57BL/6 mice.
Table S1 (B) SW IV‐134 does not induce changes in serum chemistries following treatment of tumor bearing C57BL/6 mice.
Supplementary data
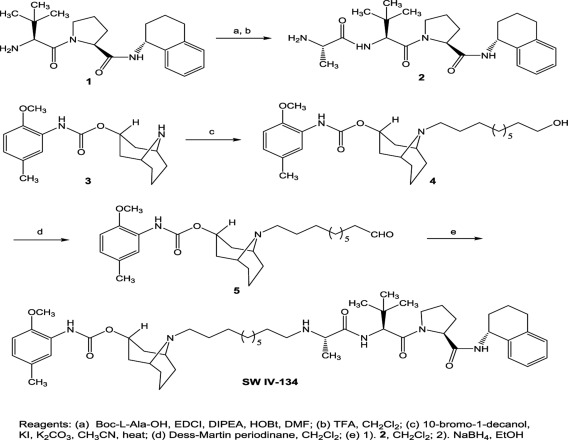
Figure S1 Schematic representation of the synthesis strategy of SW IV‐134.
{kind=link}
Figure S2 SW IV‐134 induces widespread apoptosis in pancreatic tumors in vivo.
{kind=link}
Acknowledgments
We acknowledge Dr. Pinku Mukherjee (University of North Carolina–Charlotte) for kindly providing (KCM) cell line. We also acknowledge Aixiao Lee (Washington University) for performing the sigma‐2 binding assays and Dr. Kimberly Trinkaus from the Division of Bio‐statistics (Washington University) for helping with statistical analysis.
Supplementary data 1.
Supplementary data related to this article can be found at http://dx.doi.org/10.1016/j.molonc.2014.03.005
Hashim Yassar M., Spitzer Dirk, Vangveravong Suwanna, Hornick Mary C., Garg Gunjal, Hornick John R., Goedegebuure Peter, Mach Robert H. and Hawkins William G., (2014), Targeted pancreatic cancer therapy with the small molecule drug conjugate SW IV‐134, Molecular Oncology, 8, doi: 10.1016/j.molonc.2014.03.005.
Contributor Information
Robert H. Mach, Email: rmach@mail.med.upenn.edu
William G. Hawkins, Email: hawkinsw@wustl.edu
References
- American Cancer Society, 2010. Cancer Facts & Figures 2010 American Cancer Society; Atlanta: [Google Scholar]
- Arlt, A. , Muerkoster, S.S. , Schafer, H. , 2013. Targeting apoptosis pathways in pancreatic cancer. Cancer lett.. 332, 346–358. [DOI] [PubMed] [Google Scholar]
- Besmer, D.M. , Curry, J.M. , Roy, L.D. , Tinder, T.L. , Sahraei, M. , Schettini, J. , Hwang, S.I. , Lee, Y.Y. , Gendler, S.J. , Mukherjee, P. , 2011. Pancreatic ductal adenocarcinoma mice lacking mucin 1 have a profound defect in tumor growth and metastasis. Cancer res.. 71, 4432–4442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dehdashti, F. , Laforest, R. , Gao, F. , Shoghi, K.I. , Aft, R.L. , Nussenbaum, B. , Kreisel, F.H. , Bartlett, N.L. , Cashen, A. , Wagner-Johnston, N. , Mach, R.H. , 2013. Assessment of cellular proliferation in tumors by PET using 18F-ISO-1. J. Nucl. Med. Official Publ. Soc. Nucl. Med.. 54, 350–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deveraux, Q.L. , Reed, J.C. , 1999. IAP family proteins–suppressors of apoptosis. Genes Dev.. 13, 239–252. [DOI] [PubMed] [Google Scholar]
- Deveraux, Q.L. , Takahashi, R. , Salvesen, G.S. , Reed, J.C. , 1997. X-linked IAP is a direct inhibitor of cell-death proteases. Nature. 388, 300–304. [DOI] [PubMed] [Google Scholar]
- Dineen, S.P. , Roland, C.L. , Greer, R. , Carbon, J.G. , Toombs, J.E. , Gupta, P. , Bardeesy, N. , Sun, H. , Williams, N. , Minna, J.D. , Brekken, R.A. , 2010. Smac mimetic increases chemotherapy response and improves survival in mice with pancreatic cancer. Cancer res.. 70, 2852–2861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du, C. , Fang, M. , Li, Y. , Li, L. , Wang, X. , 2000. Smac, a mitochondrial protein that promotes cytochrome c-dependent caspase activation by eliminating IAP inhibition. Cell. 102, 33–42. [DOI] [PubMed] [Google Scholar]
- Hornick, J.R. , Vangveravong, S. , Spitzer, D. , Abate, C. , Berardi, F. , Goedegebuure, P. , Mach, R.H. , Hawkins, W.G. , 2012. Lysosomal membrane permeabilization is an early event in Sigma-2 receptor ligand mediated cell death in pancreatic cancer. J. Exp. Clin. cancer Res. : CR. 31, 41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hornick, J.R. , Xu, J. , Vangveravong, S. , Tu, Z. , Mitchem, J.B. , Spitzer, D. , Goedegebuure, P. , Mach, R.H. , Hawkins, W.G. , 2010. The novel sigma-2 receptor ligand SW43 stabilizes pancreas cancer progression in combination with gemcitabine. Mol. cancer. 9, 298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, S. , Sinicrope, F.A. , 2008. BH3 mimetic ABT-737 potentiates TRAIL-mediated apoptotic signaling by unsequestering Bim and Bak in human pancreatic cancer cells. Cancer res.. 68, 2944–2951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kashiwagi, H. , McDunn, J.E. , Simon, P.O. , Goedegebuure, P.S. , Vangveravong, S. , Chang, K. , Hotchkiss, R.S. , Mach, R.H. , Hawkins, W.G. , 2009. Sigma-2 receptor ligands potentiate conventional chemotherapies and improve survival in models of pancreatic adenocarcinoma. J. Translational Med.. 7, 24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kashiwagi, H. , McDunn, J.E. , Simon, P.O. , Goedegebuure, P.S. , Xu, J. , Jones, L. , Chang, K. , Johnston, F. , Trinkaus, K. , Hotchkiss, R.S. , Mach, R.H. , Hawkins, W.G. , 2007. Selective sigma-2 ligands preferentially bind to pancreatic adenocarcinomas: applications in diagnostic imaging and therapy. Mol. cancer. 6, 48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komar, G. , Kauhanen, S. , Liukko, K. , Seppanen, M. , Kajander, S. , Ovaska, J. , Nuutila, P. , Minn, H. , 2009. Decreased blood flow with increased metabolic activity: a novel sign of pancreatic tumor aggressiveness. Clin. cancer Res. Official J. Am. Assoc. Cancer Res.. 15, 5511–5517. [DOI] [PubMed] [Google Scholar]
- Li, L. , Thomas, R.M. , Suzuki, H. , De Brabander, J.K. , Wang, X. , Harran, P.G. , 2004. A small molecule Smac mimic potentiates TRAIL- and TNFalpha-mediated cell death. Science N. Y.. 305, 1471–1474. [DOI] [PubMed] [Google Scholar]
- Mach, R.H. , Smith, C.R. , al-Nabulsi, I. , Whirrett, B.R. , Childers, S.R. , Wheeler, K.T. , 1997. Sigma 2 receptors as potential biomarkers of proliferation in breast cancer. Cancer Res.. 57, 156–161. [PubMed] [Google Scholar]
- Martin, W.R. , Eades, C.G. , Thompson, J.A. , Huppler, R.E. , Gilbert, P.E. , 1976. The effects of morphine- and nalorphine-like drugs in the nondependent and morphine-dependent chronic spinal dog. J. Pharmacol. Exp. Ther.. 197, 517–532. [PubMed] [Google Scholar]
- Oeckinghaus, A. , Ghosh, S. , 2009. The NF-kappaB family of transcription factors and its regulation. Cold Spring Harbor Perspect. Biol.. 1, a000034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogawa, K. , Shiba, K. , Akhter, N. , Yoshimoto, M. , Washiyama, K. , Kinuya, S. , Kawai, K. , Mori, H. , 2009. Evaluation of radioiodinated vesamicol analogs for sigma receptor imaging in tumor and radionuclide receptor therapy. Cancer Sci.. 100, 2188–2192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oost, T.K. , Sun, C. , Armstrong, R.C. , Al-Assaad, A.S. , Betz, S.F. , Deckwerth, T.L. , Ding, H. , Elmore, S.W. , Meadows, R.P. , Olejniczak, E.T. , Oleksijew, A. , Oltersdorf, T. , Rosenberg, S.H. , Shoemaker, A.R. , Tomaselli, K.J. , Zou, H. , Fesik, S.W. , 2004. Discovery of potent antagonists of the antiapoptotic protein XIAP for the treatment of cancer. J. Med. Chem.. 47, 4417–4426. [DOI] [PubMed] [Google Scholar]
- Ostenfeld, M.S. , Fehrenbacher, N. , Hoyer-Hansen, M. , Thomsen, C. , Farkas, T. , Jaattela, M. , 2005. Effective tumor cell death by sigma-2 receptor ligand siramesine involves lysosomal leakage and oxidative stress. Cancer res.. 65, 8975–8983. [DOI] [PubMed] [Google Scholar]
- Peng, Y. , Sun, H. , Nikolovska-Coleska, Z. , Qiu, S. , Yang, C.Y. , Lu, J. , Cai, Q. , Yi, H. , Kang, S. , Yang, D. , Wang, S. , 2008. Potent, orally bioavailable diazabicyclic small-molecule mimetics of second mitochondria-derived activator of caspases. J. Med. Chem.. 51, 8158–8162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quirion, R. , Bowen, W.D. , Itzhak, Y. , Junien, J.L. , Musacchio, J.M. , Rothman, R.B. , Su, T.P. , Tam, S.W. , Taylor, D.P. , 1992. A proposal for the classification of sigma binding sites. Trends Pharmacol. Sci.. 13, 85–86. [DOI] [PubMed] [Google Scholar]
- Rothe, M. , Pan, M.G. , Henzel, W.J. , Ayres, T.M. , Goeddel, D.V. , 1995. The TNFR2-TRAF signaling complex contains two novel proteins related to baculoviral inhibitor of apoptosis proteins. Cell. 83, 1243–1252. [DOI] [PubMed] [Google Scholar]
- Shiozaki, E.N. , Chai, J. , Rigotti, D.J. , Riedl, S.J. , Li, P. , Srinivasula, S.M. , Alnemri, E.S. , Fairman, R. , Shi, Y. , 2003. Mechanism of XIAP-mediated inhibition of caspase-9. Mol. Cell. 11, 519–527. [DOI] [PubMed] [Google Scholar]
- Siegel, R. , Naishadham, D. , Jemal, A. , 2012. Cancer statistics, 2012. CA Cancer J. Clinic.. 62, 10–29. [DOI] [PubMed] [Google Scholar]
- Spitzer, D. , Simon, P.O. , Kashiwagi, H. , Xu, J. , Zeng, C. , Vangveravong, S. , Zhou, D. , Chang, K. , McDunn, J.E. , Hornick, J.R. , Goedegebuure, P. , Hotchkiss, R.S. , Mach, R.H. , Hawkins, W.G. , 2012. Use of multifunctional sigma-2 receptor ligand conjugates to trigger cancer-selective cell death signaling. Cancer Res.. 72, 201–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su, T.P. , 1993. Delineating biochemical and functional properties of sigma receptors: emerging concepts. Crit. Rev. Neurobiol.. 7, 187–203. [PubMed] [Google Scholar]
- Sun, H. , Nikolovska-Coleska, Z. , Yang, C.Y. , Qian, D. , Lu, J. , Qiu, S. , Bai, L. , Peng, Y. , Cai, Q. , Wang, S. , 2008. Design of small-molecule peptidic and nonpeptidic Smac mimetics. Acc. Chem. Res.. 41, 1264–1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun, H. , Stuckey, J.A. , Nikolovska-Coleska, Z. , Qin, D. , Meagher, J.L. , Qiu, S. , Lu, J. , Yang, C.Y. , Saito, N.G. , Wang, S. , 2008. Structure-based design, synthesis, evaluation, and crystallographic studies of conformationally constrained Smac mimetics as inhibitors of the X-linked inhibitor of apoptosis protein (XIAP). J. Med. Chem.. 51, 7169–7180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tinder, T.L. , Subramani, D.B. , Basu, G.D. , Bradley, J.M. , Schettini, J. , Million, A. , Skaar, T. , Mukherjee, P. , 2008. MUC1 enhances tumor progression and contributes toward immunosuppression in a mouse model of spontaneous pancreatic adenocarcinoma. J. Immunol. Baltimore, Md. 1950. 181, 3116–3125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vangveravong, S. , Xu, J. , Zeng, C. , Mach, R.H. , 2006. Synthesis of N-substituted 9-azabicyclo[3.3.1]nonan-3alpha-yl carbamate analogs as sigma2 receptor ligands. Bioorg. Med. Chem.. 14, 6988–6997. [DOI] [PubMed] [Google Scholar]
- Varfolomeev, E. , Blankenship, J.W. , Wayson, S.M. , Fedorova, A.V. , Kayagaki, N. , Garg, P. , Zobel, K. , Dynek, J.N. , Elliott, L.O. , Wallweber, H.J. , Flygare, J.A. , Fairbrother, W.J. , Deshayes, K. , Dixit, V.M. , Vucic, D. , 2007. IAP antagonists induce autoubiquitination of c-IAPs, NF-kappaB activation, and TNFalpha-dependent apoptosis. Cell. 131, 669–681. [DOI] [PubMed] [Google Scholar]
- Verhagen, A.M. , Ekert, P.G. , Pakusch, M. , Silke, J. , Connolly, L.M. , Reid, G.E. , Moritz, R.L. , Simpson, R.J. , Vaux, D.L. , 2000. Identification of DIABLO, a mammalian protein that promotes apoptosis by binding to and antagonizing IAP proteins. Cell. 102, 43–53. [DOI] [PubMed] [Google Scholar]
- Vince, J.E. , Wong, W.W. , Khan, N. , Feltham, R. , Chau, D. , Ahmed, A.U. , Benetatos, C.A. , Chunduru, S.K. , Condon, S.M. , McKinlay, M. , Brink, R. , Leverkus, M. , Tergaonkar, V. , Schneider, P. , Callus, B.A. , Koentgen, F. , Vaux, D.L. , Silke, J. , 2007. IAP antagonists target cIAP1 to induce TNFalpha-dependent apoptosis. Cell. 131, 682–693. [DOI] [PubMed] [Google Scholar]
- Walker, J.M. , Bowen, W.D. , Walker, F.O. , Matsumoto, R.R. , De Costa, B. , Rice, K.C. , 1990. Sigma receptors: biology and function. Pharmacol. Rev.. 42, 355–402. [PubMed] [Google Scholar]
- Wei, Z. , Mousseau, D.D. , Dai, Y. , Cao, X. , Li, X.M. , 2006. Haloperidol induces apoptosis via the sigma2 receptor system and Bcl-XS. Pharmacogen. J.. 6, 279–288. [DOI] [PubMed] [Google Scholar]
- Wu, H. , Tschopp, J. , Lin, S.C. , 2007. Smac mimetics and TNFalpha: a dangerous liaison?. Cell. 131, 655–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, J. , Zeng, C. , Chu, W. , Pan, F. , Rothfuss, J.M. , Zhang, F. , Tu, Z. , Zhou, D. , Zeng, D. , Vangveravong, S. , Johnston, F. , Spitzer, D. , Chang, K.C. , Hotchkiss, R.S. , Hawkins, W.G. , Wheeler, K.T. , Mach, R.H. , 2011. Identification of the PGRMC1 protein complex as the putative sigma-2 receptor binding site. Nature Commun.. 2, 380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng, C. , Vangveravong, S. , McDunn, J.E. , Hawkins, W.G. , Mach, R.H. , 2013. Sigma-2 receptor ligand as a novel method for delivering a SMAC mimetic drug for treating ovarian cancer. Br. J. Cancer. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng, C. , Vangveravong, S. , Xu, J. , Chang, K.C. , Hotchkiss, R.S. , Wheeler, K.T. , Shen, D. , Zhuang, Z.P. , Kung, H.F. , Mach, R.H. , 2007. Subcellular localization of sigma-2 receptors in breast cancer cells using two-photon and confocal microscopy. Cancer Res.. 67, 6708–6716. [DOI] [PubMed] [Google Scholar]
- Zobel, K. , Wang, L. , Varfolomeev, E. , Franklin, M.C. , Elliott, L.O. , Wallweber, H.J. , Okawa, D.C. , Flygare, J.A. , Vucic, D. , Fairbrother, W.J. , Deshayes, K. , 2006. Design, synthesis, and biological activity of a potent Smac mimetic that sensitizes cancer cells to apoptosis by antagonizing IAPs. ACS Chem. Biol.. 1, 525–533. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The following are the supplementary data related to this article:
Table S1 (A) SW IV‐134 does not induce changes in blood cytology (CBC) following treatment of tumor bearing C57BL/6 mice.
Table S1 (B) SW IV‐134 does not induce changes in serum chemistries following treatment of tumor bearing C57BL/6 mice.
Supplementary data
Figure S1 Schematic representation of the synthesis strategy of SW IV‐134.
Figure S2 SW IV‐134 induces widespread apoptosis in pancreatic tumors in vivo.