

Abstract
Bees are manufacturers of relevant economical products and have a pollinator role fundamental to ecosystems. Traditionally, studies focused on the genus Melipona have been mostly based on behavioral, and social organization and ecological aspects. Only recently the evolutionary history of this genus has been assessed using molecular markers, including mitochondrial genes. Even though these studies have shed light on the evolutionary history of the Melipona genus, a more accurate picture may emerge when full nuclear and mitochondrial genomes of Melipona species become available. Here we present the assembly, annotation, and characterization of a draft mitochondrial genome of the Brazilian stingless bee Melipona scutellaris using Melipona bicolor as a reference organism. Using Illumina MiSeq data, we achieved the annotation of all protein coding genes, as well as the genes for the two ribosomal subunits (16S and 12S) and transfer RNA genes as well. Using the COI sequence as a DNA barcode, we found that M. cramptoni is the closest species to M. scutellaris.
1. Introduction
Melipona scutellaris, popularly known as uruçu bee, is a stingless bee species profusely found from Bahia to Pernambuco Brazilian states. They are present in urban and rural environments and their pollinator role is pivotal to the ecosystems in which they live [1, 2].
The genus Melipona has long been target of ecological, genetic, and especially behavioral and pollination studies. Existing molecular phylogenetic studies have shown that the Melipona genus clusters with other neotropical Meliponini. Furthermore, Melipona spp. form a well-supported monophyletic group, as determined by recent studies. Stingless bees, especially, compose an ancient group, distributed worldwide around tropics, making them important species for phylogenetic relationships studies. Thus, the elucidation of social behavioral evolution relies on a better understanding of phylogenetic relationships for eusocial insects [3, 4]. For this reason, there is an increasing interest on getting more accurate phylogenetic reconstructions, based on molecular aspects.
Phylogenetic studies have explored nuclear ribosomal genes. Since 18S and 28S subunits are short genes they are therefore easily amplified and sequenced. Besides, such genes are located in very informative regions, where conserved or variable sequences may enlighten phylogenetic relations between species [5, 6]. The mitochondrial genome has also been included in such studies, since its potential for providing evolutionary information was recognized. Metazoan mitogenomes are about 16 kb long and contain 37 genes: 22 tRNAs, 2 rRNAs (16S and 12S subunits), and 13 oxidative phosphorylation proteins—7 from complex I (ND1, ND2, ND3, ND4, ND4L, ND5, and ND6), one from complex III (cytochrome B), three from complex IV (COI, COII, and COIII), and two from complex V (ATPase6 and ATPase8) [7].
The tRNA genes are embedded in variable regions. Through evolution, these regions underwent rearrangements more often than protein coding regions. Therefore, tRNA order is a tool for comparative phylogenetic analysis between species [8]. Additionally, the high copy number per cell, low recombination rate, high mutational rate, and dominantly maternal heritance have made the mitochondrial genome a powerful tool for evolutionary studies.
DNA barcoding, or taxon identification using a standardized genomic region, was initially used in the study of animal specimens [9] and later for a wider diversity of organisms. DNA barcoding based on the cytochrome c oxidase subunit 1 (COI) gene has since then become a widely accepted molecular marker for species identification. This 650 bp long sequence is a simple and reliable tool for metazoan species identification, indicating their molecular divergences or similarities [4]. Being such a short sequence, it is useful for robust phylogeny analysis and important for tracking and measuring ecosystems biodiversity, what has lately been referred to as metabarcoding [10].
Other genes may be employed for DNA barcoding, but, especially among Arthropoda, the interest on COI over other genes is mainly due to its well conserved and single copy sequence, which makes the amplification by PCR reaction easier, allowing the usage of a small set of primers. Still, COI sequence presents faster substitution rates than nuclear genes and its variations are remarkably more interspecific than intraspecific [10, 11].
DNA barcoding applications for insects have been very successful [11]. This technique allows species identification in different life stages (eggs, larvae, nymphs, and pupae), when morphologic characteristics are not easily identified [12, 13]. Besides, it makes species identification possible with only tissues or fragmented parts of the insect [14]. Further, the importance of phylogenetic relationship analysis relies on understanding ecosystems biodiversity, taking into account that insects are important at pollination, decomposition, pest control, and even disease vectors [10].
The present study characterized the draft of M. scutellaris mitogenome annotation according to its gene order, gene conservation, and taxonomic characterization by DNA barcoding.
2. Material and Methods
2.1. Biological Material
Total DNA was extracted from a pool of five male individuals from the Meliponary UFU at Universidade Federal de Uberlândia, campus Umuarama (S 180 55′/W 450 17′). DNA extraction was performed with CTAB buffer, which consists of 2% (w/v) CTAB diluted in 100 mM Tris-HCl, 20 mM EDTA, and 1.4 M NaCl. Immediately before maceration, 0.2% (v/v) β-mercaptoethanol was added; 150 μL of CTAB buffer was added and maceration was performed manually with a pestle. Then 350 μL of CTAB buffer and 5 μL of RNase solution (100 mg/mL) were added, following incubation at 37°C for 1 hour. Later, 5 μL of proteinase K solution (20 mg/mL) was added with additional incubation at 50°C for 1 hour. For homogenate extraction, addition of 240 μL of Phenol/Chloroform/Isoamyl alcohol (25 : 24 : 1) and then centrifugation at 12,000 ×g for 10 minutes. Supernatant was transferred for a new tube and DNA was precipitated with 500 μL of absolute ethanol, following centrifugation at 12,000 ×g for 15 minutes. Ethanol 70% was used for pellet washing, with a volume of 500 μL and centrifugation at 12,000 ×g for 3 minutes. This step was repeated [15]. Pellet was dried at room temperature overnight and eluted into 100 μL of MiliQ water.
2.2. Mitochondrial Genome Sequencing
The total genome sequencing of M. scutellaris was performed at the René Rachou Research Center, Fiocruz Minas (Belo Horizonte, MG) using an Illumina platform (MiSeq) and a paired-end strategy. The library was constructed with the Nextera XT DNA Sample Preparation Kit, following the manufacturer's instructions. Fragments of 404 bp long were carried out for sequencing. The average read length was 250 bp and the final throughput was 8.4 Gbp.
2.3. Genome Assembly
In order to achieve more reliable and accurate results, two kinds of assembly software were employed: SOAPdenovo2 [16] and Velvet [17]. The first M. scutellaris mitogenome created in this work was made using contigs generated by the SOAPdenovo2 software with varying kmer parameter between 23 and 127.
The assembly by Velvet generated assemblies for different kmer values: 31, 41, 51, 61, 71, 81, 91, or 99. The MuMmer Package 3.0 [18] aligned the contigs from each pair of assemblies with the reference sequence and show-tiling was used to complete the scaffolding of the set of contigs. Combinations of the following set of parameters were used in show-tiling: -i: 50, 70, or 90; -V: 0. 5 or 10; -v: 10, 50, or 90; -c: included or not.
The generated sequences were filtered according to the following conditions: (i) sequence size between 14,000 and 17,000 base pairs; (ii) AT content greater than or equal to 84%; (iii) maximum gap tolerance of 100 nucleotides.
The remaining sequences were submitted to MITOS [19], for functional annotation. Steps for Velvet assembly and subsequent annotation with MITOS are schemed in Figure 1. The reference mitogenome of M. bicolor was also submitted in order to provide a more reliable basis for further analysis. The data from annotations were used for analysis of gene order and similarity to reference sequence.
Figure 1.
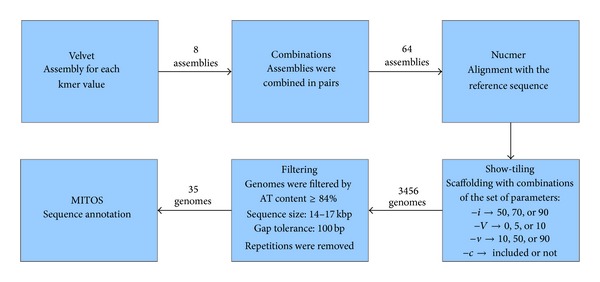
Velvet genome assembly, parameters adopted and MITOS annotation.
The genes from the reference genome of M. bicolor are listed in the first column of Table 1. Relatively to each gene, the adjacent genes were analyzed (upstream and downstream) according to their frequencies (occurrence: “OCR” in Table 1). The annotated genes from M. scutellaris were locally aligned against the reference mitogenome, highlighting identity and E-value. Transfer RNAs secondary structures were predicted using tRNASCAN-SE software [20].
Table 1.
Annotated genes in M. scutellaris mitogenome, upstream/downstream relations, and similarity analysis.
| Reference gene |
Upstream gene | Downstream gene | Similarity to reference sequence | |||||
|---|---|---|---|---|---|---|---|---|
| Name | OCR∗ | Name | OCR∗ | Name | OCR∗ | Blast algorithm | Best identity | E-value |
| trnI | 2 | trnA | 2 | trnQ | 2 | blastn | — | — |
|
| ||||||||
| trnA | 34 | trnT trnN |
27 25 |
rrnS | 30 | blastn | 92.06 | 2.00E − 021 |
|
| ||||||||
| trnK | 16 | trnQ ** | 5 | trnE trnM |
6 4 |
blastn | 93.62 | 2.00E − 016 |
|
| ||||||||
| trnM | 28 | trnX ** | 22 | nad2 | 27 | blastn | 95.71 | 2.00E − 028 |
|
| ||||||||
| nad2 | 36 | trnM | 27 | trnC | 32 | tblastx | 96.43 | 2.00E − 018 |
|
| ||||||||
| trnC | 32 | nad2 | 32 | trnW | 30 | blastn | — | — |
|
| ||||||||
| trnW | 32 | trnC | 30 | trnY | 25 | blastn | 96.92 | 6.00E − 028 |
|
| ||||||||
| trnY | 26 | trnW | 25 | cox1 | 25 | blastn | — | — |
|
| ||||||||
| cox1 | 36 | trnY | 25 | trnL2 | 20 | tblastx | 98.33 | 2.00E − 099 |
|
| ||||||||
| trnL2 | 20 | cox1 | 20 | cox2 | 20 | blastn | — | — |
|
| ||||||||
| cox2 | 36 | tnrL2 | 20 | tnrD | 32 | tblastx | 96.55 | 3.00E − 063 |
|
| ||||||||
| trnD | 32 | cox2 | 32 | atp8 | 31 | blastn | — | — |
|
| ||||||||
| atp8 | 35 | trnD | 31 | atp6 | 34 | tblastx | 93.75 | 2.00E − 005 |
|
| ||||||||
| atp6 | 36 | atp8 | 34 | cox3 | 36 | tblastx | 94.44 | 1.00E − 009 |
|
| ||||||||
| cox3 | 36 | atp6 | 36 | trnV | 17 | tblastx | 94.12 | 4.00E − 027 |
|
| ||||||||
| trnS1 | 27 | trnR nad3 |
12 10 |
trnF | 19 | blastn | 96.88 | 2.00E − 027 |
|
| ||||||||
| trnG | 3 | cox3 | 3 | nad3 atp8 |
2 1 |
blastn | — | — |
|
| ||||||||
| nad3 | 36 | trnV cox3 |
17 14 |
trnR | 24 | tblastx | 95.00 | 7.00E − 006 |
|
| ||||||||
| trnR | 24 | nad3 | 24 | trnS1 trnN |
12 9 |
blastn | — | — |
|
| ||||||||
| trnN | 34 | rrnL | 25 | trnA | 25 | blastn | 96.72 | 9.00E − 026 |
|
| ||||||||
| trnE | 12 | trnK | 6 | trnF | 7 | blastn | — | — |
|
| ||||||||
| trnF | 28 | trnS1 | 19 | nad5 | 15 | blastn | — | — |
|
| ||||||||
| nad5 | 36 | trnF | 15 | trnQ ** | 35 | tblastx | 95.83 | 1.00E − 013 |
|
| ||||||||
| trnH | 0 | — | — | |||||
|
| ||||||||
| nad4 | 36 | trnQ ** | 35 | nad4l | 28 | tblastx | 95.00 | 1.00E − 028 |
|
| ||||||||
| nad4l | 33 | nad4 | 28 | trnP | 26 | tblastx | 94.74 | 0.13 |
|
| ||||||||
| trnT | 32 | nad4l | 9 | trnA | 27 | blastn | — | — |
| trnQ ** | 7 | |||||||
|
| ||||||||
| trnP | 35 | nad4l | 26 | nad6 | 35 | blastn | — | — |
|
| ||||||||
| nad6 | 35 | trnP | 36 | cob | 35 | tblastx | 94.12 | 4.00E − 022 |
|
| ||||||||
| cob | 36 | nad6 | 35 | trnS2 | 35 | tblastx | 95.65 | 1.00E − 011 |
|
| ||||||||
| trnS2 | 35 | cob | 35 | nad1 | 35 | blastn | 100.00 | 7.00E − 032 |
|
| ||||||||
| nad1 | 36 | trnS2 | 35 | rrnL | 34 | tblastx | 92.86 | 5.00E − 021 |
|
| ||||||||
| trnL1 | 2 | trnY nad1 |
1 1 |
rrnL trnL2 |
1 1 |
blastn | — | — |
|
| ||||||||
| rrnL | 36 | nad1 | 34 | trnN | 25 | blastn | 95.26 | 3.00E − 095 |
|
| ||||||||
| trnV | 23 | cox3 | 17 | nad3 | 17 | blastn | — | — |
|
| ||||||||
| rrnS | 36 | trnA | 30 | tnrQ | 29 | blastn | 95.08 | 4.00E − 024 |
*OCR: occurrences in different assemblies.
∗∗Genes not found in M. bicolor mitogenome.
3. Results and Discussion
In total, 36 assemblies were performed, generating 36 annotations which allowed inferring that all protein-coding genes and tRNA genes were annotated. Using all annotations, the gene order analysis revealed synteny between M. scutellaris mitogenome and the reference genome. Ribosomal genes identities were also analyzed (Table 1), as well as the conservation of secondary structure for tRNA.
3.1. Organization and Partial Characterization of M. scutellaris Mitogenome
In Table 1, the “reference gene” column corresponds to the mitochondrial gene order of M. bicolor. Considering the genes that are found in the reference genome, column “OCR” (second column) shows how many times each gene is annotated for M. scutellaris, within the 36 annotations. “Upstream gene” and “downstream gene” columns refer to which genes are found in those respective positions, relative to the reference gene. Finally, “OCR” columns (fifth and sixth columns) mean how many times a gene is found in such position, relative to another gene, also considering the total of 36 annotations.
M. scutellaris mitogenome shows an overall high synteny when compared to M. bicolor mitogenome. Figure 2 is an illustrative scheme which gathers the annotated genes under the most frequent order, among all 36 assemblies. Although all genes were found, they are not all present in the following scheme, as long as some few ones must yet have their position validated.
Figure 2.
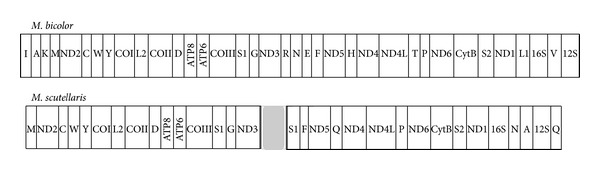
Comparison between M. bicolor's mitogenome and gene organization achieved for M. scutellaris. Genes whose position is not yet validated were omitted (in gray).
Regarding tRNA genes, aligning the annotated genes against the reference genome did not generate considerable identity values for all cases. However, submitting the M. scutellaris sequences to tRNAScan-SE, it was possible to infer that the tRNA secondary structures of M. scutellaris are viable and well conserved. An example is displayed in Figure 3.
Figure 3.
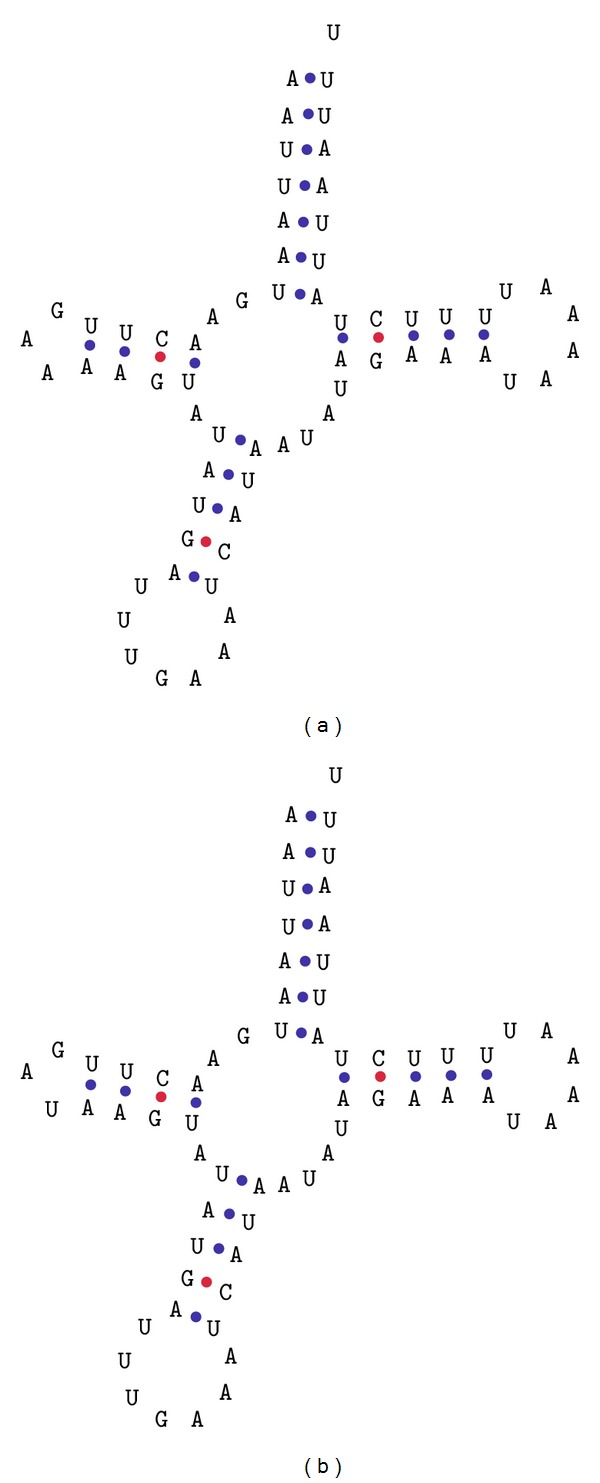
Comparison of secondary structure predictions for tRNAS2 genes of Melipona scutellaris (a) and Melipona bicolor (b).
In Table 1 it is possible to notice that in some cases there is more than one gene in the upstream and/or downstream position, providing different possibilities of gene organization. Such genes are tRNAA, tRNAK, tRNAG, tRNAR, tRNAT, and tRNAL1.
The coding-protein genes for M. scutellaris are syntenic to their homologous in M. bicolor mitogenome. Some tRNA genes are also syntenic, namely, tRNAM, tRNAW, tRNAY, tRNAL2, tRNAD, tRNAF, tRNAP, and tRNAS2. However, it is also possible to infer that some tRNA genes have underwent rearrangement, which are tRNAV, tRNAS1, tRNAN, and tRNAA. Some other tRNA genes must yet have their position validated, since their occurrence (OCR) is very low compared to the average, such as tRNAI, tRNAG, tRNAE, and tRNAL1 (not shown in Figure 2). It is known that tRNAs order is a particular feature of each insect species [8], and tRNAs distribution, copy number, and codon usage patterns are especially important in evolutionary studies [21].
Our results provided evidences of possible duplication of two genes in M. scutellaris mitogenome in comparison to M. bicolor. In Table 1 it is possible to see the occurrences for tRNAQ downstream to ND5 and 12S genes: 35/36 and 29/36, respectively. Still, tRNAA has also high occurrences for being upstream to 12S (30/36), composing the following cluster: 16S_tRNAN_tRNAA_12S. But there is also a high occurrence for being downstream to tRNAT (27/36), whose position is not yet clear, although it is already unlikely to be part of the cluster mentioned above.
A single gene found in M. bicolor mitogenome was not found in M. scutellaris, which is tRNAH. On the other hand, gene tRNAX was annotated, suggesting a tRNA that is not identified, requiring further validation.
Moreover, submitting the mitogenome sequence of M. bicolor to MITOS, as a support for further analysis, a gene that is not found in the currently available sequence was annotated, namely, tRNAC. This gene was also annotated in M. scutellaris mitogenome, in the same position. Such synteny suggests that tRNAC gene may be present in both genomes and this information provided by MITOS may have been omitted by other kinds of annotation software.
Concerning the protein-coding regions, they are probably not yet complete. Genes' lengths are not all as the expected, preventing a conclusion about the codon usage in M. scutellaris' mitogenome.
Finally, comparing both assemblies, N50 values adopted were 343 for SOAPdenovo2 and 722-362 for Velvet. The longer scaffolds achieved had close lengths, which are 15580 and 15206, for SOAPdenovo2 and Velvet, respectively. In general, all assemblies generated scaffolds around 14000–15580 bp long. However, despite the good quality of the data (phred > 30), it was not possible to achieve any significant scaffold in a first approach. For this reason, the present methodology was adopted in order to reach the evidences of genes' presence. As discussed above, all protein-coding sequences were annotated, as well as transfer RNA-coding and ribosomal subunits genes. It is assumed that using a single library of inserts 400 bp long may have influences over the results under the expected, requiring a complementary library for subsequent analyses.
3.2. DNA Barcoding for Taxonomic Identification
The cytochrome c oxidase subunit I gene (COI) sequence annotated in this study was submitted to BOLD Systems database [22] for taxonomic analysis. It was validated as belonging to the Melipona genus with 100% probability.
Melipona cramptoni had the highest identity (98.99%) to M. scutellaris COI sequence. Other closest hits were Melipona rufiventris (98.43%) and Melipona eburnea (98.40%). The reference organism, M. bicolor, was ranked at 97th position, with 95.2% identity. This observation highlights the limitations of using M. bicolor as a reference for assembly.
A higher identity between M. scutellaris and M. rufiventris was already expected. Rocha et al. [23] have demonstrated by cytogenetic studies that among Melipona species there are different heterochromatin content and distribution patterns. Two groups may be distinguished for such characteristics that may be considered for evolutionary analysis as well.
Rocha et al. [23] established M. bicolor as belonging to Group I, which comprises species with less than 50% of heterochromatin. Species belonging to such group display heterochromatin content concentrated in one portion of the chromosome. On the other hand, Group II is composed of species with heterochromatin content higher than 50%, spread along the chromosome extension. By cytogenetic studies, M. scutellaris and M. rufiventris were classified as being part of Group II. Therefore, DNA barcoding result revealing a closer relation between M. scutellaris and M. rufiventris rather than M. bicolor confirms cytogenetic studies. However, the close relation between M. scutellaris and M. cramptoni has not yet been investigated.
4. Conclusion
The results generated to the mitogenome of M. scutellaris in this study suggest a conserved character in comparison to M. bicolor. Protein coding genes order especially appears to be well conserved. Some tRNA genes underwent rearrangement, but a conservation pattern could also be analyzed as well.
Through taxonomic identification search on BOLD Systems database it was possible to assume that M. cramptoni is the closest species to M. scutellaris (98.99%), although M. bicolor is also found among closely related species (95.2% identity). The second closest species to M. scutellaris found at BOLD database is M. rufiventris, what is confirmed by cytogenetic studies.
Conflict of Interests
The authors declare that there is no conflict of interests regarding the publication of this paper.
References
- 1.Pianaro A. Ecologia química de abelhas brasileiras: Melipona rufiventris, Melipona scutellaris, Plabeia droryana, Nannotrigona testaceicornis, Tetragonisca angustula e Centris trigonoides [M.S. thesis] UNICAMP, Chemistry Institute; 2007. [Google Scholar]
- 2.Kerr WE, Carvalho GA, Nascimento VA. A Abelha Uruçu: Biologia, Manejo e Conservação. Liber Liber; 1996. [Google Scholar]
- 3.Ramírez SR, Nieh JC, Quental TB, Roubik DW, Imperatriz-Fonseca VL, Pierce NE. A molecular phylogeny of the stingless bee genus Melipona (Hymenoptera: Apidae) Molecular Phylogenetics and Evolution. 2010;56(2):519–525. doi: 10.1016/j.ympev.2010.04.026. [DOI] [PubMed] [Google Scholar]
- 4.Rasmussen C, Cameron SA. Global stingless bee phylogeny supports ancient divergence, vicariance, and long distance dispersal. Biological Journal of the Linnean Society. 2010;99(1):206–232. [Google Scholar]
- 5.Henriques JM. Identificação molecular (DNA barcode) dos peixes da Bacia do Rio Ribeira de Iguapé e dos Rios Costeiros do Estado de São Paulo [Doctorate thesis] UNESP, Biosciences Institute; 2010. [Google Scholar]
- 6.Caterino MS, Cho S, Sperling FAH. The current state of insect molecular systematics: a thriving tower of babel. Annual Review of Entomology. 2000;45:1–54. doi: 10.1146/annurev.ento.45.1.1. [DOI] [PubMed] [Google Scholar]
- 7.Sheffler WS. Mitochondria. 2nd edition. John Wiley & Sons; 2008. [Google Scholar]
- 8.Rodovalho CM. Caracterização do transcriptoma e genoma mitocondrial da formiga cortadeira Atta laevigata (FORMICIDAE: ATTINI) [Doctorate thesis] UNESP, Biosciences Institute; 2011. [Google Scholar]
- 9.Hebert PDN, Ratnasingham S, DeWaard JR. Barcoding animal life: cytochrome c oxidase subunit 1 divergences among closely related species. Proceedings of the Royal Society B: Biological Sciences. 2003;270(1):S96–S99. doi: 10.1098/rsbl.2003.0025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yu DW, Ji Y, Emerson BC. Biodiversity soup: metabarcoding of arthropods for rapid biodiversity assessment and biomonitoring. Methods in Ecology and Evolution. 2012;3:613–623. [Google Scholar]
- 11.Ander M, Troell K, Chirico J. barcoding of biting midges in the genus Culicoides: a tool for species determination. Medical and Veterinary Entomology. 2013;27(3):323–331. doi: 10.1111/j.1365-2915.2012.01050.x. [DOI] [PubMed] [Google Scholar]
- 12.Virgilio M, Backeljau T, Nevado B, De Meyer M. Comparative performances of DNA barcoding across insect orders. BMC Bioinformatics. 2010;11, article 206 doi: 10.1186/1471-2105-11-206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Park D-S, Foottit R, Maw E, Hebert PDN. Barcoding bugs: DNA-based identification of the true bugs (insecta: Hemiptera: Heteroptera) PLoS ONE. 2011;6(4) doi: 10.1371/journal.pone.0018749.e18749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Smith MA, Bertrand C, Crosby K, et al. Wolbachia and DNA barcoding insects: patterns, potential, and problems. PLoS ONE. 2012;7(5) doi: 10.1371/journal.pone.0036514.e36514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen H, Rangasamy M, Tan SY, Wang H, Siegfried BD. Evaluation of five methods for total DNA extraction from western corn rootworm beetles. PLoS ONE. 2010;5(8) doi: 10.1371/journal.pone.0011963.e11963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Luo R, Liu B, Xie Y, et al. SOAPdenovo2: an empirically improved memory-efficient short-read de novo assembler. GigaScience. 1(1):18–23. doi: 10.1186/2047-217X-1-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zerbino DR, Birney E. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Research. 2008;18(5):821–829. doi: 10.1101/gr.074492.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kurtz S, Phillippy A, Delcher AL, et al. Versatile and open software for comparing large genomes. Genome Biology. 2004;5(2, article R12) doi: 10.1186/gb-2004-5-2-r12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bernt M, Donath A, Jühling F, et al. MITOS: improved de novo metazoan mitochondrial genome annotation. Molecular Phylogenetics and Evolution. 2013;69(2):313–319. doi: 10.1016/j.ympev.2012.08.023. [DOI] [PubMed] [Google Scholar]
- 20.Schattner P, Brooks AN, Lowe TM. The tRNAscan-SE, snoscan and snoGPS web servers for the detection of tRNAs and snoRNAs. Nucleic Acids Research. 2005;33(2):W686–W689. doi: 10.1093/nar/gki366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Behura SK, Stanke M, Desjardins CA, Werren JH, Severson DW. Comparative analysis of nuclear tRNA genes of Nasonia vitripennis and other arthropods, and relationships to codon usage bias. Insect Molecular Biology. 2010;19(1):49–58. doi: 10.1111/j.1365-2583.2009.00933.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ratnasingham S, Hebert PDN. BOLD: the barcode of life data system: barcoding. Molecular Ecology Notes. 2007;7(3):355–364. doi: 10.1111/j.1471-8286.2007.01678.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rocha MP, Pompolo SG, Fernandes A, Campos LAO. Melipona—six decades of cytogenetic. Journal of Biosciences. 2007;23(1):111–117. [Google Scholar]