

Abstract
Studies have found an association between aberrant DNA methylation and arsenic-induced skin lesions. Yet, little is known about DNA methylation changes over time in people who develop arsenic-induced skin lesions. We sought to investigate epigenome-wide changes of DNA methylation in people who developed arsenic-induced skin lesions in a ten year period. In 2009–2011, we conducted a follow-up study of 900 skin lesion cases and 900 controls and identified 10 people who developed skin lesions since a baseline survey in 2001–2003. The 10 cases (“New Cases”) were matched with 10 controls who did not have skin lesions at baseline or follow-up (“Persistent Controls”). Drinking water and blood samples were collected and skin lesion was diagnosed by the same physician at both time points. We measured DNA methylation in blood using Infinium HumanMethylation450K BeadChip, followed by quantitative validation using pyrosequencing. Two-sample t-tests were used to compare changes in percent methylation between New Cases and Persistent Controls. Six CpG sites with greatest changes of DNA methylation over time among New Cases were further validated with a correlation of 93% using pyrosequencing. One of the validated CpG site (cg03333116; change of %methylation was 13.2 in New Cases versus −0.09 in Persistent Controls; P <0.001) belonged to the RHBDF1 gene, which was previously reported to be hypermethylated in arsenic-exposed cases. We examined DNA methylation changes with the development of arsenic-induced skin lesions over time but nothing was statistically significant given the small sample size of this exploratory study and the high dimensionality of data.
Keywords: Arsenic, DNA methylation, Illumina 450K, longitudinal, skin lesion
INTRODUCTION
DNA methylation silences tumor suppressor genes [Baylin, 2005]. Alterations in DNA methylation may be crucial in the pathogenesis of many skin diseases, such as basal cell carcinoma and malignant melanoma [Millington, 2008]. In the past few years several environmental chemicals have been shown to produce DNA methylation alterations [Hou et al., 2012]. Of these chemicals, arsenic has consistently demonstrated epigenetic mechanisms of actions including the ability to alter DNA methylation [Baccarelli and Bollati, 2009]. Both in vitro and in vivo experiments have shown that arsenic exposure can induce global DNA hypomethylation, as well as gene-specific hypomethylation and hypermethylation [Kile et al., 2012; Ren et al., 2011; Reichard and Puga, 2010; Sciandrello et al., 2004]. Numerous studies have shown associations between global hypomethylation with both reduced chromosome stability and altered genome function [Slotkin and Martienssen, 2007; Schulz, 2006]. There is evidence that arsenic can elicit adverse health effects in humans such as skin lesions via DNA hypomethylation [Pilsner et al., 2009].
Millions of people globally are exposed to arsenic through naturally contaminated drinking water. Bangladesh is one of the most severely affected countries, where people are highly exposed to arsenic by drinking arsenic-contaminated water from tubewells [Chowdhury et al., 2000]. The most well-characterized and first observable symptom of chronic arsenic exposure are skin lesions, which are also known to be highly correlated with skin cancers, especially basal cell carcinoma (BCC), squamous cell carcinoma (SCC) and Bowen’s disease [Centeno et al., 2002; Tseng et al., 1968]. It was estimated that at least 100,000 people have developed skin lesions caused by arsenic poisoning in Bangladesh [Smith et al., 2000]. DNA methylation could play a role in the association between arsenic exposure and skin lesions, and the eventual development of arsenic-related skin cancers.
We sought to identify differential methylation of genes that could illuminate the biological mechanisms and pathways of arsenic toxicity using epigenome-wide scans. Until now, there has only been one genome-wide study done on DNA methylation in arsenic-exposed skin lesion cases. Smeester et al. performed a cross-sectional, genome-wide site-specific DNA methylation in lymphocyte DNA of 8 female skin lesion cases and 8 female controls using the Affymetrix Human Promoter 1.0R arrays, and found 183 genes with differential patterns, of which 182 were hypermethylated in individuals with skin lesions [Smeester et al., 2011]. Many of the genes were involved in arsenic-associated diseases, such as heart disease, diabetes, and cancer.
However, DNA methylation is a dynamic process that can be modified by many factors including aging, environmental and dietary exposures [Cantone and Fisher, 2013; Feil, 2006; Fraga et al., 2005]. No studies have used epigenome-wide methods for DNA methylation analysis to screen for alterations associated with arsenic exposure with the development of arsenic-induced skin lesions over time. Therefore, we conducted a prospective study to further investigate DNA methylation changes that are associated with arsenic-associated skin lesions.
To achieve this goal, we conducted an exploratory study in Bangladesh based on a case-control follow-up study of skin lesions over a period of 10 years to evaluate epigenome-wide DNA methylation changes among individuals who were initially without skin lesions at the baseline study and developed skin lesions at follow-up (“New Cases”), and compare their methylation changes with matched individuals who remained as controls at both baseline and follow-up (“Persistent Controls”). We first measured blood DNA methylation using the Illumina Infinium HumanMethylation 450K BeadChip, which allows us to measure simultaneously the methylation levels of 485,577 CpG (cytosine-phosphate-guanine) sites. We then validated CpG sites that showed the greatest change in methylation using the highly quantitative pyrosequencing method. Our objective was to provide epigenetic insights on the development of arsenic-induced skin lesions that may eventually progress to the skin cancers. Since it may be possible to modify epigenetics through diet, this work may lead to the development of more effective prevention methods and intervention strategies in order to protect the susceptible population from developing future skin cancers.
MATERIALS AND METHODS
Study population
In 2001–2003, we recruited 1800 participants (900 cases of skin lesions and 900 controls) to examine genetic susceptibility to arsenic-induced skin lesions, as previously described by Breton et al [Breton et al., 2006]. Cases were defined as participants who have at least one type of skin lesions. Controls were age- and sex-matched healthy individuals living in the same general community as the cases. In 2009–2011, we re-contacted 1542 participants and 957 participants agreed to participate enroll in a follow up study [Seow et al., 2012]. From this populations, we identified a total of 10 people who did not have skin lesions in 2001–2003 at baseline but were diagnosed with skin lesions at follow-up (“New Cases”) and 397 people who did not have skin lesions at baseline or at follow-up (“Persistent Controls”). For this exploratory study, we included all 10 New Cases and matched them to 10 Persistent Controls. Using data collected at baseline, controls were frequency-matched on age (continuous), sex (dichotomous), smoking status (dichotomous), chewing betel nut (dichotomous) and body mass index (BMI) (continuous).
Questionnaires and interviews
Trained interviewers administered questionnaires to collect socio-demographic information, drinking water history, medical history, lifestyle factors, dietary information, water consumption (liters of water/liquid ingested per day), residential history. To minimize the potential for bias, interviewers in the follow-up study were blinded to the participants’ disease status and arsenic exposure at baseline.
Arsenic exposure assessment
We collected 45ml of water sample from each participant’s primary drinking source in a 50ml falcon tube (BD Biosciences, Franklin Lakes, NJ, USA) and added one drop (0.1 ml) of pure trace metal grade nitric acid to preserve the samples for trace metal analysis. Samples were stored at room temperature prior to analysis. Analysis of each sample for arsenic concentration was completed using Environmental Protection Agency (EPA) method 200.8 with Inductively Coupled Plasma Mass Spectroscopy (ICP-MS) (Environmental Laboratory Services, North Syracuse, New York, USA). For quality control, instrument performance was validated using repeated measurements of standard reference water (PlasmaCAL multi-element QC standard #1 solution, SCP Science, Champlain, Canada) with an average percentage recovery of 95%. Ten percent of the samples were randomly selected and analyzed in duplicate to confirm reliability. The average percentage difference between duplicates was 2.5%. The limit of detection (LOD) was 1μg As/L. Samples with below the LOD were assigned a value of 0.5μg As/L.
Skin lesion assessment
The same physician examined all participants for skin lesions at baseline and follow-up. The physician was blinded to their exposure levels. Cases were defined as participants who had at least one type of arsenic-induced skin lesion (melanosis, keratosis, hyperkeratosis or leukomelanosis). Melanosis (yes/no) was defined as any diffuse or spotted lesion characterized by dark pigmentation on the face, oral cavity, neck, upper and lower limbs, chest or back. Keratosis (yes/no) was defined as any diffuse or spotted lesion characterized by hard and roughened skin elevations observed on the palm or dorsum of the hands and/or the sole or plantar of the foot. Hyperkeratosis (yes/no) was defined as extensively thickened keratosis easily visible from a distance. Leukomelanosis (yes/no) was defined as depigmentation characterized by black and white spots present anywhere on the body.
Assessment for genome-wide DNA methylation levels
Peripheral blood samples were collected from participants both at baseline and follow-up. Skin biopsies were not available at both time points. DNA was extracted from peripheral blood samples before being bisulfite-treated with EZ-DNA Methylation Kit (Zymo Research, Orange, CA, USA) according to the manufacturer’s protocol and running them on the Illumina’s Infinium HumanMethylation450K BeadChip (Illumina, San Diego, CA, USA). A total of 40 DNA samples were used in this analysis that were collected at baseline and follow up, from the 10 New Cases (20 samples) and 10 matched Persistent Controls (20 samples). Image processing and methylation intensity data extraction were performed using Illumina’s GenomeStudio Methylation module version 1.0 (Illumina, San Diego, CA, USA). Methylation beta values were defined as the ratio of methylated intensity to the total intensities (methylated and unmethylated), using M/(M+U+100), where M is the signal from the probe corresponding to the methylated target CpG, U is the signal from the probe corresponding to the unmethylated target, and 100 was added to protect against division-by-zero. The average beta value ranged between 0 (completely unmethylated) and 1 (completely methylated) and is interpreted as the fraction of DNA molecules whose target CpG is methylated [Du et al., 2010]. We used these beta values in all our analyses and reported it as %methylation, which was expressed as the percentage of 5-methylated cytosines (%5mC) in total (methylated and unmethylated) cytosines. The average number of detected loci across all samples was 98.8%.
Subset quantile normalization
The Tost pipeline was used to normalize the raw beta values [Touleimat and Tost, 2012]. The pipeline consists of four main steps and includes adjusting for color bias, background correction and sample normalization. Briefly, the first QC step involves filtering out poorly detected samples with detection P > 0.01, followed by probe filtering by removing genetic variations (CpG sites associated with SNPs of MAF 5% based on 1000 Genomes Project [www.1000genomes.org/]) and allosomal positions, and signal correction to adjust for color bias and background subtraction, before performing subset-based quantile normalization within different CpG locations and probe type. We removed from the analyses 26,674 SNP probes, 32,476 CpG sites with a detection value of more than 0.01, 11,646 CpG sites on sex chromosomes and 45,717 sites with missing values, leaving a total of 369,064 CpG sites for analysis (Figure S1).
Pyrosequencing
To validate methylation results from the Infinium HumanMethylation450K BeadChip, we used pyrosequencing. We selected 6 CpG sites for pyrosequencing because they showed the largest %methylation change (absolute change > 10% and P < 0.05) between baseline and follow-up in New Cases [Yuen et al., 2010]. To obtain bisulfite converted DNA, one μg of genomic DNA (concentration 50 ng/μL) was treated using the EZ DNA Methylation-Gold Kit (Zymo Research, Orange, CA, USA) according to the manufacturer’s protocol. The bisulfite-treated DNA was first amplified using bisulfite-PCR and PCR products were purified and sequenced by pyrosequencing using methods as previously described [Bollati et al., 2007]. Primers for each assay are listed in Table S1. We used built-in controls to verify bisulfite conversion efficiency for all assays. In each assay, we measured the percentage of %5mC. Duplicate runs were done for each assay to ensure reproducibility of results and the average R-squared value for each CpG site is 0.93 (Table S2).
Statistical analysis
Drinking water arsenic levels between New Cases and Persistent Controls were compared using the Wilcoxon rank sum test. Batch effects across different chips were adjusted for using the ComBat function in Bioconductor’s ‘sva’ package [Johnson et al., 2007; Leek et al., 2012]. Density plots comparing chip effect and P-values before and after adjustment with ComBat showed that batch effects by chip number has been substantially removed (Figure S2). Arsenic-induced skin lesions were evaluated as a dichotomous variable (i.e. presence or absence of at least one type of skin lesions). Since the New Cases and Persistent Controls were frequency-matched on age, sex, BMI, smoking status and chewing betel nuts at baseline, these predictors of DNA methylation were very similar between the two groups and were excluded from the analyses.
We calculated a percent difference in methylation between baseline and follow-up at each CpG site for each participant. We then used two-sample t-tests to compare the mean % difference in methylation at each CpG site between New Cases and Persistent Controls. CpG sites with P <0.05 were then sorted in descending order according to their absolute %methylation difference to identify CpG sites that had the greatest change in %methylation in New Cases. The same analysis was performed for the top CpG sites using the pyrosequencing results. Spearman correlation coefficient was calculated to compare the two platforms.
All statistical analyses were conducted using R version 2.13.1 and Bioconductor ‘sva’ packages. All tests were conducted as two-sided and were considered significant with a P-value of less than 0.05. We used the false discovery rate (FDR) using Benjamini-Hochberg method [Benjamini and Hochberg, 1995] to correct for multiple testing and to control for overall type I error of 10%.
RESULTS
Demographics of New Cases and Persistent Controls
We found significantly higher baseline water arsenic levels in New Cases (12.2 ± 437 μg/L) as compared to the Persistent Controls (2.15 ± 7.2 μg/L, P < 0.001) (Table 1). A similar trend was seen for the arsenic levels at follow-up, with the New Cases having significantly higher water arsenic levels than the Persistent Controls (21 ± 125 μg/L New Cases versus 9.15 ± 19.9 μg/L Persistent Controls, P < 0.001).
Table 1.
Characteristics of New Cases (N=10) and matched Persistent Controls (N=10) at baseline (2001–2003) in Pabna, Bangladesh
| Characteristica | New Cases (N=10) |
Persistent Controls (N=10) |
P-valueb | |
|---|---|---|---|---|
| Sex | ||||
| Male | 7 (70%) | 7 (70%) | – | |
| Female | 3 (30%) | 3 (30%) | ||
| Age, years | 37.4 ± 10.2 | 37.0 ± 9.8 | – | |
| Smoking Status | ||||
| Ever | 3 (30%) | 3 (30%) | – | |
| Never | 7 (70%) | 7 (70%) | ||
| Chew Betel Nuts | ||||
| Yes | 5 (50%) | 4 (40%) | – | |
| No | 5 (50%) | 6 (60%) | ||
| BMI, kg/m2 | 19.5 ± 2.0 | 20.5 ± 2.3 | – | |
| Baseline water arsenic, μg/L | 12.2 ± 437 | 2.15 ± 7.2 | < 0.001 | |
| Follow-up water arsenic, μg/L | 21.0 ± 125 | 9.15 ± 19.9 | < 0.001 | |
Data are shown as mean ± standard deviation (SD) for age and body mass index (BMI), median ± interquatile range (IQR) for water arsenic, and n (%) for categorical variables (sex, smoking status, and chew betel nuts).
P-values comparing New Cases and Persistent Controls obtained from Wilcoxon rank sum test with continuity correction for water arsenic. Comparisons were not performed for matching factors (sex, age, smoking status, chew betel nuts, and BMI) between New Cases and Persistent Controls.
Top CpG sites with largest %methylation difference in New Cases
The top 20 CpG sites with the greatest %methylation changes between New Cases and Persistent Controls are shown in Table 2. While these CpG sites had a minimum of 11% absolute change in methylation levels between baseline and follow-up, none remained significant when accounting for multiple comparisons. This is likely due to our small sample size (N=20). Of the 20 top CpG sites, 13 CpGs in New Cases (TCEB3B, CYC1, CDH4, RHBDF1, CCDC154, JAKMIP3, AGAP2, PL-5283, CHPF, PPAP2C, PCNT, SLC6A3 and MAP3K1) increased in %methylation, and 7 CpGs (MYO3B, KIAA1683, LOC642597, C2orf81, ESRRG, PRDM9 and TNXB) decreased in %methylation between baseline and follow-up. For 14 out of the top 20 CpG sites, the %methylation differences between baseline and follow-up within New Cases were in opposite directions compared to the Persistent Controls. For example, methylation at the cg24883732 (TCEB3B) probe increased by 17% in the New Cases but decreased by 5.4% in Persistent Controls (P = 0.02). Boxplots of %methylation changes between baseline and follow-up for the top 6 CpG sites are shown in Figure 1. We also used M-values in the analyses and found no significant differences in the results (Table S3).
Table 2.
Top 20 CpG sites with the greatest differential %methylation changes in New Cases compared to Persistent Controls
| CpG sitea | Gene | Chr | Gene region | Location | New Cases %methylation
|
Persistent Controls %methylation
|
P-valuec | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Baseline | Follow-up | Changeb | Baseline | Follow-up | Changeb | ||||||
| cg24883732 | TCEB3B | 18 | 5′UTR | Island | 46 | 63 | 17 | 71 | 66 | −5.4 | 0.02 |
| cg15059639 | MYO3B | 2 | Body | – | 72 | 57 | −16 | 56 | 64 | 8.1 | 0.004 |
| cg03065888 | CYC1 | 8 | TSS1500 | N_Shore | 43 | 58 | 15 | 51 | 47 | −4.2 | 0.004 |
| cg19296405 | KIAA1683 | 19 | Body | Island | 80 | 66 | −14 | 76 | 77 | 1.2 | 0.004 |
| cg00218770 | CDH4 | 20 | Body | N_Shore | 68 | 82 | 13 | 78 | 76 | −2.5 | 0.02 |
| cg03333116 | RHBDF1 | 16 | Body | Island | 69 | 82 | 13 | 83 | 76 | −6.9 | < 0.001 |
| cg06352616 | CCDC154 | 16 | Body | Island | 33 | 45 | 12 | 46 | 43 | −3.8 | 0.05 |
| cg03371700 | JAKMIP3 | 10 | Body | Island | 65 | 78 | 12 | 78 | 79 | 0.8 | 0.03 |
| cg11511175 | AGAP2 | 12 | 3′UTR | Island | 54 | 66 | 12 | 54 | 49 | −5.5 | 0.033 |
| cg17900076 | LOC642597 | 18 | Body | N_Shore | 61 | 49 | −12 | 55 | 62 | 7.5 | 0.009 |
| cg24405174 | PL-5283 | 7 | TSS1500 | N_Shore | 39 | 51 | 12 | 38 | 40 | 1.4 | 0.045 |
| cg00028056 | CHPF | 2 | Body | N_Shore | 68 | 80 | 12 | 73 | 68 | −5.2 | 0.004 |
| cg02916425 | C2orf81 | 2 | TSS1500 | Island | 82 | 70 | −12 | 74 | 78 | 3.8 | 0.03 |
| cg10090568 | ESRRG | 1 | 5′UTR | – | 60 | 49 | −12 | 57 | 59 | 2.0 | 0.002 |
| cg27370573 | PPAP2C | 19 | TSS1500 | Island | 69 | 80 | 12 | 64 | 64 | 0.5 | 0.047 |
| cg22217449 | PCNT | 21 | Body | Island | 39 | 50 | 12 | 45 | 37 | −7.2 | 0.024 |
| cg07664579 | SLC6A3 | 5 | Body | S_Shelf | 45 | 56 | 12 | 59 | 60 | 0.5 | 0.025 |
| cg01667892 | PRDM9 | 5 | TSS200 | – | 77 | 65 | −11 | 71 | 71 | −0.6 | 0.029 |
| cg25148456 | MAP3K1 | 5 | TSS1500 | N_Shore | 64 | 75 | 11 | 72 | 73 | 0.7 | 0.028 |
| cg02749948 | TNXB | 6 | 5′UTR | – | 86 | 75 | −11 | 79 | 82 | 2.4 | 0.038 |
Abbreviations: 5′UTR, 5′-Untranslated region; TSS1500, 1500 bp upstream of transcription start site; TSS200, 200 bp upstream of transcription site; N_Shore, North shore which are regions flanking island; S_Shelf, South shelf which are regions flanking islands; Chr, Chromosome
Data were sorted by decreasing absolute methylation difference between baseline and follow-up in New Cases.
Changes of %methylation were calculated as %methylation at follow-up (2009–2011) subtracted by %methylation at baseline (2001–2003).
P-values were obtained from two-sample t-tests and none of them passed multiple comparisons at 10% FDR.
Figure 1.
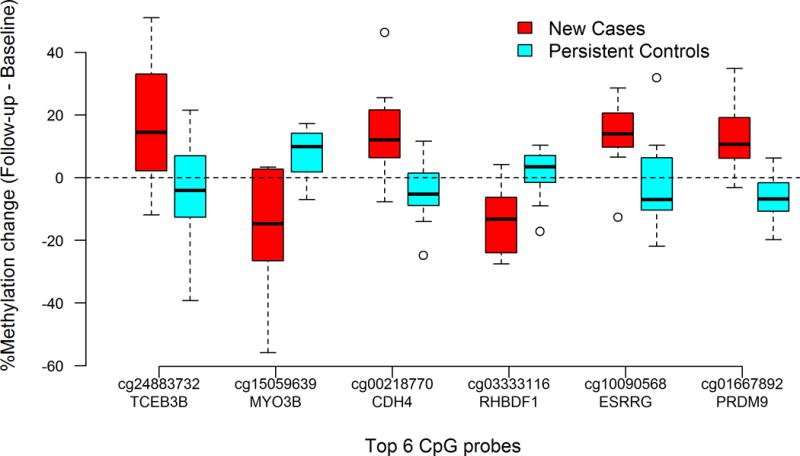
Boxplots of %methylation change between baseline and follow-up for top 6 CpG sites in New Cases (red) and Persistent Controls (blue)
Boxplots of %methylation change between baseline and follow-up were plotted for the top 6 CpG sites (on genes TCEB3B, MYO3B, CDH4, RHBDF1, ESRRG, and PRDM9) that are significantly different between New Cases and Persistent Controls. Red denotes New Cases and blue denotes Persistent Controls.
Validating quantitative capacity of Infinium 450K Beadchip
Table 3 shows the pyrosequencing results for the 6 CpG sites that showed the largest %methylation differences in New Cases. The CpG sites were located on genes TCEB3B, MYO3B, CDH4, RHBDF1, ESRRG and PRDM9. The correlation coefficients ranged from 0.39 to 0.90 for the methylation levels between the Infinium HumanMethylation450K BeadChip and pyrosequencing across all 6 CpG sites. The highest correlation was seen for CpG sites on genes PRDM9 (90% correlation), MYO3B (85% correlation) and ESRRG (64% correlation). Three genes PRDM9 (7.7% change in New Cases versus −0.05% change in Persistent Controls, P < 0.001)¸ RHBDF1 (13.2% change in New Cases versus −0.09% change in Persistent Controls, P < 0.001) and CDH4 (9.1% change in New Cases versus −0.6% change in Persistent Controls, P < 0.001) were found to have significantly different %methylation change in New Cases, as compared to Persistent Controls.
Table 3.
Pyrosequencing results of 6 CpG sites and comparison with Infinium HumanMapping450K Beadchip, in order of decreasing correlation
| CpG sitea | Gene | Probe type |
rhob | New Cases %methylation
|
Persistent Controls %methylation
|
P-valued | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Baseline (PSQ) |
Baseline (450K) |
Follow-up (PSQ) |
Follow-up (450K) |
Changec (PSQ) |
Baseline (PSQ) |
Baseline (450K) |
Follow-up (PSQ) |
Follow-up (450K) |
Changec (PSQ) |
|||||
| cg01667892 | PRDM9 | 2 | 0.90 | 75.8 | 76.7 | 68.2 | 65.3 | −7.6 | 72.9 | 71.5 | 72.9 | 70.8 | −0.04 | < 0.001 |
| cg15059639 | MYO3B | 2 | 0.85 | 64.9 | 72.3 | 56.9 | 56.7 | −8.0 | 54.9 | 56.3 | 56.7 | 64.4 | 1.8 | 0.08 |
| cg10090568 | ESRRG | 2 | 0.64 | 52.9 | 60.5 | 50.7 | 48.8 | −2.2 | 56.9 | 57.1 | 53.5 | 59.1 | −3.4 | 0.61 |
| cg24883732 | TCEB3B | 2 | 0.46 | 80.6 | 45.8 | 82.5 | 62.7 | 1.9 | 95.7 | 71.3 | 95.0 | 65.9 | −0.7 | 0.17 |
| cg03333116 | RHBDF1 | 2 | 0.39 | 78.3 | 68.8 | 91.6 | 81.6 | 13.3 | 79.3 | 82.7 | 79.2 | 75.8 | −0.09 | < 0.001 |
| cg00218770 | CDH4 | 2 | 0.39 | 82.1 | 68.2 | 91.3 | 81.7 | 9.2 | 84.2 | 78.5 | 83.7 | 76.0 | −0.5 | < 0.001 |
Abbreviations: PSQ, pyrosequencing; 450K, Infinium HumanMapping450K Beadchip
Data were sorted by decreasing Spearman correlation coefficient in New Cases.
Spearman correlation coefficients (rho) were shown to assess the concordance of %methylation levels between pyrosequencing and Infinium HumanMapping450K Beadchip.
Changes of %methylation were calculated as %methylation at follow-up (2009–2011) subtracted by %methylation at baseline (2001–2003).
P-values were obtained from two-sample t-tests for comparisons of change %methylation (PSQ) between New Cases and Persistent Controls.
DISCUSSION
We assessed longitudinal epigenome-wide changes of DNA methylation in 10 people who developed arsenic-induced skin lesions over a ten year follow up. We observed that the %methylation at different CpG sites both increased and decreased in people who developed skin lesions. This further illustrates the dynamic nature of DNA methylation. However, the differences in %methylation between people who developed skin lesions and those that did not were not statistically significantly different from people who did not develop skin lesions after considering issues of multiple comparisons. This was likely due to the small number of people who developed skin lesions in our sample population. The water arsenic concentrations are significantly different between cases and controls. Therefore, it is possible that the DNA methylation we observed was induced by water arsenic. However, we checked the association between baseline arsenic and DNA methylation of the top 6 CpG sites and none were statistically significant (data not shown).
Interestingly our longitudinal study and Smeester’s cross-sectional study observed that rhomboid 5 homolog 1 (RHBDF1) had higher methylation in people with arsenic-induced skin lesions compared to controls [Smeester et al., 2011]. In Smeester et al., rhomboid 5 homolog 1 (RHBDF1) had a 1.47-fold higher methylation between cases and controls (P = 0.075, q-value = 0.046) and we found a 1.19-fold change (equivalent to a 13% increase) in %methylation of a CpG site on the RHBDF1 gene in New Cases as compared to their baseline levels. We also observed a 6.9% decrease in %methylation of the same probe in the Persistent Controls. This indicates that the hypermethylation of the RHBDF1 gene is more likely related to arsenic exposure and the development of skin lesions than a natural progression due to aging. RHBDF1 gene regulates the secretion of several ligands of the epidermal growth factor receptor (EGFR) [Nakagawa et al., 2005]. It is an activated oncogene in many cancers that indirectly activates the EGFR signaling pathway and therefore may regulate cell survival, proliferation and migration [Yan et al., 2008; Zettl et al., 2011; Zou et al., 2009]. Studies have also found RHBDF1 to be essential to epithelial cancer cell growth [Yan et al., 2008] and to play a part in G-protein-coupled receptors-mediated transactivation of EGFR growth signals in head and neck squamous cancer cells [Zou et al., 2009]. In New Cases, the mean baseline %methylation of RHBDF1 gene appeared to be lower than the Persistent Controls, and showed an increase in %methylation at follow-up. Therefore, the %methylation at baseline could possibly serve as a predictive marker for the development of skin lesions 10 years later, and could be used to identify a sub-population at risk of arsenic-induced skin lesions.
A total of 127 CpG sites in 75 out of the 183 genes (41.0%) reported by Smeester et al. passed significance level of 0.05 in our study, but none achieved a level of significance that survived multiple comparisons. The most significant CpG in our study that lies on the gene body of NR1H2 gene (nuclear receptor subfamily 1, group H, member 2; P = 2.03 × 10−6) was also shortlisted as one of the 183 significant genes by Smeester et al. However, the other 108 genes found to be significant in the Smeester’s study did not show up as significant in our study. The main reasons for the difference between our findings might be due to the different study design (longitudinal in our study versus case-control in Smeester et al.) and different health outcomes of interest (development of new skin lesions in our study versus pre-existing arsenic-induced skin lesion cases in Smeester et al.). In addition, the ethnicity of the two study populations was different in the two studies (Bangladesh in our study versus Mexico in Smeester et al.).
Of the top 20 genes in our study, 7 of them belonged to ‘metal ion binding’ group according to Gene Ontology (GO) terms (FAT). They are AGAP2 (ArfGAP with GTPase domain, ankyrin repeat and PH domain 2), PRDM9 (PR domain containing 9), CDH4 (cadherin 4, type 1, R-cadherin (retinal)), CHPF (chondroitin polymerizing factor), CYC1 (cytochrome c-1), ESRRG (estrogen-related receptor gamma) and MAP3K1 (mitogen-activated protein kinase 1). Two genes were found to be enriched for skin diseases (MAP3K1 and TNXB), and three genes were found to be enriched for metabolic pathways (CHPF, CYC1 and PPAP2C). Interestingly, the PPAP2C protein results in decreased susceptibility to arsenic trioxide in vitro. In our study, the PPAP2C probe (cg27370573) was found to be hypermethylated in New Cases and therefore, possibly silencing the gene, leading to increased susceptibility to arsenic and subsequent development of skin lesions [Liu et al., 2010].
In addition to RHBDF1, PRDM9 and CDH4 genes also showed consistent results for both the Infinium HumanMethylation450K BeadChip and pyrosequencing, displaying differential methylation levels in the New Cases as compared to the Persistent Controls. PRDM9 is PR domain zinc finger protein 9, which has histone H3(K4) trimethyltransferase activity and is essential for proper meiotic progression [Thomas et al., 2009]. CDH4 is cadherin-4, which plays an important role in brain, kidney and muscle development. Aberrant methylation of CDH4 gene promoter has been previously observed in gastric, colorectal and nasopharyngeal cancers, suggesting that CDH4 might act as a possible tumor suppressor gene [Miotto et al., 2004; Du et al., 2011]. Therefore, hypermethylation of CDH4 gene in the New Cases might indicate early signs of carcinogenesis.
Our study was limited to 10 participants who developed skin lesions over the follow-up period. Given that that our study was small and we did not find any sites that were statistically significant after controlling for multiple comparisons, it is possible that most sites identified may have been identified by chance. We were able to match these individuals to controls on many potential confounders which improved our analysis. Because samples were not randomly distributed across chips, ComBat may not sufficiently eliminate the potential for batch effects and that we may have over- or under-estimated the associations between the top 20 CpG sites and skin lesions depending on the type of misclassifications (differential or non-differential). However, the effect size of chip effect was shown to be reduced with the use of ComBat (Figure S2). This provides evidence that the association was driven to the null due to non-differential misclassification, although we still cannot evaluate the possibility of differential misclassifications. Our study was also limited by only having whole blood available for ascertaining DNA methylation. Blood is not necessarily a target tissue for skin lesions and peripheral white blood cells (WBCs) represent a mixed cell population. WBC methylation has been reported to correlate poorly with tumor tissue methylation [van Bemmel et al., 2012]. However, because WBCs can be easily obtained from human subjects, they represent a primary tissue for the development of epigenetic biomarkers that might be amenable to use for preventive and diagnostic purposes. Moreover, we performed an additional analysis to estimate the effect sizes due to different cell types [Houseman et al., 2012] and found the largest possible effect size to be 5% (data not shown). All of our top 20 CpG sites, however, had an effect size of at least 11% in New Cases (Table 2). In addition, because methylation is a dynamic process and the time-window in which crucial methylation changes take place in the development of skin lesions are currently unclear, we cannot be absolutely sure if the significant methylation changes we observed in the New Cases are fully attributable to the development of skin lesions. RNA expression was not examined to determine the impact of DNA methylation on gene expression.
Strengths of this study include temporality by assessing longitudinal DNA methylation changes within individuals over time to account for the effects of time on DNA methylation; cross-platform validity with a two-stage design using Illumina Infinium HumanMethylation450K Beadchip followed by pyrosequencing; homogeneous study population with a similar genetic background that minimizes population stratifications; comprehensive information was also collected to adjust for potential confounders by matching cases and controls. We also compared the methylation differences in New Cases with Persistent Controls, and therefore controlled for degradation of samples and changing methylation patterns over time in the New Cases.
CONCLUSIONS
This exploratory project identified several differentially methylated genes in people who developed arsenic-induced skin lesions over a 10 year period that were also reported in another study of arsenic related skin lesions. These findings demonstrate that DNA methylation variations occurred with the development of arsenic-induced skin lesions. Larger studies that examine changes in methylation over time are required to validate these findings. In addition, the transcriptional effects of these differentially methylated genes should be evaluated to determine if they are epigenetically regulated and if they are involved in skin pathogenesis.
Supplementary Material
Acknowledgments
We thank Dr. Liming Liang and Dr. Tamar Sofer for their help during the preliminary stages of analysis.
GRANT SPONSORS
This study was funded by NIH (NIEHS) grants P30 ES00002, RO1 ES011622, P42 E016454, and the Genes and Environment Initiative from the Harvard School of Public Health.
Footnotes
STATEMENT OF AUTHOR CONTRIBUTIONS
D.C.C. contributed to the conceptualization of the project, the design, and funding of the study. Q.Q., M.R., and G.M. recruited the patients, collected the data, and supervised the field activities in Bangladesh. HM.B. performed the pyrosequencing lab work. W.J.S. designed the exploratory project, collected the 450K methylation data, analyzed the data, and led the interpretation of results. W.J.S. prepared the manuscript draft with important intellectual input from D.C.C., M.L.K., A.A.B., X.L., and WC.P. All authors approved the final manuscript.
The authors declare they have no competing financial interests.
References
- Baccarelli A, Bollati V. Epigenetics and environmental chemicals. Curr Opin Pediatr. 2009;21:243–251. doi: 10.1097/mop.0b013e32832925cc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baylin SB. DNA methylation and gene silencing in cancer. Nat Clin Pract Oncol. 2005;2(Suppl. 1):S4–S11. doi: 10.1038/ncponc0354. [DOI] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y. Controlling the False Discovery Rate – a Practical and Powerful Approach to Multiple Testing. J Roy Stat Soc B Met. 1995;57:289–300. [Google Scholar]
- Bollati V, Baccarelli A, Hou L, Bonzini M, Fustinoni S, Cavallo D, Byun HM, Jiang J, Marinelli B, Pesatori AC, Bertazzi PA, Yang AS. Changes in DNA methylation patterns in subjects exposed to low-dose benzene. Cancer Research. 2007;67:876–880. doi: 10.1158/0008-5472.CAN-06-2995. [DOI] [PubMed] [Google Scholar]
- Breton CV, Houseman EA, Kile ML, Quamruzzaman Q, Rahman M, Mahiuddin G, Christiani DC. Gender-specific protective effect of hemoglobin on arsenic-induced skin lesions. Cancer Epidemiol Biomarkers Prev. 2006;15:902–907. doi: 10.1158/1055-9965.EPI-05-0859. [DOI] [PubMed] [Google Scholar]
- Cantone I, Fisher AG. Epigenetic programming and reprogramming during development. Nature Structural & Molecular Biology. 2013;20:282–289. doi: 10.1038/nsmb.2489. [DOI] [PubMed] [Google Scholar]
- Centeno JA, Mullick FG, Martinez L, Page NP, Gibb H, Longfellow D, Thompson C, Ladich ER. Pathology related to chronic arsenic exposure. Environ Health Perspect. 2002;110(Suppl 5):883–886. doi: 10.1289/ehp.02110s5883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chowdhury UK, Biswas BK, Chowdhury TR, Samanta G, Mandal BK, Basu GC, Chanda CR, Lodh D, Saha KC, Mukherjee SK, Roy S, Kabir S, Quamruzzaman Q, Chakraborti D. Groundwater arsenic contamination in Bangladesh and West Bengal, India. Environ Health Perspect. 2000;108:393–397. doi: 10.1289/ehp.00108393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du C, Huang T, Sun D, Mo Y, Feng H, Zhou X, Xiao X, Yu N, Hou B, Huang G, Ernberg I, Zhang Z. CDH4 as a novel putative tumor suppressor gene epigenetically silenced by promoter hypermethylation in nasopharyngeal carcinoma. Cancer Letters. 2011;309:54–61. doi: 10.1016/j.canlet.2011.05.016. [DOI] [PubMed] [Google Scholar]
- Du P, Zhang X, Huang CC, Jafari N, Kibbe WA, Hou L, Lin SM. Comparison of Beta-value and M-value methods for quantifying methylation levels by microarray analysis. BMC Bioinformatics. 2010;11:587. doi: 10.1186/1471-2105-11-587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feil R. Environmental and nutritional effects on the epigenetic regulation of genes. Mutation Research. 2006;600:46–57. doi: 10.1016/j.mrfmmm.2006.05.029. [DOI] [PubMed] [Google Scholar]
- Fraga MF, Ballestar E, Paz MF, Ropero S, Setien F, Ballestar ML, Heine-Suñer D, Cigudosa JC, Urioste M, Benitez J, Boix-Chornet M, Sanchez-Aguilera A, Ling C, Carlsson E, Poulsen P, Vaag A, Stephan Z, Spector TD, Wu YZ, Plass C, Esteller M. Epigenetic differences arise during the lifetime of monozygotic twins. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:10604–10609. doi: 10.1073/pnas.0500398102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou L, Zhang X, Wang D, Baccarelli A. Environmental chemical exposures and human epigenetics. Int J Epidemiol. 2012;41:79–105. doi: 10.1093/ije/dyr154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houseman EA, Accomando WP, Koestler DC, Christensen BC, Marsit CJ, Nelson HH, Wiencke JK, Kelsey KT. DNA methylation arrays as surrogate measures of cell mixture distribution. BMC Bioinformatics. 2012;13:86. doi: 10.1186/1471-2105-13-86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson WE, Li C, Rabinovic A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics. 2007;8:118–127. doi: 10.1093/biostatistics/kxj037. [DOI] [PubMed] [Google Scholar]
- Kile ML, Baccarelli A, Hoffman E, Tarantini L, Quamruzzaman Q, Rahman M, Mahiuddin G, Mostoa G, Hsueh YM, Wright RO, Christiani DC. Prenatal arsenic exposure and DNA methylation in maternal and umbilical cord leukocytes. Environ Health Perspect. 2012;120:1061–1066. doi: 10.1289/ehp.1104173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leek JT, Johnson EW, Parker HS, Jaffe AE, Storey JD. The sva package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics. 2012;28:882–883. doi: 10.1093/bioinformatics/bts034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q, Zhang H, Smeester L, Zou F, Kesic M, Jaspers I, Pi J, Fry RC. The NRF2-mediated oxidative stress response pathway is associated with tumor cell resistance to arsenic trioxide across the NCI-60 panel. BMC Medical Genomics. 2010;3:37. doi: 10.1186/1755-8794-3-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millington GWM. Epigenetics and dermatological disease. Pharmacogenomics. 2008;9:1835–1850. doi: 10.2217/14622416.9.12.1835. [DOI] [PubMed] [Google Scholar]
- Miotto E, Sabbioni S, Veronese A, Calin GA, Gullini S, Liboni A, Gramantieri L, Bolondi L, Ferrazzi E, Gafa R, Lanza G, Negrini M. Frequent aberrant methylation of the CDH4 gene promoter in human colorectal and gastric cancer. Cancer Research. 2004;64:8156–8159. doi: 10.1158/0008-5472.CAN-04-3000. [DOI] [PubMed] [Google Scholar]
- Nakagawa T, Guichard A, Castro CP, Xiao Y, Rizen M, Zhang HZ, Hu D, Bang A, Helms J, Bier E, Derynck R. Characterization of a human rhomboid homolog, p100hRho/RHBDF1, which interacts with TGF-alpha family ligands. Dev Dyn. 2005;233:1315–1331. doi: 10.1002/dvdy.20450. [DOI] [PubMed] [Google Scholar]
- Pilsner JR, Liu X, Ahsan H, Ilievski V, Slavkovich V, Levy D, Factor-Litvak P, Graziano JH, Gamble MV. Folate deficiency, hyperhomocysteinemia, low urinary creatinine, and hypomethylation of leukocyte DNA are risk factors for arsenic-induced skin lesions. Environ Health Perspect. 2009;117:254–260. doi: 10.1289/ehp.11872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reichard JF, Puga A. Effects of arsenic exposure on DNA methylation and epigenetic gene regulation. Epigenomics. 2010;2:87–104. doi: 10.2217/epi.09.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren X, McHale CM, Skibola CF, Smith AH, Smith MT, Zhang L. An emerging role for epigenetic dysregulation in arsenic toxicity and carcinogenesis. Environ Health Perspect. 2011;119:11–19. doi: 10.1289/ehp.1002114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz WA. L1 retrotransposons in human cancers. J Biomed Biotechnol. 2006;2006:83672. doi: 10.1155/JBB/2006/83672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sciandrello G, Caradonna F, Mauro M, Barbata G. Arsenic-induced DNA hypomethylation affects chromosomal instability in mammalian cells. Carcinogenesis. 2004;25:413–417. doi: 10.1093/carcin/bgh029. [DOI] [PubMed] [Google Scholar]
- Seow WJ, Pan WC, Kile ML, Baccarelli AA, Quamruzzaman Q, Rahman M, Mahiuddin G, Mostofa G, Lin X, Christiani DC. Arsenic reduction in drinking water and improvement in skin lesions: a follow-up study in Bangladesh. Environ Health Perspect. 2012;120:1733–1738. doi: 10.1289/ehp.1205381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slotkin RK, Martienssen R. Transposable elements and the epigenetic regulation of the genome. Nat Rev Genet. 2007;8:272–285. doi: 10.1038/nrg2072. [DOI] [PubMed] [Google Scholar]
- Smeester L, Rager JE, Bailey KA, Guan XJ, Smith N, Garcia-Vargas G, Del Razo LM, Drobná Z, Kelkar H, Stýblo M, Fry RC. Epigenetic Changes in Individuals with Arsenicosis. Chemical Research in Toxicology. 2011;24:165–167. doi: 10.1021/tx1004419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith AH, Lingas EO, Rahman M. Contamination of drinking-water by arsenic in Bangladesh: a public health emergency. Bulletin of the World Health Organization. 2000;78:1093–1103. [PMC free article] [PubMed] [Google Scholar]
- Thomas JH, Emerson RO, Shendure J. Extraordinary molecular evolution in the PRDM9 fertility gene. PLoS ONE. 2009;4:e8505. doi: 10.1371/journal.pone.0008505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Touleimat N, Tost J. Complete pipeline for Infinium((R)) Human Methylation 450K BeadChip data processing using subset quantile normalization for accurate DNA methylation estimation. Epigenomics. 2012;4:325–341. doi: 10.2217/epi.12.21. [DOI] [PubMed] [Google Scholar]
- Tseng WP, Chu HM, How SW, Fong JM, Lin CS, Yeh S. Prevalence of skin cancer in an endemic area of chronic arsenicism in Taiwan. Journal of the National Cancer Institute. 1968;40:453–463. [PubMed] [Google Scholar]
- van Bemmel D, Lenz P, Liao LM, Baris D, Sternberg LR, Warner A, Johnson A, Jones M, Kida M, Schwenn M, Schned AR, Silverman DT, Rothman N, Moore LE. Correlation of LINE-1 methylation levels in patient-matched buffy coat, serum, buccal cell, and bladder tumor tissue DNA samples. Cancer Epidemiol Biomarkers Prev. 2012;21:1143–1148. doi: 10.1158/1055-9965.EPI-11-1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan Z, Zou H, Tian F, Grandis JR, Mixson AJ, Lu PY, Li LY. Human rhomboid family-1 gene silencing causes apoptosis or autophagy to epithelial cancer cells and inhibits xenograft tumor growth. Molecular cancer therapeutics. 2008;7:1355–1364. doi: 10.1158/1535-7163.MCT-08-0104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuen RK, Peñaherrera MS, von Dadelszen P, McFadden DE, Robinson WP. DNA methylation profiling of human placentas reveals promoter hypomethylation of multiple genes in early-onset preeclampsia. Eur J Hum Genet. 2010;18:1006–1012. doi: 10.1038/ejhg.2010.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zettl M, Adrain C, Strisovsky K, Lastun V, Freeman M. Rhomboid family pseudoproteases use the ER quality control machinery to regulate intercellular signaling. Cell. 2011;145:79–91. doi: 10.1016/j.cell.2011.02.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou H, Thomas SM, Yan ZW, Grandis JR, Vogt A, Li LY. Human rhomboid family-1 gene RHBDF1 participates in GPCR-mediated transactivation of EGFR growth signals in head and neck squamous cancer cells. Faseb J. 2009;23:425–432. doi: 10.1096/fj.08-112771. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.