

Abstract
Xanthium strumarium L. is a member of the Asteraceae commonly used in Cuba, mainly as diuretic. Some toxic properties of this plant have also been reported and, to date, very little is known about its genotoxic properties. The present work aims was to evaluate the potential cytotoxic and genotoxic risk of whole extract from Xanthium strumarium L. whole extract of aerial parts. No positive response was observed in a battery of four Salmonella typhimurium strains, when exposed to concentrations up to 5 mg/plate, with and without mammalian metabolic activation (liver microsomal S9 fraction from Wistar rats). In CHO cells, high concentrations (25–100 μg/mL) revealed significant reduction in cell viability. Results from sister chromatid exchanges, chromosome aberrations, and comet assay showed that X. strumarium extract is genotoxic at the highest concentration used, when clear cytotoxic effects were also observed. On the contrary, no increase in micronuclei frequency in bone marrow cells was observed when the extract was orally administered to mice (100, 500, and 2000 mg/Kg doses). The data presented here constitute the most complete study on the genotoxic potential of X. strumarium L. and show that the extract can induce in vitro DNA damage at cytotoxic concentrations.
1. Introduction
Xanthium strumarium L. (Asteraceae), a medicinal plant commonly found as a weed, is widely distributed in North America, Brazil, China, Malaysia, and hotter parts of India. The herb is traditionally used mostly in treating several aliments. In Cuba, X. strumarium is commonly called “guisazo de caballo” and it has been used as diuretic [1]. The plant has been employed for a long time in folk therapy. More than 20 properties have been attributed to the decoctions and tinctures from the leaves and roots, for example, antirheumatic, antisyphilitic, appetiser, diaphoretic, diuretic, emollient, laxative, and sedative activities. Other actions have been confirmed by experimental pharmacology: anti-inflammatory, analgesic [2, 3], antibacterial, anticancer [4, 5], antifungal [6], antihypoglycemic [7], antimitotic [8, 9], antitrypanosomal, antimalarial [10], and diuretic activities [11].
The toxicity reported for the plant suggests that potential negative effects on human health elicited by the consumption of herbal remedies based on X. strumarium should be considered. Genotoxicity is one of the most important toxic effects and its relevance is greater as we are dealing with events taking place at sublethal doses leading to long-term effects such as cancer and reproductive diseases.
The present work aims at assessing the potential cytotoxic and genotoxic risk of such extract. For this purpose, different genetic endpoints were assayed, in order to evaluate DNA damage at different genetic expression levels. We assessed the cell viability of Xanthium strumarium L. extracts on Chinese hamster ovary (CHO) cells using MTT assay. Then we tested the mutagenicity of the extract in four short-term assays: Salmonella/microsome (the Ames test) for gene mutation, cytogenetic assays on CHO cells for chromosomal damage in vitro, comet assay for primary DNA damage, and the micronucleus test in mouse bone marrow for clastogenic and aneugenic effects in vivo.
2. Materials and Methods
2.1. Plant Material and Extract Preparation
Xanthium strumarium L. was collected from the Medicinal Plants Experimental Station “Dr. Juan Tomás Roig” in Güira de Melena, Artemisa. A voucher specimen with number ROIG 4594 was deposited at the herbarium of this institution. A fluid extract was prepared from the dried material by hydroalcoholic extraction in four rounds of percolation. It was concentred under reduced pressure to obtain a raw extract (whole extract), kept in sealed containers at 4°C. The chemical characterization of the extract was performed by qualitative analysis as described by Miranda and Cuellar [12]. This analysis revealed a higher polysaccharides content, phenol, saponin, terpenes, sesquiterpene lactones, steroids, cumarin, and flavonoids.
2.2. Negative Control
In all cases (in vivo and in vitro assays) the negative control was the appropriate solvent used to dissolve plant extract and all the employed substances. Dimethyl sulfoxide (DMSO) was the solvent in in vitro assays and ethanol in in vivo test.
2.3. Positive Control
The following positive controls were used: aminofluorene (2AF), sodium azide, cyclophosphamide (CP), 9-aminoacridine (9AA), and pichrolonic acid (PA) in Ames test; CP in mouse bone marrow micronucleus; mitomycin C and X-ray in chromosome aberrations assays; hydrogen peroxide (H2O2) in the alkaline comet assay in CHO cells.
2.4. In Vitro Assays
2.4.1. Salmonella/Microsome Assay
The plate-incorporation mutagenicity assay was performed as described by Maron and Ames [13] using Salmonella typhimurium tester strains TA 1535, TA 1537, TA98, and TA100. Five concentrations of the extract were tested in triplicate plates in a single experiment, covering a range from 50 to 5000 μg of solids/plate. Liver microsomal S9 fraction was prepared from Wistar rats treated with phenobarbital and 5.6 benzoflavone. S9 mix used for exogenous metabolic activation contained a 4% S9 fraction in a solution of cofactors (NADP-regenerating system). Revertant colonies were counted manually after 72 h of incubation at 37°C.
2.4.2. Cell Culture and Cytogenetic Assays in Chinese Hamster Ovary (CHO) Cells
CHO cells were grown in Ham F10 medium supplemented with 10% foetal bovine serum, 100 IU/mL penicillin, 100 IU/mL streptomycin, and 2 mM L-glutamine in a 5% CO2 humidified atmosphere at 37°C. All experiments were performed, seeding 4 × 105 cells per 25 cm2 flask. Under these conditions, the cell cycle of this line was approximately 12 h.
(1) MTT Cytotoxicity Assay. The MTT [3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazoliumbromide] cytotoxicity assay was carried out in accordance with the protocol described by Mosmann [14] with some modifications. Added to each well of a culture microplate were approximately 1 × 104 CHO cells. The cells were exposed to extract for 24 h. At the end of this period, the cells were incubated with MTT (5 mg/mL) for 4 h. The plates were read in a microplate spectrophotometer (OMEGA) at 550 nm. The IC50 were determined from a dose-response curve by using 5 different concentrations (6.25, 12.5, 25, 50, and 100 μg/mL). Analyses were done in triplicate for each concentration.
(2) Sister Chromatid Exchanges (SCE) Assay. Cells were treated for 3 h with a recovery time of 24 h or were continuously exposed for 27 h to X. strumarium extracts. Cultures received BrdUrd at the final concentration of 1.5 μg/mL during the last 24 h of incubation. 5 × 10−7 M colchicine was always added 2 h before fixation. As previously described, FPG Hoechst- Giemsa technique modified by De Salvia et al. [15] was used for differential staining of sister chromatids. The scoring of SCE was according to Sánchez-Lamar et al. [16] and 20 metaphases per experimental point were analyzed.
(3) Chromosome Aberration (CA) Assay. The chromosome aberration test was conducted according to the international guidelines for this assay [17]. Exponentially growing CHO cells were treated according to the following experimental schedule. Two types of treatment were employed: 3 h treatment with 15 h recovery time and 18 h continuous treatment until harvesting. 5 × 10−7 M colchicine was added 2 h before harvesting. After trypsinisation, cells were treated with a hypotonic solution, fixed with acetic acid and methanol (1 : 3) solution, and stained as reported by Galloway et al. [18]. At least 150 metaphases were scored for each dose of plant extract.
(4) Alkaline Comet Assay. The alkaline comet assay was performed as described by Cornetta et al. [19] with minor modifications. Exponentially growing CHO cells were treated for 3 h with different concentrations (5, 15, and 45 μg/mL) of the extract. At the end of treatment 40 μL of cellular suspension was mixed with 5 mL trypan blue for 3 min. Cell suspension was pipetted onto a glass microscope slide and analyzed under a phase-contrast microscope, for calculating the proportion of nonstained (viable) cells. Immediately after treatment 20 μL of cells was mixed with 180 μL of 0.7% low melting point agarose in PBS (Ca and Mg free) at 37°C and immediately pipetted onto a frosted glass microscope slide precoated with a layer of 1% normal melting point agarose, similarly prepared in PBS. Two slides were prepared for each experimental point. The agarose was allowed to set at +4°C for the necessary time and the slides incubated in a lysis solution (2.5 M NaCl, CAS number 7647-14-5, 10 mMTris-HCl,CAS number 77-86-1, 100 mM Na2EDTA, CAS number 6381-92-6, NaOH, CAS number 1310-73-2, to pH 10, 1% Triton, CAS number 9002-93-1, and 10% DMSO, CAS number 67-68-5) for 50 min. After lysis, slides were placed onto a horizontal electrophoresis unit containing fresh buffer (1 mM Na2EDTA, 300 mM NaOH, pH 13) for 20 min to allow for DNA unwinding. Electrophoresis was conducted for 15 min (25 V, 300 mA) at 4°C. Subsequently, slides were gently washed in neutral buffer solution for 5 min (0.4 MTris-HCl, pH 7.5), fixed in 100% freshly methanol for 3 min, and stained with ethidium bromide (2 μg/mL, CAS number 001239458). Slides were analyzed using a fluorescence microscope (Leica) equipped with a camera. Fifty comets on each slide, coded and blindly scored, were acquired using “I.A.S.” software automatic image analysis system purchased from Delta System (Rome, Italy). To quantify the induced DNA damage the Tail DNA (TD), which is a measure of the percentage of migrated DNA in the tail, was used [20].
2.5. In Vivo Assays
Swiss mice obtained from the National Centre for Production of Laboratory Animals (CENPALAB, Cuba) were bred in our laboratory animal-care facility and employed in the present work. Adult healthy animals, 8–10 weeks old, weighing 25–30 g of either sex were used. Each experimental group consisted of five animals. All the animal studies reported in this work were carried out in accordance with the Cuban regulations on the protection of animals (Código Práctico para el uso de Animales de Laboratorio, Centro Nacional para la Producción de Animales de Laboratorio, CENPALAB) and the Declaration of Helsinki. All experimental protocols were revised by the Animal Care and Use Committee of the Faculty of Biology, University of Havana, and conducted humanely.
2.5.1. Mouse Bone Marrow Micronucleus (MN) Assay
The extract was administered p.o. in a volume of 10 mL/kg. Three dose groups (500, 1000, and 2000 mg/kg b.w) were assayed in both sexes. The usual subacute protocol of two administrations 24 h apart and a single sacrifice 24 h later was employed [21]. Mouse bone marrow was obtained from five animals for each dose of extract and controls as reported by Schmid [22]. Smears were stained with May-Grunwald and Giemsa (Merck) and analysed for the presence of MN. Genotoxicity index (GI), which is the percentage of polychromatic erythrocytes (PCE) with micronuclei (PCE-MN), was calculated from 2000 cells and the cytotoxicity index (CI) was determined as the ratio of PCE to normochromatic erythrocytes (NCE) (PCE/NCE) calculated from 250 erythrocytes perslide.
2.6. Statistical Analysis
Means and standard errors were determined using Kolmogorov-Smirnov test. Controls and treated samples were compared using the Student's t-test, the two-way ANOVA, the bi- and trifactorial ANOVA, the Dunnett's test, Fisher test, and Mann-Whitney U test, depending on the assay [23]. In in vitro cytogenetic assays, the tested concentrations of extract were considered positive when statistically significant increases in total alterations frequency (CA or SCE) exceeded the historical control mean values. In Salmonella test, the controls and treated samples were compared using SALANAL (Salmonella Assay Analysis, version 1 US Environmental Protection Agency).
3. Results
3.1. Salmonella/Microsome Assay
Reversion frequencies observed in the Salmonella/microsome mutagenicity test are presented in Tables 1 and 2. No increase of revertants per plate was observed in any of the four standard bacterial strains used in the assay, with and without extrinsic metabolic activation provided by S9 mix, in the relatively wide range of concentrations tested (50–5000 μg/plate).
Table 1.
Mutagenicity testing of Xanthium strumarium extract in the Salmonella/microsome assay (TA 1535 and TA 1537).
| Concentration (μg/placa) |
TA 1535 Mean revertants/plate ± SD |
TA 1537 Mean revertants/plate ± SD |
||
|---|---|---|---|---|
| −S9 | +S9 | −S9 | +S9 | |
| 0a | 26.00 ± 8.54 | 10.3 ± 2.52 | 9.67 ± 2.89 | 8.00 ± 0.0 |
| 50 | 21.67 ± 4.04 | 9.00 ± 1.73 | 9.33 ± 0.58 | 7.33 ± 2.08 |
| 150 | 23.67 ± 2.08 | 8.67 ± 4.16 | 8.00 ± 1.00 | 6.00 ± 2.00 |
| 500 | 28.33 ± 9.07 | 11.33 ± 2.52 | 14.00 ± 3.46 | 8.33 ± 3.21 |
| 1500 | 32.67 ± 2.08 | 10.33 ± 6.11 | 12.33 ± 3.08 | 6.67 ± 2.31 |
| 5000 | 28.67 ± 1.53 | 5.67 ± 2.52 | 16.3 ± 4.93 | 7.33 ± 4.04 |
| NaA1.5b | 405.3 ± 70.5∗∗ | |||
| CP 500b | 228.3 ± 36.2∗∗ | |||
| 9AA 100b | 460.0 ± 50.0∗∗ | |||
| 2AF 20b | 580.0 ± 34.6∗∗ | |||
+S9: with liver microsomal S9 fraction; −S9: without liver microsomal S9 fraction.
aNegative control, dimethyl sulfoxide (DMSO).
bPositive controls. NaA: sodium azide; CP: cyclophosphamide; 9AA: 9-aminoacridine; 2AF: 2-aminofluorene.
∗∗ P < 0.01 (Student's t-test).
Table 2.
Mutagenicity testing of Xanthium strumarium extract in the Salmonella/microsome assay (TA 100 and TA 98).
| Concentration (μg/placa) |
TA 100 Mean revertants/plate ± SD |
TA 98 Mean revertants/plate ± SD |
||
|---|---|---|---|---|
| −S9 | +S9 | −S9 | +S9 | |
| 0a | 58.33 ± 16.56 | 62.60 ± 15.1 | 25.67 ± 3.79 | 38.33 ± 2.52 |
| 50 | 54.33 ± 5.13 | 64.10 ± 10.5 | 26.00 ± 4.36 | 38.67 ± 11.85 |
| 150 | 45.33 ± 4.73 | 60.04 ± 13.7 | 28.67 ± 7.64 | 45.67 ± 8.33 |
| 500 | 44.00 ± 2.00 | 60.33 ± 5.77 | 22.00 ± 5.29 | 35.67 ± 20.11 |
| 1500 | 46.67 ± 2.52 | 70.70 ± 7.37 | 26.00 ± 4.36 | 40.67 ± 8.62 |
| 5000 | 51.67 ± 3.21 | 67.31 ± 2.52 | 27.00 ± 2.65 | 38.33 ± 2.52 |
| NaA1.5b | 480.0 ± 22.6∗∗ | |||
| 2AF 20b | 2280.0 ± 169.7∗∗ | 3300.0 ± 141.4∗∗ | ||
| PA 100b | 992.0 ± 11.3∗∗ | |||
+S9: with hepatic fraction S9; −S9: without hepatic fraction S9.
aNegative control, dimethyl sulfoxide (DMSO).
bPositive controls. NaA: sodium azide; 2AF: 2-aminofluorene; PA: pichrolonic acid.
**P < 0.01 (Student's t-test).
3.2. MTT Cytotoxicity Assay
Figure 1 shows the results obtained in the MTT assay in CHO cells treated with X. strumarium extract. The readings of the cytotoxicity assay determined spectrophotometrically showed that extract concentrations of 6.25, 12.5, 25, 50, and 100 μg/mL after 24 h of treatment exerted a cytotoxic effect on CHO cells in both dose-time dependent manners. Cytotoxic effect was observed in 25–100 µg/mL. IC50 value of X. strumarium extract in CHO cells was 20.5 μg/mL
Figure 1.
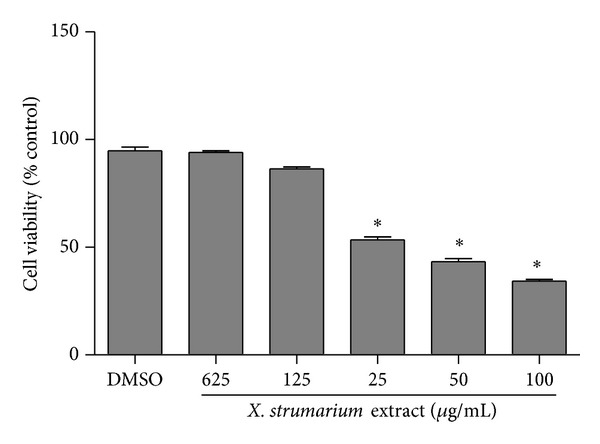
Cytotoxic effects of X. Strumarium extract in CHO cells by the MTT assay. Cells were exposed for 24 h to different concentrations (6.25, 12.5, 25, 50, and 100 μg/mL) of the extract. Negative control: DMSO (1%). Each value represents the mean ± SE of three independent experiments. Statistically significant difference compared to negative control (∗P < 0.05, ANOVA and Dunnett's test).
3.3. Sister Chromatid Exchanges (SCE) Assay
A significant increase of SCE per cell was observed at 15 μg/mL when cells were exposed to the extract for 3 h and then recovered for 24 h in fresh medium (Figure 2). The mean ± SD SCE frequency at 15 μg/mL extract was 15.2 ± 4.01 as compared with 8.95 ± 2.76 in DMSO-treated cells. When a continuous treatment of 27 h was applied, a statistically significant increase in SCE was observed at 5 μg/mL of X. strumarium extract. Higher concentrations could not be analysed since a strong antiproliferative effect of the extract was observed and no second mitoses were recorded.
Figure 2.
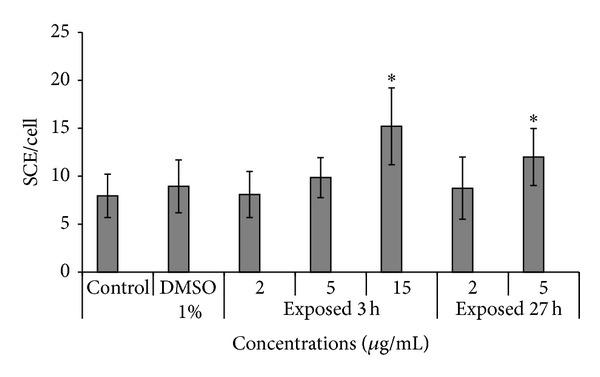
SCE assay analysis in CHO cells treated with crude extract of X. strumarium L. (2, 5 and 15 μg/mL). Cells were exposed for 3 h or 27 h to X. strumarium extracts. Cultures received BrdUrd at the final concentration of 1.5 μg/mL during the last 24 h of incubation. Negative control: DMSO (1%). Each value represents the mean ± SD of three independent experiments. Statistically significant difference compared to negative control (∗P < 0.05, ANOVA and Dunnett's test).
3.4. Chromosome Aberration (CA) Assay
The X. strumarium extract produced a significant increase in the percentage of CA at the highest tested concentration, that is, 45 μg/mL. At this concentration, the mitotic index was lower than 50% of the mitotic index observed in controls indicating occurrence of cell toxicity from the extract treatment (Table 3). A slight significant increase in the frequency of CA was observed in 25 μg/mL when CHO cells were exposed for 3 h and when cells were continuously treated for 18 h with the X. strumarium extract. No mitoses could be observed after 18 h of treatment with 45 μg/mL of extract.
Table 3.
Evaluation of chromosomal aberrations and mitotic index in CHO cells treated with whole extract of Xanthium strumarium L.
| Concentration (μg/mL) |
Number of cells | Aberrant cellsa (%) | Mitotic index (%) | Chromatid breaks |
Chromosome breaks |
bExch. | Total Ab. |
|---|---|---|---|---|---|---|---|
| DMSO 1% | 150 | 1.33 | 74 | 0.66 | 0.66 | 1.33 | |
|
| |||||||
| 3 h + 15 h recovery | |||||||
| 10 | 150 | 1.33 | 77 | 0.66 | 0.66 | 1.33 | |
| 25 | 150 | 4.00∗ | 62 | 0.66 | 3.33 | 4.00 | |
| 45 | 150 | 8.66∗∗ | 32 | 2.00 | 7.33 | 9.33 | |
|
| |||||||
| 18 h continuous | |||||||
| 10 | 150 | 2.00 | 44 | 0.66 | 1.33 | 2.00 | |
| 25 | 150 | 4.00∗ | 53 | 2.66 | 2.66 | 5.33 | |
| 45 | 150 | NM | 2 | ||||
| Mit Cc | 100 | 23.00∗∗ | 21.00 | 12.00 | 33.00 | ||
|
| |||||||
| RXd | 100 | 38.00∗∗ | 2.00 | 48.00 | 16.00 | 66.00 | |
aExcluding gaps; NM: no mitoses; bchromosome and chromatid exchanges; cmitomycin C positive control (0.05 μg/mL); dRX: positive control (2 Gy); ∗P < 0.05; ∗∗P < 0.01 versus DMSO sample.
3.5. Comet Assay
The data obtained with comet assay are shown in Figure 3. The percentage of unviable cells never exceeded 20% in each experimental point. A dose-related increase in DNA breaks was observed in treated samples when compared to DMSO-treated one. A significant result (P < 0.05) was only obtained at the highest concentration of the extract and hedgehog or apoptotic-like cells were observed.
Figure 3.
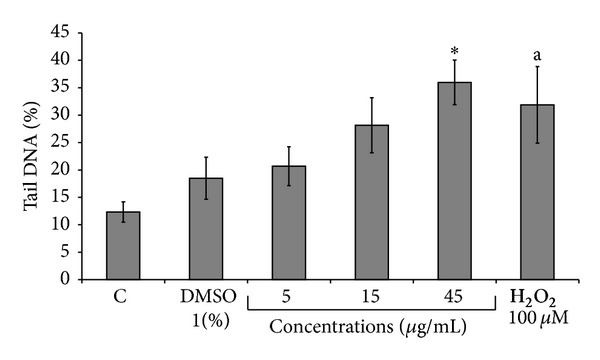
Comet assay analysis in CHO cells treated with fluid extract of Xanthium strumarium L. CHO cells were treated for 3 h with different concentrations (5, 15, and 45 μg/mL) of the extract. Positive control: hydrogen peroxide (100 μM for 20 min) and negative control: DMSO (1%). Each value represents the mean ± SD of three independent experiments. Statistically significant difference compared to negative control (*P < 0.05 versus DMSO sample, a P < 0.05 versus control at Mann-Whitney U test).
3.6. Mouse Bone Marrow Micronucleus (MN) Assay
The results obtained for mice treated with different doses of X. strumarium extract are shown in Table 4. No significant difference in the frequency of MNPCE was observed between mice treated with X. strumarium extract and the negative control for both sexes. A high increase in the frequency of MNPCE was detected in mice treated with cyclophosphamide compared to the negative control (P < 0.01). No significant differences in the PCE/NCE ratio were observed when comparing mice treated with X. strumarium extract and the respective negative control. No signs of toxicity were found in mice at dose levels of 500–2000 mg/kg body weight. There was no mortality in any of the groups; the initial and final weights of the animals were found to be similar to controls.
Table 4.
Micronucleus test results in mice treated with whole extract of Xanthium strumarium L.
| Dose (mg/kg) | Female | Male | ||
|---|---|---|---|---|
| PCE/NCEa ± SD | MNPCE/2000b ± SD | PCE/NCEa ± SD | MNPCE/2000b ± SD | |
| 0 | 0.46 ± 1.32 | 0.18 ± 0.43 | 0.35 ± 1.39 | 0.20 ± 0.24 |
| 500 | 0.07 ± 1.57 | 0.03 ± 0.08 | 0.10 ± 1.70 | 0.03 ± 0.11 |
| 1000 | 0.28 ± 1.58 | 0.06 ± 0.08 | 0.16 ± 1.51 | 0.03 ± 0.07 |
| 2000 | 0.08 ± 1.46 | 0.03 ± 0.11 | 0.07 ± 1.46 | 0.03 ± 0.11 |
| Ethanolc | 0.05 ± 1.39 | 0.01 ± 0.08 | 0.32 ± 1.45 | 0.04 ± 0.08 |
| CPd | 0.06 ± 0.63∗∗ | 1.80 ± 7.62∗∗ | 0.07 ± 0.53∗∗ | 0.75 ± 5.38∗∗ |
aPCE/NCE: polychromatic (PCE) to normochromatic (NCE) erythrocyte ratio; bMNPCE/2000: frequency of micronucleated PCE; cNegative control: ethanol 60%; dPositive control: cyclophosphamide 20 mg/Kg. ∗∗P < 0.01 (Student's t-test).
4. Discussion
Plant remedies are traditionally used in Cuba for the treatment of different illness. This knowledge has remarkably been improved through the prioritized policy of the Cuban Ministry of Public Health of developing pharmaceuticals and supplements from natural sources as well as a scientifically sustained phytotherapy. To confirm the pharmacologic properties of Xanthium strumarium L., species belonging to the Cuban flora are part of that aim.
X. strumarium L is one of the most popular herbal formulas in the world; however, evidence-based information about genotoxicity is limited. Many studies have reported pharmacological efficacies and benefits of X. strumarium [2–11], but there is little information on its risk and safety.
Data presented in this report have involved an extensive genotoxic assessment of a complex mixture, the whole extract of X. strumarium L. as part of preclinical studies; the use of in vitro and in vivo assays was decisive in order to obtain a picture of the genotoxic potential of this plant product and to identify potential risk/hazard for human health due to DNA damage and mutation induction.
The species of the genus Xanthium are an important source of sesquiterpene lactones which are responsible for most of biological activities attributed to this genus. Sesquiterpene lactones termed xanthanolides, xanthatin, 8-epi xanthatin, and 8-epi tomentosin and others phytocompounds as xanthus-strumarina, hydrochinonas, carboxy-atractyloside, albuminoids and organic acids has been obtained from X. strumarium species, which have been reported as cytotoxic substance [2, 4, 5]. Moreover, many reports evidenced that sesquiterpene lactones present in plants have cytotoxic activity [4, 24, 25]. These sesquiterpene lactones trigger mitochondrial membrane transition, loss of mitochondrial membrane potential, and release of proapoptotic mitochondrial proteins leading to caspase activation and apoptotic cell death [26]. Xanthatin also caused a programmed cell death like in trypanosomes as evidenced by a reduction in mitochondrial membrane potential [27].
Our results showed the cytotoxic capacity of X. strumarium extract when high concentrations (25–100 μg/mL) for 24 h were applied to CHO cells in in vitro experimental conditions. The decrease in cell viability at high concentrations can be related to a toxic effect on the metabolism of the cell or to DNA damage. Coincidentally, precedent studies [2, 4, 5] conducted in in vitro experimental conditions reported cytotoxic activity of extracts obtained from this vegetable species. However, acute and subacute administration of root extracts from X. strumarium did not produce any toxicity when in vivo assay was assessed [28].
The in vitro assays used in the present study cover different levels of genotoxic damage expression. In these assays we include toxic concentrations greater than the IC50 to confirm cytotoxicity-mediated genotoxicity. A first level was the induction of point mutations at specific loci in Salmonella typhimurium. The negative outcome of bacterial testing of extract seems to correspond with the absence of potential to induce mutagenic changes in DNA. The second level was the analysis of SCE in mammalian CHO cells. The X. strumarium extract produced a significant induction of SCE, which is a sensitive method for the measurement of DNA damage at primary structure level. The X. strumarium extract also proved to be clastogenic at the cytogenetic level measuring CA induction in CHO cells only at a concentration that produced severe toxic effects, as assessed by the strong decrease in the mitotic index. The positive result obtained with the highest dose of the extract in the in vitro comet assay where hedgehog or apoptotic-like cells were observed indicated that the test substance induces DNA primary damage (both single- and double-strand breaks) in cultured mammalian cells. This result is in line with that obtained with the same concentration in CA analysis.
Maybe the DNA damage can be explained on the basis of a systemic toxic effect. Today, it is known that the double bonds present in the α-methyleneγ-lactone and cyclopentanone moieties confer to sesquiterpene lactones an affinity towards thiol groups of proteins and glutathione inducing oxidative stress in the cell [29]. There is no evidence that chromosomal aberrations induced by sesquiterpene lactones result from direct interaction with DNA or the chromosome structure; rather, an indirect mechanism mediated by cytotoxicity has been suggested [24]. The issue of cell-toxicity-mediated genotoxicity has been intensively addressed and many related endpoints have been proposed [30]. Amongst them, inhibition of key enzymes of energetic metabolism (oxidative phosphorylation) and nucleic acid replication (DNA polymerase) and expression of NFkB and caspase 3, which are proapoptotic and induce DNA damage, have been observed by sesquiterpene lactones [24, 25, 31, 32].
In addition, the present study reveals that the administration of 500–2000 mg/Kg dose of X. strumarium extractexhibited no genotoxic activity in our MN in vivo assays. There was no mortality or signs of toxicity in male and female animals. The treatment, until sacrifice time, did not produce any adverse reactions or any significant changes in the body weights (data not shown) as compared to untreated controls. In a similar work realized by Diaz et al. [11] the authors did not observe increases in the micronucleated erythrocytes frequency in mouse treated orally with extracts of this plant at doses of 500, 1000, and 2000 mg/kg body weight. Another study of toxicity performed with chloroform and hexane soluble fractions of X. strumarium reveals that the administration of a very high dose (5 g/kg body weight) for acute toxicity determination could not make any abnormal changes at gross as well as histopathological levels in the treated animals [28].
In our case, observed genotoxic effects were not confirmed in vivo with micronucleus test. This experiment evidenced our postulation indicating that nontoxic doses showed neither DNA damage nor toxic effects. Besides the usual arguments invoked to explain the in vivo negative performance of in vitro genotoxins, which deal mainly with the pharmacokinetic profile of absorption-distribution-metabolism-excretion, one may wonder if genotoxic response to X. strumarium extract was related to the reported cytotoxicity, which could implicate an indirect interference with nuclear functions to arrive to cell death, as it happens for many anticancer agents [24, 25, 31, 32].
We suggest that further examination of the cytotoxic potential of these compounds should be pursued. Further studies have to be performed using various cancer models to elucidate the exact mechanism by which X. strumarium extract exerts its cytotoxic action. In addition, new molecular studies are needed in order to elucidate the interaction of this compound in cell biology and the consequences for human health.
5. Conclusions
The data presented here constitute the most complete study on the genotoxic potential the aerial parts of X. strumarium L. and show that the extract can induce in vitro DNA damage at cytotoxic concentrations and, however, does not induce micronucleus in bone marrow of mice treated orally with extracts of this plant at dose of 500–2000 mg/kg body weight in our experimental conditions.
Acknowledgments
The authors are grateful to Dipartimento di Biologia, Universita degli Studi “Roma TRE”, and Istituto di Biologia e Patologia Molecolare, CNR, for the generous support and great encouragement in carrying out the work.
Conflict of Interests
The authors declare that there is no conflict of interests regarding the publication of this paper.
References
- 1.Lizama RS, Martínez MM, Lantigua REI. Plantas medicinales de uso tradicional en pinar del Río. Estudio etnobotánico. I. Revista Cubana de Farmacia. 1998;32(1):57–62. [Google Scholar]
- 2.Han T, Li H-L, Zhang Q-Y, et al. Bioactivity-guided fractionation for anti-inflammatory and analgesic properties and constituents of Xanthium strumarium L. Phytomedicine. 2007;14(12):825–829. doi: 10.1016/j.phymed.2007.01.010. [DOI] [PubMed] [Google Scholar]
- 3.Yin MH, Kanga DG, Cho DH, Kwon TO, Lee HS. Screening of vaso relaxant activity of some medicinal plants used in oriental medicines. Journal of Ethnopharmacology. 2005;99(1):113–117. doi: 10.1016/j.jep.2005.02.013. [DOI] [PubMed] [Google Scholar]
- 4.Ramírez-Erosa I, Huang Y, Hickie RA, Sutherland RG, Barl B. Xanthatin and xanthinosin from the burs of Xanthium strumarium L. as potential anticancer agents. Canadian Journal of Physiology and Pharmacology. 2007;85(11):1160–1172. doi: 10.1139/Y07-104. [DOI] [PubMed] [Google Scholar]
- 5.Kim HS, Lee TS, Yeo SW, Seong LS, Yu TS. Isolation and characterization of antitumor agents from Xanthium strumarium L. Korean Society for Biotechnology and Bioengineering Journal. 2003;18:324–328. [Google Scholar]
- 6.Dong KK, Chang KS, Dong WB, Yeon SK, Min-Suk Y, He KK. Identification and biological characteristics of an antifungal compound extracted from Cocklebur (Xanthium strumarium) against Phytophthoradrechsleri. The Plant Pathology Journal. 2002;18:288–292. [Google Scholar]
- 7.Favier LS, María AOM, Wendel GH, et al. Anti-ulcerogenic activity of xanthanolide sesquiterpenes from Xanthium cavanillesii in rats. Journal of Ethnopharmacology. 2005;100(3):260–267. doi: 10.1016/j.jep.2005.02.042. [DOI] [PubMed] [Google Scholar]
- 8.Ferrer JP, Stoiber T, Parra AV, et al. Search of new antimitotics compounds from the Cuban flora. Boletin Latinoamericano y del Caribe de Plantas Medicinales y Aromaticas. 2011;10(1):75–82. [Google Scholar]
- 9.Menon GS, Kuchroo K, Dasgupta D. Interaction of microtubules with active principles of Xanthium strumarium . Physiological Chemistry and Physics and Medical NMR. 2001;33(2):153–162. [PubMed] [Google Scholar]
- 10.Tran QL, Tezuka Y, Ueda J-Y, et al. In vitro antiplasmodial activity of antimalarial medicinal plants used in Vietnamese traditional medicine. Journal of Ethnopharmacology. 2003;86(2-3):249–252. doi: 10.1016/s0378-8741(03)00045-x. [DOI] [PubMed] [Google Scholar]
- 11.Diaz GM, Leσn MC, Iglesias E. Evaluación del efecto genotóxico del Xhantium strumarium L., (Guisazo de caballo) Revista Cubana de Plantas Medicinales. 2004;9:27–32. [Google Scholar]
- 12.Miranda M, Cuellar A. Manual de Prácticas de Laboratorio. Farmacognosia y Productos Naturales, Universidad de La Habana, IFAL; 2000. [Google Scholar]
- 13.Maron DM, Ames BN. Revised methods for the Salmonella mutagenicity test. Mutation Research. 1983;113(3-4):173–215. doi: 10.1016/0165-1161(83)90010-9. [DOI] [PubMed] [Google Scholar]
- 14.Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. Journal of Immunological Methods. 1983;65(1-2):55–63. doi: 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]
- 15.De Salvia R, Fiore M, Aglitti T, Festa F, Ricordy R, Cozzi R. Inhibitory action of melatonin on H2O2- and cyclophosphamide-induced DNA damage. Mutagenesis. 1999;14(1):107–112. doi: 10.1093/mutage/14.1.107. [DOI] [PubMed] [Google Scholar]
- 16.Sánchez-Lamar A, Fonseca G, Fuentes JL, et al. Assessment of the genotoxic risk of Punica granatum L. (Punicaceae) whole fruit extracts. Journal of Ethnopharmacology. 2007;115(3):416–422. doi: 10.1016/j.jep.2007.10.011. [DOI] [PubMed] [Google Scholar]
- 17.ICH. Genotoxicity: Guidance on Specific Aspects of Regulatory Genotoxicity Test for Pharmaceuticals. 1995. [Google Scholar]
- 18.Galloway SM, Aardema MJ, Ishidate M, Jr., et al. Report from working group on in vitro tests for chromosomal aberrations. Mutation Research: Environmental Mutagenesis and Related Subjects Including Methodology. 1994;312(3):241–261. doi: 10.1016/0165-1161(94)00012-3. [DOI] [PubMed] [Google Scholar]
- 19.Cornetta T, Padua L, Testa A, et al. Molecular biomonitoring of a population of nurses handling antineoplastic drugs. Mutation Research: Fundamental and Molecular Mechanisms of Mutagenesis. 2008;638(1-2):75–82. doi: 10.1016/j.mrfmmm.2007.08.017. [DOI] [PubMed] [Google Scholar]
- 20.Collins AR. The comet assay for DNA damage and repair: principles, applications, and limitations. Applied Biochemistry and Biotechnology B: Molecular Biotechnology. 2004;26(3):249–261. doi: 10.1385/MB:26:3:249. [DOI] [PubMed] [Google Scholar]
- 21.Hayashi M, Tice RR, MacGregor JT, et al. In vivo rodent erythrocyte micronucleus assay. Mutation Research: Environmental Mutagenesis and Related Subjects Including Methodology. 1994;312(3):293–304. doi: 10.1016/0165-1161(94)90039-6. [DOI] [PubMed] [Google Scholar]
- 22.Schmid W. The micronucleus test for cytogenetic analysis. In: Hollaender A, editor. Chemical Mutagens: Principles and Methods for Their Detection. Vol. 4. New York, NY, USA: Plenum Press; 1976. pp. 31–53. [Google Scholar]
- 23.Kirkland DJ. Statistical Evaluation of Mutagenicity Test. Cambridge University Press; 1989. [Google Scholar]
- 24.Ramos A, Rivero R, Visozo A, Piloto J, García A. Parthenin, a sesquiterpene lactone of Parthenium hysterophorus L. is a high toxicity clastogen. Mutation Research: Genetic Toxicology and Environmental Mutagenesis. 2002;514(1-2):19–27. doi: 10.1016/s1383-5718(01)00321-7. [DOI] [PubMed] [Google Scholar]
- 25.Zhang S, Ong C-N, Shen H-M. Critical roles of intracellular thiols and calcium in parthenolide-induced apoptosis in human colorectal cancer cells. Cancer Letters. 2004;208(2):143–153. doi: 10.1016/j.canlet.2003.11.028. [DOI] [PubMed] [Google Scholar]
- 26.Rosenkranz V, Wink M. Alkaloids induce programmed cell death in bloodstream forms of trypanosomes (Trypanosoma b. brucei) Molecules. 2008;13(10):2462–2473. doi: 10.3390/molecules13102462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Marco JA, Sanz-Cervera JF, Corral J, Carda M, Jakupovic J. Xanthanolides from Xanthium: absolute configuration of xanthanol, isoxanthanol and their C-4 epimers. Phytochemistry. 1993;34(6):1569–1576. [Google Scholar]
- 28.Aranjani JM, Rao CM, Manuel A, Rao JV, Udupa N, Hebbar K. Acute and subacute toxicity of chloroform and hexaneextracts of root of Xanthium strumarium . Comparative Clinical Pathology. 2012;21(6):1223–1230. [Google Scholar]
- 29.Ahmed AA, Mahmoud AA, El-Gamal AA. A xanthanolide diol and a dimeric xanthanolide from Xanthium species. Planta Medica. 1999;65(5):470–472. doi: 10.1055/s-2006-960817. [DOI] [PubMed] [Google Scholar]
- 30.Kovacs A, Vasas A, Forgo P, Rethy B, Zupko I, Hohmann J. Xanthanolides with antitumouractivity from Xanthium italicum . Zeitschrift fur Naturforschung C. 2009;64:343–349. doi: 10.1515/znc-2009-5-607. [DOI] [PubMed] [Google Scholar]
- 31.Nibret E, Youns M, Krauth-Siegel RL, Wink M. Biological activities of xanthatin from Xanthium strumarium leaves. Phytotherapy Research. 2011;25(12):1883–1890. doi: 10.1002/ptr.3651. [DOI] [PubMed] [Google Scholar]
- 32.Zhang L, Ruan J, Yan L, et al. Xanthatin induces cell cycle arrest at G2/M checkpoint and apoptosis via disrupting NF-κB pathway in A549 non-small-cell lung cancer cells. Molecules. 2012;17(4):3736–3750. doi: 10.3390/molecules17043736. [DOI] [PMC free article] [PubMed] [Google Scholar]