

Abstract
We review the current state of membrane protein structure determination using solid-state nuclear magnetic resonance (NMR) spectroscopy. Multidimensional magic-angle-spinning correlation NMR combined with oriented-sample experiments has made it possible to measure a full panel of structural constraints of membrane proteins directly in lipid bilayers. These constraints include torsion angles, interatomic distances, oligomeric structure, protein dynamics, ligand structure and dynamics, and protein orientation and depth of insertion in the lipid bilayer. Using solid-state NMR, researchers have studied potassium channels, proton channels, Ca2+ pumps, G protein–coupled receptors, bacterial outer membrane proteins, and viral fusion proteins to elucidate their mechanisms of action. Many of these membrane proteins have also been investigated in detergent micelles using solution NMR. Comparison of the solid-state and solution NMR structures provides important insights into the effects of the solubilizing environment on membrane protein structure and dynamics.
Keywords: magic-angle spinning, multidimensional correlation, solid-state nuclear magnetic resonance, ion channels, GPCR
INTRODUCTION
Membrane proteins represent one-third of all proteins in most genomes and carry out a wide range of essential cellular functions. They transport ions and metabolites across the lipid membranes that surround cells and intracellular organelles, convert light energy to chemical energy, fuse the membranes of two cells, and transmit chemical signals across the membrane to regulate cell growth and division. The misfolding and malfunctioning of membrane proteins can cause devastating illnesses, such as heart disease, cancer, and diabetes.
Because of their natural residence in phospholipid bilayers, whose central portion is entirely hydrophobic, membrane proteins differ from globular proteins in their structural organization. Most membrane proteins have a highly hydrophobic exterior that contacts the lipids. For ion channels and transporters, this hydrophobic surface is accompanied by a hydrophilic interior that coats the passage for ions and water. Membrane proteins are usually insoluble in aqueous solution but are solubilized only in lipids and detergents. The molecular weight of these protein-detergent complexes is usually large, making it challenging to use solution nuclear magnetic resonance (NMR) spectroscopy for structure determination. Large crystals of membrane protein complexes with detergents and lipids are also difficult to grow, thus limiting the use of X-ray crystallography for atomic-resolution structure determination. Owing to these challenges, the number of unique membrane protein structures in the Protein Data Bank in early 2011 is only ~270 (http://blanco.biomol.uci.edu/Membrane_Proteins_xtal.html), which accounts for only ~1% of all unique protein structures deposited to date.
Solid-state NMR (SSNMR) is an atomic-resolution spectroscopy used to study the structure and dynamics of molecules without isotropic mobility. The systems amenable to SSNMR include not only rigid solids but also anisotropically mobile molecules such as lipid membranes. Thus membrane proteins, which have no long-range order and often exhibit anisotropic mobility, are ideal for SSNMR-based structure determination. Importantly, SSNMR allows membrane proteins to be studied in phospholipid bilayers, which represent biological membranes much more realistically than detergents do.
SSNMR yields three-dimensional (3D) structural constraints by exploiting the interactions of nuclear spin magnetic moments with static magnetic fields and radiofrequency fields and by detecting the effects of structurally sensitive nuclear spin interactions such as chemical shielding, dipolar coupling, and quadrupolar coupling in the NMR spectra. Although these fundamental aspects of SSNMR are the same as solution NMR, SSNMR has the advantages of fully detecting the orientational dependence of these nuclear spin interactions to give precise information about protein orientation and dynamics.
There are two major SSNMR approaches for studying membrane protein structure. The first involves magic-angle spinning (MAS) of the samples to obtain high-resolution spectra that reflect isotropic chemical shifts and recoupled, orientation-dependent (anisotropic), dipolar couplings and chemical shift anisotropy. MAS NMR provides a diverse array of structural information such as inter-atomic distances, backbone torsion angles, side-chain conformations, and motional amplitudes and timescales. The second approach involves aligning lipid membranes to determine the protein orientation in the phospholipid bilayer. In the past 10 years, MAS SSNMR underwent a rapid development in which a suite of 2D, 3D, and 4D correlation techniques was invented to determine the full 3D structures of proteins (1). This development coincided with increasingly sophisticated molecular biology and biochemical methods to express, isotopically label, and reconstitute proteins into lipid bilayers. These innovations resulted in the structure determination of ion channels and membrane-bound enzymes, as well as nonmembrane-associated globular proteins (2) and amyloid fibrils (3). These MAS correlation techniques also stimulated site-specific studies of ligand binding to large membrane proteins such as G protein–coupled receptors (GPCRs) (4). Finally, solution and SSNMR are increasingly used in parallel to study the same membrane proteins in detergent micelles versus lipid bilayers, thus providing opportunities for understanding the conformational dependence of membrane proteins on the environment (5).
In this article, we review the latest membrane protein structures studied by SSNMR. We describe the methodological advances in MAS NMR that enabled the comprehensive study of membrane protein structure and summarize key findings of several membrane protein systems. We emphasize membrane proteins that have been investigated using multiple NMR methods, including MAS SSNMR, oriented-membrane SSNMR, and solution NMR. This choice allows us to examine the effects of the membrane-mimetic solvent on protein structure and dynamics and highlights the increasing synergy among different approaches. More focused reviews of the application of solution NMR to membrane proteins have been recently given elsewhere (6, 7).
HIGH-RESOLUTION SOLID-STATE NMR TECHNIQUES FOR STUDYING MEMBRANE PROTEINS
Since the late 1990s, a number of multidimensional 13C-13C and 13C-15N MAS correlation experiments were introduced for resonance assignment and chemical shift determination of extensively 13C and 15N labeled proteins. These techniques were built on the previous decade’s dipolar-recoupling techniques (8). Distance-dependent internuclear dipolar couplings are normally averaged by MAS and must be selectively reintroduced with radiofrequency pulses without affecting the averaging of the undesired chemical shift anisotropy. These dipolar-recoupling sequences became the building blocks for MAS correlation experiments that transfer polarization between 13C and 15N nuclei in proteins. The concatenation of several polarization transfer steps allows the correlation of resonances within a residue and between two consecutive residues. For example, the correlation of 15N with Cα and side-chain carbons (NCACX) allows the assignment of signals belonging to a certain amino acid type, whereas the correlation of 13CO with its directly bonded 15N allows the assignment of signals belonging to two sequential residues (9-11). For these experiments, the proteins are uniformly 15N labeled, and 13C labeling can be either uniform to maximize resonance assignment or selective to enhance spectral resolution. The latter can be achieved using site-specifically labeled glucose or glycerol. For example, 2-13C-glycerol selectively labels backbone Cα and CO of many amino acids, whereas 1,3-13C-glycerol preferentially labels the side chains (12-14). To reduce spectral congestion, one can unlabel certain residue types in a protein by using natural-abundance amino acids in combination with 13C-labeled glucose (15).
Resonance assignment gives 13C and 15N isotropic chemical shifts, which constrain protein ϕ, ψ, χ torsion angles (16, 17). Modern computation methods suggest that it may be possible to use chemical shifts as the main input to determine the full 3D structure of proteins (18); thus chemical shifts will play an increasingly important role in structure determination. Complementing chemical shifts are dipolar correlation experiments to directly measure torsion angles (19, 20). For example, the correlation of N-Cα and CO-N dipolar couplings yields the ψ angle, whereas N-H and Cα-Hα dipolar correlation gives the ϕ angle. Once the chemical shifts are known, 13C and 1H spin-diffusion experiments (21-23) can be conducted to measure cross-peak intensities to determine interatomic distances. Long-range distances between nonsequential residues provide crucial constraints to the 3D fold of the protein.
These MAS correlation experiments primarily determine the monomer structure of the protein. Many membrane proteins form oligomeric assemblies in the lipid bilayer, and protein orientation and depth of insertion, protein-lipid interactions, and backbone and side-chain dynamics are integral aspects of the membrane protein structure. SSNMR is a versatile technique capable of extracting this wide range of information. To determine the oligomeric number and intermolecular packing, investigators have conducted 19F spin-diffusion (24, 25) and 1H-X dipolar-recoupling (26, 27) experiments, which exploit the high gyromagnetic ratios of 1H and 19F spins, to measure distances in the 10–15-Å range, well beyond the distance range of low-frequency nuclei such as 13C and 15N.
Membrane protein structure must be referenced to the lipid bilayer in terms of the protein orientation and depth of insertion. NMR frequencies intrinsically depend on the orientation of nuclear spins in the magnetic field. But the random orientation of typical proteoliposomes obscures the inherent orientation dependence of the frequency. To retrieve this information, one must uniaxially align the protein, by orienting the membrane either mechanically on glass plates or magnetically using liquid-crystalline media. With a uniform orientation, the dipolar coupling and chemical shift become well resolved, even without MAS, and reveal the protein orientation from the bilayer normal (28). The main hurdle in this oriented-sample solid-state NMR (OS-SSNMR) approach is the difficulty of alignment. Recently, a new approach that bypasses alignment was introduced (29). It takes advantage of the fact that when membrane proteins undergo fast rigid-body uniaxial diffusion around the bilayer normal, the order parameters that scale the anisotropic couplings depend on the orientation of the bonds relative to the motional axis. Because order parameters can be measured using MAS experiments on unoriented samples, orientation determination for uniaxially diffusive proteins amounts to mobility determination on unoriented liposomes (30). This approach has been demonstrated on several membrane peptides and proteins (30-33), and even relatively large membrane proteins such as seven transmembrane (TM) helix proteins (34) have been shown to exhibit sufficiently fast uniaxial mobility in the fluid lipid bilayer.
The depth of insertion of membrane proteins can be determined by three techniques: lipid-to-protein 1H spin diffusion (35, 36), paramagnetic relaxation enhancement by membrane-surface-bound paramagnetic ions such as Mn2+ and Gd3+ (37), and protein-lipid 13C-31P or 13C-19F distance measurements (38, 39). The probes that give a depth reference can be at the membrane surface, such as 31P and metal ions, or in the center of the membrane, such as methyl protons or CF3 groups at the lipid acyl chain ends. In most cases, it is sufficient to know the depth irrespective of the bilayer leaflet into which the protein inserts. However, for systems in which the sidedness of membrane insertion is important to know, this information can be obtained by coating only one surface of the bilayer with paramagnetic ions (40).
Elucidating the mechanisms of action of membrane proteins requires knowing not only how proteins are structured, but also how they move. Membrane proteins must change their conformations to conduct ions, bind ligands, interact with receptors, or transit from the aqueous solution to the lipid membrane. Some motions are large scale, such as rotational diffusion around the bilayer normal (30, 41), motions that change the relative orientation of two domains, and refolding to transform a water-soluble state into a membrane-bound state (42, 43). Local motions include side-chain rotameric exchanges (44), torsional fluctuations, methyl rotations, and aromatic ring flips. The effects of motion manifest in different ways in the NMR spectra. Submicrosecond motions narrow the NMR lines, whereas millisecond motions cause exchange (cross) peaks in 1D and 2D spectra. Intermediate motions between these two timescales broaden the spectra. One can determine the amplitude of fast motions from line-narrowing effects in 2H and 13C-1H dipolar spectra and extract motional rates from nuclear spin relaxation rates. Compared with solution NMR, SSNMR has the advantage that it is able to measure order parameters directly as the scaling factor between the measured couplings and the rigid-limit couplings and to convert these order parameters into motional amplitudes without making assumptions about the correlation function (45).
In addition to the above approaches for determining the structure and dynamics of membrane proteins, many SSNMR methods are in place to determine the atomic details of protein-ligand binding. Interatomic distances can be measured using both heteronuclear and homonuclear dipolar-recoupling techniques such as REDOR (46) and rotational resonance (47). Ligand dynamics, which often shed light on the surrounding protein environment, can be probed using 2H NMR. The latter has been used to good advantage, for example, to study rhodopsin-retinal interactions and drug binding to the influenza M2 protein.
Potassium Channels
Potassium channels regulate electric potentials across the membranes of muscles and neurons by conducting potassium ions down the electrochemical gradient. A number of X-ray crystal structures have revealed the potassium binding sites and protein conformational changes involved in gating (48-50). However, the structures of potassium channels in lipid bilayers and their dependence on ion concentration, pH, and channel blockers are ideally addressed by SSNMR.
MAS NMR has been used extensively to investigate the conformation and dynamics of potassium channels. By assigning the chemical shifts, one can obtain (ϕ, ψ) torsion angles and their perturbations by the environment. Baldus and coworkers examined the conformational dynamics of the potassium channel KcsA-Kv1.3 under different pH, K+ concentration, and inhibitor binding conditions using SSNMR in combination with electrophysiological experiments (51-54). The protein is a chimera between the prokaryotic pH-gated KcsA channel and the mammalian voltage-gated Kv1.3 channel. Using 2D and 3D MAS NMR, the authors assigned the majority of the protein residues (51, 52) and mapped out chemical shift changes due to channel activation, inactivation, and inhibition. When the protein changes from the high-pH closed state to the low-pH inactivated state, the selectivity filter residues show chemical shift changes that indicate a transition from the conductive to the nonconductive conformation (53). Side-chain Cδ chemical shifts indicate that Glu residues near the intracellular activation gate become protonated at low pH, opening the gate (54). High K+ concentration (50 mM) stabilizes the open state of the selectivity filter regardless of pH. The conformational changes of the selectivity filter and the intracellular activation gate are coupled: A closed selectivity filter opens the activation gate, whereas a conducting selectivity filter closes activation gate (54).
Chemical shifts combined with 1H-1H distances measured in 2D experiments showed that two channel blockers, the scorpion toxin kaliotoxin and a tetraphenylporphyrin, inhibit KcsA-Kv1.3 in different ways. Kaliotoxin perturbs the upper region of the selectivity filter (51) but leaves the filter in the conducting state, which induces the closed conformation of the intracellular activation gate (54). In contrast, tetraphenylporphyrin, with its four positively charged side chains, causes a collapsed selectivity filter and an open activation gate, similar to the effect of low pH on the channel (53, 54).
McDermott and coworkers (55, 56) studied the effects of K+ concentration at constant pH on the KcsA structure using multidimensional MAS NMR. The chemical shifts indicate two selectivity filter conformations, a low-K+ nonconducting state and a high-K+ conducting state. By monitoring the intensities of the two sets of peaks, the authors found that residue V76, which coordinates the two inner K+-selective sites 2 and 3, has a strong K+ affinity of 2–7 μM. This tight binding agrees well with electrophysiological results but differs from solution NMR results of KcsA in detergent micelles (57, 58) and suggests that one of the two inner sites is always occupied during K+ conduction. In lipid bilayers at mild low temperatures (~10°C), the low and high K+ conformers are in slow conformational exchange.
1H spin-diffusion NMR (36) has provided insights into the global conformational changes during K+ channel opening and closing. Specifically, 1H spin diffusion from water to proteins occurs at a rate that depends on the water-protein surface area. By quantifying the spin-diffusion buildup using a low-resolution lattice model of the channel structure, Baldus and coworkers (59) showed that the water-exposed surface area of the KcsA-Kv1.3 channel increased by 65% as the pH decreased to open the channel. The square root of the mixing time required to reach equilibrium, , changes with pH in a sigmoidal fashion with a mid-point at pH 6, consistent with the pH dependence of K+ channel activation.
Because KcsA is thermally stable and readily solubilized in detergents, it was also well studied by solution NMR methods. These studies focused on chemical shift perturbation and relaxation time changes due to pH- and K+-dependent channel activation and gating (57, 58, 60). Various structural signatures were identified for the high-pH and low-K+ closed state and the low-pH and high-K+ inactivated state. Information on the more transient open state was also obtained. The solution NMR studies agreed on the existence of two selectivity filter conformations and the coupled nature of the selectivity filter and activation gate changes. However, they differed on the precise timescales of the conformational change, with estimates ranging from 10−2 to 10−4 s. This discrepancy likely results from the different detergents and temperatures employed in these studies.
Proton Channels
The M2 protein of the influenza A virus forms a tetrameric pH-activated proton channel that is important for the virus life cycle (61). The TM helix of this minimalist channel contains the key residues for proton selection (His37) and channel gating (Trp41). The channel is inhibited by the antiviral drugs amantadine and rimantadine, whose efficacy was compromised by the S31N mutation in most of the recent circulating flu viruses (62). High-resolution structure of the M2 protein is important both for developing alternative antiviral drugs to target the mutant protein and for understanding fundamental aspects of proton transport. At least three states of the protein can be distinguished: the low-pH open state, the high-pH closed state, and the inhibitor-bound state. Structure determination of this protein has been intensely pursued using MAS SSNMR, oriented-sample SSNMR, and solution NMR (63).
Cross and coworkers used 15N SSNMR of glass-plate-aligned samples to determine the orientation of the M2 TM helix. These experiments showed that M2TM was tilted by ~35° in DMPC (dimyristoylphosphatidylcholine) bilayers at low, albeit uncontrolled, pH (64, 65). Subsequent experiments refined the sample preparation methods to control the pH and defined the orientations of the amantadine-bound peptide (66) as well as the orientation of the TM domain as part of the intact protein (67). Drug binding to M2TM in DMPC/DMPG (dimyristoylphosphatidylglycerol) bilayers at pH 8.8 caused a ~10° kink at Gly34 (66). When a construct including both the TM domain and its immediate C-terminal amphipathic helix was reconstituted into DOPC/DOPE (dioleoylphosphatidylcholine/dioleoylphosphatidylethanolamine) bilayers at pH 7.5, the TM helix showed a tilt angle of 32°, whereas the amphipathic helix was tilted by 105° from the bilayer normal (68); thus the latter is roughly parallel to the membrane surface (Figure 1c). The turn between the two helices, formed by only two residues, is tight and rigid.
Figure 1.

Various NMR and X-ray crystal structures of the influenza M2 protein. (a) Solid-state NMR structure of M2 (22-46) in DMPC bilayers at pH 7.5 (PDB code: 2KQT). A single amantadine (purple) drug binds to the N-terminal pore. (b) 3.5-Å crystal structure of M2 (25-46) in octylglucoside at pH 5.3 (PDB code: 3C9J). (c) Solid-state NMR orientational structure of M2 (22-62) in DOPC/DOPE bilayers at pH 7.5 (PDB code: 2L0J). (d) Solution NMR structure of M2 (18-60) in DHPC (diheptanoylphosphocholine) micelles at pH 7.5 (PDB code: 2RLF). The four rimantadine drugs (purple) bind to the C-terminal lipid-facing surface of the protein.
Hong and coworkers used MAS NMR experiments to investigate the conformation and dynamics of M2TM in lipid bilayers as a function of pH and drug binding. 2D 13C-13C and 15N-13C correlation experiments mapped out chemical shift perturbations of TM residues caused by amantadine and a spiro-piperidine inhibitor (69-71). Hot spots of conformational changes include Val27, Ser31, Gly34, and His37, most of which are sites of drug-resistant mutations. The largest chemical shift change was observed at S31 (Figure 2a), consistent with the S31N mutant being the dominant drug-resistant strain and suggesting drug binding to this site (70).
Figure 2.

Determination of the drug-binding site in the influenza M2 transmembrane domain using magic-angle-spinning solid-state NMR. (a) 2D 15N-13C correlation spectra of M2TM in DLPC (dilauroylphosphocholine) bilayers with and without amantadine (Amt). Ser31 shows a large 15N chemical shift change upon drug binding. (b) 13C-2H REDOR spectra of Ser31 and Asp44 at different drug: tetramer ratios. 13C peaks that show intensity differences (ΔS) between the control S0 and 2H-dephased S spectra are close to the deuterated amantadine. At one drug per tetramer, only Ser31 is dephased by the 2H pulses, whereas under excess drugs (four per tetramer), both Ser31 and Asp44 signals are dephased. (c) Quantitative 13C-2H REDOR decay of Ser31. The decay curve is simulated using a novel method that incorporates the four-fold symmetry of the helical bundle and the uniaxial diffusion of amantadine. The distance is parameterized in terms of the radius of the carbon from the channel axis and the height of the carbon from the middle of the two equatorial deuterium planes of the adamantane cage.
The drug-binding site of M2 became controversial in 2008, when two high-resolution structures showed very different binding sites. A 3.5-Å X-ray crystal structure (Figure 1b) showed the binding site inside the N-terminal pore, consistent with biochemical evidence (72), whereas a solution NMR study found drug-protein nuclear Overhauser effects for residues on the C-terminal surface of the TM helical bundle (73) (Figure 1d). The two studies used different drug concentrations, protein lengths, pH, and detergents, thus complicating the interpretation of the origin for the difference.
Subsequent SSNMR studies of M2 peptides in lipid bilayers as a function of drug concentration and peptide length resolved this controversy. Using 13C-2H REDOR experiments that measured intermolecular distances between 13C-labeled peptide and perdeuterated amantadine, Hong and coworkers (74) showed that the first stoichiometric equivalent of drug binds with high affinity to the N-terminal pore, closest to Ser31 (Figure 2a,b), whereas excess drugs bind to the C-terminal surface site from the membrane side with low affinity. Static 2H NMR lineshapes of perdeuterated amantadine revealed that the pore-bound drug is oriented upright while the surface-bound drug is tilted by 80° or 37° (Figure 3). The quantification of protein-drug 13C-2H distances (Figure 2c), combined with other SSNMR constraints, resulted in a 0.3-Å-resolution structure of the drug-complexed state of the TM domain (Figure 1a). Whereas previous SSNMR orientational, torsional, and interhelical distance constraints had been used to construct structure models of M2TM (70), the lack of protein-drug distances had caused these models to deviate significantly from the high-resolution structure in terms of the helix curvature and interhelical packing. The high-pH bilayer-based M2TM structure shows a tighter C-terminal half compared to the low-pH detergent-based crystal structure (72), consistent with the higher affinity of the drug for the channel at high pH.
Figure 3.

2H solid-state NMR to determine the orientation and dynamics of a ligand bound to an ion channel. The case of amantadine binding to the influenza M2 proton channel is illustrated. (a-c) 2H spectra of perdeuterated amantadine at 243 K and 303 K in DMPC bilayers with and without M2: (a) no peptide, (b) with peptide at a drug:tetramer ratio of 1:1, and (c) with peptide at a drug:tetramer ratio of 4:1. All 243-K spectra show a dominant splitting of 36 kHz, which results from the 12 equatorial C-D bonds. This coupling indicates that amantadine undergoes uniaxial diffusion around the bilayer normal with a mostly upright orientation. At 303 K, the peptide-free sample shows an 18-kHz splitting, indicating one of two tilted orientations, as shown in panel e. The 1:1 sample exhibits a dominant splitting of 36 kHz, demonstrating that the drug maintains its upright orientation, consistent with drug confinement in the pore (d). The 4:1 sample shows a dominant 18-kHz splitting, indicating that the excess drugs partition into the lipid membrane and adopt one of the tilted orientations.
13C-2H REDOR distance measurements confirmed that amantadine also binds to the N-terminal pore in the amphipathic-helix-containing M2 (75). However, the drug affinity to the surface site, Asp44, is diminished in the longer construct, which is consistent with the fact that the amphipathic helix is packed close to the TM domain (68) and partially obstructs the surface site. These findings indicate that the nonspecific surface-bound drugs in the solution NMR structure (Figure 1d) (73) result from the high drug concentration used, whereas the lack of pore-bound drugs may result from detergent-induced artifacts and the curvature-inducing effect of the amphipathic helix.
13C-2H REDOR NMR also yielded the direction of the polar amine inside the pore. CD3-deuterated rimantadine showed sizeable dipolar coupling to Gly34 but not to Val27, indicating that the polar amine points toward the C terminus of the TM helix (76). Because the drug is known to perturb the pKa of the proton-selective His37 (77), this result demonstrates that the drug inhibits the channel not only by physical occlusion but also by interfering with His37 protonation. The latter is mediated by channel dehydration, as seen in water-protein 1H spin-diffusion experiments (78).
SSNMR not only elucidated the inhibition mechanism of this proton channel but also gave atomic-level insights into its proton conduction mechanism. Two models had been proposed to explain how His37 selects protons against other monovalent cations. In the water-wire model, the His37 tetrad constricts the pore at high pH, thus blocking H+ diffusion. Upon protonation at low pH, electrostatic repulsion opens the pore at this residue, thus establishing a continuous water wire over which protons hop via the Grotthuss mechanism (79). The potential of mean force of proton hopping was calculated for different charged states of the tetrad (80) and supports this model. In the shuttle model, His37 is envisioned to undergo successive protonation and deprotonation events to shuttle protons into the virion. The process is limited by the rate of His37 conformational changes to regenerate the initial state (81).
Hong and coworkers (44) measured the His37 protonation state, rotameric structure, side-chain dynamics, and hydrogen bonding as a function of pH to understand the mechanism of proton conduction. At high pH, the neutral His37 rings pack in a CH-π stacked fashion, creating an electron-rich region that is suggested to repel water oxygens on the two sides of the tetrad (Figure 5a). The water repulsion is supported by bond-length measurements showing a short Nε2-H bond, unstretched by hydrogen bonding. At low pH, chemical shifts indicate that the His37 tetrad adopts +2 to +4 charged states, and the tt rotamers (χ1 = χ2 = 180°) keep the side chains far from each other in the spacious pore. Importantly, dipole order parameters at 303 K showed that the imidazolium rings undergo 45° two-site jumps at rates >50,000 s−1 (Figure 4), suggesting proton shuttling. Temperature-dependent dipolar couplings indicate that the energy barrier of the ring motion is >60 kJ mol−1, which is consistent with the proton conduction barrier of ~100 kJ mol−1 obtained from functional assays (82). Thus ring reorientation is the rate-limiting step of proton transport. The low-pH His37 tetrad shows bond elongation for Nε2, Cε1, and Nδ1, indicating hydrogen bonding on all three sides of the rings, supporting water-His proton transfer.
Figure 5.
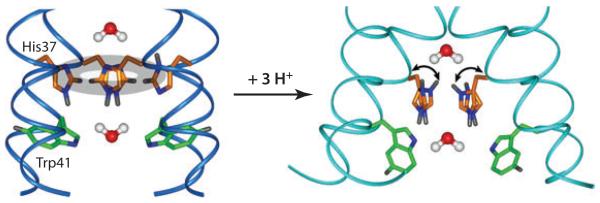
Proton-conduction mechanism of the influenza M2 channel. At high pH, the four histidine rings pack in an edge-face stacked fashion, disrupting Grotthuss hopping. Upon multiple protonation, the cationic histidines undergo microsecond ring reorientations that shuttle protons into the virion.
Figure 4.

Determination of the dynamics of the proton-selective residue His37 in the influenza M2 transmembrane peptide. (a) Cγ-Nδ1 dipolar coupling is reduced by motion at low pH, as manifested by a slower oscillation compared to the high-pH data. (b) 13Cδ2-1H dipolar coupling shows shallower dipolar dephasing at low pH, also indicating motional averaging. (c) Geometry of imidazolium motion at low pH based on the dipolar order parameters. A two-site jump around the χ2 torsional bond by 45° fits the data well.
A persistent observation in both OS- and MAS-SSNMR spectra of M2 TM is the conformational plasticity (69, 83). Although the peptide is generally α-helical, the (ϕ, ψ) torsion angles of key residues such as Val27, Gly34, and His37 and the orientation of the TM helix are affected by the lipid composition, membrane thickness, pH, and drug binding (83, 84). These residues exhibit multiple peaks per site, indicating conformational equilibrium among several states (84). The peptide shows larger disorder at low pH than at high pH, and in the absence of drugs than in their presence. Cholesterol and sphingomyelin immobilize the helical bundle (85), whereas one-component phosphocholine bilayers in the liquid-crystalline phase promote fast uniaxial diffusion of the helical bundle (86). Temperature-dependent 1H rotating-frame spin-lattice relaxation times indicate that the uniaxial diffusion is faster for the drug-complexed tetramers than the drug-free ones (87), suggesting that amantadine binding tightens the TM helical bundle, consistent with analytical ultracentrifugation results (88). Gly34 shows nonideal helical conformations in the drug-bound state and in thick lipid bilayers, and the His37 conformation deviates from an ideal helix at high pH and becomes ideal at low pH (84).
SEVEN-TRANSMEMBRANE-HELIX PROTEINS
GPCRs containing seven TM helices represent the dominant class of proteins targeted by pharmaceutical drugs. They convert external chemical and light signals to cellular responses by catalyzing the exchange of guanosine triphosphate for guanosine diphosphate in intracellular heterotrimeric G proteins, which activate subsequent cellular pathways.
Rhodopsin is a canonical member of the class A GPCR family and acts as the photoreceptor in vertebrates, converting light signals to neural responses. In the dark inactive state, the seven TM helices (H1-H7) surround the cofactor, retinal, on the extracellular side of the receptor (89, 90). The 11-cis retinal chromophore binds covalently to rhodopsin via a protonated Schiff base to the Lys296 side chain. The extracellular loop EL2, which connects H4 and H5, folds into a lid over the retinal binding site. Upon light absorption, 11-cis retinal isomerizes to the all-trans configuration within 200 fs, which causes the protein to thermally relax via a series of intermediates to the active Meta II state. Meta II then binds and activates the G protein transducin. Crystal structures of dark-adapted and several intermediate states of rhodopsin have been determined and show no large-scale changes from the dark state to the Meta I state, but there is a large conformational change from the Meta I to the Meta II state (4, 91).
2H SSNMR was used to determine the structural and dynamical changes of retinal upon light absorption (92). Based on 2H lineshapes of methyl-deuterated retinal in oriented membranes, Brown and coworkers (93, 94) extracted the relative orientations of the three planes of retinal. In the dark state, the plane for the conjugated polyene chain between the β-ionone ring and C12 (plane B) is twisted by −65° at the C6-C7 bond from the β-ionone ring and is twisted from the plane for the end of the polyene chain (plane C) by 150° around the C12-C13 bond (93). Transitioning to the Meta I state changes the C6-C7 torsion angle to 32° or 57° and makes planes B and C roughly parallel (94). Analysis of 2H T1Z and T1Q relaxation times of the methyl groups as a function of temperature yielded the activation energies (Ea) and order parameters of methyl rotations for the dark, Meta I, and Meta II states (95, 96). The methyl group attached to C5 of the β-ionone ring has the highest Ea, which is insensitive to photoactivation (95), consistent with confinement of the ring in the hydrophobic pocket of the protein. The methyl groups associated with C9 and C13 have energy barriers that change with photoactivation: Retinal isomerization increases the C9 Ea but decreases the C13 Ea. These changes were attributed to the rearrangement of the TM helices and the polyene chain protons near the methyl groups (95). Movement of the β-ionone ring was proposed to affect the H3-H5 interface and hydrogen-bonding network, whereas the C13 methyl rotation was proposed to impact EL2 and the H4-H6 hydrogen-bonding network (96).
Smith and coworkers (4, 97-100) used 2D MAS 13C correlation NMR to determine retinalopsin contacts. Using the crystal structure of the dark state as a starting point, they systematically measured retinal-protein and protein-protein distances lining the chromophore binding pocket. These residues include tyrosines, glycines, Trp265, and Ser186. The 2D 13C spin-diffusion experiment is sensitive to distances within about 5.5 Å; thus the rotation and translation of retinal manifest as the disappearance or appearance of cross peaks with protein residues. The retinal and the protein are site-specifically labeled with 13C. Multiple residues of the same type were assigned based on the crystal structure of the ground state. These studies revealed how retinal isomerization choreographed intricate changes of the TM helices and loops (4). Activation to Meta II translates retinal by 4–5Å from H7 toward H5, at the same time rotating the methyl attached to C13 by more than 90° (97). This retinal motion alters the interaction of H5 with H3, causes rigid-body rotation of H6, and affects the interaction of H7 with the H1-H4 core of the protein. Analyses of the 15N chemical shifts of key aromatic residues such as His211 on H5 and Trp265 on H7 revealed the rearrangement of interhelical hydrogen-bonding networks (98). For example, Trp265 of H6 contacts the retinal C13 methyl in the dark state but displaces to contact the C9 methyl in the Meta II state (99), suggesting that retinal movement drives an outward rotation of H6. 2D correlation experiments also provided atomic details about the effects of retinal isomerization on the EL2 loop connecting H4 and H5 (100). EL2 is displaced by the chromophore, rearranging the surrounding hydrogen-bonding networks. The EL2 changes are coupled to H5 motion, which disrupts an H3-H6 ionic lock involving Arg135 and Glu247.
In addition to mammalian rhodopsin, microbial rhodopsins such as the eubacterial green proteorhodopsin and sensory rhodopsin II (SRII) have been studied recently using MAS SSNMR (15, 101). These GPCRs act as photosensors and proton pumps and are counterparts to the well-studied haloarchaeal proton pump, bacteriorhodopsin (102). In contrast to bovine rhodopsin, the retinal cofactor of microbial rhodopsins is in the all-trans form in the ground state and converts to the 13-cis configuration upon light activation. For SRII, isomerization activates the transducer HtrII.
SSNMR studies of SRII and proteorhodopsin employed various 13C, 15N-labeled samples, including both uniform 13C labeling and reverse 13C labeling, in which a few dominant residue types were kept in natural abundance to reduce spectral congestion. Differential labeling of the receptor and transducer was employed to obtain information on SRII binding to the transducer. Using scalar-coupling-mediated 2D correlation experiments, Baldus and coworkers (15) showed that free SRII contains three highly dynamic loops out of the total six loops that connect the seven TM helices. Spectral assignment was facilitated by mobility filters that detect either rigid or dynamic residues. Polarization transfer from water to the protein was used to identify water-exposed residues (15). Upon light activation, limited chemical shift perturbations were observed in SRII, which suggest that conformational changes mainly occur in helix F. By comparing dipolar- and scalar-coupling-based correlation spectra, Baldus and coworkers found the dynamic loops to be immobilized upon receptor-transducin complexation. 2D correlation spectra of 13C-labeled receptor and 15N-labeled transducer indicate that the intermolecular interface is larger in the lipid membrane than in the crystal structure (103). Chemical shift perturbations suggest that signal transduction may be mediated by the coupled motion of helices F and G of the receptor, which causes rotations of helix TM2 of the transducer. Further information about the structural changes due to light activation and complex formation will require examination of the ~30% of the protein residues that are unlabeled in these studies.
β-SHEET-RICH OUTER MEMBRANE PROTEINS
The structures of β-barrel proteins situated in the outer membranes of gram-negative bacteria and eukaryotic mitochondria are an attractive class of membrane proteins for NMR because of the large chemical shift dispersion of β-sheet residues. The bacterial outer membrane proteins have diverse functions such as ion transport, cell-cell recognition, and cell shape maintenance (104). To date, the high-resolution structures of OmpA (104), OmpG (105), PagF (106), and VDAC have been determined in detergent micelles using solution NMR. The orientation of OmpX in phospholipid bilayers has been studied using 15N OS-SSNMR (107). An initial MAS-NMR study of OmpG has also been reported (108).
Liang & Tamm (105) determined the structures of OmpA in dodecylphosphocholine (DPC) micelles and OmpG in octylglucoside micelles. OmpA functions as a slightly anion-selective channel and as a receptor for phages and colicins (109, 110), whereas OmpG acts as a pH-regulated porin for oligosaccharide uptake. Both proteins have the characteristic architecture of a hydrogen-bonded polar core and an apolar lipid-facing surface. OmpA is an eight-stranded β-barrel connected by tight turns on the periplasmic side and mobile loops on the extracellular side (104). The NMR structure largely agrees with the crystal structure but shows more extended and more dynamic loops. The protein exhibits a dynamic gradient along the barrel axis, with the middle of the barrel being more rigid than the two ends. Interestingly, this dynamic gradient opposes the gradient of the lipid bilayer, which is the most dynamic in the center. OmpG is a larger β-barrel with 14 strands, and the solution NMR structure is shorter and rounder than the crystal structure (105). The NMR structure was solved at pH 6.3 and was likely a mixture of the open and closed conformers exchanging on the micro- to millisecond timescale.
Whereas most solution structures of outer membrane proteins assume the protein orientation, direct orientational constraints were obtained for OmpX by Mahalakshmi & Marassi (107) using OS-SSNMR. They measured 2D spectra that correlate 15N chemical shift anisotropy with 15N-1H dipolar coupling. Because of the large number of overlapped resonances they compared the experimental spectra with back-calculated spectra for different orientations of the crystal structure of OmpX. In this way, the authors determined that the β-barrel axis of OmpX was tilted by 7° from the membrane normal (107).
The voltage-gated anion channel VDAC is a eukaryotic β-barrel protein that transports metabolites, small molecules, and small proteins from the cytoplasm to the mitochondrion (111). VDAC is also involved in programmed cell death (apoptosis) through interaction with the Bcl-2 family of apoptotic proteins (112). The voltage-gating activity of VDAC is well characterized by electrophysiology (113). Two high-resolution structures of human VDAC were recently determined using solution NMR in one case (114) and NMR and X-ray diffraction in the other (115). The two structures are similar, both showing 19 β-strands traversing the membrane at an angle of ~45° with respect to the barrel axis. This result is unusual because all other known outer membrane β-barrels have an even number of strands. Strands β1 and β19 of VDAC are paired in parallel, and the N-terminal 25 residues form a partial α-helix that transverses the pore. The barrel has a diameter of ~25 Å, consistent with electron microscopy data. Detergent molecules surround the entire barrel perimeter, indicating a monomeric protein in the micelle.
Although the two high-resolution structures reached the same conclusion, Colombini (113) raised the question of whether this conformation is native, because biochemical and functional data had previously led to the prediction of a 13-stranded barrel. He contended that an artifactual conformation might have been caused by protein refolding from inclusion bodies. Countering this argument was the fact that the refolded VDAC, when reconstituted into cholesterol-containing lipid bilayers, exhibited single-channel conductance and ion selectivity similar to those of the native protein (114). It is also possible, although unlikely, that the detergent may perturb the protein conformation. Resolution of these issues will require independent structure validation in lipid bilayers using SSNMR, possibly in combination with sensitivity-enhancement techniques such as dynamic nuclear polarization.
VIRAL FUSION PROTEINS
Enveloped viruses enter cells by undergoing a large conformational change in their surface glycoproteins, which expose a hydrophobic fusion domain to the appropriate target membrane. These fusion domains represent an attractive class of membrane peptides for NMR structure determination because their hydrophobic nature has made them invisible in X-ray crystal structures of the postfusion state (116). The fusion protein of the influenza A virus hemagglutinin (HA) and the fusion protein of HIV-1 gp41 have been extensively studied using solution and SSNMR spectroscopy.
The influenza HA fuses with the endosomal membrane of epithelial cells at low pH after the protein undergoes receptor-mediated endocytosis (117). The fusion peptide (FP), located at the N terminus of the HA2 subunit, is buried at the monomer interface of the trimeric protein at neutral pH. Upon a decrease in pH, HA undergoes a dramatic refolding that translates the FP domain by ~ 100 Å, inserting it into the endosomal membrane (116). Meanwhile, a C-terminal hydrophobic domain of HA2 remains anchored in the viral membrane. Subsequent conformational changes in the rest of the protein, whose details are still not well understood, bring the two membranes together and cause lipid mixing.
Tamm and coworkers (118) determined the solution structures of a 20-residue N-terminal FP of HA and its mutants in detergent micelles. In DPC micelles at neutral and acidic pH, the HA FP shows a boomerang structure with a 105° bend between the N-terminal α-helix and the C-terminal 310 helix (Figure 6a) (119, 120). The bend is centered around Glu11 and Asn12. Electron paramagnetic resonance (EPR) of nitroxide-labeled peptide in the presence of O2 and NiEDDA yielded the depth of the peptide in POPC/POPG (palmitoyloleoylphosphocholine/palmitoyloleoylphosphoglycerol) bilayers. The Glu11-Asn12 kink lies near the membrane surface, whereas the N terminus is buried by 10Å at high pH and about 16Å at low pH. Based on residue-specific depths, the authors estimated that the N-terminal helix was tilted by 23° at pH 7.4 and 38° at pH 5 (119). The functional importance of this V-shaped structure was confirmed by structure-activity studies of several mutants. It was found that the stronger the fusogenic activity was, the tighter and more rigid the kink was. For example, a fusogenic F9A mutant has a similar boomerang structure as wild-type (WT) FP, with a 120° kink (121), whereas the inactive G1V mutant is a linear α-helix (122). The kink is stabilized by hydrophobic and hydrogen-bonding interactions between residues on the two sides. For example, mutation of Trp14 to Ala abolished fusion and resulted in a flexible kink that points the C-terminal helix to water rather than the bilayer center (120). Based on these findings, a spring-loaded boomerang model was proposed to explain the mechanism of membrane fusion (122). In this model, the coiled-coil region of HA changes its tilt angle relative to the target and viral membranes, in which the FP and the C-terminal TM peptides are embedded, thus bringing the two membranes together.
Figure 6.

NMR structures of viral fusion peptides. (a) Solution NMR structure of the influenza HA fusion peptide in DPC micelles (PDB code: 1IBN). The peptide corresponds to the N-terminal 20-residue sequence of HA2 and has a boomerang shape (118). (b) Solution NMR structure of a 23-residue construct of the HA fusion peptide in DPC micelles (PDB code: 2KXA). An antiparallel helical hairpin is observed (124). (c) Solid-state NMR structural model of the HIV gp41 fusion peptide in virus-mimetic lipid bilayers (132). Several peptides oligomerize into a curved β-sheet that inserts deeply into the lipid membrane.
The boomerang structure of the HA FP was refined by Sun & Weliky (123) using MAS SSNMR of POPC/POPG-bound peptide. They observed two sets of chemical shifts for the turn residue Glu11. The minor conformer, which was detected only at low pH at which the Glu11 side chain is protonated, is suggested to cause fusion.
Recently, Bax and coworkers (124) determined the structure of a longer, 23-residue, construct of the HA fusion domain. The longer peptide includes Trp21, Tyr22, and Gly23 residues, which are conserved across all 16 serotypes of HA genes. Inclusion of these three residues created a helical hairpin structure (Figure 6b), which is tighter than the boomerang structure of the 20-residue peptide. Six of the seven glycines lie at the interface of the hairpin, suggesting that the hairpin structure is stabilized by the ability of GxxxG and GxxG motifs to form helix-helix associations in membrane proteins (125). However, how the hairpin structure catalyzes membrane fusion is not understood.
In contrast to the influenza virus, HIV fuses with the membrane of target cells at neutral pH. Solution and SSNMR studies showed that the structure of the gp41 FP depends on both the membrane composition and peptide/lipid ratio. In detergent micelles of both anionic and zwitterionic origins at low peptide/lipid ratios (<1:100), gp41 FP is predominantly α-helical and monomeric, with larger dynamic disorder at the C terminus (126, 127). These results were obtained from residual dipolar couplings, nuclear Overhauser effects, and 15N relaxation measurements. In comparison, in cholesterol-containing lipid bilayers that mimic the HIV envelope lipid composition, 13CO chemical shifts from MAS experiments showed predominantly β-strand conformation (128). 13C-15N distance measurements indicate that these β-strands pack in both parallel and antiparallel fashions at roughly equal populations (129). Because the β-sheet conformation was obtained at peptide/lipid ratios of 1:50 to 1:200, peptide concentration does not explain the secondary structure difference between the solution NMR and SSNMR results. Instead, cholesterol appears to play the role of converting the gp41 FP from a monomeric α-helix to an oligomeric β-sheet. In cholesterol-free POPC/POPG membranes, the gp41 FP has a mixture of β-sheet and α-helix chemical shifts (130). Oligomerization is important for the fusogenic activity: Covalently bonded trimeric gp41 FP shows stronger fusogenic activity than monomeric and dimeric FPs (130, 131).
Distance measurements between 13C-labeled HIV FP and lipid 31P or 19F spins demonstrated that the stronger the fusogenic activity is, the deeper the β-sheet peptide inserts into the virus-mimetic membrane (132). The active trimeric gp41 FP inserts its middle portion all the way to the center of the bilayer (Figure 6c), whereas an inactive mutant lies on the surface of the membrane. The monomeric gp41 FP, which has partial fusogenic activity, partitions halfway into one leaflet of the membrane. These results are consistent with the notion that deeper membrane insertion causes stronger perturbation of the bilayer, thus facilitating lipid mixing between the target and viral membranes. Static 31P NMR spectra of glass-plate-aligned virus-mimetic membranes showed that gp41 FP binding increased the membrane disorder. More systematic studies of the correlation between fusogenic activity and membrane disorder will be useful to gain further insights into the fusion mechanism. Given the ability of gp41 FP to form either α-helical or β-sheet structures, it will also be important to determine the secondary structure or possibly dynamic exchanges between secondary structures that are responsible for membrane fusion.
PHOSPHOLAMBAN
Phospholamban (PLN) is a small membrane protein involved in the regulation of the cardiac cycle. Monomeric unphosphorylated PLN inhibits sarcoplasmic reticulum ATPase (SERCA) pumps and stops Ca2+ flow, whereas phosphorylated PLN reverses this effect. Pentameric PLN does not bind SERCA but acts as a storage form of PLN. Thus PLN has multiple functional states: the unphosphorylated monomeric form, the phosphorylated monomeric form, the SERCA-bound state, and the pentameric state.
Owing to its biomedical importance and its relatively small size (52 residues), PLN, similar to the influenza M2 transmembrane peptide, has been well studied by OS-SSNMR, MAS SSNMR, and solution NMR. Most studies agree on the α-helical structure of the TM domain but disagree on the cytoplasmic domain structure, dynamics, and orientation and the pentameric structure.
Veglia and coworkers used solution NMR, OS-SSNMR, and EPR to determine the structure of AFA-PLN, a functional monomeric mutant in which three Cys residues are replaced by one Phe and two Ala residues. In DPC micelles, the cytoplasmic domain of AFA-PLN exchanges between a major conformer parallel to the membrane (T state) and a minor conformer with an extended and disordered structure (R state) (133). SERCA binding shifts the equilibrium to the dynamic R state (133). 2D 15N SSNMR of oriented DOPC/DOPE bilayers shows that the TM helix of AFA-PLN is oriented by ~20° from the bilayer normal and undergoes fast uniaxial diffusion (134), whereas the cytoplasmic helix exhibits more complex motions and has a 93° tilt from the bilayer normal (134, 135). Thus the majority of monomeric AFA-PLN exhibits an L-shaped structure. In the pentameric state, WT PLN has a similar topology, except that the TM helix is less tilted (~11°) (136), giving a pinwheel assembly (137). Solution NMR of DPC-bound protein, EPR lineshapes and dynamics data, and paramagnetic relaxation enhancement results confirm the strong interaction of the cytoplasmic residues with the membrane surface (138). Intermolecular cross peaks were observed for TM residues in 2D solid-state and solution NMR spectra, indicating that the pentameric pore is very narrow (less than 2 Å); thus the pentamer is unlikely to act as an ion channel (136).
Lorigan and coworkers (139) used 2H and 15N SSNMR to probe the backbone and side-chain dynamics of pentamic WT PLN. They found that the TM residues exhibit immobilized backbones, whereas the cytoplasmic domain shows a mixture of a highly dynamic structure and an immobilized helix in POPC bilayers. The latter is manifested as a superposition of an isotropic signal and rigid-limit powder pattern in the static 15N spectra. Thus both monomeric AFA-PLN and pentameric WT PLN show coexistence of two cytoplasmic conformations. Oriented-sample 15N spectra of this pentameric PLN in POPC and DOPC/DOPE bilayers yielded a tilt angle of 11°–13° for the TM helix and an in-plane orientation for the cytoplasmic helix (140), consistent with the Veglia findings and supporting a pinwheel structure (137).
Oxenoid & Chou (141) reported a different pentamer structure for WT PLN in DPC micelles. Using primarily chemical shifts and residual dipolar couplings, they concluded that the cytoplasmic helices protrude away from the membrane plane, giving a bellflower topology. In addition, a bend at Leu37 was found in the middle of the TM helix. These two features, so far not observed by other laboratories, were suggested to be an artifact of the effects of orientational degeneracy and motional averaging on residual dipolar couplings (136). Nevertheless, the outward-pointing cytoplasmic helices present an interesting model for PLN’s interaction with SERCA, and it may be interesting to investigate whether such a structure is populated during PLN’s transition from the inactive T state to the active R state.
Baldus and coworkers (142) used MAS NMR to determine the secondary structure, dynamics, and lipid interaction of monomeric AFA-PLN in DMPC bilayers. They found the cytoplasmic residues to exhibit high disorder, as manifested by sharp signals in scalar-coupling-based 2D correlation spectra. Binding to SERCA caused the cytoplasmic domain to become more ordered and more helical (143).
The different structures of the PLN cytoplasmic domain from these studies may partly reflect the dependence of the protein structure on the lipid composition, protein/lipid ratio, and protein hydration. In general, the disordered structure of the cytoplasmic domain was absent in glass-plate-aligned samples but was readily observed in unconstrained vesicle samples used in MAS-NMR and EPR experiments. This may reflect a tendency for glass plates to enforce order to extramembrane portions of membrane proteins. The native conformational distribution of PLN may include the in-plane α-helical structure and disordered out-of-plane structure at equilibrium. Because the inactive pentameric PLN must dissociate to bind and inhibit SERCA, and because the SERCA-binding monomeric PLN involves a highly dynamic cytoplasmic domain, it will be of interest to characterize the conformational transition of the cytoplasmic domain from the in-plane state to the disordered state and to correlate this conformational change with function.
CONCLUSION
The above survey shows that SSNMR spectroscopy has advanced tremendously to make significant contributions to membrane protein structure determination both at the whole structure level and at the local level. It provides information about missing domains in crystal structures, gives atomic-resolution details about ligand structure and binding, and reveals functionally relevant protein motion. The synergy among MAS SSNMR, OS-SSNMR, and solution NMR will provide increasing opportunities to understand how membrane protein conformation depends on the solubilizing environment. As always, structure itself is not the ultimate goal. How structure dictates function and mechanism lies at the heart of structural biology. The unique ability of SSNMR spectroscopy to capture ion movements and protein dynamics, visualize protons that are invisible to crystallography, and determine native structures in lipid bilayers will make it an ever more compelling technology for membrane protein structural biology.
SUMMARY POINTS.
High-resolution magic-angle-spinning solid-state NMR gives atomic-resolution distance constraints of membrane proteins and protein-ligand binding sites.
The conformational dynamics of membrane proteins is exquisitely probed by 2H and dipolar coupling NMR.
The membrane-mimetic environment exerts significant influence on membrane protein structures. Structures of detergent-solubilized membrane proteins can be critically validated using SSNMR of bilayer-bound samples.
MAS-SSNMR studies, in combination with electrophysiology, have provided unique insights into the mechanisms of inhibition and ion conduction of potassium and proton channels.
SSNMR studies have revealed atomic details of coupled protein and retinal conformational changes of the visual receptor rhodopsin that underlie light activation.
ACKNOWLEDGMENTS
The MAS-SSNMR study of the influenza M2 proton channel was funded by NIH grant GM088204 and NSF grant MCB 0543473. The authors thank Professor David Weliky for providing the illustration in Figure 6c.
Glossary
- SSNMR
solid-state nuclear magnetic resonance
- MAS
magic-angle spinning
- GPCR
G protein–coupled receptors
- DMPC
dimyristoylphosphatidylcholine
- DOPC
dioleoylphosphatidylcholine
- DOPE
dioleoylphosphatidylethanolamine
- DPC
dodecylphosphocholine
Footnotes
DISCLOSURE STATEMENT
The authors are not aware of any affiliations, memberships, funding, or financial holdings that might be perceived as affecting the objectivity of this review.
LITERATURE CITED
- 1.McDermott AE. Structure and dynamics of membrane proteins by magic angle spinning solid-state NMR. Annu. Rev. Biophys. 2009;38:385–403. doi: 10.1146/annurev.biophys.050708.133719. [DOI] [PubMed] [Google Scholar]
- 2.McDermott AE. Structural and dynamic studies of proteins by solid-state NMR spectroscopy: rapid movement forward. Curr. Opin. Struct. Biol. 2004;14:554–61. doi: 10.1016/j.sbi.2004.09.007. [DOI] [PubMed] [Google Scholar]
- 3.Tycko R. Solid-state NMR studies of amyloid fibril structure. Annu. Rev. Phys. Chem. 2011;62:279–99. doi: 10.1146/annurev-physchem-032210-103539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Smith SO. Structure and activation of the visual pigment rhodopsin. Annu. Rev. Biophys. 2010;39:309–28. doi: 10.1146/annurev-biophys-101209-104901. [DOI] [PubMed] [Google Scholar]
- 5.Cross TA, Sharma M, Yi M, Zhou HX. Influence of solubilizing environments on membrane protein structures. Trends Biochem. Sci. 2010;36:117–25. doi: 10.1016/j.tibs.2010.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kim HJ, Howell SC, Van Horn WD, Jeon YH, Sanders CR. Recent advances in the application of solution NMR spectroscopy to multi-span integral membrane proteins. Prog. Nucl. Magn. Reson. Spectrosc. 2009;55:335–60. doi: 10.1016/j.pnmrs.2009.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Raschle T, Hiller S, Etzkorn M, Wagner G. Nonmicellar systems for solution NMR spectroscopy of membrane proteins. Curr. Opin. Struct. Biol. 2010;20:471–79. doi: 10.1016/j.sbi.2010.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Griffin RG. Dipolar recoupling in MAS spectra of biological solids. Nat. Struct. Biol. 1998;5(Suppl.):508–12. doi: 10.1038/749. [DOI] [PubMed] [Google Scholar]
- 9.Hong M. Resonance assignment of 13C/15N labeled proteins by two- and three-dimensional magic-angle-spinning NMR. J. Biomol. NMR. 1999;15:1–14. doi: 10.1023/a:1008334204412. [DOI] [PubMed] [Google Scholar]
- 10.Rienstra CM, Hohwy M, Hong M, Griffin RG. 2D and 3D 15N-13C-13C NMR chemical shift correlation spectroscopy of solids: assignment of MAS spectra of peptides. J. Am. Chem. Soc. 2000;122:10979–90. [Google Scholar]
- 11.Pauli J, Baldus M, van Rossum B, de Groot H, Oschkinat H. Backbone and side-chain 13C and 15N signal assignments of the α-spectrin SH3 domain by magic-angle spinning solid-state NMR at 17.6 Tesla. Chembiochem. 2001;2:272–81. doi: 10.1002/1439-7633(20010401)2:4<272::AID-CBIC272>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 12.Hong M. Determination of multiple ϕ-torsion angles in solid proteins by selective and extensive 13C labeling and two-dimensional solid-state NMR. J. Magn. Reson. 1999;139:389–401. doi: 10.1006/jmre.1999.1805. [DOI] [PubMed] [Google Scholar]
- 13.Hong M, Jakes K. Selective and extensive 13C labeling of a membrane protein for solid-state NMR investigation. J. Biomol. NMR. 1999;14:71–74. doi: 10.1023/a:1008334930603. [DOI] [PubMed] [Google Scholar]
- 14.Castellani F, van Rossum B, Diehl A, Schubert M, Rehbein K, Oschkinat H. Structure of a protein determined by solid-state magic-angle spinning NMR spectroscopy. Nature. 2002;420:98–102. doi: 10.1038/nature01070. [DOI] [PubMed] [Google Scholar]
- 15.Etzkorn M, Martell S, Andronesi OC, Seidel K, Engelhard M, Baldus M. Secondary structure, dynamics, and topology of a seven-helix receptor in native membranes, studied by solid-state NMR spectroscopy. Angew. Chem. Int. Ed. Engl. 2007;46:459–62. doi: 10.1002/anie.200602139. [DOI] [PubMed] [Google Scholar]
- 16.Spera S, Bax A. Empirical correlation between protein backbone conformation and Ca and Cb 13C NMR chemical shifts. J. Am. Chem. Soc. 1991;113:5490–92. [Google Scholar]
- 17.Wishart DS, Sykes BD, Richards FM. Relationship between nuclear magnetic resonance chemical shift and protein secondary structure. J. Mol. Biol. 1991;222:311–33. doi: 10.1016/0022-2836(91)90214-q. [DOI] [PubMed] [Google Scholar]
- 18.Shen Y, Lange O, Delaglio F, Rossi P, Aramini JM, et al. Consistent blind protein structure generation from NMR chemical shift data. Proc. Natl. Acad. Sci. USA. 2008;105:4685–90. doi: 10.1073/pnas.0800256105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa PR, Gross JD, Hong M, Griffin RG. Solid-state NMR measurement of ψ in peptides: a NCCN 2Q-heteronuclear local field experiment. Chem. Phys. Lett. 1997;280:95–103. [Google Scholar]
- 20.Hong M, Gross JD, Griffin RG. Site-resolved determination of peptide torsion angle ϕ from the relative orientations of backbone N-H and C-H bonds by solid-state NMR. J. Phys. Chem. B. 1997;101:5869–74. [Google Scholar]
- 21.De Paëpe G, Lewandowski JR, Loquet A, Böckmann A, Griffin RG. Proton assisted recoupling and protein structure determination. J. Chem. Phys. 2008;129:245101. doi: 10.1063/1.3036928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lange A, Luca S, Baldus M. Structural constraints from proton-mediated rare-spin correlation spectroscopy in rotating solids. J. Am. Chem. Soc. 2002;124:9704–5. doi: 10.1021/ja026691b. [DOI] [PubMed] [Google Scholar]
- 23.Takegoshi K, Nakamura S, Terao T. 13C-1H dipolar-assisted rotational resonance in magic-angle spinning NMR. Chem. Phys. Lett. 2001;344:631–37. [Google Scholar]
- 24.Buffy JJ, Waring AJ, Hong M. Determination of peptide oligomerization in lipid membranes with magic-angle spinning spin diffusion NMR. J. Am. Chem. Soc. 2005;127:4477–83. doi: 10.1021/ja043621r. [DOI] [PubMed] [Google Scholar]
- 25.Luo W, Hong M. Determination of the oligomeric number and intermolecular distances of membrane protein assemblies by anisotropic 1H-driven spin diffusion NMR spectroscopy. J. Am. Chem. Soc. 2006;128:7242–51. doi: 10.1021/ja0603406. [DOI] [PubMed] [Google Scholar]
- 26.Schmidt-Rohr K, Hong M. Measurements of carbon to amide-proton distances by C-H dipolar recoupling with 15N NMR detection. J. Am. Chem. Soc. 2003;125:5648–49. doi: 10.1021/ja0344415. [DOI] [PubMed] [Google Scholar]
- 27.Wi S, Sinha N, Hong M. Long-range 1H-19F distance measurement in peptides by solid-state NMR. J. Am. Chem. Soc. 2004;126:12754–55. doi: 10.1021/ja0462732. [DOI] [PubMed] [Google Scholar]
- 28.Opella SJ, Marassi FM. Structure determination of membrane proteins by NMR spectroscopy. Chem. Rev. 2004;104:3587–606. doi: 10.1021/cr0304121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hong M, Doherty T. Orientation determination of membrane-disruptive proteins using powder samples and rotational diffusion: a simple solid-state NMR approach. Chem. Phys. Lett. 2006;432:296–300. doi: 10.1016/j.cplett.2006.10.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cady SD, Goodman C, Tatko C, DeGrado WF, Hong M. Determining the orientation of uniaxially rotating membrane proteins using unoriented samples: a 2H, 13C, and 15N solid-state NMR investigation of the dynamics and orientation of a transmembrane helical bundle. J. Am. Chem. Soc. 2007;129:5719–29. doi: 10.1021/ja070305e. [DOI] [PubMed] [Google Scholar]
- 31.Hong M, Su Y. Structure and dynamics of cationic membrane peptides and proteins: insights from solid-state NMR. Protein Sci. 2011;20:641–55. doi: 10.1002/pro.600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Su Y, DeGrado WF, Hong M. Orientation, dynamics, and lipid interaction of an antimicrobial arylamide investigated by 19F and 31P solid-state NMR spectroscopy. J. Am. Chem. Soc. 2010;132:9197–205. doi: 10.1021/ja103658h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tang M, Waring AJ, Lehrer RI, Hong M. Effects of guanidinium-phosphate hydrogen bonding on the membrane-bound structure and activity of an arginine-rich membrane peptide from solid-state NMR. Angew. Chem. Int. Ed. Engl. 2008;47:3202–5. doi: 10.1002/anie.200705993. [DOI] [PubMed] [Google Scholar]
- 34.Lewis BA, Harbison GS, Herzfeld J, Griffin RG. NMR structural analysis of a membrane protein: bacteriorhodopsin peptide backbone orientation and motion. Biochemistry. 1985;24:4671–79. doi: 10.1021/bi00338a029. [DOI] [PubMed] [Google Scholar]
- 35.Huster D, Yao XL, Hong M. Membrane protein topology probed by 1H spin diffusion from lipids using solid-state NMR spectroscopy. J. Am. Chem. Soc. 2002;124:874–83. doi: 10.1021/ja017001r. [DOI] [PubMed] [Google Scholar]
- 36.Kumashiro KK, Schmidt-Rohr K, Murphy OJ, Ouellette KL, Cramer WA, Thompson LK. A novel tool for probing membrane protein structure: solid-state NMR with proton spin diffusion and X-nucleus detection. J. Am. Chem. Soc. 1998;120:5043–51. [Google Scholar]
- 37.Buffy JJ, Hong T, Yamaguchi S, Waring A, Lehrer RI, Hong M. Solid-state NMR investigation of the depth of insertion of protegrin-1 in lipid bilayers using paramagnetic Mn2+ Biophys. J. 2003;85:2363–73. doi: 10.1016/s0006-3495(03)74660-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tang M, Waring AJ, Hong M. Phosphate-mediated arginine insertion into lipid membranes and pore formation by a cationic membrane peptide from solid-state NMR. J. Am. Chem. Soc. 2007;129:11438–46. doi: 10.1021/ja072511s. [DOI] [PubMed] [Google Scholar]
- 39.Toke O, Maloy WL, Kim SJ, Blazyk J, Schaefer J. Secondary structure and lipid contact of a peptide antibiotic in phospholipid bilayers by REDOR. Biophys. J. 2004;87:662–74. doi: 10.1529/biophysj.103.032706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Su Y, Mani R, Hong M. Asymmetric insertion of membrane proteins in lipid bilayers by solid-state NMR paramagnetic relaxation enhancement: a cell-penetrating peptide example. J. Am. Chem. Soc. 2008;130:8856–64. doi: 10.1021/ja802383t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yamaguchi S, Huster D, Waring A, Lehrer RI, Tack BF, et al. Orientation and dynamics of an antimicrobial peptide in the lipid bilayer by solid-state NMR. Biophys. J. 2001;81:2203–14. doi: 10.1016/S0006-3495(01)75868-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Slatin SL, Qiu XQ, Jakes KS, Finkelstein A. Identification of a translocated protein segment in a voltage-dependent channel. Nature. 1994;371:158–61. doi: 10.1038/371158a0. [DOI] [PubMed] [Google Scholar]
- 43.Luo W, Yao XL, Hong M. Large structure rearrangement of colicin Ia channel domain after membrane binding from 2D 13C spin diffusion NMR. J. Am. Chem. Soc. 2005;127:6402–8. doi: 10.1021/ja0433121. [DOI] [PubMed] [Google Scholar]
- 44.Hu F, Luo W, Hong M. Mechanisms of proton conduction and gating by influenza M2 proton channels from solid-state NMR. Science. 2010;330:505–9. doi: 10.1126/science.1191714. Reports the conformation, dynamics, and hydrogen bonding of the proton-selective histidine in M2, supporting the shuttle mechanism of proton conduction. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lipari G, Szabo A, Levy RM. Protein dynamics and NMR relaxation: comparison of simulations with experiment. Nature. 1982;300:197–98. [Google Scholar]
- 46.Gullion T, Schaefer J. Rotational-echo double-resonance NMR. J. Magn. Reson. 1989;81:196–200. doi: 10.1016/j.jmr.2011.09.003. [DOI] [PubMed] [Google Scholar]
- 47.Raleigh DP, Levitt MH, Griffin RG. Rotational resonance in solid state NMR. Chem. Phys. Lett. 1988;146:71–76. [Google Scholar]
- 48.Doyle DA, Cabral JM, Pfuetzner RA, Kuo A, Gulbis JM, et al. The structure of the potassium channel: molecular basis of K+ conduction and selectivity. Science. 1998;280:69–77. doi: 10.1126/science.280.5360.69. [DOI] [PubMed] [Google Scholar]
- 49.Zhou Y, Morais-Cabral JH, Kaufman A, MacKinnon R. Chemistry of ion coordination and hydration revealed by a K+ channel-Fab complex at 2.0Å resolution. Nature. 2001;414:43–48. doi: 10.1038/35102009. [DOI] [PubMed] [Google Scholar]
- 50.Long SB, Tao X, Campbell EB, MacKinnon R. Atomic structure of a voltage-dependent K+ channel in a lipid membrane-like environment. Nature. 2007;450:376–83. doi: 10.1038/nature06265. [DOI] [PubMed] [Google Scholar]
- 51.Lange A, Giller K, Hornig S, Martin-Eauclaire MF, Pongs O, et al. Toxin-induced conformational changes in a potassium channel revealed by solid-state NMR. Nature. 2006;440:959–62. doi: 10.1038/nature04649. MAS SSNMR study of the conformational changes of a chimeric potassium channel due to toxin binding. [DOI] [PubMed] [Google Scholar]
- 52.Schneider R, Ader C, Lange A, Giller K, Hornig S, et al. Solid-state NMR spectroscopy applied to a chimeric potassium channel in lipid bilayers. J. Am. Chem. Soc. 2008;130:7427–35. doi: 10.1021/ja800190c. [DOI] [PubMed] [Google Scholar]
- 53.Ader C, Schneider R, Hornig S, Velisetty P, Wilson EM, et al. A structural link between inactivation and block of a K+ channel. Nat. Struct. Mol. Biol. 2008;15:605–12. doi: 10.1038/nsmb.1430. [DOI] [PubMed] [Google Scholar]
- 54.Ader C, Schneider R, Hornig S, Velisetty P, Vardanyan V, et al. Coupling of activation and inactivation gate in a K+ channel: potassium and ligand sensitivity. EMBO J. 2009;28:2825–34. doi: 10.1038/emboj.2009.218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Varga K, Tian L, McDermott AE. Solid-state NMR study and assignments of the KcsA potassium ion channel of S. lividans. Biochim. Biophys. Acta. 2007;1774:1604–13. doi: 10.1016/j.bbapap.2007.08.029. [DOI] [PubMed] [Google Scholar]
- 56.Bhate MP, Wylie BJ, Tian L, McDermott AE. Conformational dynamics in the selectivity filter of KcsA in response to potassium ion concentration. J. Mol. Biol. 2010;401:155–66. doi: 10.1016/j.jmb.2010.06.031. Chemical shift correlation NMR reveals high-affinity potassium binding to selectivity-filter residues and suggests the mechanism of ion transmission. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chill JH, Louis JM, Miller C, Bax A. NMR study of the tetrameric KcsA potassium channel in detergent micelles. Protein Sci. 2006;15:684–98. doi: 10.1110/ps.051954706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Imai S, Osawa M, Takeuchi K, Shimada I. Structural basis underlying the dual gate properties of KcsA. Proc. Natl. Acad. Sci. USA. 2010;107:6216–21. doi: 10.1073/pnas.0911270107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ader C, Schneider R, Seidel K, Etzkorn M, Becker S, Baldus M. Structural rearrangements of membrane proteins probed by water-edited solid-state NMR spectroscopy. J. Am. Chem. Soc. 2009;131:170–76. doi: 10.1021/ja806306e. [DOI] [PubMed] [Google Scholar]
- 60.Baker KA, Tzitzilonis C, Kwiatkowski W, Choe S, Riek R. Conformational dynamics of the KcsA potassium channel governs gating properties. Nat. Struct. Mol. Biol. 2007;14:1089–95. doi: 10.1038/nsmb1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Pinto LH, Lamb RA. The M2 proton channels of influenza A and B viruses. J. Biol. Chem. 2006;281:8997–9000. doi: 10.1074/jbc.R500020200. [DOI] [PubMed] [Google Scholar]
- 62.Bright RA, Medina MJ, Xu X, Perez-Oronoz G, Wallis TR, et al. Incidence of adamantane resistance among influenza A (H3N2) viruses isolated worldwide from 1994 to 2005: a cause for concern. Lancet. 2005;366:1175–81. doi: 10.1016/S0140-6736(05)67338-2. [DOI] [PubMed] [Google Scholar]
- 63.Cady SD, Luo WB, Hu FH, Hong M. Structure and function of the influenza A M2 proton channel. Biochemistry. 2009;48:7356–64. doi: 10.1021/bi9008837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Song Z, Kovacs FA, Wang J, Denny JK, Shekar SC, et al. Transmembrane domain of M2 protein from influenza A virus studied by solid-state 15N polarization inversion spin exchange at magic angle NMR. Biophys. J. 2000;79:767–75. doi: 10.1016/S0006-3495(00)76334-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wang J, Kim S, Kovacs F, Cross TA. Structure of the transmembrane region of the M2 protein H+ channel. Prot. Sci. 2001;10:2241–50. doi: 10.1110/ps.17901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hu J, Asbury T, Achuthan S, Li C, Bertram R, et al. Backbone structure of the amantadine-blocked trans-membrane domain M2 proton channel from influenza A virus. Biophys. J. 2007;92:4335–43. doi: 10.1529/biophysj.106.090183. 15N SSNMR of oriented membranes reveals a drug-induced kink of the M2 transmembrane helix. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Tian C, Gao PF, Pinto LH, Lamb RA, Cross TA. Initial structural and dynamic characterization of the M2 protein transmembrane and amphipathic helices in lipid bilayers. Protein Sci. 2003;12:2597–605. doi: 10.1110/ps.03168503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sharma M, Yi M, Dong H, Qin H, Peterson E, et al. Atomistic mechanism of the influenza A proton channel from a structure solved in a lipid bilayer. Science. 2010;330:509–12. doi: 10.1126/science.1191750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Cady SD, Hong M. Amantadine-induced conformational and dynamical changes of the influenza M2 transmembrane proton channel. Proc. Natl. Acad. Sci. USA. 2008;105:1483–88. doi: 10.1073/pnas.0711500105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Cady SD, Mishanina TV, Hong M. Structure of amantadine-bound M2 transmembrane peptide of influenza A in lipid bilayers from magic-angle-spinning solid-state NMR: the role of Ser31 in amantadine binding. J. Mol. Biol. 2009;385:1127–41. doi: 10.1016/j.jmb.2008.11.022. Chemical shift perturbation from 2D MAS spectra implicates Ser31 as the drug binding site of the influenza M2 transmembrane domain. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wang J, Cady SD, Balannik V, Pinto LH, DeGrado WF, Hong M. Discovery of spiro-piperidine inhibitors and their modulation of the dynamics of the M2 proton channel from influenza A virus. J. Am. Chem. Soc. 2009;131:8066–76. doi: 10.1021/ja900063s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Stouffer AL, Acharya R, Salom D, Levine AS, Di Costanzo L, et al. Structural basis for the function and inhibition of an influenza virus proton channel. Nature. 2008;451:596–99. doi: 10.1038/nature06528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Schnell JR, Chou JJ. Structure and mechanism of the M2 proton channel of influenza A virus. Nature. 2008;451:591–95. doi: 10.1038/nature06531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Cady SD, Schmidt-Rohr K, Wang J, Soto CS, DeGrado WF, Hong M. Structure of the amantadine binding site of influenza M2 proton channels in lipid bilayers. Nature. 2010;463:689–92. doi: 10.1038/nature08722. Protein-drug distance measurements elucidate the pharmacologically relevant drug binding site and the high-resolution structure of the influenza M2 transmembrane domain. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Cady SD, Wang T, Hong M. Membrane-dependent effects of a cytoplasmic helix on the structure and drug binding of the influenza virus M2 protein. J. Am. Chem. Soc. 2011;133:11572–79. doi: 10.1021/ja202051n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Cady SD, Wang J, Wu Y, DeGrado WF, Hong M. Specific binding of adamantane drugs and direction of their polar amines in the pore of the influenza M2 transmembrane domain in lipid bilayers and dodecylphosphocholine micelles determined by NMR spectroscopy. J. Am. Chem. Soc. 2011;133:4274–84. doi: 10.1021/ja102581n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hu J, Fu R, Cross TA. The chemical and dynamical influence of the anti-viral drug amantadine on the M2 proton channel transmembrane domain. Biophys. J. 2007;93:276–83. doi: 10.1529/biophysj.106.102103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Luo W, Hong M. Conformational changes of an ion channel detected through water-protein interactions using solid-state NMR spectroscopy. J. Am. Chem. Soc. 2010;132:2378–84. doi: 10.1021/ja9096219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Sansom MSP, Kerr ID, Smith GR, Son HS. The influenza A virus M2 channel: a molecular modeling and simulation study. Virology. 1997;233:163–73. doi: 10.1006/viro.1997.8578. [DOI] [PubMed] [Google Scholar]
- 80.Chen H, Wu Y, Voth GA. Proton transport behavior through the influenza A M2 channel: insights from molecular simulation. Biophys. J. 2007;93:3470–79. doi: 10.1529/biophysj.107.105742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Pinto LH, Dieckmann GR, Gandhi CS, Papworth CG, Braman J, et al. A functionally defined model for the M2 proton channel of influenza A virus suggests a mechanism for its ion selectivity. Proc. Natl. Acad. Sci. USA. 1997;94:11301–6. doi: 10.1073/pnas.94.21.11301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lin TI, Schroeder C. Definitive assignment of proton selectivity and attoampere unitary current to the M2 ion channel protein of influenza A virus. J. Virol. 2001;75:3647–56. doi: 10.1128/JVI.75.8.3647-3656.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Li C, Qin H, Gao FP, Cross TA. Solid-state NMR characterization of conformational plasticity within the transmembrane domain of the influenza A M2 proton channel. Biochim. Biophys. Acta. 2007;1768:3162–70. doi: 10.1016/j.bbamem.2007.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Hu F, Luo W, Cady SD, Hong M. Conformational plasticity of the influenza A M2 transmembrane peptide in lipid bilayers under varying pH, drug binding and membrane thickness. Biochim. Biophys. Acta. 2011;1808:415–23. doi: 10.1016/j.bbamem.2010.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Luo W, Cady SD, Hong M. Immobilization of the influenza A M2 transmembrane peptide in virus-envelope mimetic lipid membranes: a solid-state NMR investigation. Biochemistry. 2009;48:6361–68. doi: 10.1021/bi900716s. A virus-mimetic lipid membrane immobilizes rotational diffusion of the M2 helical bundle and gives high-resolution spectra at physiological temperature. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Cady SD, Hong M. Simultaneous extraction of multiple orientational constraints of membrane proteins by 13C-detected N-H dipolar couplings under magic angle spinning. J. Magn. Reson. 2008;191:219–25. doi: 10.1016/j.jmr.2008.01.001. [DOI] [PubMed] [Google Scholar]
- 87.Cady SD, Hong M. Effects of amantadine binding on the dynamics of bilayer-bound influenza A M2 transmembrane peptide studied by NMR relaxation. J. Biomol. NMR. 2009;45:185–96. doi: 10.1007/s10858-009-9352-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Cristian L, Lear JD, DeGrado WF. Use of thiol-disulfide equilibria to measure the energetics of assembly of transmembrane helices in phospholipid bilayers. Proc. Natl. Acad. Sci. USA. 2003;100:14772–77. doi: 10.1073/pnas.2536751100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Li J, Edwards PC, Burghammer M, Villa C, Schertler GF. Structure of bovine rhodopsin in a trigonal crystal form. J. Mol. Biol. 2004;343:1409–38. doi: 10.1016/j.jmb.2004.08.090. [DOI] [PubMed] [Google Scholar]
- 90.Okada T, Sugihara M, Bondar AN, Elstner M, Entel P, Buss V. The retinal conformation and its environment in rhodopsin in light of a new 2.2Å crystal structure. J. Mol. Biol. 2004;342:571–83. doi: 10.1016/j.jmb.2004.07.044. [DOI] [PubMed] [Google Scholar]
- 91.Palczewski K, Kumasaka T, Hori T, Behnke CA, Motoshima H, et al. Crystal structure of rhodopsin: a G protein-coupled receptor. Am. J. Ophthalmol. 2000;289:739–45. doi: 10.1126/science.289.5480.739. [DOI] [PubMed] [Google Scholar]
- 92.Brown MF, Salgado GF, Struts AV. Retinal dynamics during light activation of rhodopsin revealed by solid-state NMR spectroscopy. Biochim. Biophys. Acta. 2010;1798:177–93. doi: 10.1016/j.bbamem.2009.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Salgado GF, Struts AV, Tanaka K, Fujioka N, Nakanishi K, Brown MF. Deuterium NMR structure of retinal in the ground state of rhodopsin. Biochemistry. 2004;43:12819–28. doi: 10.1021/bi0491191. [DOI] [PubMed] [Google Scholar]
- 94.Salgado GF, Struts AV, Tanaka K, Krane S, Nakanishi K, Brown MF. Solid-state 2H NMR structure of retinal in metarhodopsin I. J. Am. Chem. Soc. 2006;128:11067–71. doi: 10.1021/ja058738+. [DOI] [PubMed] [Google Scholar]
- 95.Struts AV, Salgado GF, Brown MF. Solid-state 2H NMR relaxation illuminates functional dynamics of retinal cofactor in membrane activation of rhodopsin. Proc. Natl. Acad. Sci. USA. 2011;108:8263–68. doi: 10.1073/pnas.1014692108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Struts AV, Salgado GF, Martinez-Mayorga K, Brown MF. Retinal dynamics underlie its switch from inverse agonist to agonist during rhodopsin activation. Nat. Struct. Mol. Biol. 2011;18:392–94. doi: 10.1038/nsmb.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Patel AB, Crocker E, Eilers M, Hirshfeld A, Sheves M, Smith SO. Coupling of retinal isomerization to the activation of rhodopsin. Proc. Natl. Acad. Sci. USA. 2004;101:10048–53. doi: 10.1073/pnas.0402848101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Patel AB, Crocker E, Reeves PJ, Getmanova EV, Eilers M, et al. Changes in interhelical hydrogen bonding upon rhodopsin activation. J. Mol. Biol. 2005;347:803–12. doi: 10.1016/j.jmb.2005.01.069. [DOI] [PubMed] [Google Scholar]
- 99.Crocker E, Eiler M, Ahuja S, Hornak V, Hirshfeld A, et al. Location of Trp265 in metarhodopsin II: implications for the activation mechanism of the visual receptor rhodopsin. J. Mol. Biol. 2006;357:163–72. doi: 10.1016/j.jmb.2005.12.046. [DOI] [PubMed] [Google Scholar]
- 100.Ahuja S, Hornak V, Yan EC, Syrett N, Goncalves JA, et al. Helix movement is coupled to displacement of the second extracellular loop in rhodopsin activation. Nat. Struct. Mol. Biol. 2009;16:168–75. doi: 10.1038/nsmb.1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Shi L, Ahmed MA, Zhang W, Whited G, Brown LS, Ladizhansky V. Three-dimensional solid-state NMR study of a seven-helical integral membrane proton pump: structural insights. J. Mol. Biol. 2009;386:1078–93. doi: 10.1016/j.jmb.2009.01.011. [DOI] [PubMed] [Google Scholar]
- 102.Griffiths JM, Bennett AE, Engelhard M, Siebert F, Raap J, et al. Structural investigation of the active site in bacteriorhodopsin: geometric constraints on the roles of Asp-85 and Asp-212 in the proton-pumping mechanism from solid state NMR. Biochemistry. 2000;39:362–71. doi: 10.1021/bi991106d. [DOI] [PubMed] [Google Scholar]
- 103.Etzkorn M, Seidel K, Li L, Martell S, Geyer M, et al. Complex formation and light activation in membrane-embedded sensory rhodopsin II as seen by solid-state NMR spectroscopy. Structure. 2010;18:293–300. doi: 10.1016/j.str.2010.01.011. [DOI] [PubMed] [Google Scholar]
- 104.Arora A, Abildgaard F, Bushweller JH, Tamm LK. Structure of outer membrane protein A transmembrane domain by NMR spectroscopy. Nat. Struct. Biol. 2001;8:334–38. doi: 10.1038/86214. [DOI] [PubMed] [Google Scholar]
- 105.Liang B, Tamm LK. Structure of outer membrane protein G by solution NMR spectroscopy. Proc. Natl. Acad. Sci. USA. 2007;104:16140–45. doi: 10.1073/pnas.0705466104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Hwang PM, Choy WY, Lo EI, Chen L, Forman-Kay JD, et al. Solution structure and dynamics of the outer membrane enzyme PagP by NMR. Proc. Natl. Acad. Sci. USA. 2002;99:13560–65. doi: 10.1073/pnas.212344499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Mahalakshmi R, Marassi FM. Orientation of the Escherichia coli outer membrane protein OmpX in phospholipid bilayer membranes determined by solid-state NMR. Biochemistry. 2008;47:6531–38. doi: 10.1021/bi800362b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Hiller M, Krabben L, Vinothkumar KR, Castellani F, van Rossum BJ, et al. Solid-state magic-angle spinning NMR of outer-membrane protein G from Escherichia coli. Chembiochem. 2005;6:1679–84. doi: 10.1002/cbic.200500132. [DOI] [PubMed] [Google Scholar]
- 109.Arora A, Rinehart D, Szabo G, Tamm LK. Refolded outer membrane protein A of Escherichia coli forms ion channels with two conductance states in planar lipid bilayers. J. Biol. Chem. 2000;275:1594–600. doi: 10.1074/jbc.275.3.1594. [DOI] [PubMed] [Google Scholar]
- 110.Tamm LK, Abildgaard F, Arora A, Blad H, Bushweller JH. Structure, dynamics and function of the outer membrane protein A (OmpA) and influenza hemagglutinin fusion domain in detergent micelles by solution NMR. FEBS Lett. 2003;555:139–43. doi: 10.1016/s0014-5793(03)01127-x. [DOI] [PubMed] [Google Scholar]
- 111.Malia TJ, Wagner G. NMR structural investigation of the mitochondrial outer membrane protein VDAC and its interaction with antiapoptotic Bcl-xL. Biochemistry. 2007;46:514–25. doi: 10.1021/bi061577h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Hiller S, Abramson J, Mannella C, Wagner G, Zeth K. The 3D structures of VDAC represent a native conformation. Trends Biochem. Sci. 2010;35:514–21. doi: 10.1016/j.tibs.2010.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Colombini M. The published 3D structure of the VDAC channel: native or not? Trends Biochem.Sci. 2009;34:382–89. doi: 10.1016/j.tibs.2009.05.001. [DOI] [PubMed] [Google Scholar]
- 114.Hiller S, Garces RG, Malia TJ, Orekhov VY, Colombini M, Wagner G. Solution structure of the integral human membrane protein VDAC-1 in detergent micelles. Science. 2008;321:1206–10. doi: 10.1126/science.1161302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Bayrhuber M, Meins T, Habeck M, Becker S, Giller K, et al. Structure of the human voltage-dependent anion channel. Proc. Natl. Acad. Sci. USA. 2008;105:15370–75. doi: 10.1073/pnas.0808115105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Bullough PA, Hughson FM, Skehel JJ, Wiley DC. Structure of influenza haemagglutinin at the pH of membrane fusion. Nature. 1994;371:37–43. doi: 10.1038/371037a0. [DOI] [PubMed] [Google Scholar]
- 117.Harrison SC. Viral membrane fusion. Nat. Struct. Mol. Biol. 2008;15:690–98. doi: 10.1038/nsmb.1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Tamm LK, Lai AL, Li Y. Combined NMR and EPR spectroscopy to determine structures of viral fusion domains in membranes. Biochim. Biophys. Acta. 2007;1768:3052–60. doi: 10.1016/j.bbamem.2007.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Han X, Bushweller JH, Cafiso DS, Tamm LK. Membrane structure and fusion-triggering conformational change of the fusion domain from influenza hemagglutinin. Nat. Struct. Biol. 2001;8:715–20. doi: 10.1038/90434. [DOI] [PubMed] [Google Scholar]
- 120.Lai AL, Park H, White JM, Tamm LK. Fusion peptide of influenza hemagglutinin requires a fixed angle boomerang structure for activity. J. Biol. Chem. 2006;281:5760–70. doi: 10.1074/jbc.M512280200. [DOI] [PubMed] [Google Scholar]
- 121.Lai AL, Tamm LK. Locking the kink in the influenza hemagglutinin fusion domain structure. J. Biol. Chem. 2007;282:23946–56. doi: 10.1074/jbc.M704008200. [DOI] [PubMed] [Google Scholar]
- 122.Tamm LK. Hypothesis: spring-loaded boomerang mechanism of influenza hemagglutinin-mediated membrane fusion. Biochim. Biophys. Acta. 2003;1614:14–23. doi: 10.1016/s0005-2736(03)00159-7. [DOI] [PubMed] [Google Scholar]
- 123.Sun Y, Weliky DP. 13C-13C correlation spectroscopy of membrane-associated influenza virus fusion peptide strongly supports a helix-turn-helix motif and two turn conformations. J. Am. Chem. Soc. 2009;131:13228–29. doi: 10.1021/ja905198q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Lorieau JL, Louis JM, Bax A. The complete influenza hemagglutinin fusion domain adopts a tight helical hairpin arrangement at the lipid:water interface. Proc. Natl. Acad. Sci. USA. 2010;107:11341–46. doi: 10.1073/pnas.1006142107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Russ WP, Engelman DM. The GxxxG motif: a framework for transmembrane helix-helix association. J. Mol. Biol. 2000;296:911–19. doi: 10.1006/jmbi.1999.3489. [DOI] [PubMed] [Google Scholar]
- 126.Jaroniec CP, Kaufman JD, Stahl SJ, Viard M, Blumenthal R, et al. Structure and dynamics of micelle-associated human immunodeficiency virus gp41 fusion domain. Biochemistry. 2005;44:16167–80. doi: 10.1021/bi051672a. [DOI] [PubMed] [Google Scholar]
- 127.Li Y, Tamm LK. Structure and plasticity of the human immunodeficiency virus gp41 fusion domain in lipid micelles and bilayers. Biophys. J. 2007;93:876–85. doi: 10.1529/biophysj.106.102335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Yang J, Gabrys CM, Weliky DP. Solid-state nuclear magnetic resonance evidence for an extended β strand conformation of the membrane-bound HIV-1 fusion peptide. Biochemistry. 2001;40:8126–37. doi: 10.1021/bi0100283. [DOI] [PubMed] [Google Scholar]
- 129.Yang J, Weliky DP. Solid-state nuclear magnetic resonance evidence for parallel and antiparallel strand arrangements in the membrane-associated HIV-1 fusion peptide. Biochemistry. 2003;42:11879–90. doi: 10.1021/bi0348157. [DOI] [PubMed] [Google Scholar]
- 130.Qiang W, Weliky DP. HIV fusion peptide and its cross-linked oligomers: efficient syntheses, significance of the trimer in fusion activity, correlation of βstrand conformation with membrane cholesterol, and proximity to lipid headgroups. Biochemistry. 2009;48:289–301. doi: 10.1021/bi8015668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Yang R, Prorok M, Castellino FJ, Weliky DP. A trimeric HIV-1 fusion peptide construct which does not self-associate in aqueous solution and which has 15-fold higher membrane fusion rate. J. Am. Chem. Soc. 2004;126:14722–23. doi: 10.1021/ja045612o. [DOI] [PubMed] [Google Scholar]
- 132.Qiang W, Sun Y, Weliky DP. A strong correlation between fusogenicity and membrane insertion depth of the HIV fusion peptide. Proc. Natl. Acad. Sci. USA. 2009;106:15314–19. doi: 10.1073/pnas.0907360106. Protein-lipid distance measurements by MAS experiments show deeper insertion of the active HIV fusion peptide into the membrane. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Zamoon J, Nitu F, Karim C, Thomas DD, Veglia G. Mapping the interaction surface of a membrane protein: unveiling the conformational switch of phospholamban in calcium pump regulation. Proc. Natl. Acad. Sci. USA. 2005;102:4747–52. doi: 10.1073/pnas.0406039102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Traaseth NJ, Buffy JJ, Zamoon J, Veglia G. Structural dynamics and topology of phospholamban in oriented lipid bilayers using multidimensional solid-state NMR. Biochemistry. 2006;45:13827–34. doi: 10.1021/bi0607610. [DOI] [PubMed] [Google Scholar]
- 135.Traaseth NJ, Shi L, Verardi R, Mullen DG, Barany G, Veglia G. Structure and topology of monomeric phospholamban in lipid membranes determined by a hybrid solution and solid-state NMR approach. Proc. Natl. Acad. Sci. USA. 2009;106:10165–70. doi: 10.1073/pnas.0904290106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Verardi R, Shi L, Traaseth NJ, Walsh N, Veglia G. Structural topology of phospholamban pentamer in lipid bilayers by a hybrid solution and solid-state NMR method. Proc. Natl. Acad. Sci. USA. 2011;108:9101–6. doi: 10.1073/pnas.1016535108. Reports a high-resolution pinwheel structure of the phospholamban pentamer, where the cytoplasmic helix is parallel to the membrane plane. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Robia SL, Flohr NC, Thomas DD. Phospholamban pentamer quaternary conformation determined by in-gel fluorescence anisotropy. Biochemistry. 2005;44:4302–11. doi: 10.1021/bi0478446. [DOI] [PubMed] [Google Scholar]
- 138.Traaseth NJ, Verardi R, Torgersen KD, Karim CB, Thomas DD, Veglia G. Spectroscopic validation of the pentameric structure of phospholamban. Proc. Natl. Acad. Sci. USA. 2007;104:14676–81. doi: 10.1073/pnas.0701016104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Abu-Baker S, Lu JX, Chu S, Brinn CC, Makaroff CA, Lorigan GA. Side chain and backbone dynamics of phospholamban in phospholipid bilayers utilizing 2H and 15N solid-state NMR spectroscopy. Biochemistry. 2007;46:11695–706. doi: 10.1021/bi700749q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Abu-Baker S, Lu JX, Chu S, Shetty KK, Gor’kov PL, Lorigan GA. The structural topology of wild-type phospholamban in oriented lipid bilayers using 15N solid-state NMR spectroscopy. Protein Sci. 2007;16:2345–49. doi: 10.1110/ps.072977707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Oxenoid K, Chou JJ. The structure of phospholamban pentamer reveals a channel-like architecture in membranes. Proc. Natl. Acad. Sci. USA. 2005;102:10870–75. doi: 10.1073/pnas.0504920102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Andronesi OC, Becker S, Seidel K, Heise H, Young HS, Baldus M. Determination of membrane protein structure and dynamics by magic-angle-spinning solid-state NMR spectroscopy. J. Am. Chem. Soc. 2005;127:12965–74. doi: 10.1021/ja0530164. [DOI] [PubMed] [Google Scholar]
- 143.Seidel K, Andronesi OC, Krebs J, Griesinger C, Young HS, et al. Structural characterization of Ca2+-ATPase-bound phospholamban in lipid bilayers by solid-state nuclear magnetic resonance (NMR) spectroscopy. Biochemistry. 2008;47:4369–76. doi: 10.1021/bi7024194. [DOI] [PubMed] [Google Scholar]