

Abstract
Integrated ferritin protein cage function is the reversible synthesis of protein-caged, solid Fe2O3•H2O minerals from Fe2+, for metabolic iron concentrates and oxidant protection; biomineral order varies in different ferritin proteins. The conserved 4, 3, 2 geometric symmetry of ferritin protein cages, parallels subunit dimer, trimer and tetramer interfaces, and coincides with function at several cage axes. Multiple subdomains distributed in the self- assembling ferritin nanocages have functional relationships to cage symmetry such as Fe2+ transport though ion channels (3-fold symmetry), biomineral nucleation/order (4-fold symmetry) and mineral dissolution (3-fold symmetry) studied in ferritin variants. Cage subunit dimers (2-fold symmetry) influence iron oxidation and mineral dissolution, based on effects of natural or synthetic subunit dimer crosslinks. 2Fe2+/O2 catalysis in ferritin occurs in single subunits, but with cooperativity (n=3) that is possibly related to the structure/function of the ion channels, which are constructed from segments of 3 subunits. Here, we study 2Fe2+ + O2 protein catalysis (diferric peroxo formation) and dissolution of ferritin Fe2O3•H2O biominerals in variants with altered subunit interfaces for trimers (ion channels), E130I, and external dimer surfaces (E88A) as controls, and altered tetramer subunit interfaces (L165I and H169F). The results extend observations on the functional importance of structure at ferritin protein 2-fold and 3-fold cage axes to show function at ferritin 4-fold cage axes. Here, conserved amino acids facilitate dissolution of ferritin protein-caged iron biominerals. Biological and nanotechnological uses of ferritin protein cage 4-fold symmetry and solid state mineral properties remain largely unexplored.
Keywords: Ferritin, protein nanocage, ferric oxo, di-iron protein, nanobiomineral, mineral dissolution, ion channel protein
Introduction
Ferritin protein cages are containers for mineralized iron concentrates that are used in biology as reservoirs for iron-cofactor synthesis in heme, iron-sulfur clusters, and iron bound directly to protein [1]; the substrates for ferritin minerals, Fe2+ and O2 or H2O2, also confer antioxidant properties on ferritin. Ferritin biosynthesis rates are controlled by oxidants targeted to an antioxidant response segment in the gene (DNA-ARE) [2] and by Fe2+ binding to a noncoding riboregulator (IRE-RNA) in the ferritin messenger RNA [3]. The supramolecular ferritin proteins, ~480 or ~240 kDa, self-assemble in solution and possibly in vivo, from multiple, polypeptide subunits (~ 20 kDa), each folded into 4-α-helix bundles. Ferritin protein cages are hollow and studded with multiple, functional protein subdomains such as Fe2+ ion channels penetrating the protein cage around the C3 axes, Fe2+/O oxidoreductase sites in the centers of ferritin protein cage subunits, and in 24 subunit ferritins, nucleation channels between the catalytic centers and the mineral growth cavity [1, 4]. The products of ferritin catalysis, multinuclear, [Fe3+O]x mineral precursors, move through the protein cage from the multiple, catalytic sites to the central, mineral growth cavity [5]. Ferritin protein cages with 24 subunits display remarkable symmetry at the interfaces of subunit dimers, trimers, tetramers, (Figure 1).
Figure 1. The ferritin protein cage viewed from different cage symmetry axes.
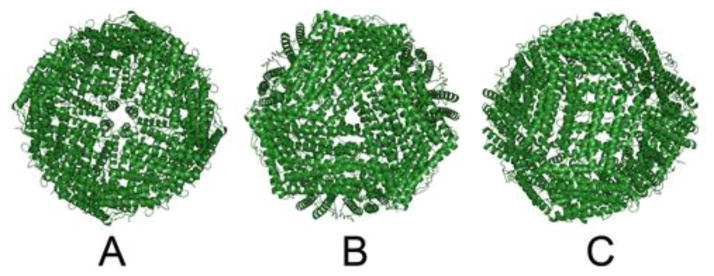
A. The fourfold (C4), symmetry axes and tetrameric subunit interface. B. The three fold (C3) symmetry axes and trimeric subunit interfaces that form ion channels for Fe2+ entry and exit. C. The twofold (C2) symmetry axes and dimeric interfaces which, when crosslinked naturally or synthetically, alter function [12, 13]. Drawn using Pymol software and PDB file 1MFR.
Relationships between ferritin protein cage dimer interfaces and Fe2+ oxidation and mineral dissolution were observed in earlier studies [6, 7], though they remain incompletely explored. The role of ferritin 3-fold protein cage interfaces around the ion channels, where Fe2+ enters and exits from the protein cage has been studied more extensively as exemplified in reference [5, 8].
Focus on the function of structural domains at the 4-fold symmetry axes of ferritin cages (Figures 1,2), which is characteristic of the larger (24 subunit) ferritin nanocages, developed more recently from the conversations and experiments among us, Elizabeth Theil, Paola Turano and Ivano Bertini. Bioinorganic activities, both scientific (International Conference on Biological Inorganic Chemistry, ICBIC Meetings, Metals in Biology Gordon Research Conferences) and professional activities SBIC (Society of Biological Inorganic Chemistry) gave Liz, Ivano and Paola the opportunities to develop a collaboration using solution kinetics, NMR spectroscopy (solution and solid state) and protein crystallography to solve several puzzles about ferritin iron traffic through the protein cage [5, 8]. One result was the development of a novel combination of methods to study such a large protein [9, 10]. An unexpected outcome, however, was the observation of ferric oxo products accumulating in channels between the active sites and the cavity of the ferritin cage; such results suggested the possibility that postcatalytic [Fe3+O]x products emerged from the nucleation channels and protein cage around the 4-fold symmetry axes formed by tetramers of ferritin subunit helix 5 in 24 subunit ferritin cages. Thus the 4-fold cage axes may have a functional role in 24 subunit ferritins [5].
Figure 2. Modified sites at C2, C3 and C4 symmetry axes in ferritin protein cages.
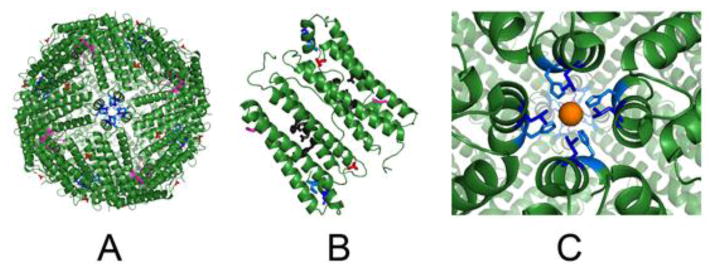
Drawn from XRD data in PDB file 3RDG, using Pymol. Ferritin protein-metal complexes were obtained by soaking apoferritin crystals in Fe2+ solutions. A. Ferritin protein cage viewed from the C4 axes (outside view); residue substitutions are shown in the 24 subunit cage. B. A pair of subunits in a 24 subunit ferritin protein cage (the subunit dimer interface is in the center). Active site residues are black sticks. C. Close up of C4 axes, in ferritin which form from the short, fifth helices, which are outside the 4-α-helix bundles and have axes almost ⊥ to the bundle axes; hydrated iron (rust) is weakly coordinating to Nε2 of H169 and in close proximity to L165. C2 loops/cage surface, E88 - red; C3 ion channels, E130 - magenta, C4 tetrameric subunit junctions of 4-helix bundles with the fifth helix, L165 - blue, H169 - blue marine.1
The striking symmetry at the junction of four ferritin, polypeptide subunits, around the ferritin cage 4-fold symmetry axes (Figure 1A; Figure 2A, C), was first observed in a protein crystal structure of horse spleen ferritin protein cages, thirty-five years ago [11]. But it was only three years ago, using novel NMR methodologies we developed specifically to study large molecules like ferritin, that we observed Fe3+O multimers emerging from the protein cage and destined for the mineral growth cavity [5, 12]. Such observations indicate that ferritin protein-based catalysis is linked by the nucleation channels to protein-controlled mineral nucleation and mineral growth [5, 13]. Here, we compare function in solution at the 4-fold ferritin cages by modifying a residue that binds metals inside the C4 channel in protein crystals, H169, and nearby, hydrophobic residue L165. As controls, we made amino acid substitutions with hydrophobic residues E88 on the cage surface, near 2-fold cage symmetry axes that are known to be sensitive to modification [6,14], and E130 in the ion channels around the 3-fold cage symmetry axes, known to regulate Fe2+ entry and exit [1] (Figure 1A, Figure 2). The 4-fold cages axes of ferritin have only been studied with intrusive deletion of the entire helix 5 [15], and are characteristic of 24 subunit ferritins. The results affecting the C2 axes surface and C3 axes of ferritin protein cage axes (E88A and E130I) confirm earlier studies and complement the new results that show a functional role of ferritin C4 axes in ferritin mineral dissolution.
Materials and Methods
Mutagenesis
Site directed amino acid substitutions in frog-M, ferritin protein cages were generated by PCR, with expression plasmid pET-3a frog-M DNA as the template, using the Quik Change II® site-directed mutagenesis kit (Stratagene). The DNA in the coding regions in all the protein expression vectors was analyzed for sequence confirmation (Primm srl, Milan, Italy).
Protein Expression
pET-3a constructs encoding wild-type frog-M ferritin and mutants were transformed into Escherichia coli BL21(DE3) pLysS cells and subsequently cultured in LB medium containing ampicillin (0.1 mg/ml) and chloramphenicol (34 μg/ml). Cells were grown at37 °C, until A600 nm reached 0.6–0.8 and subsequently induced with isopropyl 1-thio-β-D-galactopyranoside (1 mM final concentration) for 4 h, before being harvested; recombinant ferritins were purified as described previously [5, 8]. Briefly, cells were broken by sonication, and the cell free extract obtained after centrifugation (40 min, 40000 rpm, 4 °C) was incubated for 15 min at 65 °C as first purification step. After removal of the aggregated proteins (15 min, 40000 rpm, 4 °C), ferritin was dialyzed against Tris Cl 20 mM pH 7.5. Next, the sample was loaded onto a Q-Sepharose column and eluted with a linear NaCl gradient of 0–1 M in Tris 20 mM, pH 7.5. Fractions containing ferritin were identified by SDS-PAGE, combined and further purified by size exclusion chromatography using a Superdex 200 16/60 column. All variants had WT elution patterns.
The solid, ferritin [Fe2O3•H2O]x biominerals remain soluble as long as the protein cage remains in its native state. Damaged ferritin protein cages occur inside living cells that have sustained abnormal oxidative damage or incorporated abnormally large amounts of iron, e.g. 3–4000 Fe/cage; they are called hemosiderin, which is defined as damaged ferritin [13]. In this study to assure highly active ferritin protein cages, as before [16,17], we used freshly prepared solutions of ferritin protein and kept the amount of iron entering each cage relatively low (480 Fe2+).
Fe2+/O2 Catalysis and Fe3+O Mineralization
Single turnover catalysis (48 Fe2+/ferritin cage, 2 Fe2+/subunit), in frog-M ferritin, wild type or with amino acid substitutions, was monitored as the change in A650 nm (diferric peroxo or DFP) or A350 nm (Fe3+O), after rapid mixing (<10msec) equal volumes of 100 μM protein subunits (4.16 μM protein cages) in 200 mM MOPS, 200 mM NaCl, pH 7.0, with a freshly prepared solutions of 200 μM ferrous sulfate in 1 mM HCl, in a UV/visible, stopped-flow spectrophotometer (SX.18MV stopped-flow reaction analyzer, Applied Photophysics, UK). Routinely, 4000 data points were collected during the first 10 seconds. Initial rates of DFP and [Fe3+O] species formation were determined from the linear fitting of the initial phases of 650 nm and 350 nm trace (0.01–0.03 s). The kinetics of biomineral formation were followed, after addition of 480 Fe2+/cage (20 Fe2+/subunit), as the change of A350 nm (Fe3+O), using rapid mixing (<10msec) of 50 μM protein subunits (2.08 μM protein cages) in 200 mM MOPS, 200 mM NaCl, pH 7.0, with an equal volume of freshly prepared 1 mM ferrous sulfate in 1 mM HCl; the same UV/visible, stopped-flow spectrophotometer was used and 4000 data points were routinely collected during 1000 seconds.
Fe3+O Mineral Dissolution/Chelation
Recombinant ferritin protein cages were mineralized with ferrous sulfate (480 Fe2+/cage), in 100 mM MOPS, 100 mM NaCl, pH 7.0. After mixing, the solutions were incubated for 2 hours at room temperature and then overnight at 4 °C to complete the iron mineralization reaction. Fe2+ exit from caged ferritin minerals was initiated by reducing the ferritin mineral with added NADH (2.5 mM) and FMN (2.5 mM) and trapping the reduced and dissolved Fe2+ as the [Fe(2, 2′-bipyridyl)3]2+ complex, outside the protein cage. Fe2+ release from the protein cage was measured as the absorbance of [Fe(2, 2′-bipyridyl)3]2+ at the max of A522 nm. The experiments were performed at two different iron and protein concentrations: 2.08 μM cage and 1.0 mM iron, and 1.04 μM cages and 0.5 mM iron, respectively, with similar results. Initial rates (μmol liter−1 s−1) were calculated using the molar extinction coefficient for [Fe(2, 2′-bipyridyl)3]2+, 8430 M−1 cm−1, from the slope of the linear plot (R2 = 0.98–0.99) of the data related to the first minute of the linear phase.
Statistical analysis
The data were analyzed by Student’s t test and P < 0.05 was considered significant.
Results
Ferritin protein cage C3 (ion channel) and C4 cage axes have different roles in ferritin catalysis and mineral formation
The overall function of ferritin protein is the reversible formation of protein-caged biomineral, Fe2O3•H2O. We used newly prepared solutions of ferritin protein and freshly prepared Fe2+ solutions to compare initial rates of catalysis in a series of variant ferritin protein cages; the amount of Fe2+ substrate added was sufficient for the formation of relatively small iron biominerals (480 Fe2+/cage) and minimized damage from radical chemistry side reactions [18]. Frog ferritin M cages were used as models because of the extensive characterization of Fe2+/O2 reactions in the wild type and variant protein by UV-vis, NMR, VTVH-MCD, EXAFS spectroscopies and XRD of protein crystals [1, 8, 17, 19, 20].
Effects of modifying the 2-, 3-, and 4-fold ferritin protein cages axes were explored among four different ferritin variants, using both functional and structural metal-protein interactions as a guide. We selected for study of ferritin symmetry axes: 1. E130I in the Fe2+ entry channels (3-fold cage axes). 2. E88A on the cage surface at the subunit dimer interfaces (2-fold cage axes); 3. L165I and H169F around the 4-fold symmetry axes; ferritin protein crystal structures show metal ions bound in ion channels and at the 4-fold axes at H169 (Fig 2C), as exemplified in [8, 16]. For comparisons with the E88A substitution, glutamate 130 was substituted by an isoleucine residue because we wanted to analyze the effect of replacing the C3, ion channel carboxylate with a bulky, neutral amino acid. In wild type (WT) and variant ferritins rates of formation of the specific catalytic intermediate (diferric peroxo, A650 nm), detectable in eukaryotic ferritins, were compared. The mixed [Fe3+O]x species (A350 nm), which include contributions from the diferric peroxo intermediate, the decay product(s), mineral nuclei and the mineral itself, were also compared. Solution measurements to monitor [Fe3+O]x species, both bound to ferritin protein cages and in the solid, caged ferritin biomineral, as used here, probe natural properties of ferritin protein cages.
Ferritin E130I at the ferritin protein cage 3-fold channels provided a control for ferritin 4-fold channels, since we already knew that modifying E130 by Ala prevented any catalytic activity measured as DFP [17] or the less specific A350 nm, in both frog and human ferritin [17, 18]. E130I inhibited ferritin catalysis (DFP formation) as did ferritins E127A and D122R [17, 19] but contrasted with E130A, in which DFP formation was completely absent [17]. Fe2+ oxidation activity in ferritin E130I was 8% compared that of WT, measured at the specific DFP intermediate, and 5% measuring the less specific [Fe3+O]x at A350 nm (Figure 3, Table 1).
Figure 3. Selectivity for ferritin catalysis of residues at the Fe2+ entry sites (3-fold channels) and subunit dimer interfaces.

Amino acid substitution at the 4-fold ferritin cage axes had no effect on ferritin protein cage catalysis (A650 nm transient diferric peroxo enzymatic intermediate), contrasting with 2-fold (E88A) and 3-fold cage axes (E130I) axes. The absorbance maximum of A. DFP (A650 nm) and B. (Fe3+O)x species (A350 nm) were measured with rapid mixing, UV-vis spectroscopy and shown here for a set of representative curves (1 out of 3 independent analyses for each mutant). The rates (Table 1, data from 3 independent analyses) were calculated as described in the Methods. WT-wild type ferritin protein cages; E130I, ion entry channels, at 3-fold axes, E88A cage surface, near 2-fold axes, and L165I and H169F, on 4 – fold cage axes. *Significantly different than WT: P < 0.05.
Table 1.
Control of the catalytic reaction
| Protein | Change location (cage axis) | Initial rate of DFP formation (ΔA650nm/s) | Initial rate of Fe3+O formation (ΔA350nm/s) |
|---|---|---|---|
| WT | none | 1.33 ± 0.04 | 2.13 ± 0.07 |
| E88A* | surface charge, 2-fold | 1.05 ± 0.07 | 1.70 ± 0.26 |
| E130I* | 3-fold | 0.11 ± 0.001 | 0.11 ± 0.02 |
| L165I | 4-fold | 1.51 ± 0.08 | 2.25 ± 0.05 |
| H169F | 4-fold | 1.44 ± 0.17 | 2.06 ± 0.05 |
Residue 88 is on the ferritin cage surface at the center of the two fold symmetry axes in the protein cages. It is in the long loop that connects helices 2 and 3 of ferritin subunit 4-α-helix bundles (Figure 2B). Usually residue 88 is a negatively charged E or D amino acid. In eukaryotic ferritins residue 88, on the cage surface near the subunit dimer axes, will contribute to the general electrostatics of the protein surface. The catalytic activity of ferritin E88A was relatively robust, but significantly (P<0.05) lower (20%) than that of WT ferritin (Figure 3, Table 1), which complements the older data showing that making the dimer interface rigid alters the iron content of ferritin [6, 14].
The ferritin 4-fold cage symmetry axes of 24 subunit ferritins are created by helix 5 segments, contributed by four subunits (Fig 1A, 2C). In contrast to the C3 ferritin protein cage axes where the Fe2+ ion channels are located, the function of the C4 ferritin protein cage axes has been little studied. Helix 5 is short and distant from the catalytic di-iron sites, which are buried in the center of each subunit (Figure 2B). Rather than a simple extension of the 4-α-helix bundle in each subunit helix 5 is at an angle of about 60° from the axis of each subunit bundle. To probe a functional role of residues in helix 5, ferritin variants H169F, and L165I were created; H169 and L165 side chains point into the interior of the cage, at the C4 cage axes; in some ferritin protein crystal structures, metal ions bind in the C4 cage axes at H169 [5, 16]. Ferritin catalytic activity and mineral precursor formation were unaffected by the L165I and H169F amino acid substitutions (Figure 3, Table 1 and Figure 4).
Figure 4. Changing ferritin C4 channel variants L165I and H169F had no effect on biomineral formation (480 Fe2+/cage).
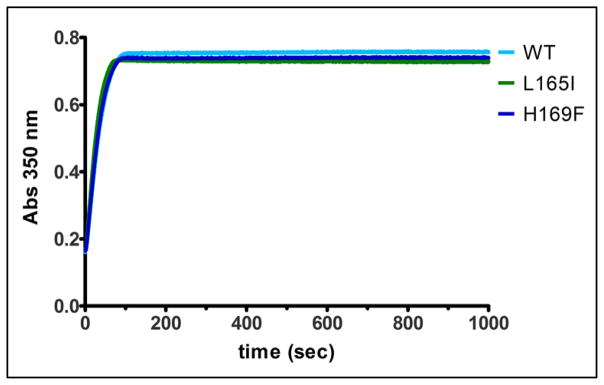
Amino acid substitution at the 4-fold ferritin cage axes had no effect on biomineral formation (A350 nm, (Fe3+O)x species). The absorbance maximum for L165I, H169F and WT was measured with rapid mixing, UV-vis spectroscopy and shown here for a set of representative curves (1 out of 2 independent analyses for each mutant).
Protein cage effects on ferritin mineral dissolution/chelation
The dissolution of the ferritin minerals is, like the initiation of ferritin mineral formation, dependent on properties of the ferritin protein cage and is controlled by the folded state of ferritin protein the ion channel cage entrances and exits around the 3-fold cage axes) [11]. Changes in ion channel (C3 axes) residues D122R and L134P, studied earlier, caused large increases in ferritin mineral dissolution, initiated by adding the reductant species[20, 21]. Insertion of small residues, such as in ferritin E130A, have little effect [17]. Because ferritin ion channels contain segments of three subunits, at each C3 ions channel there are three glutamates replaced by three alanines in ferritin E130A [17] or by three isoleucine residues in E130I, studied here. Insertion of the three bulky, hydrophobic isoleucine residues into the ferritin ion channels around the C3 axes, similarly to alanine insertion E130A [17], reduced the amount of dissolved caged mineral significantly (18% at 50 min, P<0.05) (Figure 5, Table 2).
Figure 5. Altered ferritin protein cages change dissolution/chelation of iron from the caged, ferritin biomineral.
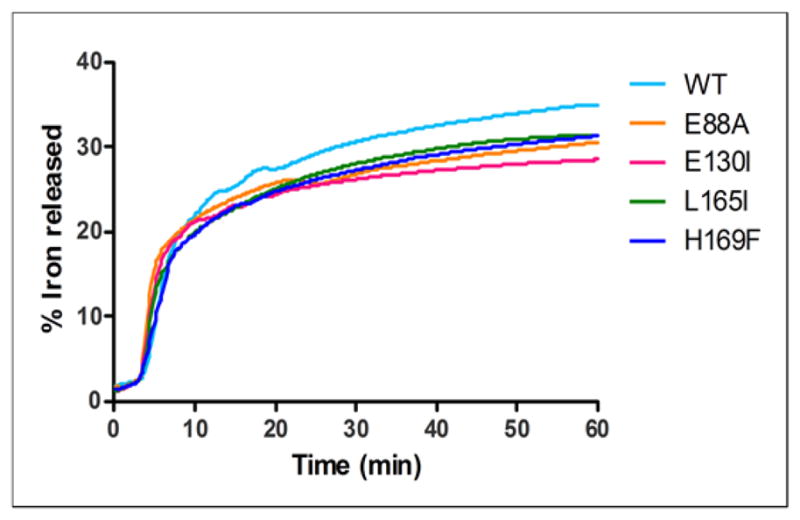
Amino acid substitution at the 4-fold cage axes (nucleation channel exits), L165I or H169F slows ferritin mineral dissolution/chelation, without effects on Fe2+ entry, contrasting with substitutions at the 3-fold axes (ion entry/exit channels), E130I, and on the surface near 2-fold axes subunit dimer interfaces (E88A), which alter both Fe2+ entry and mineral dissolution. Iron was recovered from caged minerals of hydrated ferric oxide, synthesized by and inside of ferritin protein cages, by adding reductant (NADH and FMN) in the presence of a chelator, bipyridyl, and measuring the rate for formation of [Fe (bipyridyl)3]2+ outside the protein cage as described in the Methods [6]. One representative curve for each mutant out of 5 independent experiments is reported in the figure; initial rates and % of Fe2+ released at 50 min calculated from 5 independent experiments are reported in Table 2. * Significantly different than WT: P<0.05.
Table 2.
Control of Ferritin Mineral Dissolution
| Protein | Change location (cage axis) | Initial rate of Fe dissolution as Fe(II)-bipyridyl (ΔμM/s) | % Fe2+ released in 50 min |
|---|---|---|---|
| WT | none | 0.74 ± 0.13 | 31 ± 1.6 |
| E88A | surface charge, 2-fold | 0.82 ± 0.14 | 27.2 ± 1.3* |
| E130I | 3-fold | 0.75 ± 0.09 | 26.1 ± 12* |
| L165I | 4-fold | 0.70 ± 0.09 | 28.6 ± 1.5* |
| H169F | 4-fold | 0.62 ± 0.09* | 27.8 ± 1.9* |
A role for the C4 axes in ferritin mineral dissolution was demonstrated, for the first time in ferritin H165F and L165I (Figure 5). Here mineral dissolution was inhibited (Figure 5) with no effect on mineral formation (Figure 4). The initial rate of ferritin mineral dissolution in H169F, a nonconservative substitution, was significantly slower than WT (~ 16%), P< 0.05. Significant (P<0.05) changes were also seen in the amount of dissolved caged mineral at 50 min for both the nonconservative, C4 substitution of ferritin H169F and the conservative C4 substitution of ferritin L165I (Figure 5).
Changes in the C3 ion channel ferritin, E130I, and the surface, ferritin E88A (Figure 5, Table 2), also displayed significant decreases in ferritin mineral dissolution. In an earlier study, a change in ferritin mineral dissolution was also caused by an amino acid substitution, L154G, near the C4 cage axes, but in the “unstructured” loop that connects helix 5, to helix 4 in the subunit bundle [22]. This result emphasizes the sensitivity of mineral dissolution to protein structure at and near the C4 cage axes. However, mineral dissolution rates in ferritin L154G were faster than in H169F or L165I.
Discussion
Ferritin protein cages are self-assembling, highly symmetrical, multi-subunit proteins, mainly cytoplasmic, in almost all cells, ranging from single-celled, anaerobic Archaea to multicellular, air-breathing humans and green plants; in addition, a distinct, catalytically active (H), ferritin is synthesized in the cytoplasm with targeting sequences that transport the protein to into mitochondria. The concentrations of ferritin inside cells reflect environmental iron availability and, in differentiated cells, the specialized, metabolic program. Ferritin protein cages have multiple reaction sites and subdomain structures specific, for processing iron at various stages in the reversible formation of the Fe2O3•H2O caged mineral, which are arrayed within the multi-subunit protein cage. The caged, ferritin, iron mineral is a source of concentrated, metabolic iron. During the process of ferritin iron mineralization, Fe2+ and O2 or in some case H2O2, are converted into the caged mineral by the ferritin protein cage itself. The consumption of Fenton chemistry species by ferritin protein cages during protein-caged, iron mineral synthesis confers an additional, antioxidant role on ferritins. Genetic regulation of ferritin protein synthesis by oxidants that target ARE-DNA promoters, emphasize the antioxidant function of ferritins [3, 13].
One of the difficulties in assessing functional metal-ferritin interactions from metal binding studies in ferritin protein co-crystals, soaked ferritin protein crystals, solution titrations of added iron (NMR), Fe2+ in ferritin frozen glasses (MCD-CD), etc, is the number and types of metal–protein interactions. Such interactions include: (1) Fe2+ transport and movement through cage ion channels, (2) Fe2+ as a substrate in protein catalysis, (3) Fe2+ dissolving from the caged mineral after mineral reduction and rehydration, and (4) exiting the cage, Fe3+-O-Fe3+ as an intra-cage catalytic product, (4) [Fe3+O]x as a mineral precursor, and (5) Fe2O3•H2O in a caged iron mineral. The multiple sites of Fe-protein interactions in ferritins contrast sharply with the single Fe-protein interaction site in many well-defined iron protein cofactors, exemplified by di-iron ribonucleotide reductase or methane monooxygenase. If the complexity and sophistication of ferritin protein cage functions are minimized, misinterpretation of earlier data can occur resulting in oversimplification [23], and possible confusion.
Structural and functional identification of activity sites in ferritin protein cages has depended on integrating the results of protein crystallography, protein engineering, and measurements of events related to ferritin biomineral formation and dissolution (“Fe2+ exit”) [13], [24–26]. Among the functions of ferritin protein cages are: 1. Fe2+ transport through ferritin protein cage ion channels (residues 110–134); 2. Fe2+ to di-iron sites, through the protein cage to di-iron, Fe2+/O2 redox enzymatic sites (residues E23, E58, H61, E103, Q137, D/A/S140); and 3. [Ferric- oxo]x nucleation and transport, through the protein cage to the internal mineral growth cavity. Residues for [Fe3+O]X mineral nucleation are incompletely identified, but include A26, V42 and T149 [5, 17]. When ferritin Fe2+ transport or catalysis is inhibited at single sites, caged biominerals form inside ferritin protein cages, albeit very, very slowly (~1/1000th the normal rate). Control of mineralization rates, and possibly biomineral order and dissolution rates are associated with protein-dependent Fe2+ interactions in ferritin protein cages (ion channels, catalytic centers, and through nucleation channels to the ferritin protein mineral growth cavity). After growth and dissolution of the caged ferritin mineral by electrons (provided, in solution at least, by external electron donors such as NADH/FMN) dissolved Fe2+ passes to and through the same channels used for Fe2+ entry (residues 110–134) and out of the protein cage to the cytoplasm in vivo or solution in vitro), based on effects of disrupting ion channel structure [16, 21]. By monitoring the specific catalytic intermediate in mineralization, diferric peroxo, and the mineral dissolution rates (Fe2+ bound to bipyridyl outside the protein cage [21]), we confirm a relationship between conserved amino acids on the 3, and 2 fold symmetry axes and observe a novel functions of residues at the C4 axes of ferritin protein cages. Here we observed that conserved amino acids the C4 axes of ferritin protein cages also contribute to dissolution of the caged iron mineral.
Structure at the two-fold cage symmetry axes, the subunit dimer interfaces of ferritin protein cages, influence ferritin cage assembly [14] and change both iron oxidation and mineral dissolution rates (short biological or chemical crosslinks) [6, 7]. The effect on ferritin function of localized charges at cage surface and dimer interface is illustrated here, by a new ferritin variant, E88A. The inhibition in 2Fe2+/O2 catalysis and the enhancement of mineral dissolution in E88A, with the localized changes in electrostatics at the dimer interface, may be related to the unidentified paths of proton shuttling out of the protein cage. While proton exit is required for, and observed experimentally in solution during iron mineral formation, the proton escape paths are little studied. Moreover, H2O coordinated to each Fe2+ substrate at the di-iron sites [27], and released when hydrated Fe2+ ions from [Fe3+O]x multimers also moves along currently undefined pathways, although ordered water is observed in all, high resolution ferritin protein crystal structures. Formation, and aging (dehydration) of the caged ferritin mineral, involves hydrolysis and proton release. E88, on the surface might be the terminus of currently unidentified, proton exit path in ferritin protein cages.
The three-fold symmetry axes of ferritin cages, coincide with the subunit triple junctions that form the Fe2+ entry/exit channels; they are more extensively characterized subdomains in ferritin protein cages than the others. The ferritin Fe2+ channels, members of the large ion channel family that includes membrane ion channels, are 15 Ǻ long, with a 5 Ǻ constriction in the middle and pores 7–9 Å in diameter at each end; the pores are on the external and internal surfaces of the protein cage [16]. A number of divalent cations, including Mg, Fe, Co, Cu, and Zn, have been observed bound to ion channel residues, in either cocrystallization or crystal soaking experiments [8, 16, 28]. The external gate/pore structure of ferritin ion channels is stabilized by a network of hydrogen bonds and ionic bonds among conserved amino acids between the subunit N-terminal extension (“gate”) and the ion channel walls [19]. Several synthetic peptides as well as low concentrations of urea (1 mM) have functional effects on ferritin pore gating [29, 30], but the biological partners that regulate ferritin ion channel gating remain unidentified. Conserved carboxylate residues that alter ferritin pore gating include D122, E130 and D127, studied as D122R, E130A and E127A [17, 19]. In this study E130I contrasts with another E130 variant, E130A, because E301I has a large side chain (Figure 3, Table 1) and retains some catalytic activity [17, 18]. Ferritin minerals form in E130A, albeit very slowly [17]. When rates of dissolving/chelation of ferritin minerals in E130A and E130I are compared to WT, Fe2+ exit is similar to WT (Figure 5, Table 2) [17], which illustrates that the electrostatic and size properties of ferritin ion channels associated with the constriction around E130, selectively control Fe2+ entry and distribution to the multiple catalytic centers (three-ion-channel). However, when ferritin ion channel structure is extensively disrupted, both Fe2+ entry and exit are altered [19, 20].
Structure at the C4 ferritin cage axes, which are also the junctions of four subunits, are specific to 24 subunit ferritins, are associated with resistance to protein denaturation and retention of iron inside the protein cage [15], and also influences mineral dissolution (Figure 5, Table 2). H169 in the short, fifth helix that forms the four- fold symmetry axes in 24 subunit ferritin protein cages (Figure 2C), binds a variety of metal atoms in protein crystals [8, 16, 28] and L165 provides hydrophobic interactions in the in 4-fold axes. However, neither conservative amino acid substitution, L165I nor nonconservative substitution H169F had any effect on ferritin catalysis and mineral formation (Figure 3, Table 1). Inserting large hydrophobic residues (L165I and H169F) into the C4 axis structure of ferritin protein cages, created by subunit helix 5 contributed by four ferritin subunits, inhibited ferritin mineral dissolution (Figure 5, Table 2) Inserting a smaller residue (L154G) near L165 or H169, in the loop between helix 4 and helix 5, enhances Fe2+ release [22]. Such observations emphasize the influence of ferritin protein structure at the C4 cage axes on the complex process of ferritin mineral dissolution.
Many mechanistic features of ferritin nanomineral dissolution, such as how external reductants and protons reach the protein-caged nanomineral surfaces and how dissolved Fe2+ ions reach the ion channels at the C3 protein cage axes, remain unknown, even though they were posited as much as thirty –five years ago [31]. Answers will require a number of targeted, nanochemical studies in the future. However, the current studies reveal the significant role of ferritin protein cage structure around the 4 four-fold symmetry axes in modulating dissolution of the caged mineral. Not only are such observations important for understanding the well known roles of ferritin in basic iron cell biology, but they can be exploited in nanotechnology [21] to control the turnover of synthetic materials made inside ferritin protein cages. While the structural symmetry around the four-fold axes of ferritin 24 subunit protein cages is visually stunning, and is often at the center of graphic illustrations of the ferritin protein cage, it is now clear that the four-fold symmetry axes in 24 subunit ferritin protein cages are more than “just a pretty face”. The conservation of C4 axis structure in ferritin protein cages not only relates to Fe2O3•H2O dissolution, but may also reflect binding sites for biological reductants or intracellular protein chaperones. Understanding macromolecular interactions between ferritin and other cellular molecules is key to iron biology and to using ferritin cages in medicinal chemistry as drug delivery vessels and in nanotechnology for syntheses of novel nanomaterials [32].
Acknowledgments
This work was supported by the CHORI Partners, the National Institutes of Health Grant DK20251 (ECT) and MIUR PRIN 2009 “Biologia strutturale meccanicistica: avanzamenti metodologici e biologici” (PT).
Footnotes
The residue numbering scheme was developed for horse spleen ferritin, the first ferritin protein for which the protein crystal structure and natural (not predicted) amino acid sequence was obtained.
All authors contributed to the work; the majority of the experimental work was contributed by CB; manuscript writing was mainly by ECT and PT; all authors have read the submitted manuscript.
References
- 1.Theil EC, Behera RK, Tosha T. Coordination chemistry reviews. 2013;257:579–586. doi: 10.1016/j.ccr.2012.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hintze KJ, Theil EC. Proc Natl Acad Sci USA. 2005;102:15048–15052. doi: 10.1073/pnas.0505148102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Goss DJ, Theil EC. Acc Chem Res. 2011;44:1320–1328. doi: 10.1021/ar2001149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Le Brun NE, Crow A, Murphy ME, Mauk AG, Moore GR. Biochimica et biophysica acta. 2010;1800:732–744. doi: 10.1016/j.bbagen.2010.04.002. [DOI] [PubMed] [Google Scholar]
- 5.Turano P, Lalli D, Felli IC, Theil EC, Bertini I. Proc Natl Acad Sci USA. 2010;107:545–550. doi: 10.1073/pnas.0908082106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mertz JR, Theil EC. J Biol Chem. 1983;258:11719–11726. [PubMed] [Google Scholar]
- 7.McKenzie RA, Yablonski MJ, Gillespie GY, Theil EC. Arch Biochem Biophys. 1989;272:88–96. doi: 10.1016/0003-9861(89)90198-7. [DOI] [PubMed] [Google Scholar]
- 8.Bertini I, Lalli D, Mangani S, Pozzi C, Rosa C, Theil EC, Turano P. J Am Chem Soc. 2012;134:6169–6176. doi: 10.1021/ja210084n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Matzapetakis M, Turano P, Theil EC, Bertini I. J Biomol NMR. 2007;38:237–242. doi: 10.1007/s10858-007-9163-9. [DOI] [PubMed] [Google Scholar]
- 10.Bermel W, Bertini I, Felli IC, Matzapetakis M, Pierattelli R, Theil EC, Turano P. J Mag Reson. 2007;188:301–310. doi: 10.1016/j.jmr.2007.07.004. [DOI] [PubMed] [Google Scholar]
- 11.Banyard SH, Stammers DK, Harrison PM. Nature. 1978;271:282–284. doi: 10.1038/271282a0. [DOI] [PubMed] [Google Scholar]
- 12.Lalli D, Turano P. Acc Chem Res. 2013 doi: 10.1021/ar4000983. [DOI] [PubMed] [Google Scholar]
- 13.Theil EC. Curr Opin Chem Biol. 2011;15:304–311. doi: 10.1016/j.cbpa.2011.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Huard DJ, Kane KM, Tezcan FA. Nature chemical biology. 2013;9:169–176. doi: 10.1038/nchembio.1163. [DOI] [PubMed] [Google Scholar]
- 15.Levi S, Luzzago A, Franceschinelli F, Santambrogio P, Cesareni G, Arosio P. Biochem J. 1989;264:381–388. doi: 10.1042/bj2640381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tosha T, Ng HL, Bhattasali O, Alber T, Theil EC. J Am Chem Soc. 2010;132:14562–14569. doi: 10.1021/ja105583d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Haldar S, Bevers LE, Tosha T, Theil EC. J Biol Chem. 2011;286:25620–25627. doi: 10.1074/jbc.M110.205278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yang X, Arosio P, Chasteen ND. Biophysical journal. 2000;78:2049–2059. doi: 10.1016/S0006-3495(00)76752-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tosha T, Behera RK, Theil EC. Inorg Chem. 2012;51:11406–11411. doi: 10.1021/ic3010135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tosha T, Behera RK, Ng HL, Bhattasali O, Alber T, Theil EC. J Biol Chem. 2012;287:13016–13025. doi: 10.1074/jbc.M111.332734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Takagi H, Shi D, Ha Y, Allewell NM, Theil EC. J Biol Chem. 1998;273:18685–18688. doi: 10.1074/jbc.273.30.18685. [DOI] [PubMed] [Google Scholar]
- 22.Haldar S, Tosha T, Theil EC. Indian Journal of Chemistry. 2011;50A:414–419. [Google Scholar]
- 23.Honarmand Ebrahimi K, Bill E, Hagedoorn PL, Hagen WR. Nature chemical biology. 2012;8:941–948. doi: 10.1038/nchembio.1071. [DOI] [PubMed] [Google Scholar]
- 24.Arosio P, Ingrassia R, Cavadini P. Biochimica et biophysica acta. 2009;1790:589–599. doi: 10.1016/j.bbagen.2008.09.004. [DOI] [PubMed] [Google Scholar]
- 25.Salgado EN, Radford RJ, Tezcan FA. Acc Chem Res. 2010;43:661–672. doi: 10.1021/ar900273t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Uchida M, Kang S, Reichhardt C, Harlen K, Douglas T. Biochimica et biophysica acta. 2010;1800:834–845. doi: 10.1016/j.bbagen.2009.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schwartz JK, Liu XS, Tosha T, Theil EC, Solomon EI. J Am Chem Soc. 2008;130:9441–9450. doi: 10.1021/ja801251q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Toussaint L, Bertrand L, Hue L, Crichton RR, Declercq JP. J Mol Biol. 2007;365:440–452. doi: 10.1016/j.jmb.2006.10.010. [DOI] [PubMed] [Google Scholar]
- 29.Liu X, Jin W, Theil EC. Proc Natl Acad Sci USA. 2003;100:3653–3658. doi: 10.1073/pnas.0636928100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu XS, Patterson LD, Miller MJ, Theil EC. J Biol Chem. 2007;282:31821–31825. doi: 10.1074/jbc.C700153200. [DOI] [PubMed] [Google Scholar]
- 31.Jones T, Spencer R, Walsh C. Biochemistry. 1978;17:4011–4017. doi: 10.1021/bi00612a021. [DOI] [PubMed] [Google Scholar]
- 32.Theil EC, Behera RK. The Chemistry of Nature’s Iron Biomineral in Ferritin Protein Nanocages. Wiley; 2013. [Google Scholar]