

Abstract
Chronic overnutrition creates chronic hyperglycemia that can gradually induce insulin resistance and insulin secretion impairment. These disorders, if not intervened, will eventually be followed by appearance of frank diabetes. The mechanisms of this chronic pathogenic process are complex but have been suggested to involve production of reactive oxygen species (ROS) and oxidative stress. In this review, I highlight evidence that reductive stress imposed by overflux of NADH through the mitochondrial electron transport chain is the source of oxidative stress, which is based on establishments that more NADH recycling by mitochondrial complex I leads to more electron leakage and thus more ROS production. The elevated levels of both NADH and ROS can inhibit and inactivate glyceraldehyde 3-phosphate dehydrogenase (GAPDH), respectively, resulting in blockage of the glycolytic pathway and accumulation of glycerol 3-phospate and its prior metabolites along the pathway. This accumulation then initiates all those alternative glucose metabolic pathways such as the polyol pathway and the advanced glycation pathways that otherwise are minor and insignificant under euglycemic conditions. Importantly, all these alternative pathways lead to ROS production, thus aggravating cellular oxidative stress. Therefore, reductive stress followed by oxidative stress comprises a major mechanism of hyperglycemia-induced metabolic syndrome.
1. Introduction
Type 2 diabetes is generally an overnutritional disease [1–3]. It is caused by insulin resistance and insulin secretion impairment induced gradually and mainly by high blood glucose in conjunction with other factors such as obesity, aging, genetic predisposition, and physical inactivity [4–9]. Persistent overnutrition creates a steady level of high blood glucose that is toxic to macrovascular and microvascular systems [10–12], an effect known as glucotoxicity [13–17]. While oxidative stress is thought to contribute to the pathogenesis of glucotoxicity during the development of diabetes and diabetic complications [18–26], reductive stress due to excess NADH [27–33] generated by high blood glucose has attracted less attention. In this review, by following the mechanisms of NADH production and recycling, I highlight evidence that reductive stress followed by oxidative stress comprises the fundamental pathogenic mechanisms of chronic hyperglycemia in the development of diabetes and diabetic complications.
2. Euglycemia
A normal level of blood glucose below 100 mg/dL is tightly maintained, regulated, and achieved by rate of glucose uptake by all tissues and rate of glucose synthesis by the liver [34] and to a less magnitude by the kidney [35]. Approximately, 75% of the body's total glucose is consumed by insulin-insensitive tissues including the brain, red blood cells, the liver, and the gut, while the rest is consumed by insulin-sensitive tissues including muscle [36]. Postprandially, a rapid increase in blood glucose content stimulates insulin secretion, resulting in a temporary increase in blood insulin concentration known as hyperinsulinemia. The increases in blood concentrations of both glucose and insulin coordinately inhibit glucose production by the liver and facilitate glucose uptake by insulin-insensitive tissues [37]. Therefore, euglycemia is strictly maintained, which is highly dependent not only on proper insulin secretion from the β-cells upon nutritional stimulation but also on insulin action in the liver and peripheral tissues [37].
3. NADH and Reductive Stress
Electrons from aerobic breakdown of glucose are mainly stored in NADH for oxygen reduction and ATP production. Therefore, NADH is a reducing compound and an excessive amount of it can cause reductive stress [30, 32, 38–40]. Overproduction of NADH or lack of NAD+ can induce the accumulation of NADH, leading to imbalance between NADH and NAD+ and creating a condition known as pseudohypoxia [29, 41–44]. This is a condition under which oxygen cannot be effectively consumed. This would cause metabolic stress or metabolic syndrome as it often occurs in diabetes [44–47]. It should be noted that GSH and NADPH accumulation, tightly linked to NADH metabolism [48], can also induce reductive stress [39, 49–54]. As mitochondrial complex I is the major enzyme responsible for NADH recycling, impairment of complex I function can thus induce NADH accumulation and reductive stress [55] that could be linked to inhibition of insulin release by β-cells [56, 57].
4. Hyperglycemia, Elevated Levels of NADH, and Mitochondrial Electron Pressure
The glycolytic pathway breaks down nearly 80%–90% of the body's glucose, while the pentose phosphate pathway consumes the remaining 10%–20% under physiological condition [58, 59]. Under hyperglycemic condition, more glucose will flux through the glycolytic pathway that produces more pyruvate and acetyl-CoA, leading to more NADH production. As NADH is an electron carrier, excess amount of it will cause an electron pressure on the mitochondrial electron transport chain [40, 60–62]. This is particularly true for hepatocytes and pancreatic β-cells in that glucokinase (hexokinase D) is a supply-driven enzyme [63], and this enzyme is not inhibited by glucose-6-phosphate (G6P) [64–66]. Therefore, the more glucose the more G6P produced that will be broken down through glycolysis and Krebs cycle, leading to more NADH production. Figure 1 shows the major conventional pathways that can generate more NADH when glucokinase is used to phosphorylate glucose for glucose breakdown in tissues such as pancreas and liver [67–70].
Figure 1.
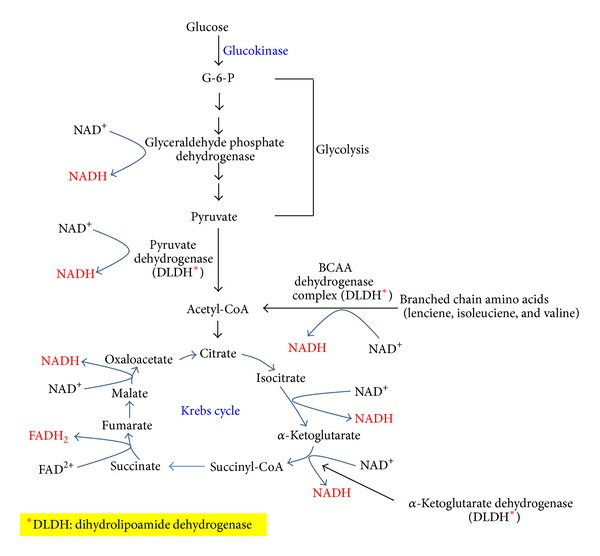
The conventional pathways that generate NADH by breaking down glucose via glycolysis and the Krebs cycle. The enzymes involved in NADH/NAD+ recycling are shown. ∗DLDH stands for dihydrolipoamide dehydrogenase and is the component in each given enzyme complex that actually makes NADH from NAD+ [191].
5. NADH-Imposed Electron Pressure and Mitochondrial Superoxide Production
The electron pressure induced by overproduced NADH will place a heavy burden on mitochondrial complex I that is the major site for NADH recycling (Figure 2). Under this condition, complex I will respond within its capacity to oxidize more NADH to NAD+, in an attempt to ameliorate the pseudohypoxic condition. An inherent nature of NADH flux through complex I is that more superoxide will also be made when more NADH is oxidized by complex I as this complex is also involved in proton pumping [71–73], leading to a proportional increase in electron leakage that will partially reduce oxygen to yield superoxide [71, 74–77]. This scenario could get worse under pseudohypoxic conditions as less NAD+ is available for transporting electrons to oxygen [55], leaving more oxygen available for partial reduction by the leaked electrons from complex I and complex III, the latter being also involved in proton pumping [78–80]. It should be noted that complex II and dihydrolipoamide dehydrogenase could also produce superoxide [81–83].
Figure 2.
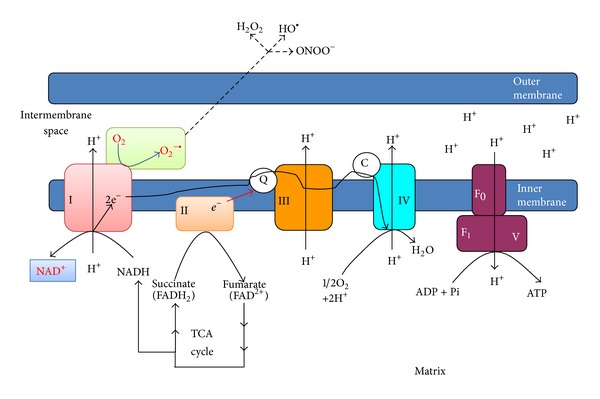
NADH oxidation by complex I in the electron transport chain. Electrons from NADH are transported via CoQ and cytochrome c to molecular oxygen. This process involves proton pumping that is tightly linked to superoxide production. ATP synthesis by complex V driven by the proton gradient is also shown.
6. Superoxide and Oxidative Stress
Superoxide is the precursor of all reactive oxygen species that at elevated levels can cause oxidative stress [84, 85]. As has been established, superoxide can be converted to hydrogen peroxide by superoxide dismutase; hydrogen peroxide can then be converted to form hydroxyl radical by metal ions [84, 86, 87]. In the meantime, superoxide can also react with nitric oxide to produce peroxynitrite (ONOO−) [88, 89]. All these reactive species can cause oxidation of proteins, lipids, and DNA [90]. Consequently, an oxidative stress condition has fully developed due to a high level of NADH, achieving the transition from reductive stress to oxidative stress [43, 91–93]. Therefore, reductive stress is not the reverse of oxidative stress; it actually leads to oxidative stress [94, 95].
7. Inhibition of Glyceraldehyde 3-Phosphate Dehydrogenase and Alternative Glucose Metabolic Pathways
As has been discussed above, an oversupply of NADH can lead to overproduction of mitochondrial superoxide and other forms of ROS. These ROS can then impair the activity of glyceraldehyde 3-phosphate dehydrogenase (GAPDH) [22, 96] that is very sensitive to oxidative modifications [21, 97–103] due to a redox-sensitive cysteine residue at its active center [104, 105]. Additionally, high level of NADH would also inhibit GAPDH activity [106]. Such impairments would collectively decrease the efficiency of glucose metabolism via glycolysis and Krebs cycle, inducing accumulation of glyceraldehyde 3-phosphate (G3P). Therefore, all the intermediate products above and including G3P will have to be disposed by pathways that branch off the glycolytic pathways (Figure 3) [107, 108].
Figure 3.
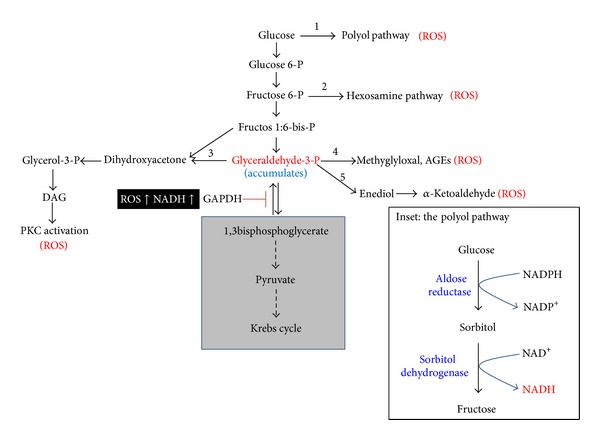
The branch-off pathways that are activated to dispose excess glucose when glyceraldehyde 3-phosphate dehydrogenase (GAPDH) is inactivated by ROS. These five alternative pathways [21, 115], in addition to the electron transport chain shown in Figure 2, are linked to ROS production, thus further exacerbating oxidative stress. Inset shows the polyol pathway. Pathways in the grey area would no longer efficiently break down glucose when GAPDH is inactivated by posttranslational modifications.
8. The Branching-Off Pathways and Oxidative Stress
There have been five pathways [21] that can branch off the glycolytic pathway under chronic hyperglycemic conditions (Figure 3). These pathways are minor and insignificant in glucose metabolism under normoglycemic conditions, but can become major pathways to flux high level glucose. As will be discussed below, all the five pathways have been linked to ROS production, oxidative stress, and the pathogenesis of diabetes and diabetic complications [21, 109–115].
8.1. The Polyol Pathway
When blood glucose level is high, cellular metabolic pathways change, which usually leads to deleterious effects [5]. A major pathway that is activated in response to hyperglycemia is the polyol pathway [44, 116–118], in which glucose is reduced by aldose reductase to form sorbitol, and the formed sorbitol is then converted to fructose by sorbitol dehydrogenase. This pathway, as shown in Figure 3 (Inset), converts NADPH to NADH using two step reactions and leads to redox imbalance between NADH and NAD+. As the ratio of NAD+/NADH decreases due to an increase in NADH content, reductive stress can ensue. Because aldose reductase has a very high Km for glucose [119], it can only be activated by a high level of glucose. Hence, this enzyme could also be considered as a supply-driven enzyme [120, 121]. Under hyperglycemic conditions, the polyol pathway has been estimated to utilize more than 30% of the body's glucose [101]. Therefore, this pathway can also contribute significantly to reductive stress [32, 119] and has been thought to play an important role in the pathogenesis of diabetic complications [122–125].
Additionally, in the first reaction of the polyol pathway (Figure 3 inset), NADPH is consumed and, when NADPH level goes lower, so does reduced form of glutathione (GSH). This is because glutathione reductase needs NADPH to regenerate GSH from GSSG (oxidized form of glutathione) [126]. As GSH level goes lower, cellular antioxidant capacity can be compromised, resulting in elevated levels of reactive oxygen species that can attack macromolecules and induce oxidative damage [126]. Therefore, the polyol pathway is also a source of oxidative stress [127–129]. It should also be pointed out that activation of the polyol pathway in return will further decrease glucose consumption by the glycolytic pathway as sorbitol dehydrogenase competes with GAPDH for NAD+ [130, 131]. Moreover, as nitric oxide synthase also uses NADPH as a cofactor, a lowered level of NADPH can lead to a decrease in nitric oxide production, thereby facilitating vasoconstriction and platelet aggregation [132].
8.2. The Hexosamine Pathway
This pathway branches off from fructose 6-phosphate in the glycolytic pathway. Fructose 6-phosphate is the substrate of the enzyme glutamine-fructose 6-P amidotransferase (GFAT), which is the rate-limiting enzyme for this pathway. GFAT makes glucosamine 6-P from fructose 6-P and the former is further converted to UDP-N-acetylglucosamine, which is the substrate for specific O-GlcNAc transferase that catalyzes posttranslational modifications of proteins via O-GlcNAc on serine and threonine residues [133–135]. Increased glucose flux through this pathway has been shown to be involved in ROS generation and oxidative stress [136–138] and has been implicated in diabetic complications [139–142].
8.3. The Protein Kinase C Activation Pathway
Fructose 1:6-bisphosphate can break down to form dihydroxyacetone phosphate and glyceraldehyde 3-phosphate with the former being readily isomerized to glyceraldehyde 3-phosphate under the action of triose phosphate isomerase. Accumulation of glyceraldehyde 3-phosphate can increase the synthesis of diacylglycerol that is an activator of protein kinase C (PKC). PKC activation is known to be involved in elevating the content of TGF-β-1, endothelin-1, NF-κB, and vascular endothelial growth factor [22, 143, 144] and is also known to induce ROS production by NADPH oxidase that catalyzes one electron reduction of molecular oxygen to form superoxide [145–147]. Mechanistically, it has been established that PKC activates NADPH oxidase by phosphorylating the p47phox subunit, triggering the translocation of this subunit from cytosol to membrane whereby it assembles with other components to form an active NADPH oxidase that is capable of making superoxide from oxygen [148, 149]. PKC activation can also induce insulin resistance by inhibiting Akt-dependent nitric oxide synthase function [150].
8.4. Advanced Glycation End Products (AGEs)
In addition to the polyol pathway, this pathway has also been thought to be a major mechanism of oxidative stress under hyperglycemic condition [151, 152]. High level of glucose can induce formation of methylglyoxal from glyceraldehyde 3-phosphate when GAPDH function is impaired. Methylglyoxal can modify proteins via glycation of amino groups on proteins [153, 154]. One of the major products is glycated hemoglobin (HbA1c) that has been used as a biomarker for diabetes [155, 156]. Therefore, this nonenzymatic process can greatly impair protein function. Moreover, this glycation pathway is known to liberate ROS [157, 158] and upregulate the expression of cell surface receptor for AGEs, leading to activation of the NF-κB signaling pathway and chronic inflammation [159–161].
8.5. The Glyceraldehyde Autoxidation Pathway
This pathway also branches off from glyceraldehyde 3-phosphate in the glycolytic pathway. Glyceraldehyde 3-phosphate is formed from fructose 1:6-bisphospate by the enzyme aldose. Under certain conditions, glyceraldehyde 3-phosphate can undergo autoxidation [162], a process that can generate hydrogen peroxide and α-ketoaldehydes in diabetes mellitus [21, 163].
9. Oxidative Stress, Diabetes, and Diabetic Complications
As discussed above, all the sources of ROS and oxidative stress can be traced back to high blood glucose and NADH overproduction. Therefore, chronic hyperglycemia would inevitably cause chronic reductive stress that leads to oxidative stress. As ROS production is a common feature of the above described pathways [119, 164], chronic oxidative stress certainly plays a central role in the development of diabetes and diabetic complications [22, 165, 166]. Indeed, it has been reported that ROS can induce insulin resistance [74, 167], impair insulin synthesis [168], and impair beta cell insulin secretion [97, 169]. Additionally, oxidative stress biomarkers have been shown to be increased in individuals who exhibit insulin resistance [170–173] or insulin secretion impairment [174–177], indicating a positive correlation between oxidative stress and insulin resistance and insulin secretion impairment. Moreover, numerous studies have also established that ROS are involved in the etiology of diabetic complications including retinopathy, neuropathy, cardiomyopathy, and nephropathy [123, 178–182]. Given that oxidative stress originates from NADH-imposed reductive stress [31, 183], attenuating hyperglycemia-triggered reductive stress may provide potential therapeutic approaches for preventing the development of diabetes and diabetic complications.
10. Conclusion
Persistent high blood glucose is highly toxic [16, 112]. It not only induces insulin resistance but also impairs insulin secretion by pancreatic β-cells [184]. Over time, hyperglycemia will produce detrimental effects on macrovascular and microvascular systems [185, 186]. Figure 4 summarizes schematically the pathways discussed in this review and their pathogenic roles in chronic hyperglycemia via NADH, ROS, and oxidative stress. As hyperglycemia results in excessive production of acetyl-CoA that feeds into the Krebs cycle, making excess NADH, mitochondrial electron transport chain is thus under heavy electron pressure [40, 60, 61]. Therefore, oxidation of the overproduced NADH by mitochondria will inevitably lead to production of more superoxide and hence more ROS [187, 188], which can in turn attack and inactivate GAPDH. This would trigger the accumulation of glycolytic metabolites upstream of glyceraldehyde 3-phosphate and activate the alternative glucose disposal pathways that all are linked to ROS production and hence increase the magnitude of oxidative stress [21, 189, 190]. Therefore, reductive stress followed by oxidative stress could serve as the major mechanism of glucotoxicity under chronic hyperglycemic conditions. An increase in NADH oxidation by mitochondria without an accompanying increase in ROS production may be a potential therapeutic approach for diabetes and diabetic complications.
Figure 4.
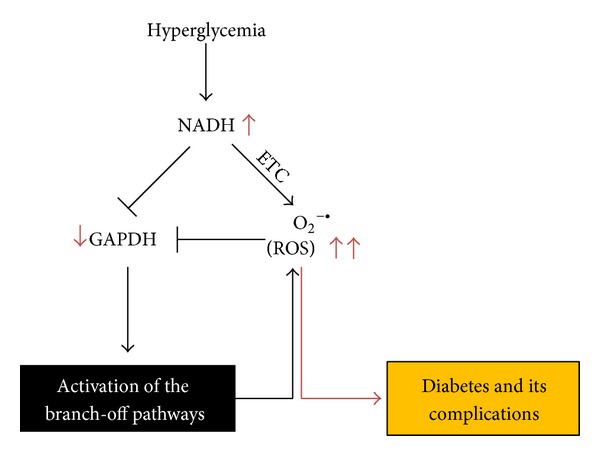
Hyperglycemia induces overproduction of NADH and mitochondrial ROS that inhibit GAPDH activity. This inhibition then activates the alternative glucose metabolic pathways, which further produce ROS involved in glucotoxicity that is responsible for the development of diabetes and diabetic complications. ETC: electron transport chain.
Acknowledgment
Liang-Jun Yan is supported in part by a Grant from the National Institute of Neurological Disorders and Stroke (R01NS079792).
Conflict of Interests
The author declares that there is no conflict of interests regarding the publication of this paper.
References
- 1.Prentki M, Nolan CJ. Islet β cell failure in type 2 diabetes. Journal of Clinical Investigation. 2006;116(7):1802–1812. doi: 10.1172/JCI29103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gupta D, Krueger CB, Lastra G. Over-nutrition, obesity and insulin resistance in the development of β-cell dysfunction. Current Diabetes Reviews. 2012;8(2):76–83. doi: 10.2174/157339912799424564. [DOI] [PubMed] [Google Scholar]
- 3.Cai D. Neuroinflammation and neurodegeneration in overnutrition-induced diseases. Trends in Endocrinology and Metabolism. 2013;24(1):40–47. doi: 10.1016/j.tem.2012.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.DeFronzo RA. Insulin resistance: a multifaceted syndrome responsible for NIDDM, obesity, hypertension, dyslipidaemia and atherosclerosis. Netherlands Journal of Medicine. 1997;50(5):191–197. doi: 10.1016/s0300-2977(97)00012-0. [DOI] [PubMed] [Google Scholar]
- 5.Tuch B, Dunlop M, Proietto J. Diabetes Research: A Guide for Postgraduates. Harwood Academic Publishers; 2000. [Google Scholar]
- 6.Barnett AH. Type 2 Diabetes. 2nd edition. Oxford University Press; 2012. [Google Scholar]
- 7.DeFronzo RA. Pathogenesis of type 2 diabetes mellitus. Medical Clinics of North America. 2004;88(4):787–835. doi: 10.1016/j.mcna.2004.04.013. [DOI] [PubMed] [Google Scholar]
- 8.Kahn SE, Cooper ME, Del Prato S. Pathophysiology and treatment of type 2 diabetes: perspectives on the past, present, and future. The Lancet. 2014;383:1068–1083. doi: 10.1016/S0140-6736(13)62154-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Szkudelski T. Streptozotocin-nicotinamide-induced diabetes in the rat. Characteristics of the experimental model. Experimental Biology and Medicine. 2012;237(5):481–490. doi: 10.1258/ebm.2012.011372. [DOI] [PubMed] [Google Scholar]
- 10.Heagerty AM, Aalkjaer C, Bund SJ, Korsgaard N, Mulvany MJ. Small artery structure in hypertension: dual processes of remodeling and growth. Hypertension. 1993;21(4):391–397. doi: 10.1161/01.hyp.21.4.391. [DOI] [PubMed] [Google Scholar]
- 11.Forbes JM, Cooper ME. Mechanisms of diabetic complications. Physiological Reviews. 2013;93(1):137–188. doi: 10.1152/physrev.00045.2011. [DOI] [PubMed] [Google Scholar]
- 12.Stirban A, Gawlowski T, Roden M. Vascular effects of advanced glycation endproducts: clinical effects and molecular mechanisms. Molecular Metabolism. 2013;3:94–108. doi: 10.1016/j.molmet.2013.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Leibowitz G, Bachar E, Shaked M, et al. Glucose regulation of β-cell stress in type 2 diabetes. Diabetes, Obesity and Metabolism. 2010;12(2):66–75. doi: 10.1111/j.1463-1326.2010.01280.x. [DOI] [PubMed] [Google Scholar]
- 14.Somesh BP, Verma MK, Sadasivuni MK, et al. Chronic glucolipotoxic conditions in pancreatic islets impair insulin secretion due to dysregulated calcium dynamics, glucose responsiveness and mitochondrial activity. BMC Cell Biology. 2013;14(1, article 31) doi: 10.1186/1471-2121-14-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dedoussis GVZ, Kaliora AC, Panagiotakos DB. Genes, diet and type 2 diabetes mellitus: a review. Review of Diabetic Studies. 2007;4(1):13–24. doi: 10.1900/RDS.2007.4.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Del Prato S. Role of glucotoxicity and lipotoxicity in the pathophysiology of Type 2 diabetes mellitus and emerging treatment strategies. Diabetic Medicine. 2009;26(12):1185–1192. doi: 10.1111/j.1464-5491.2009.02847.x. [DOI] [PubMed] [Google Scholar]
- 17.Bensellam M, Laybutt DR, Jonas J-C. The molecular mechanisms of pancreatic β-cell glucotoxicity: recent findings and future research directions. Molecular and Cellular Endocrinology. 2012;364(1-2):1–27. doi: 10.1016/j.mce.2012.08.003. [DOI] [PubMed] [Google Scholar]
- 18.Rösen P, Nawroth PP, King G, Möller W, Tritschler H-J, Packer L. The role of oxidative stress in the onset and progression of diabetes and its complications: a summary of a congress series sponsored by UNESCO-MCBN, the American diabetes association and the German diabetes society. Diabetes/Metabolism Research and Reviews. 2001;17(3):189–212. doi: 10.1002/dmrr.196. [DOI] [PubMed] [Google Scholar]
- 19.Bandeira SDM, da Fonseca LJS, Guedes GDS, Rabelo LA, Goulart MOF, Vasconcelos SML. Oxidative stress as an underlying contributor in the development of chronic complications in diabetes mellitus. International Journal of Molecular Sciences. 2013;14(2):3265–3284. doi: 10.3390/ijms14023265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stadler K. Oxidative stress in diabetes. Advances in Experimental Medicine and Biology. 2012;771:272–287. doi: 10.1007/978-1-4614-5441-0_21. [DOI] [PubMed] [Google Scholar]
- 21.Robertson AP. Chronic oxidative stress as a central mechanism for glucose toxicity in pancreatic islet beta cells in diabetes. Journal of Biological Chemistry. 2004;279(41):42351–42354. doi: 10.1074/jbc.R400019200. [DOI] [PubMed] [Google Scholar]
- 22.Giacco F, Brownlee M. Oxidative stress and diabetic complications. Circulation Research. 2010;107(9):1058–1070. doi: 10.1161/CIRCRESAHA.110.223545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Arora MK, Singh UK. Oxidative stress: meeting multiple targets in pathogenesis of diabetic nephropathy. Current Drug Targets. 2014;15:531–538. doi: 10.2174/1389450115666140321120635. [DOI] [PubMed] [Google Scholar]
- 24.Fiorentino TV, Prioletta A, Zuo P, Folli F. Hyperglycemia-induced oxidative stress and its role in diabetes mellitus related cardiovascular diseases. Current Pharmaceutical Design. 2013;19:5695–5703. doi: 10.2174/1381612811319320005. [DOI] [PubMed] [Google Scholar]
- 25.Mortuza R, Chakrabarti S. Glucose-induced cell signaling in the pathogenesis of diabetic cardiomyopathy. Heart Failure Reviews. 2014;19(1):75–86. doi: 10.1007/s10741-013-9381-z. [DOI] [PubMed] [Google Scholar]
- 26.Ruderman NB, Carling D, Prentki M, Cacicedo JM. AMPK, insulin resistance, and the metabolic syndrome. Journal of Clinical Investigation. 2013;123(7):2764–2772. doi: 10.1172/JCI67227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Williamson JR, Ido Y. Understanding retinal cytosolic reductive stress. Investigative Ophthalmology & Visual Science. 1998;39(7):1295–1296. [PubMed] [Google Scholar]
- 28.Williamson JR, Kilo C, Ido Y. The role of cytosolic reductive stress in oxidant formation and diabetic complications. Diabetes Research and Clinical Practice. 1999;45(2-3):81–82. doi: 10.1016/s0168-8227(99)00034-0. [DOI] [PubMed] [Google Scholar]
- 29.Ido Y, Williamson JR. Hyperglycemic cytosolic reductive stress “pseudohypoxia”: implications for diabetic retinopathy. Investigative Ophthalmology & Visual Science. 1997;38(8):1467–1470. [PubMed] [Google Scholar]
- 30.Ghyczy M, Boros M. Electrophilic methyl groups present in the diet ameliorate pathological states induced by reductive and oxidative stress: a hypothesis. British Journal of Nutrition. 2001;85(4):409–414. doi: 10.1079/bjn2000274. [DOI] [PubMed] [Google Scholar]
- 31.Teodoro JS, Rolo AP, Palmeira CM. The NAD ratio redox paradox: why does too much reductive power cause oxidative stress? Toxicology Mechanisms and Methods. 2013;23(5):297–302. doi: 10.3109/15376516.2012.759305. [DOI] [PubMed] [Google Scholar]
- 32.Tilton RG. Diabetic vascular dysfunction: links to glucose-induced reductive stress and VEGF. Microscopy Research and Technique. 2002;57(5):390–407. doi: 10.1002/jemt.10092. [DOI] [PubMed] [Google Scholar]
- 33.Bassi R, Trevisani A, Tezza S, et al. Regenerative therapies for diabetic microangiopathy. Experimental Diabetes Research. 2012;2012:11 pages. doi: 10.1155/2012/916560.916560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Klover PJ, Mooney RA. Hepatocytes: critical for glucose homeostasis. International Journal of Biochemistry and Cell Biology. 2004;36(5):753–758. doi: 10.1016/j.biocel.2003.10.002. [DOI] [PubMed] [Google Scholar]
- 35.Gerich JE, Woerle HJ, Meyer C, Stumvoll M. Renal gluconeogenesis: its importance in human glucose homeostasis. Diabetes Care. 2001;24(2):382–391. doi: 10.2337/diacare.24.2.382. [DOI] [PubMed] [Google Scholar]
- 36.Gerich JE. Control of glycaemia. Bailliere’s Clinical Endocrinology and Metabolism. 1993;7(3):551–586. doi: 10.1016/s0950-351x(05)80207-1. [DOI] [PubMed] [Google Scholar]
- 37.DeFronzo RA, Ferrannini E. Regulation of hepatic glucose metabolism in humans. Diabetes/Metabolism Reviews. 1987;3(2):415–459. doi: 10.1002/dmr.5610030204. [DOI] [PubMed] [Google Scholar]
- 38.Valadi H, Valadi Å, Ansell R, et al. NADH-reductive stress in Saccharomyces cerevisiae induces the expression of the minor isoform of glyceraldehyde-3-phosphate dehydrogenase (TDH1) Current Genetics. 2004;45(2):90–95. doi: 10.1007/s00294-003-0469-1. [DOI] [PubMed] [Google Scholar]
- 39.Farhana A, Guidry L, Srivastava A, Singh A, Hondalus MK, Steyn AJC. Reductive stress in microbes: implications for understanding mycobacterium tuberculosis disease and persistence. Advances in Microbial Physiology. 2010;57:43–117. doi: 10.1016/B978-0-12-381045-8.00002-3. [DOI] [PubMed] [Google Scholar]
- 40.Lipinski B, Ghyczy M, Boros M. Evidence in support of a concept of reductive stress (multiple letters) British Journal of Nutrition. 2002;87(1):93–94. doi: 10.1079/BJN2001435. [DOI] [PubMed] [Google Scholar]
- 41.Gomes AP, Price NL, Ling AJ, et al. Declining NAD(+) induces a pseudohypoxic state disrupting nuclear-mitochondrial communication during aging. Cell. 2013;155:1624–1638. doi: 10.1016/j.cell.2013.11.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Obrosova IG, Stevens MJ, Lang H-J. Diabetes-induced changes in retinal NAD-redox status: pharmacological modulation and implications for pathogenesis of diabetic retinopathy. Pharmacology. 2001;62(3):172–180. doi: 10.1159/000056091. [DOI] [PubMed] [Google Scholar]
- 43.Van den Enden MK, Nyengaard JR, Ostrow E, Burgan JH, Williamson JR. Elevated glucose levels increase retinal glycolysis and sorbitol pathway metabolism: implications for diabetic retinopathy. Investigative Ophthalmology and Visual Science. 1995;36(8):1675–1685. [PubMed] [Google Scholar]
- 44.Williamson JR, Chang K, Frangos M, et al. Hyperglycemic pseudohypoxia and diabetic complications. Diabetes. 1993;42(6):801–813. doi: 10.2337/diab.42.6.801. [DOI] [PubMed] [Google Scholar]
- 45.Karunanayake EH, Hearse DJ, Mellows G. The synthesis of [14C]streptozotocin and its distribution and excretion in the rat. Biochemical Journal. 1974;142(3):673–683. doi: 10.1042/bj1420673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Virág L, Szabó C. The therapeutic potential of poly(ADP-ribose) polymerase inhibitors. Pharmacological Reviews. 2002;54(3):375–429. doi: 10.1124/pr.54.3.375. [DOI] [PubMed] [Google Scholar]
- 47.Chiu J, Xu BY, Chen S, Feng B, Chakrabarti S. Oxidative stress-induced, poly(ADP-ribose) polymerase-dependent upregulation of ET-1 expression in chronic diabetic complications. Canadian Journal of Physiology and Pharmacology. 2008;86(6):365–372. doi: 10.1139/Y08-033. [DOI] [PubMed] [Google Scholar]
- 48.Oka S-I, Hsu C-P, Sadoshima J. Regulation of cell survival and death by pyridine nucleotides. Circulation Research. 2012;111(5):611–627. doi: 10.1161/CIRCRESAHA.111.247932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bánhegyi G, Mandl J, Csala M. Redox-based endoplasmic reticulum dysfunction in neurological diseases. Journal of Neurochemistry. 2008;107(1):20–34. doi: 10.1111/j.1471-4159.2008.05571.x. [DOI] [PubMed] [Google Scholar]
- 50.Rajasekaran NS, Connell P, Christians ES, et al. Human αB-crystallin mutation causes oxido-reductive stress and protein aggregation cardiomyopathy in mice. Cell. 2007;130(3):427–439. doi: 10.1016/j.cell.2007.06.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yu Q, Lee CF, Wang W, et al. Elimination of NADPH oxidase activity promotes reductive stress and sensitizes the heart to ischemic injury. Journal of the American Heart Association. 2014;3 doi: 10.1161/JAHA.113.000555.e000555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jones DP, Go Y-M. Redox compartmentalization and cellular stress. Diabetes, Obesity and Metabolism. 2010;12(2):116–125. doi: 10.1111/j.1463-1326.2010.01266.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lugrin J, Rosenblatt-Velin N, Parapanov R, Liaudet L. The role of oxidative stress during inflammatory processes. Biological Chemistry. 2014;395:203–230. doi: 10.1515/hsz-2013-0241. [DOI] [PubMed] [Google Scholar]
- 54.Naviaux RK. Oxidative shielding or oxidative stress? Journal of Pharmacology and Experimental Therapeutics. 2012;342(3):608–618. doi: 10.1124/jpet.112.192120. [DOI] [PubMed] [Google Scholar]
- 55.He Q, Wang M, Petucci C, Gardell SJ, Han X. Rotenone induces reductive stress and triacylglycerol deposition in C2C12 cells. International Journal of Biochemistry and Cell Biology. 2013;45:2749–2755. doi: 10.1016/j.biocel.2013.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Esposti MD, Ngo A, Myers MA. Inhibition of mitochondrial complex I may account for IDDM induced by intoxication with the rodenticide vacor. Diabetes. 1996;45(11):1531–1534. doi: 10.2337/diab.45.11.1531. [DOI] [PubMed] [Google Scholar]
- 57.Lamson DW, Plaza SM. Mitochondrial factors in the pathogenesis of diabetes: a hypothesis for treatment. Alternative Medicine Review. 2002;7(2):94–111. [PubMed] [Google Scholar]
- 58.Abdul-Ghani MA, DeFronzo RA. Pathogenesis of insulin resistance in skeletal muscle. Journal of Biomedicine and Biotechnology. 2010;2010:19 pages. doi: 10.1155/2010/476279.476279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wamelink MMC, Struys EA, Jakobs C. The biochemistry, metabolism and inherited defects of the pentose phosphate pathway: a review. Journal of Inherited Metabolic Disease. 2008;31(6):703–717. doi: 10.1007/s10545-008-1015-6. [DOI] [PubMed] [Google Scholar]
- 60.Ola MS, Berkich DA, Xu Y, et al. Analysis of glucose metabolism in diabetic rat retinas. The American Journal of Physiology: Endocrinology and Metabolism. 2006;290(6):E1057–E1067. doi: 10.1152/ajpendo.00323.2005. [DOI] [PubMed] [Google Scholar]
- 61.Rosca MG, Vazquez EJ, Chen Q, Kerner J, Kern TS, Hoppel CL. Oxidation of fatty acids is the source of increased mitochondrial reactive oxygen species production in kidney cortical tubules in early diabetes. Diabetes. 2012;61(8):2074–2083. doi: 10.2337/db11-1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Rebolledo OR, Actis Dato SM. Postprandial hyperglycemia and hyperlipidemia-generated glycoxidative stress: its contribution to the pathogenesis of diabetes complications. European Review for Medical and Pharmacological Sciences. 2005;9(4):191–208. [PubMed] [Google Scholar]
- 63.Reyes A, Cardenas ML. All hexokinase isoenzymes coexist in rat hepatocytes. Biochemical Journal. 1984;221(2):303–309. doi: 10.1042/bj2210303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Otaegui PJ, Ferre T, Pujol A, Riu E, Jimenez R, Bosch F. Expression of glucokinase in skeletal muscle: a new approach to counteract diabetic hyperglycemia. Human Gene Therapy. 2000;11(11):1543–1552. doi: 10.1089/10430340050083270. [DOI] [PubMed] [Google Scholar]
- 65.Pal M. Recent advances in glucokinase activators for the treatment of type 2 diabetes. Drug Discovery Today. 2009;14(15-16):784–792. doi: 10.1016/j.drudis.2009.05.013. [DOI] [PubMed] [Google Scholar]
- 66.Matschinsky FM. Regulation of pancreatic β-cell glucokinase: from basics to therapeutics. Diabetes. 2002;51(3):S394–S404. doi: 10.2337/diabetes.51.2007.s394. [DOI] [PubMed] [Google Scholar]
- 67.Matschinsky F, Liang Y, Kesavan P, et al. Glucokinase as pancreatic β cell glucose sensor and diabetes gene. Journal of Clinical Investigation. 1993;92(5):2092–2098. doi: 10.1172/JCI116809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Postic C, Shiota M, Magnuson MA. Cell-specific roles of glucokinase in glucose homeostasis. Recent Progress in Hormone Research. 2001;56:195–217. doi: 10.1210/rp.56.1.195. [DOI] [PubMed] [Google Scholar]
- 69.Larion M, Miller BG. Homotropic allosteric regulation in monomeric mammalian glucokinase. Archives of Biochemistry and Biophysics. 2012;519(2):103–111. doi: 10.1016/j.abb.2011.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lenzen S. A fresh view of glycolysis and glucokinase regulation: history and current status. Journal of Biological Chemistry. 2014;289(18):12189–12194. doi: 10.1074/jbc.R114.557314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hirst J, King MS, Pryde KR. The production of reactive oxygen species by complex I. Biochemical Society Transactions. 2008;36(5):976–980. doi: 10.1042/BST0360976. [DOI] [PubMed] [Google Scholar]
- 72.Pryde KR, Hirst J. Superoxide is produced by the reduced flavin in mitochondrial complex I: a single, unified mechanism that applies during both forward and reverse electron transfer. Journal of Biological Chemistry. 2011;286(20):18056–18065. doi: 10.1074/jbc.M110.186841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hirst J. Mitochondrial complex I. Annual Review of Biochemistry. 2013;82:551–575. doi: 10.1146/annurev-biochem-070511-103700. [DOI] [PubMed] [Google Scholar]
- 74.Kim J-A, Wei Y, Sowers JR. Role of mitochondrial dysfunction in insulin resistance. Circulation Research. 2008;102(4):401–414. doi: 10.1161/CIRCRESAHA.107.165472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Yan LJ. Protein redox modification as a cellular defense mechanism against tissue ischemic injury. Oxidative Medicine and Cellular Longevity. 2014;2014:12 pages. doi: 10.1155/2014/343154.343154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Esterházy D, King MS, Yakovlev G, Hirst J. Production of reactive oxygen species by complex I (NADH:ubiquinone oxidoreductase) from Escherichia coli and comparison to the enzyme from mitochondria. Biochemistry. 2008;47(12):3964–3971. doi: 10.1021/bi702243b. [DOI] [PubMed] [Google Scholar]
- 77.Ceriello A. Postprandial hyperglycemia and diabetes complications: is it time to treat? Diabetes. 2005;54(1):1–7. doi: 10.2337/diabetes.54.1.1. [DOI] [PubMed] [Google Scholar]
- 78.St-Pierre J, Buckingham JA, Roebuck SJ, Brand MD. Topology of superoxide production from different sites in the mitochondrial electron transport chain. Journal of Biological Chemistry. 2002;277(47):44784–44790. doi: 10.1074/jbc.M207217200. [DOI] [PubMed] [Google Scholar]
- 79.Murphy MP. How mitochondria produce reactive oxygen species. Biochemical Journal. 2009;417(1):1–13. doi: 10.1042/BJ20081386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Dröse S, Brandt U. Molecular mechanisms of superoxide production by the mitochondrial respiratory chain. Advances in Experimental Medicine and Biology. 2012;748:145–169. doi: 10.1007/978-1-4614-3573-0_6. [DOI] [PubMed] [Google Scholar]
- 81.Siebels I, Drose S. Q-site inhibitor induced ROS production of mitochondrial complex II is attenuated by TCA cycle dicarboxylates. Biochimica et Biophysica Acta. 2013;1827:1156–1164. doi: 10.1016/j.bbabio.2013.06.005. [DOI] [PubMed] [Google Scholar]
- 82.Yan L-J, Sumien N, Thangthaeng N, Forster MJ. Reversible inactivation of dihydrolipoamide dehydrogenase by mitochondrial hydrogen peroxide. Free Radical Research. 2013;47(2):123–133. doi: 10.3109/10715762.2012.752078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Quinlan CL, Goncalves RL, Hey-Mogensen M, Yadava N, Bunik VI, Brand MD. The 2-oxoacid dehydrogenase complexes in mitochondria can produce superoxide/hydrogen peroxide at much higher rates than complex I. Journal of Biological Chemistry. 2014 doi: 10.1074/jbc.M113.545301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Jackson MJ, Papa S, Bolaños J, et al. Antioxidants, reactive oxygen and nitrogen species, gene induction and mitochondrial function. Molecular Aspects of Medicine. 2002;23(1–3):209–285. doi: 10.1016/s0098-2997(02)00018-3. [DOI] [PubMed] [Google Scholar]
- 85.Yan LJ. Positive oxidative stress in aging and aging-related disease tolerance. Redox Biology. 2014;2:165–169. doi: 10.1016/j.redox.2014.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Turrens JF, Alexandre A, Lehninger AL. Ubisemiquinone is the electron donor for superoxide formation by complex III of heart mitochondria. Archives of Biochemistry and Biophysics. 1985;237(2):408–414. doi: 10.1016/0003-9861(85)90293-0. [DOI] [PubMed] [Google Scholar]
- 87.Turrens JF. Superoxide production by the mitochondrial respiratory chain. Bioscience Reports. 1997;17(1):3–8. doi: 10.1023/a:1027374931887. [DOI] [PubMed] [Google Scholar]
- 88.Cai Z, Yan LJ. Protein oxidative modifications: beneficial roles in disease and health. Journal of Biochemical and Pharmacological Research. 2013;1:15–26. [PMC free article] [PubMed] [Google Scholar]
- 89.Shah S, Iqbal M, Karam J, Salifu M, McFarlane SI. Oxidative stress, glucose metabolism, and the prevention of type 2 diabetes: pathophysiological insights. Antioxidants and Redox Signaling. 2007;9(7):911–929. doi: 10.1089/ars.2007.1629. [DOI] [PubMed] [Google Scholar]
- 90.Yan LJ. Current Protocols in Protein Science. chapter 14, unit 14. 2009. Analysis of oxidative modification of proteins. [DOI] [PubMed] [Google Scholar]
- 91.Stadtman ER. Biochemical markers of aging. Experimental Gerontology. 1988;23(4-5):327–347. doi: 10.1016/0531-5565(88)90036-8. [DOI] [PubMed] [Google Scholar]
- 92.Yan L-J, Levine RL, Sohal RS. Oxidative damage during aging targets mitochondrial aconitase. Proceedings of the National Academy of Sciences of the United States of America. 1997;94:11168–11172. doi: 10.1073/pnas.94.21.11168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Dawson TL, Gores GJ, Nieminen A-L, Herman B, Lemasters JJ. Mitochondria as a source of reactive oxygen species during reductive stress in rat hepatocytes. The American Journal of Physiology: Cell Physiology. 1993;264(4):C961–C967. doi: 10.1152/ajpcell.1993.264.4.C961. [DOI] [PubMed] [Google Scholar]
- 94.Pung YF, Chilian WM. Corruption of coronary collateral growth in metabolic syndrome: role of oxidative stress. World Journal of Cardiology. 2010;2:421–427. doi: 10.4330/wjc.v2.i12.421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Pall ML. Explaining “Unexplained Illnesses”: Disease Paradigm for Chronic Fatigue Syndrome, Multiple Chemical Sensitivity, Fibromyalgia, Post-Traumatic Stress Disorder, and Gulf War Syndrome. Harrington Park Press; 2007. [Google Scholar]
- 96.Brownlee M. Biochemistry and molecular cell biology of diabetic complications. Nature. 2001;414(6865):813–820. doi: 10.1038/414813a. [DOI] [PubMed] [Google Scholar]
- 97.Sakai K, Matsumoto K, Nishikawa T, et al. Mitochondrial reactive oxygen species reduce insulin secretion by pancreatic β-cells. Biochemical and Biophysical Research Communications. 2003;300(1):216–222. doi: 10.1016/s0006-291x(02)02832-2. [DOI] [PubMed] [Google Scholar]
- 98.Du X, Matsumura T, Edelstein D, et al. Inhibition of GAPDH activity by poly(ADP-ribose) polymerase activates three major pathways of hyperglycemic damage in endothelial cells. Journal of Clinical Investigation. 2003;112(7):1049–1057. doi: 10.1172/JCI18127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Zhang T, Gong Y, Zhou H, Xie P, Guan S, Yi W. Oxidative stress-the key mechanisms of diabetic peripheral neuropathy. North The American Journal of Medicine and Science. 2013;6:87–90. [Google Scholar]
- 100.Madsen-Bouterse S, Mohammad G, Kowluru RA. Glyceraldehyde-3-phosphate dehydrogenase in retinal microvasculature: implications for the development and progression of diabetic retinopathy. Investigative Ophthalmology and Visual Science. 2010;51(3):1765–1772. doi: 10.1167/iovs.09-4171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Fantus IG. The pathogenesis of the chronic complications of the diabetes mellitus. Endocrinology Rounds. 2002;2:1–8. [Google Scholar]
- 102.Puthanveetil P, Zhang D, Wang Y, et al. Diabetes triggers a PARP1 mediated death pathway in the heart through participation of FoxO1. Journal of Molecular and Cellular Cardiology. 2012;53(5):677–686. doi: 10.1016/j.yjmcc.2012.08.013. [DOI] [PubMed] [Google Scholar]
- 103.Hwang NR, Yim S-H, Kim YM, et al. Oxidative modifications of glyceraldehyde-3-phosphate dehydrogenase play a key role in its multiple cellular functions. Biochemical Journal. 2009;423(2):253–264. doi: 10.1042/BJ20090854. [DOI] [PubMed] [Google Scholar]
- 104.Trentham DR. Aspects of the chemistry of D-glyceraldehyde 3-phosphate dehydrogenase. Biochemical Journal. 1968;109(4):603–612. doi: 10.1042/bj1090603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Rivera-Nieves J, Thompson WC, Levine RL, Moss J. Thiols mediate superoxide-dependent NADH modification of glyceraldehyde-3-phosphate dehydrogenase. Journal of Biological Chemistry. 1999;274(28):19525–19531. doi: 10.1074/jbc.274.28.19525. [DOI] [PubMed] [Google Scholar]
- 106.Ussher JR, Jaswal JS, Lopaschuk GD. Pyridine nucleotide regulation of cardiac intermediary metabolism. Circulation Research. 2012;111(5):628–641. doi: 10.1161/CIRCRESAHA.111.246371. [DOI] [PubMed] [Google Scholar]
- 107.Funk SD, Yurdagul A, Orr AW. Hyperglycemia and endothelial dysfunction in atherosclerosis: lessons from type 1 diabetes. International Journal of Vascular Medicine. 2012;2012:19 pages. doi: 10.1155/2012/569654.569654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Rask-Madsen C, King GL. Vascular complications of diabetes: mechanisms of injury and protective factors. Cell Metabolism. 2013;17(1):20–33. doi: 10.1016/j.cmet.2012.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Abdul-Ghani MA, DeFronzo RA. Oxidative stress in type 2 diabetes. In: Miwa S, Beckman KB, Muller FL, editors. Oxidative Stress in Aging. Humana Press; 2008. pp. 191–212. [Google Scholar]
- 110.Davidson MB, Bate G, Kirkpatrick P. Fresh from the pipeline: exenatide. Nature Reviews Drug Discovery. 2005;4(9):713–714. doi: 10.1038/nrd1828. [DOI] [PubMed] [Google Scholar]
- 111.Poitout V, Robertson RP. Minireview: secondary β-cell failure in type 2 diabetes: a convergence of glucotoxicity and lipotoxicity. Endocrinology. 2002;143(2):339–342. doi: 10.1210/endo.143.2.8623. [DOI] [PubMed] [Google Scholar]
- 112.Poitout V, Robertson RP. Glucolipotoxicity: fuel excess and β-cell dysfunction. Endocrine Reviews. 2008;29(3):351–366. doi: 10.1210/er.2007-0023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Yang Y, Hayden MR, Sowers S, Bagree SV, Sowers JR. Retinal redox stress and remodeling in cardiometabolic syndrome and diabetes. Oxidative Medicine and Cellular Longevity. 2010;3(6) doi: 10.4161/oxim.3.6.14786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Callaghan MJ, Ceradini DJ, Gurtner GC. Hyperglycemia-induced reactive oxygen species and impaired endothelial progenitor cell function. Antioxidants and Redox Signaling. 2005;7(11-12):1476–1482. doi: 10.1089/ars.2005.7.1476. [DOI] [PubMed] [Google Scholar]
- 115.Kassab A, Piwowar A. Cell oxidant stress delivery and cell dysfunction onset in type 2 diabetes. Biochimie. 2012;94(9):1837–1848. doi: 10.1016/j.biochi.2012.01.020. [DOI] [PubMed] [Google Scholar]
- 116.Yasunari K, Kohno M, Kano H, Minami M, Yoshikawa J. Aldose reductase inhibitor improves insulin-mediated glucose uptake and prevents migration of human coronary artery smooth muscle cells induced by high glucose. Hypertension. 2000;35(5):1092–1098. doi: 10.1161/01.hyp.35.5.1092. [DOI] [PubMed] [Google Scholar]
- 117.Ng TF, Lee FK, Song ZT, et al. Effects of sorbitol dehydrogenase deficiency on nerve conduction in experimental diabetic mice. Diabetes. 1998;47:961–966. doi: 10.2337/diabetes.47.6.961. [DOI] [PubMed] [Google Scholar]
- 118.Lee AYW, Chung SSM. Contributions of polyol pathway to oxidative stress in diabetic cataract. The FASEB Journal. 1999;13(1):23–30. doi: 10.1096/fasebj.13.1.23. [DOI] [PubMed] [Google Scholar]
- 119.Brownlee M. The pathobiology of diabetic complications: a unifying mechanism. Diabetes. 2005;54(6):1615–1625. doi: 10.2337/diabetes.54.6.1615. [DOI] [PubMed] [Google Scholar]
- 120.Yabe-Nishimura C. Aldose reductase in glucose toxicity: a potential target for the prevention of diabetic complications. Pharmacological Reviews. 1998;50(1):21–33. [PubMed] [Google Scholar]
- 121.Dunlop M. Aldose reductase and the role of the polyol pathway in diabetic nephropathy. Kidney International, Supplement. 2000;58(77):S3–S12. doi: 10.1046/j.1523-1755.2000.07702.x. [DOI] [PubMed] [Google Scholar]
- 122.Sena LA, Chandel NS. Physiological roles of mitochondrial reactive oxygen species. Molecular Cell. 2012;48(2):158–166. doi: 10.1016/j.molcel.2012.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Li Q, Hwang YC, Ananthakrishnan R, Oates PJ, Guberski D, Ravichandran R. Polyol pathway and modulation of ischemia-reperfusion injury in Type 2 diabetic BBZ rat hearts. Cardiovascular Diabetology. 2008;7, article 33 doi: 10.1186/1475-2840-7-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Gabbay KH. The sorbitol pathway and the complications of diabetes. The New England Journal of Medicine. 1973;288(16):831–836. doi: 10.1056/NEJM197304192881609. [DOI] [PubMed] [Google Scholar]
- 125.Kador PF, Kinoshita JH. Role of aldose reductase in the development of diabetes-associated complications. The American Journal of Medicine A. 1985;79(5):8–12. doi: 10.1016/0002-9343(85)90504-2. [DOI] [PubMed] [Google Scholar]
- 126.Yan L-J, Christians ES, Liu L, Xiao X, Sohal RS, Benjamin IJ. Mouse heat shock transcription factor 1 deficiency alters cardiac redox homeostasis and increases mitochondrial oxidative damage. The EMBO Journal. 2002;21(19):5164–5172. doi: 10.1093/emboj/cdf528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Ohmura C, Watada H, Azuma K, et al. Aldose reductase inhibitor, epalrestat, reduces lipid hydroperoxides in type 2 diabetes. Endocrine Journal. 2009;56(1):149–156. doi: 10.1507/endocrj.k08e-237. [DOI] [PubMed] [Google Scholar]
- 128.Yeung C-M, Lo ACY, Cheung AKH, Chung SSM, Wong D, Chung SK. More severe type 2 diabetes-associated ischemic stroke injury is alleviated in aldose reductase-deficient mice. Journal of Neuroscience Research. 2010;88(9):2026–2034. doi: 10.1002/jnr.22349. [DOI] [PubMed] [Google Scholar]
- 129.Tang WH, Martin KA, Hwa J. Aldose reductase, oxidative stress, and diabetic mellitus. Frontiers in Pharmacology. 2012;3, article 87 doi: 10.3389/fphar.2012.00087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Tang WH, Wu S, Wong TM, Chung SK, Chung SSM. Polyol pathway mediates iron-induced oxidative injury in ischemic-reperfused rat heart. Free Radical Biology and Medicine. 2008;45(5):602–610. doi: 10.1016/j.freeradbiomed.2008.05.003. [DOI] [PubMed] [Google Scholar]
- 131.Hwang YC, Bakr S, Ellery CA, Oates PJ, Ramasamy R. Sorbitol dehydrogenase: a novel target for adjunctive protection of ischemic myocardium. The FASEB Journal. 2003;17(15):2331–2333. doi: 10.1096/fj.03-0128fje. [DOI] [PubMed] [Google Scholar]
- 132.Cameron NE, Cotter MA. The relationship of vascular changes to metabolic factors in diabetes mellitus and their role in the development of peripheral nerve complications. Diabetes/Metabolism Reviews. 1994;10(3):189–224. doi: 10.1002/dmr.5610100302. [DOI] [PubMed] [Google Scholar]
- 133.Issad T, Masson E, Pagesy P. O-GlcNAc modification, insulin signaling and diabetic complications. Diabetes and Metabolism. 2010;36(6):423–435. doi: 10.1016/j.diabet.2010.09.001. [DOI] [PubMed] [Google Scholar]
- 134.Ma J, Hart GW. Protein O-GlcNAcylation in diabetes and diabetic complications. Expert Review of Proteomics. 2013;10:365–380. doi: 10.1586/14789450.2013.820536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Fardini Y, Masson E, Boudah O, et al. O-GlcNAcylation of FoxO1 in pancreatic beta cells promotes Akt inhibition through an IGFBP1-mediated autocrine mechanism. The FASEB Journal. 2014;28:1010–1021. doi: 10.1096/fj.13-238378. [DOI] [PubMed] [Google Scholar]
- 136.Gupta Rajapakse A, Ming X-F, Carvas JM, Yang Z. O-linked β-N-acetylglucosamine during hyperglycemia exerts both anti-inflammatory and pro-oxidative properties in the endothelial system. Oxidative Medicine and Cellular Longevity. 2009;2(3):172–175. doi: 10.4161/oxim.2.3.8482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Goldberg H, Whiteside C, George Fantus I. O-linked β-N-acetylglucosa mine supports p38 MAPK activation by high glucose in glomerular mesangial cells. The American Journal of Physiology: Endocrinology and Metabolism. 2011;301(4):E713–E726. doi: 10.1152/ajpendo.00108.2011. [DOI] [PubMed] [Google Scholar]
- 138.Lima VV, Spitler K, Choi H, Webb RC, Tostes RC. O-glcnacylation and oxidation of proteins: is signalling in the cardiovascular system becoming sweeter? Clinical Science. 2012;123(8):473–486. doi: 10.1042/CS20110638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.McLarty JL, Marsh SA, Chatham JC. Post-translational protein modification by O-linked N-acetyl-glucosamine: its role in mediating the adverse effects of diabetes on the heart. Life Sciences. 2013;92(11):621–627. doi: 10.1016/j.lfs.2012.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Gurel Z, Sieg KM, Shallow KD, Sorenson CM, Sheibani N. Retinal O-linked N-acetylglucosamine protein modifications: implications for postnatal retinal vascularization and the pathogenesis of diabetic retinopathy. Molecular Vision. 2013;19:1047–1059. [PMC free article] [PubMed] [Google Scholar]
- 141.Schleicher ED, Weigert C. Role of the hexosamine biosynthetic pathway in diabetic nephropathy. Kidney International, Supplement. 2000;58(77):S13–S18. doi: 10.1046/j.1523-1755.2000.07703.x. [DOI] [PubMed] [Google Scholar]
- 142.Semba RD, Huang H, Lutty GA, Van Eyk JE, Hart GW. The role of O-GlcNAc signaling in the pathogenesis of diabetic retinopathy. Proteomics: Clinical Applications. 2014;8:218–231. doi: 10.1002/prca.201300076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Feng B, Ruiz MA, Chakrabarti S. Oxidative-stress-induced epigenetic changes in chronic diabetic complications. Canadian Journal of Physiology and Pharmacology. 2013;91(3):213–220. doi: 10.1139/cjpp-2012-0251. [DOI] [PubMed] [Google Scholar]
- 144.Xia L, Wang H, Munk S, et al. Reactive oxygen species, PKC-α1, and PKC-ζ mediate high-glucose-induced vascular endothelial growth factor expression in mesangial cells. The American Journal of Physiology: Endocrinology and Metabolism. 2007;293(5):E1280–E1288. doi: 10.1152/ajpendo.00223.2007. [DOI] [PubMed] [Google Scholar]
- 145.Sasaki S, Inoguchi T. The role of oxidative stress in the pathogenesis of diabetic vascular complications. Diabetes and Metabolism Journal. 2012;36(4):255–261. doi: 10.4093/dmj.2012.36.4.255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Xia L, Wang H, Munk S, et al. High glucose activates PKC-ζ and NADPH oxidase through autocrine TGF-β1 signaling in mesangial cells. The American Journal of Physiology: Renal Physiology. 2008;295(6):F1705–F1714. doi: 10.1152/ajprenal.00043.2008. [DOI] [PubMed] [Google Scholar]
- 147.Teshima Y, Takahashi N, Nishio S, et al. Production of reactive oxygen species in the diabetic heart: roles of mitochondria and NADPH oxidase. Circulation Journal. 2014;78:300–306. doi: 10.1253/circj.cj-13-1187. [DOI] [PubMed] [Google Scholar]
- 148.Fontayne A, Dang PM-C, Gougerot-Pocidalo M-A, El Benna J. Phosphorylation of p47phox sites by PKC α, βII, δ, and ζ: effect on binding to p22phox and on NADPH oxidase activation. Biochemistry. 2002;41(24):7743–7750. doi: 10.1021/bi011953s. [DOI] [PubMed] [Google Scholar]
- 149.Bey EA, Xu B, Bhattacharjee A, et al. Protein kinase Cδ is required for p47phox phosphorylation and translocation in activated human monocytes. Journal of Immunology. 2004;173(9):5730–5738. doi: 10.4049/jimmunol.173.9.5730. [DOI] [PubMed] [Google Scholar]
- 150.Naruse K, Rask-Madsen C, Takahara N, et al. Activation of vascular protein kinase C-beta; inhibits Akt-dependent endothelial nitric oxide synthase function in obesity-associated insulin resistance. Diabetes. 2006;55(3):691–698. doi: 10.2337/diabetes.55.03.06.db05-0771. [DOI] [PubMed] [Google Scholar]
- 151.Mellor KM, Brimble MA, Delbridge LM. Glucose as an agent of post-translational modification in diabetes: new cardiac epigenetic insights. Life sciences. 2014 doi: 10.1016/j.lfs.2014.03.020. [DOI] [PubMed] [Google Scholar]
- 152.Lovestone S, Smith U. Advanced glycation end products, dementia, and diabetes. Proceedings of the National Academy of Sciences of the United States of America. 2014;111:4743–4744. doi: 10.1073/pnas.1402277111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Queisser MA, Yao D, Geisler S, et al. Hyperglycemia impairs proteasome function by methylglyoxal. Diabetes. 2010;59(3):670–678. doi: 10.2337/db08-1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Thornalley PJ, Langborg A, Minhas HS. Formation of glyoxal, methylglyoxal and 8-deoxyglucosone in the glycation of proteins by glucose. Biochemical Journal. 1999;344(1):109–116. [PMC free article] [PubMed] [Google Scholar]
- 155.Koga M, Murai J, Morita S, Saito H, Kasayama S. Comparison of annual variability in HbA1c and glycated albumin in patients with type 1 vs. type 2 diabetes mellitus. Journal of Diabetes and its Complications. 2013;27(3):211–213. doi: 10.1016/j.jdiacomp.2012.12.001. [DOI] [PubMed] [Google Scholar]
- 156.Gholap NN, Davies MJ, Mostafa SA, Khunti K. Diagnosing type 2 diabetes and identifying high-risk individuals using the new glycated haemoglobin (HbA1c) criteria. British Journal of General Practice. 2013;63(607):e165–e167. doi: 10.3399/bjgp13X663244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Wells-Knecht KJ, Brinkmann E, Wells-Knecht MC, et al. New biomarkers of Maillard reaction damage to proteins. Nephrology Dialysis Transplantation. 1996;11(5):41–47. doi: 10.1093/ndt/11.supp5.41. [DOI] [PubMed] [Google Scholar]
- 158.Wells-Knecht KJ, Zyzak DV, Litchfield JE, Thorpe SR, Baynes JW. Mechanism of autoxidative glycosylation: identification of glyoxal and arabinose as intermediates in the autoxidative modification of proteins by glucose. Biochemistry. 1995;34(11):3702–3709. doi: 10.1021/bi00011a027. [DOI] [PubMed] [Google Scholar]
- 159.Muñoz A, Costa M. Nutritionally mediated oxidative stress and inflammation. Oxidative Medicine and Cellular Longevity. 2013;2013:11 pages. doi: 10.1155/2013/610950.610950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Hayden MR, Tyagi SC. Islet redox stress: the manifold toxicities of insulin resistance, metabolic syndrome and amylin derived islet amyloid in type 2 diabetes mellitus. Journal of the Pancreas. 2002;3(4):86–108. [PubMed] [Google Scholar]
- 161.Vlassara H, Striker GE. Advanced glycation endproducts in diabetes and diabetic complications. Endocrinology and Metabolism Clinics of North America. 2013;42:697–719. doi: 10.1016/j.ecl.2013.07.005. [DOI] [PubMed] [Google Scholar]
- 162.Mira ML, Martinho F, Azevedo MS, Manso CF. Oxidative inhibition of red blood cell ATPases by glyceraldehyde. Biochimica et Biophysica Acta: Bioenergetics. 1991;1060(3):257–261. doi: 10.1016/s0005-2728(05)80315-9. [DOI] [PubMed] [Google Scholar]
- 163.Wolff SP, Dean RT. Glucose autoxidation and protein modification. The potential role of “autoxidative glycosylation” in diabetes. Biochemical Journal. 1987;245(1):243–250. doi: 10.1042/bj2450243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Eiselein L, Schwartz HJ, Rutledge JC. The challenge of type 1 diabetes mellitus. ILAR Journal. 2004;45(3):231–236. doi: 10.1093/ilar.45.3.231. [DOI] [PubMed] [Google Scholar]
- 165.Bocci V, Zanardi I, Huijberts MS, Travagli V. An integrated medical treatment for type-2 diabetes. Diabetes and Metabolic Syndrome. 2014;8:57–61. doi: 10.1016/j.dsx.2013.10.004. [DOI] [PubMed] [Google Scholar]
- 166.Ye J. Mechanisms of insulin resistance in obesity. Frontiers of Medicine in China. 2013;7(1):14–24. doi: 10.1007/s11684-013-0262-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Yang X, Feng L, Li C, Li Y. Tranilast alleviates endothelial dysfunctions and insulin resistance via preserving glutathione peroxidase 1 in rats fed a high-fat emulsion. Journal of Pharmacological Sciences. 2014;124:18–30. doi: 10.1254/jphs.13151fp. [DOI] [PubMed] [Google Scholar]
- 168.Robertson RP, Zhang H-J, Pyzdrowski KL, Walseth TF. Preservation of insulin mRNA levels and insulin secretion in HIT cells by avoidance of chronic exposure to high glucose concentrations. Journal of Clinical Investigation. 1992;90(2):320–325. doi: 10.1172/JCI115865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Karunakaran U, Park K-G. A systematic review of oxidative stress and safety of antioxidants in diabetes: focus on islets and their defense. Diabetes and Metabolism Journal. 2013;37(2):106–112. doi: 10.4093/dmj.2013.37.2.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Katsuki A, Sumida Y, Urakawa H, et al. Increased oxidative stress is associated with serum levels of triglyceride, insulin resistance, and hyperinsulinemia in Japanese metabolically obese, normal-weight men. Diabetes Care. 2004;27(2):631–632. doi: 10.2337/diacare.27.2.631. [DOI] [PubMed] [Google Scholar]
- 171.Wittmann I, Nagy J, Ceriello A, Pirisi M. Are insulin resistance and atherosclerosis the consequences of oxidative stress? Diabetologia. 1996;39(8):1002–1003. doi: 10.1007/BF00403924. [DOI] [PubMed] [Google Scholar]
- 172.Paolisso G, D’Amore A, Volpe C, et al. Evidence for a relationship between oxidative stress and insulin action in non-insulin-dependent (type II) diabetic patients. Metabolism: Clinical and Experimental. 1994;43(11):1426–1429. doi: 10.1016/0026-0495(94)90039-6. [DOI] [PubMed] [Google Scholar]
- 173.Urakawa H, Katsuki A, Sumida Y, et al. Oxidative stress ts associated with adiposity and insulin resistance in men. Journal of Clinical Endocrinology and Metabolism. 2003;88(10):4673–4676. doi: 10.1210/jc.2003-030202. [DOI] [PubMed] [Google Scholar]
- 174.Tanaka Y, Gleason CE, Tran POT, Harmon JS, Robertson RP. Prevention of glucose toxicity in HIT-T15 cells and Zucker diabetic fatty rats by antioxidants. Proceedings of the National Academy of Sciences of the United States of America. 1999;96(19):10857–10862. doi: 10.1073/pnas.96.19.10857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Robertson RP, Tanaka Y, Takahashi H, Tran POT, Harmon JS. Prevention of oxidative stress by adenoviral overexpression of glutathione-related enzymes in pancreatic islets. Annals of the New York Academy of Sciences. 2005;1043:513–520. doi: 10.1196/annals.1333.058. [DOI] [PubMed] [Google Scholar]
- 176.Matsuoka T-A, Kajimoto Y, Watada H, et al. Glycation-dependent, reactive oxygen species-mediated suppression of the insulin gene promoter activity in HIT cells. Journal of Clinical Investigation. 1997;99(1):144–150. doi: 10.1172/JCI119126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Butler AE, Janson J, Bonner-Weir S, Ritzel R, Rizza RA, Butler PC. β-cell deficit and increased β-cell apoptosis in humans with type 2 diabetes. Diabetes. 2003;52(1):102–110. doi: 10.2337/diabetes.52.1.102. [DOI] [PubMed] [Google Scholar]
- 178.Pennathur S, Wagner JD, Leeuwenburgh C, Litwak KN, Heinecke JW. A hydroxyl radical-like species oxidizes cynomolgus monkey artery wall proteins in early diabetic vascular disease. Journal of Clinical Investigation. 2001;107(7):853–860. doi: 10.1172/JCI11194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Rindler PM, Crewe CL, Fernandes J, Kinter M, Szweda LI. Redox regulation of insulin sensitivity due to enhanced fatty acid utilization in the mitochondria. The American Journal of Physiology: Heart and Circulatory Physiology. 2013;305(5):H634–H643. doi: 10.1152/ajpheart.00799.2012. [DOI] [PubMed] [Google Scholar]
- 180.Henriksen EJ, Diamond-Stanic MK, Marchionne EM. Oxidative stress and the etiology of insulin resistance and type 2 diabetes. Free Radical Biology and Medicine. 2011;51(5):993–999. doi: 10.1016/j.freeradbiomed.2010.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Katakam PVG, Snipes JA, Steed MM, Busija DW. Insulin-induced generation of reactive oxygen species and uncoupling of nitric oxide synthase underlie the cerebrovascular insulin resistance in obese rats. Journal of Cerebral Blood Flow and Metabolism. 2012;32(5):792–804. doi: 10.1038/jcbfm.2011.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Fisher-Wellman KH, Neufer PD. Linking mitochondrial bioenergetics to insulin resistance via redox biology. Trends in Endocrinology and Metabolism. 2012;23(3):142–153. doi: 10.1016/j.tem.2011.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Lemasters JJ, Nieminen A-L. Mitochondrial oxygen radical formation during reductive and oxidative stress to intact hepatocytes. Bioscience Reports. 1997;17(3):281–291. doi: 10.1023/a:1027332611839. [DOI] [PubMed] [Google Scholar]
- 184.Cernea S, Dobreanu M. Diabetes and beta cell function: from mechanisms to evaluation and clinical implications. Biochemical Medicine. 2013;23:266–280. doi: 10.11613/BM.2013.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Roseman HM. Progression from obesity to type 2 diabetes: lipotoxicity, glucotoxicity, and implications for management. Journal of Managed Care Pharmacy. 2005;11(6, supplement):S3–S11. [PubMed] [Google Scholar]
- 186.Johnson EL. Glycemic variability in type 2 diabetes mellitus: oxidative stress and macrovascular complications. Advances in Experimental Medicine and Biology. 2012;771:139–154. doi: 10.1007/978-1-4614-5441-0_13. [DOI] [PubMed] [Google Scholar]
- 187.Tiganis T. Reactive oxygen species and insulin resistance: the good, the bad and the ugly. Trends in Pharmacological Sciences. 2011;32(2):82–89. doi: 10.1016/j.tips.2010.11.006. [DOI] [PubMed] [Google Scholar]
- 188.Rains JL, Jain SK. Oxidative stress, insulin signaling, and diabetes. Free Radical Biology and Medicine. 2011;50(5):567–575. doi: 10.1016/j.freeradbiomed.2010.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Piconi L, Quagliaro L, Ceriello A. Oxidative stress in diabetes. Clinical Chemistry and Laboratory Medicine. 2003;41(9):1144–1149. doi: 10.1515/CCLM.2003.177. [DOI] [PubMed] [Google Scholar]
- 190.Ceriello A. New insights on oxidative stress and diabetic complications may lead to a “causal” antioxidant therapy. Diabetes Care. 2003;26(5):1589–1596. doi: 10.2337/diacare.26.5.1589. [DOI] [PubMed] [Google Scholar]
- 191.Yan L-J, Yang S-H, Shu H, Prokai L, Forster MJ. Histochemial staining and qualification of dihydrolipoamide dehydrogenase diaphorase activity using blue native PAGE. Electrophoresis. 2007;28(7):1036–1045. doi: 10.1002/elps.200600574. [DOI] [PubMed] [Google Scholar]