

Abstract
Staphylococcus aureus is capable of causing a spectrum of human illnesses. During serious S. aureus infections, the staphylococcal pathogen-associated molecular patterns (PAMPs) such as peptidoglycan, lipoteichoic acid, and lipoproteins and even intact S. aureus, are believed to act in conjunction with the staphylococcal superantigens (SSAg) to activate the innate and adaptive immune system, respectively, and cause immunopathology. However, recent studies have shown that staphylococcal PAMPs could suppress inflammation by several mechanisms and protect from staphylococcal toxic shock syndrome, a life-threatening systemic disease caused by toxigenic S. aureus. Given the contradictory pro- and anti-inflammatory roles of staphylococcal PAMPs, we examined the effects of S. aureus-derived molecular patterns on immune responses driven by SSAg in vivo using HLA-DR3 and HLA-DQ8 transgenic mice. Our study showed that neither S. aureus-derived peptidoglycans (PGN), lipoteichoic acid (LTA), nor heat-killed Staphylococcus aureus (HKSA) inhibited SSAg-induced T cell proliferation in vitro. They failed to antagonize the immunostimulatory effects of SSAg in vivo as determined by their inability to attenuate systemic cytokine/chemokine response and reduce SSAg-induced T cell expansion. These staphylococcal PAMPs also failed to protect HLA-DR3 as well as HLA-DQ8 transgenic mice from either SSAg-induced toxic shock or pneumonia induced by a SSAg-producing strain of S. aureus.
1. Introduction
Staphylococcus aureus is a highly successful opportunistic pathogen. A surprisingly high percentage of healthy humans (up to 40%) can be asymptomatically colonized with S. aureus [1]. At the same time, S. aureus can cause a wide variety of diseases, from relatively benign skin infections, such as folliculitis and furunculosis, to life-threatening conditions, including erysipelas, deep-seated abscesses, osteomyelitis, pneumonia, endocarditis, sepsis, and toxic shock syndrome (TSS) [2]. Recent epidemiological studies indicate that, at least in the US, the annual mortality caused by the methicillin-resistant strains of S. aureus is higher than the mortality caused by HIV/AIDS [3, 4]. Therefore, it is important to understand the immunopathogenesis of invasive diseases caused by S. aureus.
The pathogenicity and virulence of invasive S. aureus are determined by several exotoxins. Staphylococcal superantigens (SSAg) are one such family of exotoxins produced by S. aureus. SSAg are the most potent, naturally occurring biological activators of T lymphocytes [5]. Unlike conventional antigens, SSAg bind directly to cell surface MHC class II molecules outside of the peptide-binding groove without undergoing any processing. Subsequently, they bind to certain T cell receptor β chain variable region (TCR Vβ) families and robustly activate the T cells expressing them. The specificity of the SSAg to certain TCR Vβ regions alone, but not the TCR per se, results in activation of a large pool of CD4+ as well as CD8+ T cells (30% to 70% of the total T cells) [5]. SSAg can also activate the antigen presenting cells (APC), such as B cells, monocyte/macrophages, and dendritic cells (DC), by several mechanisms either directly, by crosslinking their MHC class II molecules [6], or indirectly, through cytokines/chemokines. This results in a profound elevation in systemic levels of several cytokines/chemokines which lead to clinical syndromes such as systemic inflammatory response syndrome (SIRS) and multiple organ dysfunction syndrome (MODS), which can culminate in the death of the afflicted patients [7]. As SSAg are produced readily in vivo, they play a major role in the pathogenesis of serious infections such as sepsis, pneumonia, and infective endocarditis (reviewed in [8]).
While the SSAg predominantly activate T cells of the adaptive immune system through TCR, certain other staphylococcal components such as the cell wall peptidoglycans, lipoteichoic acid, lipoproteins, unmethylated bacterial DNA containing the CpG motifs and even the intact bacteria, collectively called pathogen-associated molecular patterns (PAMPs), can activate the immune system through pathogen recognition receptors (PRR) comprising the toll-like receptors (TLR) and the NOD-like receptors (NLR), expressed by cells of both innate and adaptive arms of the immune system [9–11]. This leads to production of several proinflammatory cytokines, chemokines, and other inflammatory mediators [6, 12–16]. Some studies have shown that staphylococcal PAMPs can even directly activate T cells to a proinflammatory phenotype in a TLR2-Myd88 dependent manner [17], while others have demonstrated that S. aureus and staphylococcal PAMPs are able to override the immune regulatory functions of T regulatory cells, thereby indirectly promoting inflammation [18]. Overall, staphylococcal PAMPs can also promote inflammation and immunopathology similar to SSAg by several different mechanisms.
Given that both the SSAg and the staphylococcal PAMPs elicit a predominantly proinflammatory type of immune response [16, 19, 20], it is widely believed that, during invasive S. aureus infections, the PAMPs would act additively or synergistically with SSAg leading to a more pronounced inflammatory response and inflicting severe immunopathology [7, 21]. On the contrary, some recent studies have shown that the staphylococcal cell wall components and certain other staphylococcal PAMPs downregulate the immune response to SSAg as well as dampen antistaphylococcal immunity [22–24]. Thus, the staphylococcal PAMPs appear to have opposing immunological properties according to different studies. Given the significance of S. aureus infections [2, 25], it is important to understand the interaction between staphylococcal PAMPs and SSAg in vivo using appropriate animal models. As transgenic mice expressing human MHC class II molecules closely mimic the human immune response to infections caused by toxigenic staphylococci as well as streptococci producing superantigens [26–29], we examined the modulatory role of staphylococcal PAMPs in immune response to SSAg and the outcome of S. aureus pneumonia using HLA-DR3 and HLA-DQ8 transgenic mice.
2. Materials and Methods
2.1. Mice
HLA-DR3 transgenic mice expressing HLA-DRA1∗0101 and HLA-DRB1∗0301 transgenes and HLA-DQ8 transgenic mice expressing DQA1∗0301 and DQB1∗0302 on the endogenous MHC class II-null background were used in this study [30, 31]. Hereafter, they are referred to as HLA-DR3 or HLA-DQ8 mice, respectively. Mice were bred within the barrier facility of Mayo Clinic Immunogenetics Mouse Colony (Rochester, MN) and moved to a conventional facility after weaning. All experiments were approved by the Institutional Animal Care and Use Committee.
2.2. Antibodies and Reagents
The following antibodies were used for flow cytometry: CD4 (clone: GK1.5), CD8 (clone: 53–6.7), TCR Vβ6 (clone: RR4-7), TCR Vβ8 (clone: F23.1), CD86 (clone: GL-1), and class II (clone: Tu39). All antibodies were from BD Biosciences (San Jose, CA). Highly purified, endotoxin-reduced, staphylococcal enterotoxin B (SEB, Toxin Laboratories, Sarasota, FL, USA) was dissolved in PBS at 1 mg/mL and stored frozen at −80°C in aliquots. Peptidoglycans (PGN) purified from S. aureus, lipoteichoic acid (LTA) derived from S. aureus, and heat-killed Staphylococcus aureus (HKSA) were purchased from Invivogen (San Diego, CA), dissolved or resuspended in endotoxin-free PBS, respectively, and stored frozen at −80°C. The above-mentioned TLR agonists from this vendor have been extensively used in biomedical research.
2.3. In Vitro Cultures
For T cell proliferation, single-cell suspensions of splenocytes from HLA-DR3 and HLA-DQ8 transgenic mice were depleted of red blood cells by buffered ammonium chloride lysis. Cells were cultured in HEPES-buffered RPMI 1640 containing 5% fetal calf serum, serum supplement, streptomycin, and penicillin, at a concentration of 1 × 105 cells/well in 100 μL volumes in 96-well round-bottomed tissue culture plates. SEB, PGN, and HKSA were added at indicated concentrations at the same time. After 24 hours, the cells were pulsed with tritiated thymidine (1 μg/well). Cells were harvested 18 hours later and the extent of cell proliferation was determined by a standard thymidine incorporation assay.
To study the effect of staphylococcal PAMPs on expression of MHC class II molecules and costimulatory molecules, splenocytes from HLA-DR3 transgenic mice were cultured with medium alone or with HKSA (108 bacteria/mL), SEB (1 μg/mL), or SEB + HKSA (108 bacteria/mL + 1 μg/mL, resp.) in 24-well plates. Twenty-four hours later, the cells were obtained, washed, stained with indicated antibodies, and analyzed by flow cytometry. Flow data were analyzed using FlowJo X V10.0.6. Mononuclear cells were first gated based on forward and side scatter profiles. Other analyses were performed on cells within this gate.
2.4. In Vivo Studies
For serum cytokine analyses, HLA-DR3 mice were challenged with SEB (50 μg), PGN (50 μg), HKSA (108 bacteria), or SEB plus PGN or HKSA at these concentrations. The doses of these reagents were determined from previously published studies [22, 32–34]. Given the rapidity at which SSAg cause immune activation, these staphylococcal PAMPs were administered immediately after SEB. After 3 hrs, blood was collected in serum separation tubes (BD Biosciences), and sera were separated and stored frozen at −80°C in aliquots. The cytokine concentrations in the sera were determined in duplicate using a multiplex bead assay, per the manufacturer's protocol, and using their software and hardware (Bio-Plex and Bio-Rad).
For flow cytometric studies, HLA-DR3 mice were challenged with SEB (10 μg), PGN (50 μg), LTA (50 μg), or SEB plus PGN or LTA at the above indicated concentrations. Mice were killed 3 days later and distribution of CD4+ and CD8+ T cells bearing TCR Vβ8 and Vβ6 in the spleens was determined by flow cytometry as per standard protocol.
To study the modulatory role of staphylococcal PAMPs in SEB-induced mortality without D-gal sensitization, HLA-DR3 mice were challenged with SEB (50 μg), PGN (50 μg), HKSA (108 bacteria), or SEB plus PGN or HKSA. Animals were monitored closely and the moribund animals were euthanized as per IACUC recommendations. To study the modulatory role of staphylococcal PAMPs in SEB-induced mortality with D-gal sensitization, HLA-DR3 and HLA-DQ8 transgenic mice were pretreated with D-gal (30 mg/mouse). One hour later, animals were challenged with SEB (5–10 μg), PGN (50 μg), HKSA (108 bacteria), or SEB plus PGN or HKSA.
2.5. Induction of Pneumonia with Toxigenic S. aureus
We have recently demonstrated that HLA-DR3 transgenic mice are susceptible to pneumonia induced by a toxigenic strain of S. aureus, IDRL-7971, producing the SSAg, SEA, and SEB as tested by PCR and ELISA [29, 35]. Therefore, to study the modulatory role of staphylococcal PAMPs in pneumonia induced by a superantigen-producing strain of S. aureus, HLA-DR3 transgenic mice were intratracheally challenged as described previously with the S. aureus strain, IDRL-7971 (1.3–2.5 × 108 cfu/mouse). Immediately following bacterial inoculation, mice were left untreated or injected with HKSA intraperitoneally (108 bacteria/mouse). Animals were monitored closely for symptoms and moribund animals were removed from the study.
2.6. Statistics
Statistical analyses, charts, and generation of survival curves were done using GraphPad Prism, version 4.03.
3. Results
3.1. Modulation of HLA Class II Expression by Staphylococcal PAMPs
Direct binding to cell surface MHC class II molecules without undergoing processing is the first step in the series of events by which SSAg causes immune activation [5]. Therefore, any alterations in the cell surface expression of MHC class II molecules on APCs could modulate the response elicited by SSAg. In this context, some studies have shown that staphylococcal PAMPs upregulate the expression of MHC class II as well as costimulatory molecules on APCs [36], while others have shown that staphylococcal PAMPs rather downregulate the expression of MHC class II molecules, particularly on monocytes [22–24]. Since many types of APCs are involved in the presentation of SSAg in addition to monocytes, we investigated the effect of staphylococcal PAMPs on the expression of MHC class II molecules on different APCs. As HKSA would encompass all staphylococcal PAMPs, splenocytes from HLA-DR3 transgenic mice were cultured with HKSA or SEB or both and the abilities of these agents to modulate the cell surface expression of HLA-DR3 and CD86 on B220+ cells (predominantly B cells), CD11b+ cells (predominantly monocyte/macrophages), and CD11c+ cells (predominantly DC) were determined.
We first studied the effects of HKSA on MHC class II expression on CD11b+ splenocytes. Interestingly, unlike splenocytes cultured with medium alone, two distinct subsets of CD11b+ cells could be appreciated in splenocytes cultured with HKSA based on the expression profile of CD11b, with one expressing high levels of CD11b (CD11bhi) and the other expressing lower levels, CD11blo (Figure 1). In cells cultured with medium alone, CD11bhi was the dominant phenotype (nearly 80–90% of the CD11b+ cells were of the CD11bhi phenotype). However, in cells cultured with HKSA, 50–60% of the CD11b+ cells were of the CD11blo phenotype. In a similar manner, while 80–90% of the CD11b+ cells cultured with SEB were CD11bhi, >50% of the CD11b+ cells were CD11blo when cultured with SEB + HKSA (Figure 1). Therefore, we studied the expression profiles of HLA-DR3 and CD86 on CD11bhi as well as CD11blo subsets. HKSA had very little suppressive effect on the expression of HLA-DR3 within the CD11bhi subset either when cultured alone or with SEB (Figure 1). However, the expression of HLA-DR3 was significantly reduced in the CD11blo subsets that were generated in the presence of HKSA either alone or along with SEB. Similar pattern was seen with respect to expression of CD86. Overall, HKSA lowered the expression of HLA-DR3 and CD86 only on a subset of CD11b+ cells (Figure 1).
Figure 1.
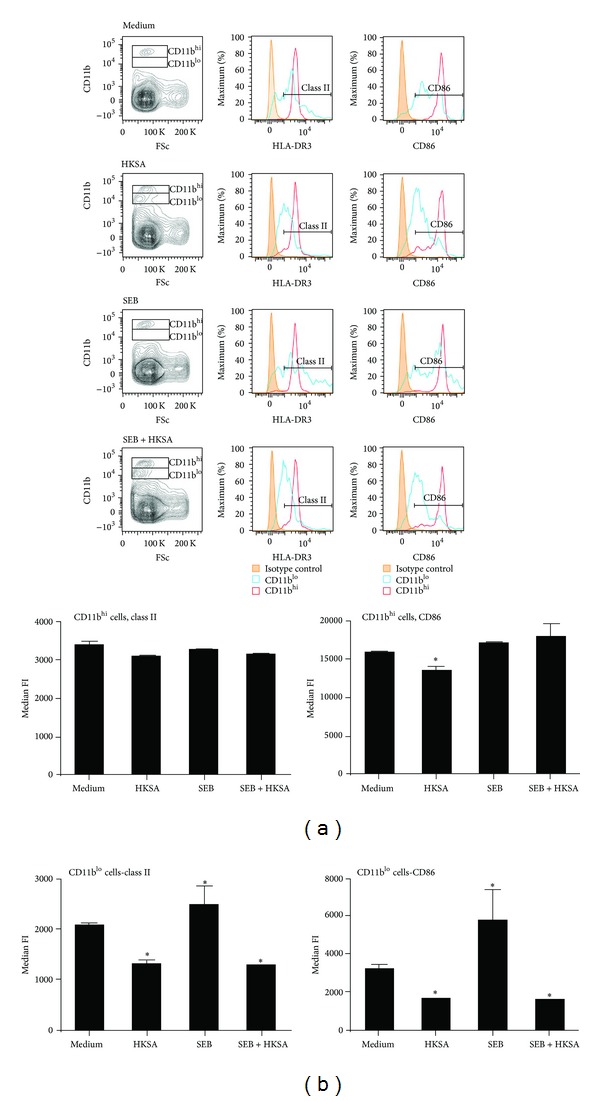
Modulation of expression of HLA-DR3 and CD86 on CD11b+ cells by HKSA. Splenic mononuclear cells from HLA-DR3 transgenic mice were cultured with medium, HKSA (108 bacteria/mL), SEB (1 μg/mL), or SEB + HKSA. 24 hours later, the cells were harvested and washed and expression of HLA-DR3 and CD86 on CD11b+ cells was analyzed by flow cytometry. Mononuclear cells were first gated based on forward and side scatter profiles. Other analyses were performed on cells within this gate. Representative histogram profiles and bar charts depicting median fluorescent intensity (MFI) are given. Each bar represents mean ± SE from 2 different experiments, each performed in triplicate. *P < 0.05 compared to cells cultured with medium.
We next studied the effect of HKSA on the expression of HLA-DR3 and CD86 on B220+ splenocytes. Unlike in CD11b+ cells, culturing with HKSA did not skew the B220+ cells to either B220hi or B220lo phenotype (Figure 2). Therefore, the entire B220+ population was studied. Unlike in CD11b+ cells, HKSA significantly increased the expression of HLA-DR3 molecules on B220+ cells over the basal level (median fluorescent intensity or MFI; 2185 ± 36 versus 2451 ± 151 in medium versus HKSA treated cells, respectively; P < 0.05; Figure 2). Culturing with SEB also significantly increased the expression of HLA-DR3 molecules on B220+ cells (MFI in SEB treated cells, 2581 ± 127 and P < 0.05, compared to untreated cells). Notably, SEB induced a much stronger upregulation of HLA-DR3 than HKSA. However, HKSA failed to suppress SEB-induced upregulation of HLA-DR3 on B220+ cells. On the contrary, expression of HLA-DR3 further increased when cultured with SEB + HKSA (MFI in SEB + HKSA treated cells, 2695 ± 205 and P < 0.05, compared to medium) (Figure 2). Similar pattern was noticed with respect to CD86 expression on B220+ cells. Culturing with HKSA caused a significant (P < 0.05) upregulation of CD86 levels on B220+ cells compared to untreated cells, while SEB caused a much stronger upregulation of CD86 compared to HKSA. However, culturing with HKSA and SEB resulted in the highest upregulation of CD86 compared to either of these agents alone. These results indicated that HKSA failed to suppress SEB-induced upregulation of HLA-DR3 and CD86 on B220+ cells. Similar phenomenon was seen in CD11c+ cells. HKSA, SEB, and their combination increased the expression of HLA-DR3 and CD86 expression on CD11c+ cells (see Supplementary Figure 1 in Supplementary Material available online at http://dx.doi.org/10.1155/2014/468285). Overall, these results suggested that HKSA augmented the expression of MHC class II and CD86 on B220+ cells and DC. While HKSA had minimal or no modulatory effect on CD11bhi cells in the presence of SEB, it reduced the expression of MHC class II and CD86 on the CD11blo cells, which were seen in the presence of HKSA.
Figure 2.
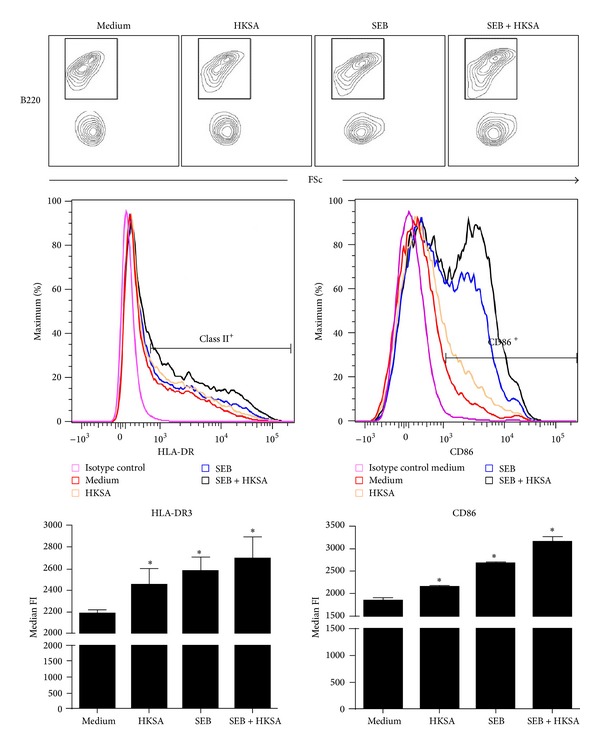
Modulation of expression of HLA-DR3 and CD86 on B220+ cells by HKSA. Splenic mononuclear cells from HLA-DR3 transgenic mice were cultured with medium, HKSA (108 bacteria/mL), SEB (1 μg/mL), or SEB + HKSA. 24 hours later, the cells were harvested and washed and expression of HLA-DR3 and CD86 on B220+ cells was analyzed by flow cytometry. Mononuclear cells were first gated based on forward and side scatter profiles. Other analyses were performed on cells within this gate. Representative histogram profiles and bar charts depicting median fluorescent intensity (MFI) are given. Each bar represents mean ± SE from 2 different experiments, each performed in triplicate. *P < 0.05 compared to cells cultured with medium.
3.2. Modulation of SSAg-Induced T Cell Activation by Staphylococcal PAMPs In Vitro
We next investigated how modulation in the expression of MHC class II molecules and CD86 induced by staphylococcal PAMPs translated into changes in SSAg-mediated T cell activation in vitro. For this, splenocytes from HLA-DR3 transgenic mice were stimulated with SEB in the presence or absence of certain staphylococcal PAMPs. First of all, HKSA by itself was weekly mitogenic to splenocytes. Most importantly, HKSA failed to suppress SEB-induced splenocyte proliferation in vitro (Figure 3(a)). We next tested the modulatory effects of PGN. PGN by itself was significantly mitogenic to splenocytes from HLA-DR3 mice (Stimulation Index ranging from 2.5 to 12 depending on the dose of PGN; P < 0.05; Student's t-test; Figure 3(b)). However, as would be expected of a superantigen, SEB was more mitogenic to splenocytes than PGN. More importantly, addition of PGN did not suppress SEB-induced splenocyte proliferation. Conversely, PGN, especially at 10 and 50 μg/mL, consistently augmented SEB-induced splenocyte proliferation (P < 0.05; Student's t-test). Similar results were obtained with HLA-DQ8 transgenic mice using PGN and HKSA (data not shown). Overall, PGN and HKSA failed to suppress SEB-induced splenocyte proliferation in vitro. On the contrary, PGN augmented SEB-induced splenocyte proliferation at higher concentration.
Figure 3.
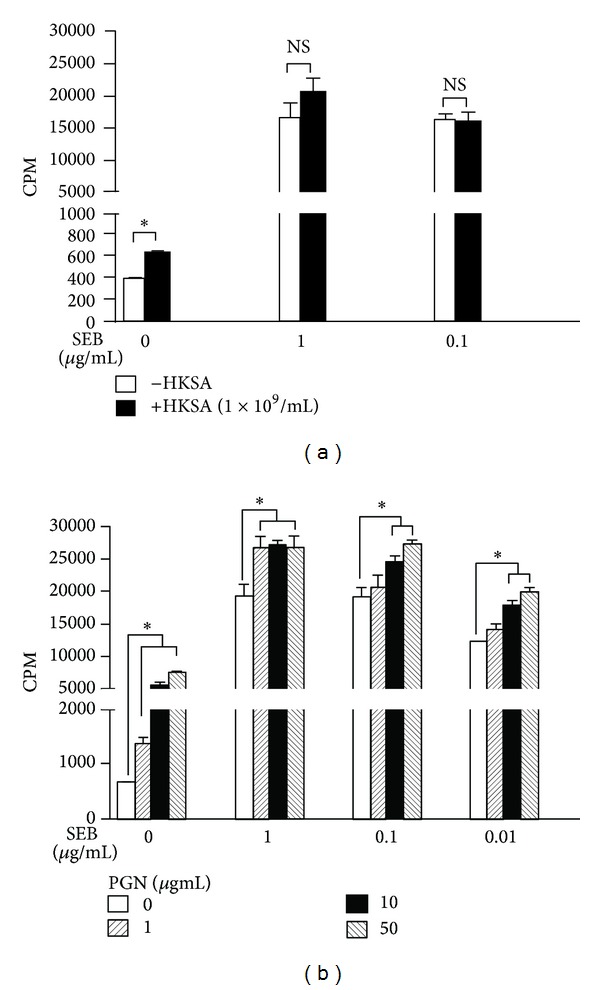
Effect of S. aureus-derived PAMPs on SEB-induced T cell proliferation in vitro. Splenic mononuclear cells from HLA-DR3 transgenic mice were cultured with indicated concentrations of SEB in the presence or absence of HKSA, 108 bacteria/mL (a), or PGN (b). T cell proliferation was determined by thymidine incorporation. *P < 0.05 compared to cells cultured without HKSA or PGN within that group. Each bar represents mean ± SE from triplicate wells. Representative data from one out of 3 similar experiments are given.
It should be noted that SSAg cause a systemic inflammatory disease characterized by failure of several vital organs that often leads to death [37], and in vitro studies with isolated splenocytes reflect very little about this systemic process. Therefore, to accurately determine the modulatory role of staphylococcal PAMPs in vivo, we subsequently performed a series of in vivo studies using our robust HLA-DR3 and HLA-DQ8 transgenic mice, which closely mimic the human immune responses to SSAg [38].
3.3. Modulation of SSAg-Induced Cytokine/Chemokine Elevation by Staphylococcal PAMPs In Vivo
Challenging HLA-DR3 transgenic mice with SSAg, such as SEB, elicits a massive and rapid elevation in systemic levels of various cytokines and chemokines leading to SIRS, multiple organ failure, and death, analogous to TSS in humans [38]. Therefore, we next investigated the modulatory effect of staphylococcal PAMPs on SEB-induced systemic inflammatory response syndrome. Serum cytokine analysis showed that naïve HLA-DR3 mice had very low levels of cytokines/chemokines (Figure 4). As expected, the serum levels of several cytokines (such as IL-6, IL12p40, IL-12p70, IL-10, IFN-γ, and TNF-α) and chemokines (MCP-1, KC, MIP-1α, MIP-1β, eotaxin, and G-CSF) were significantly elevated in mice challenged with PGN or HKSA compared to naïve mice, likely resulting from activation of the innate immune system. Given the biological property of SEB, it is not surprising to find that sera from HLA-DR3 mice challenged with SEB had profoundly elevated levels of all the cytokines and chemokines tested (Figure 4), consistent with our prior reports [30]. Moreover, the serum levels of all cytokines and chemokines tested were significantly higher in SEB treated mice compared to PGN or HKSA treated mice. This is also expected because SSAg are the most potent biological activators of the immune system. More importantly, when PGN and HKSA were injected along with SEB, they failed to suppress SEB-induced cytokine/chemokine production. There were no significant differences in the levels of various cytokines and chemokines tested between mice challenged with SEB alone compared to SEB + PGN or SEB + HKSA. Previous studies have shown that staphylococcal cell wall derivatives are more potent inducers of IL-10 than SSAg [22]. On the contrary, our studies show that SEB is more potent than staphylococcal PAMPs in eliciting IL-10. Overall, our results show that the staphylococcal PAMPs do elicit an immune response. However, they are less potent than SSAg and they failed to attenuate SEB-induced cytokine production in vivo.
Figure 4.
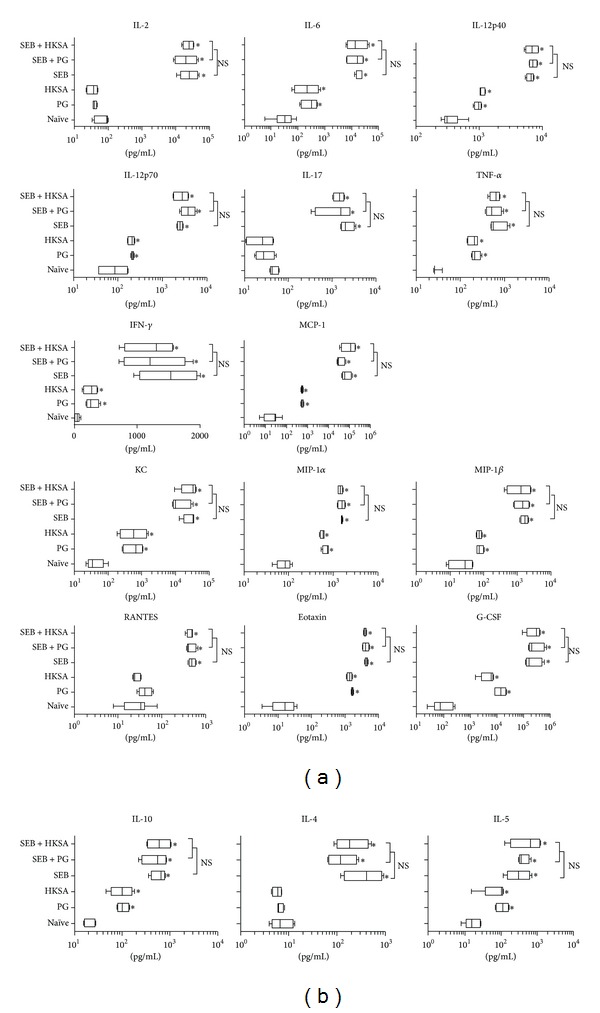
Effect of S. aureus-derived PAMPs on SEB-induced systemic inflammatory response syndrome in vivo. HLA-DR3 transgenic mice were challenged with staphylococcal enterotoxin B (SEB) alone (50 μg), staphylococcal peptidoglycan (PGN) alone (50 μg), or heat-killed Staphylococcus aureus (HKSA) alone (108 bacteria), SEB with PGN or SEB with HKSA. Animals bled 4 hours after injection and serum cytokines were quantified using Bioplex assays (Bio-Rad). Mean ± SE from 4–6 mice in each group. *P < 0.05 compared to naïve mice.
3.4. Modulation of SSAg-Induced Peripheral T Cell Expansion by Staphylococcal PAMPs In Vivo
Administration of SSAg, such as SEB, into HLA class II transgenic mice results in the expansion of CD4+ as well as CD8+ T cells expressing certain TCR Vβ families. It is known that this process is dependent on expression of MHC class II molecules and is cytokine driven. Therefore, by comparing the changes in T cell repertoire in mice challenged with SEB alone or SEB along with staphylococcal PAMPs, we were able to study the immunomodulatory functions of staphylococcal PAMPs. Compared to naïve mice, there was a 2-fold increase of CD4+ and CD8+ T cells expressing TCR Vβ8, which specifically binds to SEB, in mice challenged with SEB (Figure 5). As expected, PGN and LTA did not have this ability to induce expansion of T cells bearing specific TCR Vβ. More importantly, neither PGN nor LTA was able to suppress SEB-induced expansion of TCR Vβ8-bearing T cells (Figure 5; p = NS when comparing SEB with SEB + PGN or SEB with SEB + LTA). This corroborated with the in vivo cytokine data.
Figure 5.
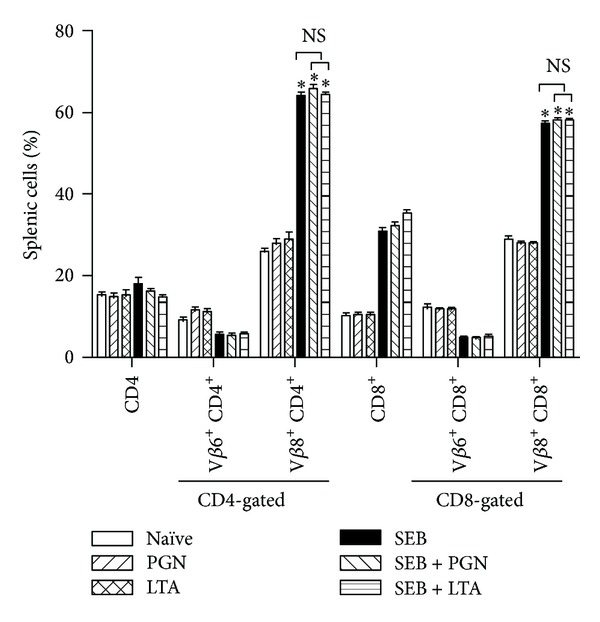
Effect of S. aureus-derived PAMPs on SEB-induced T cell expansion in vivo. HLA-DR3 transgenic mice were challenged with staphylococcal enterotoxin B (SEB) alone (10 μg), staphylococcal peptidoglycan (PGN) alone (50 μg), or lipoteichoic acid (LTA, 50 μg), SEB with PGN or SEB with LTA. Animals were killed 3 days later and distribution of T cells expressing TCR Vβ families were determined by flow cytometry. Mean ± SE from 5–8 mice in each group. *P < 0.05 compared to naïve mice. p = NS between SEB versus SEB + PGN, SEB versus SEB + LTA, and SEB + LTA versus SEB + PGN.
3.5. Modulation of SSAg-Induced TSS by Staphylococcal PAMPs
We next investigated the ability of staphylococcal PAMPs to protect from SSAg-induced TSS and death. Whereas all mice challenged with PGN or HKSA alone remained healthy, all HLA-DR3 mice challenged with SEB (50 μg) alone either succumbed to TSS or became very sick that they needed to be removed from the study (P < 0.005; the Log-rank (Mantel-Cox) test; Figure 6(a); 4–8 mice in each group). Interestingly, unlike in the previous report [22], administration of either PGN or HKSA failed to protect HLA-DR3 transgenic mice from SEB-induced TSS. Even though mortality in SEB + HKSA treated mice was delayed initially compared to SEB or SEB + PGN groups, all mice in this group succumbed earlier to TSS compared to other groups. Overall, there were no statistical differences in the survival between SEB, SEB + PGN, and SEB + HKSA groups (p = NS; the Log-rank (Mantel-Cox) test). However, the survival of all SEB-challenged mice (SEB alone, SEB + PGN, or SEB + HKSA) was significantly reduced compared to naïve and PGN or HKSA challenged mice (P < 0.005; Figure 6(a); the Log-rank (Mantel-Cox) test).
Figure 6.
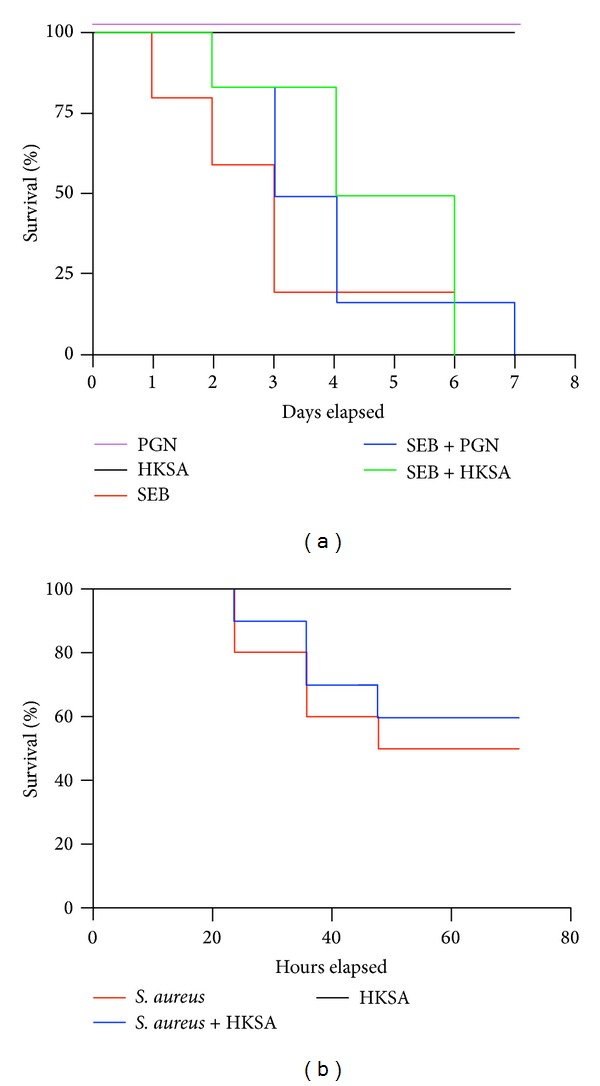
Effect of S. aureus-derived PAMPs on SEB-induced TSS and S. aureus-induced pneumonia. (a) HLA-DR3 transgenic mice were challenged with staphylococcal enterotoxin B (SEB) (50 μg), staphylococcal peptidoglycan (PGN) (50 μg), or heat-killed Staphylococcus aureus (HKSA) (108 bacteria), SEB with PGN or SEB with HKSA. Animals were monitored every 6 hours (4–8 mice per group). (b) HLA-DR3 transgenic mice were challenged intratracheally with a toxigenic strain of S. aureus capable of producing the SSAg, SEA, and SEB (1.3–2.5 × 108 cfu/mouse). Immediately following bacterial inoculation, mice were left untreated or injected with HKSA intraperitoneally (108 bacteria/mouse). Animals were monitored closely for symptoms. All moribund animals were removed from the study (6–8 mice per group).
It should be noted that the above experiments were done without D-gal sensitization. As conventional laboratory mouse strains are resistant to SSAg-induced shock, they are often presensitized with D-gal. D-gal is a hepatotoxic agent and sensitizes hepatocytes to TNF-α-mediated cell death [32, 39]. Therefore, in the next set of experiments, we pretreated HLA-DR3 mice with D-gal and one hour later mice were challenged with SEB (5–10 μg), PGN (50 μg), HKSA (108 bacteria), SEB plus PGN, or SEB plus HKSA, similar to the study by Chau et al. [22]. While all mice treated with D-gal alone, D-gal plus PGN, or D-gal plus HKSA remained healthy, all animals treated with D-gal and SEB (either alone or along with PGN or HKSA) rapidly became hypothermic and lethargic and failed to survive beyond 9 hours (6–8 mice in each group; Table 1). There were no significant differences in survival between D-gal plus SEB, D-gal plus SEB plus PGN, and D-gal plus SEB plus HKSA groups. To rule out the possibility that the lack of protection from TSS by PGN and HKSA may be specific for HLA-DR3, we repeated this study with HLA-DQ8 transgenic mice. HLA-DQ8 transgenic mice were pretreated with D-gal as described earlier and subsequently challenged with SEB (10 μg), PGN (10 or 50 μg), HKSA (108 bacteria), SEB plus PGN, or SEB plus HKSA. As seen with HLA-DR3 transgenic mice, PGN and HKSA failed to protect from SEB-induced TSS in HLA-DQ8 transgenic mice (4–6 mice in each group; Table 1). Overall, staphylococcal PAMPs conferred little protection from SSAg-induced TSS.
Table 1.
Modulation of SEB-induced TSS by staphylococcal PAMPs.
| HLA class II type | Treatmenta | D-gal | Mortality |
|---|---|---|---|
| HLA-DR3 | None | + | 0/4 |
| HLA-DR3 | PGN | + | 0/4 |
| HLA-DR3 | HKSA | + | 0/4 |
| HLA-DR3 | SEB (10 μg) | + | 8/8 |
| HLA-DR3 | SEB (5 μg) | + | 6/6 |
| HLA-DR3 | SEB (10 μg) + PGN | + | 5/5 |
| HLA-DR3 | SEB (5 μg) + PGN | + | 4/4 |
| HLA-DR3 | SEB (10 μg) + HKSA | + | 3/3 |
| HLA-DR3 | SEB (5 μg) + HKSA | + | 4/4 |
| HLA-DQ8 | None | + | 0/4 |
| HLA-DQ8 | PGN | + | 0/4 |
| HLA-DQ8 | HKSA | + | 0/4 |
| HLA-DQ8 | SEB | + | 6/6 |
| HLA-DQ8 | SEB (10 μg) + PGN | + | 6/6 |
| HLA-DQ8 | SEB (10 μg) + HKSA | + | 6/6 |
HLA-DR3 and HLA-DQ8 transgenic mice were pretreated with D-galactosamine and an hour later challenged with the following agents. Animals were followed for 9 hours.
aTreatments given
PGN: peptidoglycan—50 μg/mouse
HKSA: heat-killed S. aureus—108 bacteria/mouse
SEB: staphylococcal enterotoxin B—10 μg/mouse or 5 μg/mouse
D-gal: D-galactosamine—30 mg/mouse 1 hour prior to being challenged with PGN, HKSA, SEB or at the same time.
3.6. Role of Staphylococcal PAMPS in Outcome of S. aureus-Induced Lethal Pneumonia
In the final in vivo study, we investigated the immunomodulatory effects of staphylococcal PAMPs on pneumonia induced superantigen-producing S. aureus [29, 35]. Since HKSA would encompass all the staphylococcal PAMPs, infected animals were treated with HKSA. As expected, HKSA by itself was not lethal (Figure 6(b)). As shown by us earlier, there was significantly higher mortality (P < 0.01; the Log-rank (Mantel-Cox) test) in HLA-DR3 mice infected with the toxigenic S. aureus strain IDRL-7971 compared to mice treated with HKSA alone [29, 35]. Moreover, the mortality remained significantly high in HLA-DR3 transgenic mice that were infected with IDRL-7971 and treated with HKSA. The survival curves of mice with IDRL-7971-induced pneumonia and those with IDRL-7971-induced pneumonia treated with HKSA were not significantly different (P = 0.62; the Log-rank (Mantel-Cox) test). Overall, these results suggested that staphylococcal PAMPs do not confer significant protection in certain staphylococcal diseases such as TSS and pneumonia.
4. Discussion
The pattern recognition receptors (PRR) expressed by the cells of the innate immune system rapidly sense PAMPs [10] and mount an inflammatory response to contain infection [40, 41]. PAMPs associated with S. aureus are no exceptions [9, 20, 42]. However, the superantigen exotoxins produced by S. aureus (SSAg) predominantly activate the T cell arm of adaptive immunity by directly binding to MHC class II molecules and subsequently stimulating the T lymphocytes expressing certain TCR Vβ families [5]. Given that both staphylococcal PAMPs and SSAg activate the immune system, exposure to both of these staphylococcal entities, as might occur in many clinical conditions such as staphylococcal sepsis, pneumonia, or toxic shock syndrome (TSS), is believed to result in synergistic activation of the innate and adaptive immune system and a worse disease outcome [7, 21, 43]. However, some recent studies have suggested that staphylococcal PAMPs may actually suppress the immune response to SSAg and cause immune tolerance or even immune deviation following S. aureus infections [22–24]. Given these conflicting reports, we investigated the immunomodulatory role of staphylococcal PAMPs in immune responses elicited by SSAg during TSS and in staphylococcal pneumonia. Our studies, which used the robust HLA class II transgenic mouse model, showed that staphylococcal PAMPs do not suppress SSAg-induced T cell proliferation, do not dampen SSAg-induced systemic inflammatory response, and fail to protect from SSAg-induced TSS or staphylococcal pneumonia.
One of the major hypotheses put forth by the previous in vitro studies addressing the immunomodulatory properties of staphylococcal PAMPs is that they cause immunomodulation by downregulating the expression of MHC class II on macrophages [22, 24]. These studies demonstrated that the staphylococcal cell wall components bind to TLR2, which are constitutively expressed on monocyte/macrophages and downregulate their MHC class II molecule expression [22, 24]. This diminishes the ability of monocyte/macrophages to present SSAg, causing reduced T cell activation, immunosuppression/immune deviation, and protection from TSS [22, 24]. However, these studies did not take into consideration the role of other APCs that are present in vivo.
It is well known that, in addition to monocytes, HLA class II molecules are constitutively expressed at very high levels on several APC types, including DC and B cells. Particularly, B cells express high levels of MHC class II molecules and can efficiently present SSAg. While we were able to reproduce the findings of Frodermann et al. and Wang et al. that HKSA could downregulate the expression of MHC class II on a subset of CD11b+ cells [23, 24], surprisingly HKSA had little or no suppressive effect on the expression of MHC class II molecules on B220+ cells (Figures 1 and 2) and CD11c+ (Supplementary Figure 1). Rather HKSA augmented the expression of MHC class II as well as CD86, an important costimulatory molecule on these APC subsets. Since B cells and DC vastly outnumber the monocyte/macrophages in the spleen, it is not surprising that staphylococcal PAMPs had no net immunosuppressive effect in vitro and in vivo. The discrepancy between previous studies and our study could be attributed to the use of human cells versus cells from HLA class II transgenic mice, respectively.
In addition to professional APC, even human T cells are known to express HLA class II molecules upon activation. In a similar manner, in addition to the conventional APCs, MHC class II (HLA-DR3) molecules are also expressed on activated T cells in our transgenic mice, as shown by RT-PCR and flow cytometry. T cells from HLA-DR3 transgenic mice can present antigens and SSAg to other T cells [44]. Thus, HLA class II molecules are widely expressed by several professional and nonprofessional APCs. Given the abundance of HLA class II molecules in vivo, the affinities of SSAg for HLA class II molecules, the rapidity (SSAg can elicit an immune response within minutes) and robustness with which SSAg elicit an immune response, and the presentation of SSAg by professional and nonprofessional APCs (including T lymphocytes) might rapidly activate the immune system and override the regulatory effects of TLR2. Our in vitro and in vivo experiments support this hypothesis.
Another mechanism by which staphylococcal PAMPs mediate immune modulation is through induction of IL-10, specifically by the monocytes [23, 24]. However, staphylococcal PAMP-driven, IL-10-mediated immunomodulation has been shown to be abolished by INF-γ [23, 24]. We and others have consistently shown that SSAg readily induce IFN-γ and systemic levels of IFN-γ are elevated (both IFN-γ mRNA and protein) in mice (even in the current study) and humans undergoing TSS [27, 32, 38]. Therefore, while staphylococcal PAMPs could suppress the immune response through monocyte/macrophage-derived IL-10, elevated levels of proinflammatory cytokines (such as IL-12, IFN-γ, and IL-17) might overcome this suppressive effect. In this context, other studies have shown that even human PBMCs stimulated with SSAg and exposed to staphylococcal PAMPs or HKSA produced significantly higher levels of proinflammatory cytokines [45]. Even staphylococcal PAMPs by themselves can induce the production of proinflammatory cytokines by human PBMC and promote immunopathology [34, 46–48].
Finally, the animal model used to investigate the interaction between staphylococcal PAMPs and SSAg also plays a major role in drawing apt conclusions. We have shown that our HLA class II transgenic mice respond robustly to SSAg and are highly susceptible to S. aureus-induced pneumonia as well as SSAg-induced TSS without requiring any sensitizing agents such as LPS or D-galactosamine [38]. Therefore, the heightened potency of SSAg in HLA class II transgenic mice (and in humans) may be beyond the realm of immunoregulation by staphylococcal PAMPs. Taken together, our results suggest that staphylococcal PAMPs offer little protection during staphylococcal TSS and pneumonia conditions wherein higher amounts of SSAg are present. High mortality rates associated with invasive S. aureus infections underscore the fact that even in humans staphylococcal PAMPs are unable to effectively mitigate SIRS and MODS. However, as suggested in a recent study [49], it is possible that staphylococcal PAMPs might play an immunosuppressive or immune deviatory role during S. aureus colonization where elevated levels of SSAg are not expected to be present. This needs further investigation.
Supplementary Material
Splenic mononuclear cells from HLA-DR3 transgenic mice were cultured with medium, HKSA (108 bacteria/ml), SEB (1μg/ml) or SEB+HKSA. 24 hours later, the cells were harvested, washed and expression of HLA-DR3 and CD86 on CD11c+ was analyzed by flow cytometry. Charts depict mean fluorescent intensity (MFI). Each bar represents mean ± SE from 2 different experiments.
Acknowledgments
We thank Julie Hanson and her crew for excellent mice husbandry and Michelle Smart for characterizing the transgenic mice. This study was funded by NIH grants AI101172 (GR) and AI68741 (GR and CSD).
Abbreviations
- PAMPs:
Pathogen-associated molecular patterns
- SSAg:
Staphylococcal superantigen
- PGN:
Peptidoglycans
- LTA:
Lipoteichoic acid
- HKSA:
heat-killed Staphylococcus aureus
- SEB:
Staphylococcal enterotoxin B.
Conflict of Interests
The authors declare that there is no conflict of interests regarding the publication of this paper.
References
- 1.Kluytmans J, van Belkum A, Verbrugh H. Nasal carriage of Staphylococcus aureus: epidemiology, underlying mechanisms, and associated risks. Clinical Microbiology Reviews. 1997;10:505–520. doi: 10.1128/cmr.10.3.505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Klevens RM, Morrison MA, Nadle J, et al. Invasive methicillin-resistant Staphylococcus aureus infections in the United States. The Journal of the American Medical Association. 2007;298(15):1763–1771. doi: 10.1001/jama.298.15.1763. [DOI] [PubMed] [Google Scholar]
- 3.Otto M. Understanding the epidemic of community-associated MRSA and finding a cure: are we asking the right questions? Expert Review of Anti-Infective Therapy. 2009;7(2):141–143. doi: 10.1586/14787210.7.2.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Boucher HW, Corey GR. Epidemiology of methicillin-resistant Staphylococcus aureus . Clinical Infectious Diseases. 2008;46(supplement 5):S344–S349. doi: 10.1086/533590. [DOI] [PubMed] [Google Scholar]
- 5.Fraser JD, Proft T. The bacterial superantigen and superantigen-like proteins. Immunological Reviews. 2008;225(1):226–243. doi: 10.1111/j.1600-065X.2008.00681.x. [DOI] [PubMed] [Google Scholar]
- 6.Duffy D, Rouilly V, Libri V, et al. Functional analysis via standardized whole-blood stimulation systems defines the boundaries of a healthy immune response to complex stimuli. Immunity. 2014;40(3):436–450. doi: 10.1016/j.immuni.2014.03.002. [DOI] [PubMed] [Google Scholar]
- 7.Lappin E, Ferguson AJ. Gram-positive toxic shock syndromes. The Lancet Infectious Diseases. 2009;9(5):281–290. doi: 10.1016/S1473-3099(09)70066-0. [DOI] [PubMed] [Google Scholar]
- 8.Spaulding AR, Salgado-Pabón W, Kohler PL, Horswill AR, Leung DYM, Schlievert PM. Staphylococcal and streptococcal superantigen exotoxins. Clinical Microbiology Reviews. 2013;26(3):422–447. doi: 10.1128/CMR.00104-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fournier B, Philpott DJ. Recognition of Staphylococcus aureus by the innate immune system. Clinical Microbiology Reviews. 2005;18(3):521–540. doi: 10.1128/CMR.18.3.521-540.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mogensen TH. Pathogen recognition and inflammatory signaling in innate immune defenses. Clinical Microbiology Reviews. 2009;22(2):240–273. doi: 10.1128/CMR.00046-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.de Kimpe SJ, Kengatharan M, Thiemermann C, Vane JR. The cell wall components peptidoglycan and lipoteichoic acid from Staphylococcus aureus act in synergy to cause shock and multiple organ failure. Proceedings of the National Academy of Sciences of the United States of America. 1995;92(22):10359–10363. doi: 10.1073/pnas.92.22.10359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cho JS, Pietras EM, Garcia NC, et al. IL-17 is essential for host defense against cutaneous Staphylococcus aureus infection in mice. The Journal of Clinical Investigation. 2010;120(5):1762–1773. doi: 10.1172/JCI40891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Reynolds JM, Pappu BP, Peng J, et al. Toll-like receptor 2 signaling in CD4+ T lymphocytes promotes T helper 17 responses and regulates the pathogenesis of autoimmune disease. Immunity. 2010;32(5):692–702. doi: 10.1016/j.immuni.2010.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Khare S, Dorfleutner A, Bryan NB, et al. An NLRP7-containing inflammasome mediates recognition of microbial lipopeptides in human macrophages. Immunity. 2012;36(3):464–476. doi: 10.1016/j.immuni.2012.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kapetanovic R, Parlato M, Fitting C, Quesniaux V, Cavaillon J-M, Adib-Conquy M. Mechanisms of TNF induction by heat-killed Staphylococcus aureus differ upon the origin of mononuclear phagocytes. The American Journal of Physiology-Cell Physiology. 2011;300(4):C850–C859. doi: 10.1152/ajpcell.00187.2010. [DOI] [PubMed] [Google Scholar]
- 16.Sethi S, Chakraborty T. Role of TLR- / NLR-signaling and the associated cytokines involved in recruitment of neutrophils in murine models of Staphylococcus aureus infection. Virulence. 2011;2(4):316–328. doi: 10.4161/viru.2.4.16142. [DOI] [PubMed] [Google Scholar]
- 17.Schmaler M, Jann NJ, Ferracin F, Landmann R. T and B cells are not required for clearing Staphylococcus aureus in systemic infection despite a strong TLR2-ÄìMyD88-dependent T cell activation. The Journal of Immunology. 2011;186(1):443–452. doi: 10.4049/jimmunol.1001407. [DOI] [PubMed] [Google Scholar]
- 18.Nyirenda MH, Sanvito L, Darlington PJ, et al. TLR2 stimulation drives human naive and effector regulatory T cells into a Th17-like phenotype with reduced suppressive function. The Journal of Immunology. 2011;187(5):2278–2290. doi: 10.4049/jimmunol.1003715. [DOI] [PubMed] [Google Scholar]
- 19.Tsujimoto H, Ono S, Efron PA, Scumpia PO, Moldawer LL, Mochizuki H. Role of toll-like receptors in the development of sepsis. Shock. 2008;29(3):315–321. doi: 10.1097/SHK.0b013e318157ee55. [DOI] [PubMed] [Google Scholar]
- 20.Volz T, Nega M, Buschmann J, et al. Natural Staphylococcus aureus-derived peptidoglycan fragments activate NOD2 and act as potent costimulators of the innate immune system exclusively in the presence of TLR signals. The FASEB Journal. 2010;24(10):4089–4102. doi: 10.1096/fj.09-151001. [DOI] [PubMed] [Google Scholar]
- 21.van der Poll T, Opal SM. Host-pathogen interactions in sepsis. The Lancet Infectious Diseases. 2008;8(1):32–43. doi: 10.1016/S1473-3099(07)70265-7. [DOI] [PubMed] [Google Scholar]
- 22.Chau TA, McCully ML, Brintnell W, et al. Toll-like receptor 2 ligands on the staphylococcal cell wall downregulate superantigen-induced T cell activation and prevent toxic shock syndrome. Nature Medicine. 2009;15(6):641–648. doi: 10.1038/nm.1965. [DOI] [PubMed] [Google Scholar]
- 23.Frodermann V, Chau TA, Sayedyahossein S, Toth JM, Heinrichs DE, Madrenas J. A modulatory interleukin-10 response to staphylococcal peptidoglycan prevents Th1/Th17 adaptive immunity to Staphylococcus aureus . Journal of Infectious Diseases. 2011;204(2):253–262. doi: 10.1093/infdis/jir276. [DOI] [PubMed] [Google Scholar]
- 24.Wang J, Roderiquez G, Norcross MA. Control of adaptive immune responses by Staphylococcus aureus through IL-10, PD-L1, and TLR2. Scientific Reports. 2012;2, article 606 doi: 10.1038/srep00606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Otto M. Basis of virulence in community-associated methicillin-resistant Staphylococcus aureus . Annual Review of Microbiology. 2010;64:143–162. doi: 10.1146/annurev.micro.112408.134309. [DOI] [PubMed] [Google Scholar]
- 26.DaSilva L, Welcher BC, Ulrich RG, Aman MJ, David CS, Bavari S. Humanlike immune response of human leukocyte antigen-DR3 transgenic mice to staphylococcal enterotoxins: a novel model for superantigen vaccines. Journal of Infectious Diseases. 2002;185(12):1754–1760. doi: 10.1086/340828. [DOI] [PubMed] [Google Scholar]
- 27.Sriskandan S, Unnikrishnan M, Krausz T, et al. Enhanced susceptibility to superantigen-associated streptococcal sepsis in human leukocyte antigen-DQ transgenic mice. Journal of Infectious Diseases. 2001;184(2):166–173. doi: 10.1086/322018. [DOI] [PubMed] [Google Scholar]
- 28.Roy CJ, Warfield KL, Welcher BC, et al. Human leukocyte antigen-DQ8 transgenic mice: a model to examine the toxicity of aerosolized staphylococcal enterotoxin B. Infection and Immunity. 2005;73(4):2452–2460. doi: 10.1128/IAI.73.4.2452-2460.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Karau MJ, Tilahun A, Schmidt S, Clark CR, Patel R, Rajagopalan G. Linezolid is superior to vancomycin in experimental pneumonia caused by superantigen-producing Staphylococcus aureus in HLA class II transgenic mice. Antimicrobial Agents and Chemotherapy. 2012;56(10):5401–5405. doi: 10.1128/AAC.01080-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rajagopalan G, Tilahun AY, Asmann YW, David CS. Early gene expression changes induced by the bacterial superantigen, staphylococcal enterotoxin B and its modulation by a proteasome inhibitor. Physiological Genomics. 2009;37(3):279–293. doi: 10.1152/physiolgenomics.90385.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rajagopalan G, Smart MK, Krco CJ, David CS. Expression and function of transgenic HLA-DQ molecules and lymphocyte development in mice lacking invariant chain. The Journal of Immunology. 2002;169(4):1774–1783. doi: 10.4049/jimmunol.169.4.1774. [DOI] [PubMed] [Google Scholar]
- 32.Tilahun AY, Holz M, Wu T-T, David CS, Rajagopalan G. Interferon gamma-dependent intestinal pathology contributes to the lethality in bacterial superantigen-induced toxic shock syndrome. PLoS ONE. 2011;6(2) doi: 10.1371/journal.pone.0016764.e16764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang X, Meng X, Kuhlman JR, et al. Knockout of Mkp-1 enhances the host inflammatory responses to gram-positive bacteria. The Journal of Immunology. 2007;178(8):5312–5320. doi: 10.4049/jimmunol.178.8.5312. [DOI] [PubMed] [Google Scholar]
- 34.Leemans JC, Heikens M, van Kessel KPM, Florquin S, van der Poll T. Lipoteichoic acid and peptidoglycan from Staphylococcus aureus synergistically induce neutrophil influx into the lungs of mice. Clinical and Diagnostic Laboratory Immunology. 2003;10(5):950–953. doi: 10.1128/CDLI.10.5.950-953.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tilahun AY, Karau MJ, Clark CR, Patel R, Rajagopalan G. The impact of tacrolimus on the immunopathogenesis of staphylococcal enterotoxin-induced systemic inflammatory response syndrome and pneumonia. Microbes and Infection. 2012;14(6):528–536. doi: 10.1016/j.micinf.2012.01.001. [DOI] [PubMed] [Google Scholar]
- 36.Schmaler M, Jann NJ, Ferracin F, Landmann R. T and B cells are not required for clearing Staphylococcus aureus in systemic infection despite a strong TLR2-MyD88-dependent T cell activation. The Journal of Immunology. 2011;186(1):443–452. doi: 10.4049/jimmunol.1001407. [DOI] [PubMed] [Google Scholar]
- 37.Fry DE. Sepsis, systemic inflammatory response, and multiple organ dysfunction: the mystery continues. The American Surgeon. 2012;78(1):1–8. [PubMed] [Google Scholar]
- 38.Tilahun AY, Marietta EV, Wu T-T, Patel R, David CS, Rajagopalan G. Human leukocyte antigen class II transgenic mouse model unmasks the significant extrahepatic pathology in toxic shock syndrome. The American Journal of Pathology. 2011;178(6):2760–2773. doi: 10.1016/j.ajpath.2011.02.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Silverstein R. D-galactosamine lethality model: scope and limitations. Journal of Endotoxin Research. 2004;10(3):147–162. doi: 10.1179/096805104225004879. [DOI] [PubMed] [Google Scholar]
- 40.Lee MS, Kim Y-J. Signaling pathways downstream of pattern-recognition receptors and their cross talk. Annual Review of Biochemistry. 2007;76:447–480. doi: 10.1146/annurev.biochem.76.060605.122847. [DOI] [PubMed] [Google Scholar]
- 41.Takeuchi O, Hoshino K, Akira S. Cutting edge: TLR2-deficient and MyD88-deficient mice are highly susceptible to Staphylococcus aureus infection. The Journal of Immunology. 2000;165(10):5392–5396. doi: 10.4049/jimmunol.165.10.5392. [DOI] [PubMed] [Google Scholar]
- 42.Wardenburg JB, Williams WA, Missiakas D. Host defenses against Staphylococcus aureus infection require recognition of bacterial lipoproteins. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(37):13831–13836. doi: 10.1073/pnas.0603072103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sriskandan S, Altmann DM. The immunology of sepsis. Journal of Pathology. 2008;214(2):211–223. doi: 10.1002/path.2274. [DOI] [PubMed] [Google Scholar]
- 44.Mangalam A, Rodriguez M, David C. Role of MHC class II expressing CD4+ T cells in proteolipid protein91-110-induced EAE in HLA-DR3 transgenic mice. European Journal of Immunology. 2006;36(12):3356–3370. doi: 10.1002/eji.200636217. [DOI] [PubMed] [Google Scholar]
- 45.Hopkins PA, Pridmore AC, Ellmerich S, et al. Increased surface toll-like receptor 2 expression in superantigen shock. Critical Care Medicine. 2008;36(4):1267–1276. doi: 10.1097/CCM.0b013e31816a0a78. [DOI] [PubMed] [Google Scholar]
- 46.Kang S-S, Kim HJ, Jang MS, et al. Gene expression profile of human peripheral blood mononuclear cells induced by Staphylococcus aureus lipoteichoic acid. International Immunopharmacology. 2012;13(4):454–460. doi: 10.1016/j.intimp.2012.05.010. [DOI] [PubMed] [Google Scholar]
- 47.Han SH, Kim JH, Martin M, Michalek SM, Nahm MH. Pneumococcal lipoteichoic acid (LTA) is not as potent as staphylococcal LTA in stimulating toll-like receptor 2. Infection and Immunity. 2003;71(10):5541–5548. doi: 10.1128/IAI.71.10.5541-5548.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schröder NWJ, Morath S, Alexander C, et al. Lipoteichoic acid (LTA) of streptococcus pneumoniae and Staphylococcus aureus activates immune cells via toll-like receptor (TLR)-2, lipopolysaccharide-binding protein (LBP), and CD14, whereas TLR-4 and MD-2 are not involved. The Journal of Biological Chemistry. 2003;278(18):15587–15594. doi: 10.1074/jbc.M212829200. [DOI] [PubMed] [Google Scholar]
- 49.Wang J, Li F, Sun R, et al. Bacterial colonization dampens influenza-mediated acute lung injury via induction of M2 alveolar macrophages. Nature Communications. 2013;4, article 2106 doi: 10.1038/ncomms3106. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Splenic mononuclear cells from HLA-DR3 transgenic mice were cultured with medium, HKSA (108 bacteria/ml), SEB (1μg/ml) or SEB+HKSA. 24 hours later, the cells were harvested, washed and expression of HLA-DR3 and CD86 on CD11c+ was analyzed by flow cytometry. Charts depict mean fluorescent intensity (MFI). Each bar represents mean ± SE from 2 different experiments.