

R loops are three-stranded nucleic acid structures that comprise nascent RNA hybridized with the DNA template, leaving the nontemplate DNA single-stranded. These structures form naturally during transcription even though their persistent formation can have deleterious effects on genome integrity. Interestingly, an increasing number of studies also suggest that R loops function as potential gene expression regulators. Here, Skourti-Stathaki and Proudfoot review the most recent findings about R loops, highlighting their opposite roles in cellular fitness.
Keywords: R loops, gene expression, genome integrity
Abstract
R loops are three-stranded nucleic acid structures that comprise nascent RNA hybridized with the DNA template, leaving the nontemplate DNA single-stranded. R loops form naturally during transcription even though their persistent formation can be a risky outcome with deleterious effects on genome integrity. On the other hand, over the last few years, an increasingly strong case has been built for R loops as potential regulators of gene expression. Therefore, understanding their function and regulation under these opposite situations is essential to fully characterize the mechanisms that control genome integrity and gene expression. Here we review recent findings about these interesting structures that highlight their opposite roles in cellular fitness.
The R-loop structure was first characterized >38 years ago (Thomas et al. 1976), and the first demonstration that R loops exist in vivo came in 1995 with studies described by Crouch and colleagues (Drolet et al. 1995). They showed that R-loop formation occurs in a bacterial cell and is a consequence of the transcription process (Drolet et al. 1995). Since then and especially over the last decade, the R-loop field has become an increasingly expanded area of research, placing these structures as a potential regulator of gene expression but also as a major threat to genome stability.
Transcription-mediated R-loop formation
In general, where R loops have been described in vivo, the RNA strand is generated by RNA polymerase II (Pol II) transcribing a C-rich DNA template so that a G-rich transcript is generated. Interestingly, several studies have shown that R loops are formed preferentially when the nontemplate strand is G-rich (Reaban et al. 1994; Li and Manley 2005; Ginno et al. 2012, 2013). The increased thermodynamic stability of a G-rich RNA strand bound to C-rich DNA could be a reason for this sequence specificity (Sugimoto et al. 1995). However, it is as yet unclear how R loops are generated. According to the “extended RNA/DNA hybrid” model, the RNA/DNA hybrid duplex could be the result of an extension of the usual 8-base-pair (bp) RNA/DNA hybrid (Westover et al. 2004) within the transcription bubble as Pol II elongates. This model, however, is inconsistent with the crystallographic structure of Pol II that demonstrates the exit of DNA and RNA molecules through different channels (Westover et al. 2004), strongly arguing against it (Aguilera and Garcia-Muse 2012). A more plausible model suggests that the RNA/DNA hybrid could arise by threading back the RNA before the two strands of the DNA duplex reanneal (called the “thread back model”). According to extensive in vitro studies from the Lieber laboratory (Roy and Lieber 2009), R loops depend on three features: high G density, negative supercoiling, and DNA nicks (Roy et al. 2010). Initial R-loop formation is favored by G clusters and DNA nicks downstream from the promoter on the nontemplate DNA strand, whereas subsequent RNA/DNA hybrid extension and stabilization are enhanced by high G density and negative supercoiling (Aguilera and Garcia-Muse 2012).
Once formed, R loops are particularly stable, as RNA/DNA associations are thermodynamically more stable than DNA/DNA interactions (Roberts and Crothers 1992). This may be due to the structure of the RNA/DNA hybrid, which is thought to adopt a conformation that is an intermediate between the A form of a dsRNA and the B form of a DNA duplex (Shaw and Arya 2008). Another possibility is that a G quadruplex (G4) formed on the single-stranded exposed strand, such as in the case of the immunoglobulin (Ig) class switch region (Duquette et al. 2004), stabilizes the R-loop structure.
R loops have been reported in vivo at prokaryotic origins of replication (Masukata and Tomizawa 1984, 1990; Baker and Kornberg 1988; Lee and Clayton 1996; Carles-Kinch and Kreuzer 1997), the mitochondrial origin of replication (Xu and Clayton 1996), and the mammalian Ig class switch region in activated B lymphocytes (Yu et al. 2003). In the latter case, R-loop formation is involved in facilitating class switch recombination (CSR) that generates diverse antibody isotypes.
R loops are not restricted to Pol II transcripts. Very highly transcribed Pol I rDNA repeats also form R loops (El Hage et al. 2010). Although Pol II transcripts were not thought to be associated with R loops, recent genomic analysis of R loops in Saccharomyces cerevisiae showed the presence of R loops over Pol III transcribed tRNA genes in various mutant backgrounds (Chan et al. 2014). This implies that Pol III transcripts can also form R loops in normal cells. In this review, we focus only on R loops formed in Pol II transcripts.
R loops and genomic instability
Transcription can be a “risky” process. R loops can lead to DNA damage by the exposure of ssDNA formed as a result of the RNA/DNA hybrizidation. Being more unstable, the ssDNA would then be susceptible to lesions and transcription-associated mutagenesis (TAM) or transcription-associated recombination (TAR) (see Fig. 1). However, the mechanism leading from an R loop to genomic instability still remains largely unknown. Possibly, the unpaired DNA strand resulting from R-loop formation is more susceptible to DNA damage such as spontaneous deamination of dC to dU, leading to double-strand breaks (DSBs) and recombination (Aguilera 2002; Li and Manley 2006; Aguilera and Garcia-Muse 2012). Thus, in one possible model, accumulation of R loops could make certain regions of the genome more prone to DNA-damaging agents by increasing the occurrence of single-stranded regions. In a second potential model, a protein recognizing R-loop structures could be involved in initiating the generation of mutagenesis. One possible candidate is the activation-induced cytidine deaminase (AID). AID is an enzyme that promotes Ig heavy chain CSR and hypermutation in B lymphocytes. It functions by deaminating cytosines into uracils on single-strand target DNA sequences (Muramatsu et al. 2000; Revy et al. 2000). R loops forming behind elongating Pol II can provide the ssDNA substrate for this enzyme (Yu et al. 2003). The generated U:G mismatch may then be replicated, creating two daughter species, one of which will undergo a C → T transition mutation. However, dU could also be processed by base excision repair (BER) components such as uracil–DNA glycosylase and abasic endonuclease (APE). These enzymes remove the uracil, creating a DNA nick or leaving an abasic site (a site of base loss). Replication past the abasic site will result in random incorporation of any of the four nucleotides, possibly leading to further mutations. DNA nicks could also be converted to DNA DSBs that are recognized by the recombination-mediated repair machinery, ultimately leading to CSR for antibody genes (Di Noia and Neuberger 2002) or, more generally, DNA translocations. However, AID is specifically expressed in activated B cells and in chicken DT40 cells (which are B-cell-derived). This raises the question of how R-loop formation leads to genomic instability in cells that lack AID. DSBs have also been observed in HeLa cells upon depletion of SRSF1 (Li and Manley 2005), suggesting that other proteins could function analogously to AID in different cell types to initiate R-loop-induced genomic instability. Alternatively, spontaneous dC → dU mutation may occur at low levels.
Figure 1.

R loops as a source of DNA damage. Nascent transcripts behind elongating Pol II can invade the DNA duplex and hybridize with the DNA template strand. The RNA/DNA hybrid so formed displaces the nontemplate strand, and this three-stranded structure constitutes an R loop. R loops can cause genomic instability in different ways. First, the displaced ssDNA can act as a substrate to DNA-damaging agents, deaminases (AID), and repair enzymes (APE and BER), leading to DNA lesions and nicks. Second, G4 structures forming on the G-rich nontemplate strand can generate susceptible sites for nucleases. Finally, transcription elongation machinery impeded by stable R loops can cause replication–transcription collisions, leading to DNA recombination and DSBs. Points of contact between the DNA strand and nascent RNA indicate R-loop formation, whereas points of contact within the ssDNA indicate G4 structures. Pol II is shown as a blue icon, with an arrow indicating transcription direction. Nucleosomes are shown in green. The diagram is not drawn to scale.
An additional possible scenario suggests that transcriptional R loops induce genomic instability by interfering with DNA replication (Aguilera 2002; Gan et al. 2011; Houlard et al. 2011; Aguilera and Garcia-Muse 2012). Thus, replication fork collisions with blocked Pol II have been shown to induce TAR or DNA breaks in budding yeast and mammals (Prado and Aguilera 2005; Gottipati et al. 2008; Boubakri et al. 2010). Unrepaired DNA lesions formed on the ssDNA of the R loop or the RNA/DNA hybrids themselves forming behind elongating Pol II may somehow restrict transcription, which in turn may block replication forks. Such blocked replication forks could then generate DNA lesions and DSBs in the newly synthesized DNA. These would induce recombination-mediated repair, which in turn could lead to chromosome rearrangements and genomic instability (Aguilera and Garcia-Muse 2012). R loops are prevalent in meiosis, at least in S. cerevisiae and Caenorhabditis elegans, and their accumulation leads to replication impairment and genomic instability (Castellano-Pozo et al. 2012). In mammalian cells, replication–transcription collisions in long human genes (≥800 kb) have been shown to be associated with R-loop accumulation, which in turn causes instability at so-called common fragile sites (CFSs) (Helmrich et al. 2011). Instability at CFSs may also relate to slower or incomplete replication in areas of chromatin compaction (Debatisse et al. 2012).
Additional factors leading from a transient R-loop structure to deleterious genomic instability have only recently been identified. R-loop accumulation in S. cerevisiae, C. elegans, and human cells has been linked to histone 3 Ser10 phosphorylation (H3S10P), a mark of chromatin compaction (Castellano-Pozo et al. 2013). It is proposed that R loops trigger formation of the H3S10P mark, which in turn could cause replication fork stalling, transcription–replication collisions, and, ultimately, DSBs in the newly synthesized DNA (Castellano-Pozo et al. 2013).
Surveillance mechanisms: What protects us from R loops?
The deleterious effects of R loops formed during transcription on genome integrity have been variously documented (Aguilera 2002). Given that formation of R loops is an evolutionarily conserved mechanism (Li and Manley 2005; Aguilera and Garcia-Muse 2012), it is perhaps not surprising that different organisms have used diverse mechanisms to protect their genomes (Li and Manley 2006; Aguilera and Garcia-Muse 2012). So far, five different mechanisms are thought to regulate R-loop formation (see Fig. 2): (1) RNase H enzyme, which specifically degrades the RNA in RNA/DNA hybrid (for review, see Cerritelli and Crouch 2009); (2) RNA/DNA helicases such as the yeast Sen1 or homologous human Senataxin (Mischo et al. 2011; Skourti-Stathaki et al. 2011) and the human DHX9 helicase, which also acts on G4 structures (Chakraborty and Grosse 2011); (3) topoisomerases, which relax DNA-negative supercoiling that otherwise causes persistent R-loop formation (Drolet et al. 1994, 1995; Tuduri et al. 2009; El Hage et al. 2010; Yang et al. 2014); (4) proteins that prevent R-loop formation, such as mRNA biogenesis (Huertas and Aguilera 2003; Dominguez-Sanchez et al. 2011; Castellano-Pozo et al. 2012) and processing proteins (Li and Manley 2005; Paulsen et al. 2009; Wahba et al. 2011; Stirling et al. 2012; Santos-Pereira et al. 2013); and (5) suppressors of proteins that promote R-loop formation (i.e., Rad51 and AtNDX) (Sun et al. 2013; Wahba et al. 2013).
Figure 2.
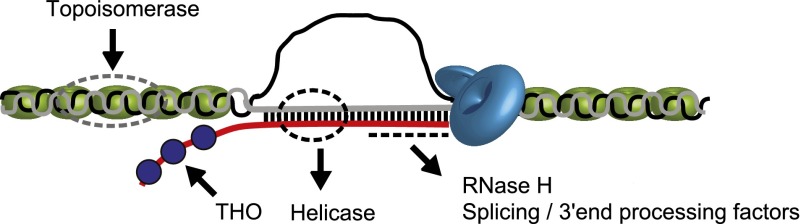
Diverse protection mechanisms against R-loop formation. Two types of surveillance factors have been identified: factors that prevent formation of R loops and factors that actively remove them. DNA topoisomerase enzymes suppress R-loop formation by relaxing the negative supercoiling behind elongating Pol II. The THO complex (blue circle) facilitates efficient packaging of nascent RNA into messenger ribonucleotide proteins (mRNPs), preventing R-loop formation. Splicing and 3′ end processing factors associate with nascent RNA and prevent R loops. RNA/DNA helicases and RNase H enzymes remove R loops once formed. DNA is shown as gray and black lines, and RNA is shown as a red line. Dotted lines indicate the site of action of different factors. The diagram is not drawn to scale.
Factors that prevent R-loop formation
In S. cerevisiae, transcription-induced R loops were first documented with the characterization of the THO complex, suggested to be involved in transcriptional elongation (Aguilera 2002). This complex consists of four nuclear proteins (Hrp1, Tho2, Mft1, and Thp2) and is associated with the transcription–export complex (TREX) containing Tex1 and the mRNA export factors Sub2 and Yra1 (Chavez et al. 2000; Strasser et al. 2002). The physical interaction of the THO complex with TREX directly links mRNA packaging with RNA export. Mutations affecting THO/TREX have been shown to affect transcriptional elongation, proper mRNA export, and recombination (Strasser et al. 2002; Rondon et al. 2003). A distinctive phenotype of yeast THO mutants (first identified in the HRP1 and THO2 genes) is their transcription-associated hyperrecombination phenotype (Aguilera and Klein 1990; Piruat and Aguilera 1998), and this is directly associated with R-loop formation (Huertas and Aguilera 2003).
The THO/TREX complex is responsible for packaging of pre-mRNA with RNA-binding proteins. Recently, another factor involved in mRNA export and processing, the yeast Npl3, also prevented R-loop induced transcription–replication collisions and genome instability (Santos-Pereira et al. 2013), suggesting a functional link between RNA metabolism and R-loop-associated genomic instability. As the nascent transcript emerges from elongating Pol II, it may have two immediate fates. It either is cotranscriptionally packaged into messenger ribonucleotide proteins (mRNPs) and exported through the nuclear pore or may invade the DNA duplex behind the elongating Pol II to form R-loop structures. R loops could then interfere with DNA replication, induce ssDNA breaks, or become recombination intermediates. Consequently, it could be argued that efficient mRNA packaging into mRNPs prevents R-loop formation and in turn restricts TAR and DNA damage (Huertas and Aguilera 2003; Moore and Proudfoot 2009; Mischo et al. 2011; Aguilera and Garcia-Muse 2012; Santos-Pereira et al. 2013). However, it has been reported that even with normal mRNP biogenesis, accumulation of R loops can still occur (Mischo et al. 2011), suggesting that R loops form at a much higher frequency than once thought.
DNA TOP1 is an evolutionarily conserved factor that suppresses R-loop formation (Drolet et al. 1995; Tuduri et al. 2009; El Hage et al. 2010). This is possibly due to the ability of TOP1 to relax negative DNA supercoiling. In the absence of TOP1, negative supercoils accumulate behind elongating Pol II to promote opening of DNA, which in turn facilitates annealing between the nascent RNA and DNA template strand with subsequent R-loop formation in bacteria (Drolet et al. 1995), S. cerevisiae (El Hage et al. 2010), and human cells (Tuduri et al. 2009). Very recently, TOP3B, another member of this subfamily, was found to reduce negative supercoiling and R-loop formation (Yang et al. 2014). The interesting point here is that TOP3B is recruited to human and mouse gene loci by recognizing arginine methylation histone marks through its interaction with the methyl-arginine effector tudor domain-containing protein 3 (TDRD3). According to the proposed model, the TDRD3–TOP3B complex is recruited to regions of active transcription; TDRD3 recognizes the methyl-arginine histone marks and the methylated C-terminal domain (CTD) of Pol II, whereas TOP3B resolves negative supercoiling that forms behind elongating Pol II and by doing so restricts R-loop formation (Yang et al. 2014). Interestingly, in this study, formation of R loops is controlled by the presence of a chromatin mark as opposed to the alternative scenario suggested by the Aguilera laboratory (Castellano-Pozo et al. 2013), where accumulated R loops cause formation of H3S10P. In the future, it will be intriguing to test under which conditions R loops regulate chromatin structure (and consequently genome dynamics). Indeed, are they a cause or consequence of the epigenetic microenvironment?
Genes of higher vertebrates are significantly longer, and the presence of introns is almost ubiquitous; therefore, they have adapted some additional mechanisms to protect their genomes. The SRSF1, a serine–arginine-rich (SR) protein that regulates the first steps of splicing, appears to interconnect pre-mRNA processing and genomic instability (Li and Manley 2005). Li and Manley (2005) demonstrated in chicken DT40 cells and human HeLa cells that depletion of SRSF1 causes the nascent transcript to form R loops, which in turn promote DNA rearrangements mediated by DSBs. In essence, the model proposed is that SRSF1 is cotranscriptionally loaded onto the nascent pre-mRNA via the phosphorylated CTD of Pol II to not only promote splicing but also prevent R-loop formation and subsequent genomic instability (Aguilera 2005; Li and Manley 2005). This is a clear example of the connections between transcription-induced R loops, pre-mRNA processing, the Pol II CTD, and transcription-associated genomic instability.
The cotranscriptional R-loop-induced genomic instability observed in DT40 cells with the loss of SRSF1 factor (Li and Manley 2005, 2006) resembles the phenotype of yeast THO mutants (Huertas and Aguilera 2003). Until recently, the involvement of the human THO/TREX complex in genomic instability was not clearly identified. A study from the Aguilera laboratory (Dominguez-Sanchez et al. 2011), however, revealed that the interplay between mRNP biogenesis and genomic instability is indeed conserved from yeast to humans. In essence, depletion of the human THO complex results in accumulated DNA breaks and consequent genomic instability, which is dependent on R-loop formation (Dominguez-Sanchez et al. 2011). In addition to this, genome-wide data suggest that RNA processing factors help prevent genomic instability (Paulsen et al. 2009), further pointing toward a powerful interplay between pre-mRNA processing and R-loop-dependent genomic instability.
Factors that remove R loops
As mentioned above, apart from the active prevention of R loops, cells also use a range of dedicated factors to actively remove them once formed. First of all, the RNase H enzymes act to cleave the RNA of RNA/DNA hybrids (Stein and Hausen 1969; Hausen and Stein 1970; Cerritelli and Crouch 2009). In most organisms, there are two types of RNase H. Eukaryotic RNase H1 consists of a single polypeptide, with the N-terminal domain being responsible for binding to the RNA/DNA hybrid (hybrid-binding domain or HBD), and the CTD containing the RNase H active site (Cerritelli and Crouch 2009). RNase H2 is composed of three different polypeptides, with the RNase 2A being the catalytic subunit. RNase H1 and RNase H2 endonucleolytically cleave the RNA within the RNA/DNA hybrid in a sequence-independent manner. However, they may have different in vivo substrates due to their differences in hybrid hydrolysis.
RNase H1 is present in the nucleus and mitochondria and is essential for mitochondrial replication (Cerritelli et al. 2003). Overexpression of nuclear RNase H1 (Cerritelli et al. 2003) has been widely used to experimentally remove R loops and so far is perhaps the only well-studied and efficient way to diminish the cellular levels of R loops. RNase H2 is not as well defined due to its multisubunit composition and low abundance. It is believed to be mostly a repair enzyme due to its substrate specificity. Unlike RNase H1, RNase H2 can recognize and cleave a single ribonucleotide inserted in a DNA duplex, and therefore it was suggested that RNase H2 removes ribonucleotides misincorporated into DNA (Eder et al. 1993; for review, see Cerritelli and Crouch 2009). RNase H2 is also responsible for removing the Okazaki fragment RNA primers from the newly synthesized lagging strand during DNA replication (Murante et al. 1998; for review, see Cerritelli and Crouch 2009). Recently, another very interesting function of RNase H2 was shown: It can uniquely process R loops that are generated during DNA replication/repair (Chon et al. 2013). It was also suggested in this same study that RNase H1 and RNase H2 have some overlapping specificities in R-loop resolution; however, RNase H1 is mainly responsible for the resolution of transcription-associated R loops (Chon et al. 2013).
Second, the yeast Sen1, a superfamily I RNA/DNA helicase (Kim et al. 1999), acts to remove R loops and prevent genomic instability by R-loop-mediated DNA damage (Mischo et al. 2011). R loops accumulate in a sen1-1 strain that carries a mutation in the helicase domain of Sen1. Furthermore, SEN1 genetically interacts with genes involved in homologous recombination (HR). Sen1 is also a termination factor for coding and noncoding genes (Ursic et al. 1997; Steinmetz et al. 2006; Kawauchi et al. 2008). Sen1, Nrd1, and Nab3 proteins comprise the NRD complex, the major factor in promoting sno/snRNA termination (Ursic et al. 1997; Steinmetz et al. 2001). Sen1 binds to the CTD phosphorylated on Ser2 (Ursic et al. 2004; Chinchilla et al. 2012), which possibly facilitates its recruitment to multiple coding as well as noncoding genes, where it tends to accumulate toward the 3′ end (Chinchilla et al. 2012). Sen1 may also play a role in coordinating transcription and replication, since it is associated with replication forks across Pol II active genes (Alzu et al. 2012).
Senataxin is the human homolog of Sen1 and has also been implicated in transcriptional termination (Suraweera et al. 2009; Skourti-Stathaki et al. 2011; Padmanabhan et al. 2012). Senataxin was initially identified when mutations causing ataxia oculomotor apraxia 2 (AOA2) and amyotrophic lateral sclerosis type 4 (ALS4) were mapped to the SETX gene (James and Talbot 2006; Palau and Espinos 2006). AOA2 mutations include both missense and nonsense mutations leading to senataxin loss of function, whereas mutations linked to ALS4 appear to be missense, dominant mutations resulting in gain of function (Chen at al. 2004; Arning et al. 2013). These diseases are associated with the progressive degeneration of motor neurons in the brain and spinal cord, progressive muscle weakness, and atrophy. SETX encodes a 302.8-kD widely expressed protein containing an N-terminal putative protein–protein interaction domain and a C-terminal DEAD-box helicase domain followed by a nuclear localization signal (NLS) (Chen et al. 2004). Most senataxin mutations found in AOA2/ALS4 families either cause premature translational termination or interfere with the function of the helicase or N-terminal protein interaction domains (Chen et al. 2004; Moreira et al. 2004; Duquette et al. 2005; Criscuolo et al. 2006; Fogel and Perlman 2006). However, the precise mechanism of toxicity caused by these mutations and manifested in AOA2/ALS4 patients remains to be elucidated. Similar to its yeast counterpart, senataxin interacts with Pol II and other RNA processing factors, such as poly(A)-binding proteins 1 and 2 (PABP1/2), hnRNPs, SAP155, and SMN, pointing toward a role for senataxin in pre-mRNA processing as well as transcriptional termination (Suraweera et al. 2009).
Increasing evidence identifies senataxin as a DNA repair enzyme (Becherel at al. 2013; Yüce and West 2013) in addition to its role in the resolution of R loops arising at G-rich termination pause sites (Skourti-Stathaki et al. 2011). Yüce and West (2013) revealed that senataxin forms increased nuclear foci in S/G2 phase in response to DNA damage and impaired DNA replication. Importantly, these foci decreased significantly after R-loop resolution or transcriptional inhibition (Yuce and West 2013). The role of senataxin in DNA damage response, particularly during mouse male meiosis (spermatogenesis), has been highlighted by analysis of mouse strains with SETX gene knockouts. This study led to a model in which senataxin is proposed to resolve R loops to ensure genome integrity during meiotic recombination (Becherel et al. 2013). THO mutants from C. elegans and S. cerevisiae also show defective meiosis and increased DNA damage (Castellano-Pozo et al. 2012), pointing toward an evolutionarily conserved mechanism to maintain genome stability during meiosis by preventing formation of these potentially harmful structures.
It is of note that defects in DNA repair enzymes are strongly linked with neurodegenerative disorders (McKinnon 2009). In the future, it will be of vital importance to understand why defects in senataxin, a ubiquitously expressed protein, particularly affect neuronal cells. AOA2 disorder manifests primarily in post-replication neurons where DNA repair could rely mostly on nonhomologous end-joining (NHEJ) rather than HR. This could possibly increase genomic instability in these neurons. A recent study suggests a novel role for the exosome in senataxin-mediated DNA damage response (Richard et al. 2013). In essence, the exosome interacts with sumoylated senataxin, which is in turn targeted to DNA damage regions. Importantly, both sumoyaltion and the interaction are disrupted in AOA2, but not ALS4, disorder. In agreement, it has also been shown that deletion of TRF4 in S. cerevisiae, a component of the TRAMP complex that activates the exosome, leads to accumulation of R loops and genomic instability (Gavaldá et al. 2013). Significantly, the exosome also associates with AID in the R-loop-enriched switch regions of B cells (Basu et al. 2011). The physiological role of the exosome–senataxin interaction has yet to be established. Future research is necessary to understand why and how this mechanism is disrupted in AOA2 disorder.
Rad51 and trans-induced R loops
So far, we described here what is known about the factors that prevent or resolve R loops. However, do some factors actively promote R-loop formation? One particular factor has been shown to promote in vivo R-loop formation in S. cerevisiae: the Rad51 protein (see Fig. 3; Wahba et al. 2013). Eukaryotic Rad51 protein that is homologous to the bacterial RecA plays a major role in HR during DNA repair of DSBs. Its loading on “damaged” DNA is thought to be stimulated by the ssDNA protein RPA. Rad51 then promotes strand exchange (invasion of ssDNA into duplex DNA) by forming nucleoprotein filaments (Benson et al. 1994; Baumann et al. 1996). The bacterial RecA was also shown to promote RNA/DNA hybrid formation in vitro (Kasahara et al. 2000; Zaitsev and Kowalczykowski 2000). The Koshland laboratory (Wahba et al. 2013) demonstrated that in S. cerevisiae, deletion of Rad51 results in reduced formation of R loops and subsequent genomic instability, especially when R loops are enhanced by inactivation of RNA processing activities. Furthermore, Rad51 colocalizes with R loops prior to formation of any DSBs. Paradoxically, Rad51 is not only a repair factor but also promotes R-loop-mediated DNA damage and genomic instability. This is particularly important in cancer cells where Rad51 could directly promote tumor biogenesis. Two regulators of Rad51 have been described: Rad52, which is required for binding of Rad51 to ssDNA, and Srs2, which acts as an antagonist and consequently prevents R-loop formation (Wahba at al. 2013). Therefore, Srs2 can be added to the list of proteins that prevent R-loop formation and subsequent induced genomic instability.
Figure 3.

Rad51 can promote cis and trans R loops. The HR factor Rad51 can promote strand exchange, ultimately leading to cotranscriptional R-loop formation (cis R-loop). Trans R loops can also be mediated by Rad51. As shown in the diagram, trans RNA may target the ssDNA as part of a pre-existing R loop. Alternatively, trans RNA could target dsDNA if local unwinding of the DNA duplex occurs by mechanisms such as DNA replication. Such trans R loops are associated with the popular CRISPR–Cas9 system in which CRISPR guide RNA hybridizes with target DNA loci generating targeted DNA breaks. Trans R loops can also occur between ncRNAs and homologous DNA, ultimately leading to transcriptional gene silencing. DNA is shown as gray and black lines, and RNA is shown as a red line. The diagram is not drawn to scale.
Interestingly, in the absence of Rad51, transcription itself fails to lead to accumulation of R loops and genomic instability (Wahba et al. 2013). A yeast strain was generated with two copies of a particular human DNA sequence: one in a yeast artificial chromosome (YAC) and the other in one of yeast’s own chromosomes but under induced transcription regulation. When transcription was activated, the induced transcript invaded the homologous DNA in the YAC, resulting in the formation of an R loop in trans (away from the initial transcription point). The formation of this trans R loop was promoted by Rad51 (Wahba et al. 2013). This finding challenges for the first time the dogma in the field that R loops form only cotranscriptionally (in cis). It also raises important questions of R-loop-induced genomic instability. Trans-induced R loops could be a bigger threat to genome integrity than those formed in cis. Upon transcription of highly repetitive elements, in cis R loops would only occur at that region, whereas in trans-induced R loops could occur in many places across the genome, creating multiple “hot spots” for genomic instability.
Trans-induced R loops have also recently been shown to enable a broad range of applications, including fast generation of genetically modified cells and animals (Gratz et al. 2013; Hwang et al. 2013; Wang et al. 2013; Yang et al. 2013) and genetic screening at a genomic level (Shalem et al. 2014; Wang et al. 2014). The so-called CRISPR (clustered regularly interspaced short palindromic repeat)–Cas9 system is a naturally occurring microbial immune system for protection against phage and other genetic elements. (for review, see Terns and Terns 2011). Short DNA fragments from infecting phage genomes are incorporated into the host genome within the CRISPR locus. Transcription of this CRISPR locus then gives rise to CRISPR RNA, which in turn is processed into short RNAs consisting of phage sequence and repeat elements from the CRISPR locus. These small RNAs complexed with Cas9 act as guides to target the homologous DNA locus, creating a trans R loop. Cas9 then cuts the target locus on each strand and ultimately silences the target DNA (Jinek et al. 2012). Very recently, the crystal structure of Cas9 in complex with guide RNA and target DNA was reported, revealing the key functional interactions, including a bona fide RNA/DNA hybrid (Nishimasu et al. 2014). By analogy to CRISPR–Cas9, it is tempting to speculate that trans R loops could explain how noncoding RNAs (ncRNAs) generally target homologous DNA, which is an underlying principle of transcriptional gene silencing by the RNAi machinery.
Even though the precise targeting of CRISPR guide RNA to target loci (Jinek et al. 2012) has made this system very popular for genome editing, little is known about the actual molecular mechanism. Given that the target DNA is in a duplex, how does guide RNA hybridize with DNA? Does Cas9 itself or another factor first denature the DNA to allow RNA/DNA hybridization? An alternative, interesting scenario could be that cis R loops must form prior to CRISPR targeting and so enable hybridization of the now single-stranded target DNA with the CRISPR guide RNA. In this case, targeting would be dependent on the formation of cis R loops and consequently on active transcription.
So far, we discussed here R loops as precursors of chromosomal rearrangements in yeast and mammalian cells with deleterious consequences to cell integrity. But is the formation of R loops always unprogrammed and potentially harmful?
The new era of R loops: from threats of genomic instability to powerful regulators of gene expression
Over the last decade and particularly the last 3 years, a new era has emerged for the R-loop field, identifying these structures as powerful regulators of gene expression. So far, we documented the unprogrammed formation of R loops as a rare outcome of the transcriptional process with potential harmful consequences—in effect, an enemy to cellular fitness (Aguilera and Garcia-Muse 2012). However, the “other side of the coin” is more positive. An ever-increasing body of evidence has shed light on a number of biological processes controlled by the programmed formation of R loops.
The first beneficial function of R loops to be uncovered was CSR at the Ig heavy chain locus in activated B cells (Yu et al. 2003), as mentioned above. This study is of particular importance, as it firstly demonstrated the in vivo formation of R loops over switch regions that undergo CSR and revealed that these R-loop structures facilitate CSR only under specific conditions. R loops forming at the CSR locus are quite long (>1 kb) and very stable, as opposed to R loops at replication origins. The length of the observed stable R loop led to the view that the DNA sequence itself might play a vital role in R-loop formation and stabilization. Indeed, these switch regions are highly repetitive, GC-rich regions. Formation of transcription-dependent R loops over these regions leaves the G-rich nontemplate DNA strand displaced. Based on an analysis using bisulfite treatment to target ssDNA, it was also shown that stable R loops occur only in the physiological orientation (with G-rich transcripts) (Yu et al. 2003).
Controlling gene expression requires the definition of gene boundaries: the 5′ end promoter and 3′ end terminator. R loops have been recently shown to form at both gene ends (see Fig. 4; Skourti-Stathaki et al. 2011; Ginno et al. 2012, 2013). However, do they control gene expression in these cases? The Chedin laboratory (Ginno et al. 2012, 2013) has recently presented evidence for widespread R-loop formation over the 5′ regions downstream from CpG promoters in the human genome. These genomic regions are GC-rich and have a strong positive GC skew (template strand having an excess of C vs. G residues). Even though a direct association of R loops in the maintenance of the unmethylated state of CpG promoters has not been established, this study suggested that R loops forming at promoter regions could potentially lead to the activation of genes by recruiting either the protective histone 3 Lys4 trimethylation (H3K4me3) mark or the DNA demethylation complex (Ginno et al. 2012). Consistent with this model, the AID complex is also found at H3K4me3-enriched promoter-proximal chromatin, including CpG islands (Yamane et al. 2011). The key factor here could be the displaced ssDNA in the R loops, which may directly recruit histone methyltransferases or DNA demethylases. Paradoxically, the ssDNA is also suggested to link R loops and DNA damage events. How cells sense these two opposite situations and act accordingly remains an important but still unanswered question.
Figure 4.

R loops are enriched at both gene ends. In human protein-coding genes, R loops form over unmethylated CpG island promoters with positive GC skew and G-rich termination regions. Promoter-enriched R loops could activate gene expression, whereas terminator-enriched R loops promote transcriptional termination by facilitating Pol II pausing downstream from the poly(A) signal. Transcription start site (TSS), transcription termination site (TTS), and poly(A) (pA) signal are shown. Colored shading indicates peaks of R loops over 5′ and 3′ gene ends. The diagram is not drawn to scale.
R loops are directly implicated in transcriptional termination of some human genes by studies from our laboratory (Skourti-Stathaki et al. 2011). R loops formed over G-rich termination regions facilitate Pol II pausing downstream from the poly(A) signal prior to transcriptional termination. However, in this situation, a very fine balance of R loops is required for efficient termination. Once formed, these R loops must then be resolved by the helicase senataxin to release the nascent RNA and so allow its Xrn2-mediated degradation, which ultimately leads to efficient Pol II transcriptional termination (Skourti-Stathaki et al. 2011). This is a situation in which, even in the same gene, R loops can have a dual role: They are required for efficient termination, but their accumulation (followed by senataxin knockdown) inhibits this process. In the future, it will be interesting to investigate how, within one gene, cells can prevent the deleterious effects of R loops (resolved by senataxin) but at the same time allow their positive function (in this case, efficient termination).
Another interesting observation further supports the connection of R loops with Pol II termination. Genome-wide analysis has previously revealed that G-rich sequences immediately downstream from the poly(A) signal are relatively common in mammalian genes (Salisbury et al. 2006). Recent genome-wide bioinformatic analysis has also shown that promoters and 3′ regions of genes are enriched in G4-forming sequences (Huppert et al. 2008). Interestingly, 3′ untranslated region (UTR) G4s are particularly prevalent in cases in which a second gene is placed in close proximity, suggesting that G4s may be involved in transcriptional termination (Huppert et al. 2008). However, it still remains to be established whether genes that form R loops at their G-rich 3′ ends or at CpG island promoters also form G4 structures. Finally, a second R-loop genomic analysis strikingly suggested that a subset of Pol II terminators with a positive GC skew corresponds to R-loop regions genome-wide (Ginno et al. 2013). As in G4s (Huppert et al. 2008), genes with R loops at their 3′ ends are located in gene-dense regions, further reinforcing the role of R loops in efficient termination.
Altogether, we suggest that R loops are a common feature of G-rich pause terminator elements in human genes (Skourti-Stathaki et al. 2011; Ginno et al. 2013). How do they promote termination? Stopping Pol II is not an easy task. Once in a processive elongation mode, Pol II elongates at 4.3 kb/min (70 bp/sec) (Darzacq et al. 2007) over a diverse sequence landscape that may extend to >1 Mb in vertebrates. Also, given the fact that Pol II is a very large protein, one could argue that such transient structures such as R loops are inadequate to anchor Pol II. Identifying the molecular mechanism by which R loops promote termination is likely to provide new insights into the regulation of gene expression at the level of transcription.
R loops and ncRNAs
R loops have also been shown recently to play a role in the regulation of ncRNA (see Fig. 5; Powell et al. 2013; Sun et al. 2013). In Arabidopsis thaliana, COOLAIR is the antisense long ncRNA (lncRNA) that regulates the expression of the FLC gene, a key repressor of flower development. Upon prolonged cold conditions, COOLAIR becomes transcriptionally active and represses FLC transcription. Until recently, the transcriptional regulation of COOLAIR itself remained uncharacterized. However, compelling new evidence on the role of R loops in this process recently came to light (Sun et al. 2013). In essence, R loops are shown to form over the promoter region of COOLAIR, and a ssDNA-binding homeodomain protein, AtNDX, binds and stabilizes these R loops. This ultimately leads to COOLAIR transcriptional repression (Sun et al. 2013). This study provides a clear example of how regulatory lncRNAs can themselves be regulated but also raises intriguing questions. How does AtNDX maintain R-loop formation in the presence of surveillance mechanisms? Does it act faster than helicases and RNase H enzymes, or do these cells somehow protect R loops and ensure COOLAIR repression and proper flowering patterns? A comparison with Rad51 may be relevant, as this protein also binds ssDNA and promotes R-loop formation in vivo in S. cerevisiae (Wahba et al. 2013). In this case, however, Rad51 promotes “unprogrammed” R-loop formation and potential genomic instability. Given the fact that both AtDNX and Rad51 recognize unpaired ssDNA derived from an R loop, how does Rad51 on the one hand restrict potentially “deleterious” R loops, while AtNDX promotes “regulatory” R loops? Regardless of the exact mechanism, it is clear that discovering how AtDNX and Rad51 regulation occurs will provide a powerful new tool to understand how cells distinguish between the two “types” of R loops.
Figure 5.

R loops transcriptionally regulate ncRNAs. (A) In plants, COOLAIR antisense lncRNA controls the expression of the FLC gene. R loops form over the promoter region of COOLAIR and are stabilized by the ssDNA-binding protein AtNDX. This causes transcriptional repression of COOLAIR and, ultimately, activation of the FLC gene. (B) In human neuronal cells, topoisomerase inhibitor topotecan causes accumulation of R loops in the G-rich termination region of the Snord116 gene. This causes chromatin decondensation and blocks read-through transcription that otherwise forms the Ube3a antisense transcript. This activates the expression of the Ube3a sense transcript. Arrows indicate the direction of transcription. For simplicity, nucleosomes are omitted. The diagram is not drawn to scale.
R loops have recently been linked with the molecular mechanism of a cancer drug, topotecan, that reactivates the expression of the imprinted silenced gene Ube3a (Powell et al. 2013). Angelman syndrome (AS) is an autism-related disorder that is caused by mutations or deletions of the maternal copy of the Ube3a gene (Kishino et al. 1997; Matsuura et al. 1997). Normally, neurons express only the maternal copy of this gene and silence the paternal copy via the Ube3a antisense transcript. So, Ube3a mutations in the maternal copy result in a complete loss of the protein, a brain-specific ubiquitin E3 ligase. Ube3a antisense is located immediately downstream from the Snord116 gene, mutations of which cause a second disorder, Prader-Willi syndrome. The cancer drug topotecan was found to reactivate the paternal copy of Ube3a by reducing the antisense Ube3a transcript in neurons and therefore could be potentially used to treat AS (Huang et al. 2011). Even though topotecan holds promise for AS treatment, it still remains unknown how it targets specifically Ube3a and no other genes within this locus. Importantly, topotecan is an inhibitor of topoisomerase, which, as mentioned above, relaxes negative supercoiling. It is now revealed that R-loop formation plays a role in the topotecan effect (Powell et al. 2013). In essence, R loops form over the G-rich Snord116 gene, which in turn causes nucleosome depletion and chromatin decondensation in the paternal allele.
Under physiological conditions, Ube3a antisense transcription silences Ube3a in cis. Upon topotecan treatment, these R loops are stabilized and so accumulate. According to this model, this R-loop accumulation causes excessive chromatin decondensation, stalling of the transcriptional machinery, and inhibition of Ube3a antisense expression. This in turn activates paternal Ube3a expression (Powell et al. 2013). This suggests that topotecan can also be used as a powerful regulator of R-loop formation in other contexts and so provides a new tool in the characterization of R-loop biology.
Two important points arise from this study: First, in this case, R loops are shown to induce nucleosome depletion and chromatin decondensation, although it is not clear how this occurs. It could also be argued that chromatin decondensation/nucleosome depletion facilitates R-loop formation. Cause cannot easily be distinguished from the effect in this case as in other R-loop-associated processes. As mentioned above, the accumulation of R loops can also induce chromatin condensation (Castellano-Pozo et al. 2013) in contrast to the Snord116 gene. Even though, in these two studies, R-loop formation may have opposite outcomes on chromatin structure, it is tempting to speculate that a more general link exists between R loops and chromatin dynamics. Second, topotecan is the first R-loop targeting drug used as a therapeutic agent in genetic disorders. Camptothecin (CPT), another topoisomerase inhibitor that is used as an anti-cancer drug, has also been shown to stabilize R loops. Interestingly, CPT causes accumulation of Pol II antisense transcripts over CpG island promoters (Marinello et al. 2013). It is evident that, more than ever, research on R loops is vital to understand how these structures could be disrupted in cancer and other diseases. This would also strengthen the likelihood that R-loop formation is tightly controlled, as its dysregulation and/or accumulation can compromise genome dynamics and function.
Finally, R loops formed over the centromeric repeats in S. pombe have been shown to mediate RNAi-dependent heterochromatin formation (Nakama et al. 2012). This study is of particular interest, since it shows that R loops are potentially involved in silencing centromeric DNA. Heterochromatic ncRNA have been suggested to remain on chromatin and function as a binding platform for the RNAi apparatus (Cam et al. 2009). Following this study, two important questions remain unanswered. How do ncRNAs remain bound to chromatin? Do RNAi factors (especially the RITS complex) target the gene transcript or the DNA strand? Nakama et al. (2012) suggested that ncRNA transcribed from heterochromatin remains bound to chromatin via the formation of an R loop and that the so-formed RNA/DNA hybrid itself is involved in the heterochromatin formation. In essence, RNA/DNA hybrid foci colocalize with centromeric heterochromatic regions, and overexpression of RNase H decreases these foci, which in turn disrupts heterochromatin formation. Given that, upon R-loop formation, the single-strand nontemplate DNA remains unpaired, Nakama et al. (2012) predicted that the RITS complex could target the single-stranded “unprotected” DNA or, alternatively, the chromatin-associated RNA to generate heterochromatin formation.
Even though this study did not discuss whether R-loop formation is the trigger of heterochromatin formation rather than its consequence, these observations again tightly connected the fields of transcription and heterochromatin, raising intriguing questions for further investigation. Do R loops facilitate transcriptional gene silencing in other regions of the genome? Is this phenomenon conserved in mammalian cells? Could R-loop “hot spots” be regions of RNAi-dependent heterochromatin assembly? In any case, this study suggested that R loops control an epigenetic mark directly or indirectly. Future studies on this potential connection will shed light on the “RNA-guided pathway for the epigenome” (Jenuwein 2002) and might place R loops as key players of the critical interrelationship between transcription and chromatin.
R loops and neurodegenerative disorders
The mechanistic connection between R loops and neurodegenerative disorders remains unclear even though examples and associations are growing. Apart from the connections presented for the Snord116 study, R loops are often associated with neurodegenerative disease caused by abnormal expansion of repeated DNA sequences (the so-called repeat expansion disorders) (Lin et al. 2010; McIvor et al. 2010; Reddy et al. 2011; Wongsurawat et al. 2012). Very recently, R loops were shown to form over the promoter of the fragile X mental retardation 1 (Fmr1) gene and coincide with its epigenetic silencing in fragile X syndrome (Colak et al. 2014). A similar mechanism has also recently been shown to occur in another trinucleotide repeat expansion disease, Friedriech’s ataxia (Groh et al. 2014). In this study, expanded disease alleles were shown to accumulate R loops, resulting in a transcriptional block and heterochromatin formation. G4 structures were also found to form in a hexanucleotide repeat expansion of the C9orf72 gene, which causes ALS and frontotemporal dementia (FTD). G4 in C9orf72 DNA promotes the formation of stable R loops, which in turn impedes transcriptional elongation and leads to production of short, abortive transcripts (Haeusler et al. 2014).
Furthermore, it was recently shown that topoisomerases promote transcription of human and mouse long genes linked to autism (King et al. 2013). Interestingly, Snord116–Ube3a antisense is an extremely long transcription unit, implying that topotecan might reduce the expression of other long genes. Indeed, topotecan has also been shown to reduce the expression of other long genes in human and mouse neurons in a dose-dependent manner (King et al. 2013). Transcription of very long genes has also been shown to cause replication/transcription collisions and accumulation of R loops at the CFSs (Helmrich et al. 2011). Some of these genes have been shown to be down-regulated in neurological diseases, such as Alzheimer’s disease (Sze et al. 2004). Additionally, defects in DNA repair proteins (which, as mentioned above, are connected to R loops) cause neurodegenerative syndromes (McKinnon 2009). Defective DNA repair in mature neuronal tissues has also been linked to aging and neurodegenerative disorders, such as Parkinson’s disease and Alzheimer’s disease. Senataxin has recently been suggested to act as a DNA repair protein (Yüce and West 2013) by resolving R loops at human genes (Skourti-Stathaki et al. 2011). Given that mutations in senataxin cause specific neurodegenerative disorders (James and Talbot 2006; Palau and Espinos 2006), perhaps senataxin, despite being a ubiquitously expressed protein, has a special role in neuronal genes by controlling the transcription of some fragile sites present in long genes. Altogether, these studies reveal a complex and coordinated network in neurons between transcription, DNA repair, and R loops. Indeed, the general physiological relevance of R loops as transcriptional regulators seems more and more likely.
Conclusions and perspectives
The last decade has seen a significant expansion of our knowledge of R-loop biology and function. For years, R loops were considered a threat to cells as a rare transcriptional by-product (Aguilera and Garcia-Muse 2012). It is only now that we start to realize that they may have a major regulatory role in gene function. They can be the “two sides of a coin,” deleterious structures but also fine-tuners of gene expression. Given their involvement in multiple cellular processes, understanding how cells prevent the negative functions of R loops yet allow their positive ones is a challenge for the years to come. Perhaps the key to this question is the unpaired ssDNA derived from these structures. It can be the trigger for genomic instability but can also provide base-pairing for trans RNAs or act as a binding scaffold for enzymes that control the transcription cycle.
A great deal has been learned in recent years about factors that prevent or resolve R loops. Research should now aim to discover more factors (in addition to Rad51 and AtNDX) that actively promote R-loop formation. Is there an evolutionarily conserved protein that is generally responsible for R-loop formation? Answering this fundamental question will perhaps allow us to better understand the dual functions of R loops and also link R loops to hitherto unanticipated cellular processes.
As mentioned above, R loops are thought to play a role in neurodegenerative disorders even though strong evidence for this association has yet to be established. R loops could offer a novel angle on regulation of transcription, and it is now the time to unravel their possible links with cancer and neurodegenerative disease. From the examples mentioned in this review, it is evident that R loops lie at the interphase of different fields: transcription, RNA processing, DNA damage, and chromatin. More than ever, we need to interconnect these fields to fully understand how R loops modulate genome dynamics.
Acknowledgments
We thank Ricardo Nunes Bastos (Department of Biochemistry, University of Oxford) for kindly generating the figures. We also thank Eleanor White (N.J.P.’s laboratory) for critical reading of this manuscript. Research in N.J.P.’s laboratory is supported by a Wellcome Trust program grant (091805/Z/10/Z) and a European Research Council advanced grant (339270-polyloop).
Footnotes
Article is online at http://www.genesdev.org/cgi/doi/10.1101/gad.242990.114.
Freely available online through the Genes & Development Open Access option.
References
- Aguilera A 2002. The connection between transcription and genomic instability. EMBO J 21: 195–201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aguilera A 2005. mRNA processing and genomic instability. Nat Struct Mol Biol 12: 737–738 [DOI] [PubMed] [Google Scholar]
- Aguilera A, Garcia-Muse T 2012. R loops: from transcription byproducts to threats to genome stability. Mol Cell 46: 115–124 [DOI] [PubMed] [Google Scholar]
- Aguilera A, Klein HL 1990. HPR1, a novel yeast gene that prevents intrachromosomal excision recombination, shows carboxy-terminal homology to the Saccharomyces cerevisiae TOP1 gene. Mol Cell Biol 10: 1439–1451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alzu A, Bermejo R, Begnis M, Lucca C, Piccini D, Carotenuto W, Saponaro M, Brambati A, Cocito A, Foiani M, et al. 2012. Senataxin associates with replication forks to protect fork integrity across RNA-polymerase-II-transcribed genes. Cell 151: 835–846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arning L, Epplen JT, Rahikkala E, Hendrich C, Ludolph AC, Sperfeld AD 2013. The SETX missense variation spectrum as evaluated in patients with ALS4-like motor neuron diseases. Neurogenetics 14: 53–61 [DOI] [PubMed] [Google Scholar]
- Baker TA, Kornberg A 1988. Transcriptional activation of initiation of replication from the E. coli chromosomal origin: an RNA–DNA hybrid near oriC. Cell 55: 113–123 [DOI] [PubMed] [Google Scholar]
- Basu U, Meng FL, Keim C, Grinstein V, Pefanis E, Eccleston J, Zhang T, Myers D, Wasserman CR, Wesemann DR, et al. 2011. The RNA exosome targets the AID cytidine deaminase to both strands of transcribed duplex DNA substrates. Cell 144: 353–363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumann P, Benson FE, West SC 1996. Human Rad51 protein promotes ATP-dependent homologous pairing and strand transfer reactions in vitro. Cell 87: 757–766 [DOI] [PubMed] [Google Scholar]
- Becherel OJ, Yeo AJ, Stellati A, Heng EY, Luff J, Suraweera AM, Woods R, Fleming J, Carrie D, McKinney K, et al. 2013. Senataxin plays an essential role with DNA damage response proteins in meiotic recombination and gene silencing. PLoS Genet 9: e1003435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benson FE, Stasiak A, West SC 1994. Purification and characterization of the human Rad51 protein, an analogue of E. coli RecA. EMBO J 13: 5764–5771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boubakri H, de Septenville AL, Viguera E, Michel B 2010. The helicases DinG, Rep and UvrD cooperate to promote replication across transcription units in vivo. EMBO J 29: 145–157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cam HP, Chen ES, Grewal SI 2009. Transcriptional scaffolds for heterochromatin assembly. Cell 136: 610–614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carles-Kinch K, Kreuzer KN 1997. RNA–DNA hybrid formation at a bacteriophage T4 replication origin. J Mol Biol 266: 915–926 [DOI] [PubMed] [Google Scholar]
- Castellano-Pozo M, Garcia-Muse T, Aguilera A 2012. R-loops cause replication impairment and genome instability during meiosis. EMBO Rep 13: 923–929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castellano-Pozo M, Santos-Pereira JM, Rondon AG, Barroso S, Andujar E, Perez-Alegre M, Garcia-Muse T, Aguilera A 2013. R loops are linked to histone H3 S10 phosphorylation and chromatin condensation. Mol Cell 52: 583–590 [DOI] [PubMed] [Google Scholar]
- Cerritelli SM, Crouch RJ 2009. Ribonuclease H: the enzymes in eukaryotes. FEBS J 276: 1494–1505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerritelli SM, Frolova EG, Feng C, Grinberg A, Love PE, Crouch RJ 2003. Failure to produce mitochondrial DNA results in embryonic lethality in Rnaseh1 null mice. Mol Cell 11: 807–815 [DOI] [PubMed] [Google Scholar]
- Chakraborty P, Grosse F 2011. Human DHX9 helicase preferentially unwinds RNA-containing displacement loops (R-loops) and G-quadruplexes. DNA Repair 10: 654–665 [DOI] [PubMed] [Google Scholar]
- Chan YA, Aristizabal MJ, Lu PY, Luo Z, Hamza A, Kobor MS, Stirling PC, Hieter P 2014. Genome-wide profiling of yeast DNA:RNA hybrid prone sites with DRIP-chip. PLoS Genet 10: e1004288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chavez S, Beilharz T, Rondon AG, Erdjument-Bromage H, Tempst P, Svejstrup JQ, Lithgow T, Aguilera A 2000. A protein complex containing Tho2, Hpr1, Mft1 and a novel protein, Thp2, connects transcription elongation with mitotic recombination in Saccharomyces cerevisiae. EMBO J 19: 5824–5834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YZ, Bennett CL, Huynh HM, Blair IP, Puls I, Irobi J, Dierick I, Abel A, Kennerson ML, Rabin BA, et al. 2004. DNA/RNA helicase gene mutations in a form of juvenile amyotrophic lateral sclerosis (ALS4). Am J Hum Genet 74: 1128–1135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chinchilla K, Rodriguez-Molina JB, Ursic D, Finkel JS, Ansari AZ, Culbertson MR 2012. Interactions of Sen1, Nrd1, and Nab3 with multiple phosphorylated forms of the Rpb1 C-terminal domain in Saccharomyces cerevisiae. Eukaryot Cell 11: 417–429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chon H, Sparks JL, Rychlik M, Nowotny M, Burgers PM, Crouch RJ, Cerritelli SM 2013. RNase H2 roles in genome integrity revealed by unlinking its activities. Nucleic Acids Res 41: 3130–3143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colak D, Zaninovic N, Cohen MS, Rosenwaks Z, Yang WY, Gerhardt J, Disney MD, Jaffrey SR 2014. Promoter-bound trinucleotide repeat mRNA drives epigenetic silencing in fragile X syndrome. Science 343: 1002–1005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Criscuolo C, Chessa L, Di Giandomenico S, Mancini P, Sacca F, Grieco GS, Piane M, Barbieri F, De Michele G, Banfi S, et al. 2006. Ataxia with oculomotor apraxia type 2: a clinical, pathologic, and genetic study. Neurology 66: 1207–1210 [DOI] [PubMed] [Google Scholar]
- Darzacq X, Shav-Tal Y, de Turris V, Brody Y, Shenoy SM, Phair RD, Singer RH 2007. In vivo dynamics of RNA polymerase II transcription. Nat Struct Mol Biol 14: 796–806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debatisse M, Le Tallec B, Letessier A, Dutrillaux B, Brison O 2012. Common fragile sites: mechanisms of instability revisited. Trends Genet 28: 22–32 [DOI] [PubMed] [Google Scholar]
- Di Noia J, Neuberger MS 2002. Altering the pathway of immunoglobulin hypermutation by inhibiting uracil-DNA glycosylase. Nature 419: 43–48 [DOI] [PubMed] [Google Scholar]
- Dominguez-Sanchez MS, Barroso S, Gomez-Gonzalez B, Luna R, Aguilera A 2011. Genome instability and transcription elongation impairment in human cells depleted of THO/TREX. PLoS Genet 7: e1002386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drolet M, Bi X, Liu LF 1994. Hypernegative supercoiling of the DNA template during transcription elongation in vitro. J Biol Chem 269: 2068–2074 [PubMed] [Google Scholar]
- Drolet M, Phoenix P, Menzel R, Masse E, Liu LF, Crouch RJ 1995. Overexpression of RNase H partially complements the growth defect of an Escherichia coli ΔtopA mutant: R-loop formation is a major problem in the absence of DNA topoisomerase I. Proc Natl Acad Sci 92: 3526–3530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duquette ML, Handa P, Vincent JA, Taylor AF, Maizels N 2004. Intracellular transcription of G-rich DNAs induces formation of G-loops, novel structures containing G4 DNA. Genes Dev 18: 1618–1629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duquette A, Roddier K, McNabb-Baltar J, Gosselin I, St-Denis A, Dicaire MJ, Loisel L, Labuda D, Marchand L, Mathieu J, et al. 2005. Mutations in senataxin responsible for Quebec cluster of ataxia with neuropathy. Ann Neurol 57: 408–414 [DOI] [PubMed] [Google Scholar]
- Eder PS, Walder RY, Walder JA 1993. Substrate specificity of human RNase H1 and its role in excision repair of ribose residues misincorporated in DNA. Biochimie 75: 123–126 [DOI] [PubMed] [Google Scholar]
- El Hage A, French SL, Beyer AL, Tollervey D 2010. Loss of topoisomerase I leads to R-loop-mediated transcriptional blocks during ribosomal RNA synthesis. Genes Dev 24: 1546–1558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fogel BL, Perlman S 2006. Novel mutations in the senataxin DNA/RNA helicase domain in ataxia with oculomotor apraxia 2. Neurology 67: 2083–2084 [DOI] [PubMed] [Google Scholar]
- Gan W, Guan Z, Liu J, Gui T, Shen K, Manley JL, Li X 2011. R-loop-mediated genomic instability is caused by impairment of replication fork progression. Genes Dev 25: 2041–2056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavaldá S, Gallardo M, Luna R, Aguilera A 2013. R-loop mediated transcription-associated recombination in trf4Δ mutants reveals new links between RNA surveillance and genome integrity. PLoS ONE 8: e65541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginno PA, Lott PL, Christensen HC, Korf I, Chedin F 2012. R-loop formation is a distinctive characteristic of unmethylated human CpG island promoters. Mol Cell 45: 814–825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginno PA, Lim YW, Lott PL, Korf I, Chedin F 2013. GC skew at the 5′ and 3′ ends of human genes links R-loop formation to epigenetic regulation and transcription termination. Genome Res 23: 1590–1600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottipati P, Cassel TN, Savolainen L, Helleday T 2008. Transcription-associated recombination is dependent on replication in Mammalian cells. Mol Cell Biol 28: 154–164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gratz SJ, Cummings AM, Nguyen JN, Hamm DC, Donohue LK, Harrison MM, Wildonger J, O’Connor-Giles KM 2013. Genome engineering of Drosophila with the CRISPR RNA-guided Cas9 nuclease. Genetics 194: 1029–1035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groh M, Lufino MMP, Wade-Martins R, Gromak N 2014. R-loops associated with triplet repeat expansions promote gene silencing in Friedrick Ataxia and fragile X syndrome. PLoS Genet 10: e1004318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haeusler AR, Donnelly CJ, Periz G, Simko EA, Shaw PG, Kim MS, Maragakis NJ, Troncoso JC, Pandey A, Sattler R, et al. 2014. C9orf72 nucleotide repeat structures initiate molecular cascades of disease. Nature 507: 195–200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hausen P, Stein H 1970. Ribonuclease H. An enzyme degrading the RNA moiety of DNA–RNA hybrids. Eur J Biochem 14: 278–283 [DOI] [PubMed] [Google Scholar]
- Helmrich A, Ballarino M, Tora L 2011. Collisions between replication and transcription complexes cause common fragile site instability at the longest human genes. Mol Cell 44: 966–977 [DOI] [PubMed] [Google Scholar]
- Houlard M, Artus J, Leguillier T, Vandormael-Pournin S, Cohen-Tannoudji M 2011. DNA–RNA hybrids contribute to the replication dependent genomic instability induced by Omcg1 deficiency. Cell Cycle 10: 108–117 [DOI] [PubMed] [Google Scholar]
- Huang HS, Allen JA, Mabb AM, King IF, Miriyala J, Taylor-Blake B, Sciaky N, Dutton JW Jr, Lee HM, Chen X, et al. 2011. Topoisomerase inhibitors unsilence the dormant allele of Ube3a in neurons. Nature 481: 185–189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huertas P, Aguilera A 2003. Cotranscriptionally formed DNA:RNA hybrids mediate transcription elongation impairment and transcription-associated recombination. Mol Cell 12: 711–721 [DOI] [PubMed] [Google Scholar]
- Huppert JL, Bugaut A, Kumari S, Balasubramanian S 2008. G-quadruplexes: the beginning and end of UTRs. Nucleic Acids Res 36: 6260–6268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang WY, Fu Y, Reyon D, Maeder ML, Tsai SQ, Sander JD, Peterson RT, Yeh JR, Joung JK 2013. Efficient genome editing in zebrafish using a CRISPR–Cas system. Nat Biotechnol 31: 227–229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- James PA, Talbot K 2006. The molecular genetics of non-ALS motor neuron diseases. Biochim Biophys Acta 1762: 986–1000 [DOI] [PubMed] [Google Scholar]
- Jenuwein T 2002. Molecular biology. An RNA-guided pathway for the epigenome. Science 297: 2215–2218 [DOI] [PubMed] [Google Scholar]
- Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E 2012. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337: 816–821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasahara M, Clikeman JA, Bates DB, Kogoma T 2000. RecA protein-dependent R-loop formation in vitro. Genes Dev 14: 360–365 [PMC free article] [PubMed] [Google Scholar]
- Kawauchi J, Mischo H, Braglia P, Rondon A, Proudfoot NJ 2008. Budding yeast RNA polymerases I and II employ parallel mechanisms of transcriptional termination. Genes Dev 22: 1082–1092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HD, Choe J, Seo YS 1999. The sen1+ gene of Schizosaccharomyces pombe, a homologue of budding yeast SEN1, encodes an RNA and DNA helicase. Biochemistry 38: 14697–14710 [DOI] [PubMed] [Google Scholar]
- King IF, Yandava CN, Mabb AM, Hsiao JS, Huang HS, Pearson BL, Calabrese JM, Starmer J, Parker JS, Magnuson T, et al. 2013. Topoisomerases facilitate transcription of long genes linked to autism. Nature 501: 58–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kishino T, Lalande M, Wagstaff J 1997. UBE3A/E6-AP mutations cause Angelman syndrome. Nat Genet 15: 70–73 [DOI] [PubMed] [Google Scholar]
- Lee DY, Clayton DA 1996. Properties of a primer RNA–DNA hybrid at the mouse mitochondrial DNA leading-strand origin of replication. J Biol Chem 271: 24262–24269 [DOI] [PubMed] [Google Scholar]
- Li X, Manley JL 2005. Inactivation of the SR protein splicing factor ASF/SF2 results in genomic instability. Cell 122: 365–378 [DOI] [PubMed] [Google Scholar]
- Li X, Manley JL 2006. Cotranscriptional processes and their influence on genome stability. Genes Dev 20: 1838–1847 [DOI] [PubMed] [Google Scholar]
- Lin Y, Dent SY, Wilson JH, Wells RD, Napierala M 2010. R loops stimulate genetic instability of CTG.CAG repeats. Proc Natl Acad Sci 107: 692–697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marinello J, Chillemi G, Bueno S, Manzo SG, Capranico G 2013. Antisense transcripts enhanced by camptothecin at divergent CpG-island promoters associated with bursts of topoisomerase I–DNA cleavage complex and R-loop formation. Nucleic Acids Res 41: 10110–10123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masukata H, Tomizawa J 1984. Effects of point mutations on formation and structure of the RNA primer for ColE1 DNA replication. Cell 36: 513–522 [DOI] [PubMed] [Google Scholar]
- Masukata H, Tomizawa J 1990. A mechanism of formation of a persistent hybrid between elongating RNA and template DNA. Cell 62: 331–338 [DOI] [PubMed] [Google Scholar]
- Matsuura T, Sutcliffe JS, Fang P, Galjaard RJ, Jiang YH, Benton CS, Rommens JM, Beaudet AL 1997. De novo truncating mutations in E6-AP ubiquitin–protein ligase gene (UBE3A) in Angelman syndrome. Nat Genet 15: 74–77 [DOI] [PubMed] [Google Scholar]
- McIvor EI, Polak U, Napierala M 2010. New insights into repeat instability: role of RNA•DNA hybrids. RNA Biol 7: 551–558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKinnon PJ 2009. DNA repair deficiency and neurological disease. Nat Rev Neurosci 10: 100–112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mischo HE, Gomez-Gonzalez B, Grzechnik P, Rondon AG, Wei W, Steinmetz L, Aguilera A, Proudfoot NJ 2011. Yeast Sen1 helicase protects the genome from transcription-associated instability. Mol Cell 41: 21–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore MJ, Proudfoot NJ 2009. Pre-mRNA processing reaches back to transcription and ahead to translation. Cell 136: 688–700 [DOI] [PubMed] [Google Scholar]
- Moreira MC, Klur S, Watanabe M, Nemeth AH, Le Ber I, Moniz JC, Tranchant C, Aubourg P, Tazir M, Schols L, et al. 2004. Senataxin, the ortholog of a yeast RNA helicase, is mutant in ataxia–ocular apraxia 2. Nat Genet 36: 225–227 [DOI] [PubMed] [Google Scholar]
- Muramatsu M, Kinoshita K, Fagarasan S, Yamada S, Shinkai Y, Honjo T 2000. Class switch recombination and hypermutation require activation-induced cytidine deaminase (AID), a potential RNA editing enzyme. Cell 102: 553–563 [DOI] [PubMed] [Google Scholar]
- Murante RS, Henricksen LA, Bambara RA 1998. Junction ribonuclease: an activity in Okazaki fragment processing. Proc Natl Acad Sci 95: 2244–2249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakama M, Kawakami K, Kajitani T, Urano T, Murakami Y 2012. DNA–RNA hybrid formation mediates RNAi-directed heterochromatin formation. Genes Cells 17: 218–233 [DOI] [PubMed] [Google Scholar]
- Nishimasu H, Ran FA, Hsu PD, Konermann S, Shehata SI, Dohmae N, Ishitani R, Zhang F, Nureki O 2014. Crystal structure of cas9 in complex with guide RNA and target DNA. Cell 156: 935–949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padmanabhan K, Robles MS, Westerling T, Weitz CJ 2012. Feedback regulation of transcriptional termination by the mammalian circadian clock PERIOD complex. Science 337: 599–602 [DOI] [PubMed] [Google Scholar]
- Palau F, Espinos C 2006. Autosomal recessive cerebellar ataxias. Orphanet J Rare Dis 1: 47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulsen RD, Soni DV, Wollman R, Hahn AT, Yee MC, Guan A, Hesley JA, Miller SC, Cromwell EF, Solow-Cordero DE, et al. 2009. A genome-wide siRNA screen reveals diverse cellular processes and pathways that mediate genome stability. Mol Cell 35: 228–239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piruat JI, Aguilera A 1998. A novel yeast gene, THO2, is involved in RNA pol II transcription and provides new evidence for transcriptional elongation-associated recombination. EMBO J 17: 4859–4872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell WT, Coulson RL, Gonzales ML, Crary FK, Wong SS, Adams S, Ach RA, Tsang P, Yamada NA, Yasui DH, et al. 2013. R-loop formation at Snord116 mediates topotecan inhibition of Ube3a-antisense and allele-specific chromatin decondensation. Proc Natl Acad Sci 110: 13938–13943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prado F, Aguilera A 2005. Impairment of replication fork progression mediates RNA polII transcription-associated recombination. EMBO J 24: 1267–1276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reaban ME, Lebowitz J, Griffin JA 1994. Transcription induces the formation of a stable RNA.DNA hybrid in the immunoglobulin α switch region. J Biol Chem 269: 21850–21857 [PubMed] [Google Scholar]
- Reddy K, Tam M, Bowater RP, Barber M, Tomlinson M, Nichol Edamura K, Wang YH, Pearson CE 2011. Determinants of R-loop formation at convergent bidirectionally transcribed trinucleotide repeats. Nucleic Acids Res 39: 1749–1762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Revy P, Muto T, Levy Y, Geissmann F, Plebani A, Sanal O, Catalan N, Forveille M, Dufourcq-Labelouse R, Gennery A, et al. 2000. Activation-induced cytidine deaminase (AID) deficiency causes the autosomal recessive form of the hyper-IgM syndrome (HIGM2). Cell 102: 565–575 [DOI] [PubMed] [Google Scholar]
- Richard P, Feng S, Manley JL 2013. A SUMO-dependent interaction between Senataxin and the exosome, disrupted in the neurodegenerative disease AOA2, targets the exosome to sites of transcription-induced DNA damage. Genes Dev 27: 2227–2232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts RW, Crothers DM 1992. Stability and properties of double and triple helices: dramatic effects of RNA or DNA backbone composition. Science 258: 1463–1466 [DOI] [PubMed] [Google Scholar]
- Rondon AG, Jimeno S, Garcia-Rubio M, Aguilera A 2003. Molecular evidence that the eukaryotic THO/TREX complex is required for efficient transcription elongation. J Biol Chem 278: 39037–39043 [DOI] [PubMed] [Google Scholar]
- Roy D, Lieber MR 2009. G clustering is important for the initiation of transcription induced R-loops in vitro, whereas high G density without clustering is sufficient thereafter. Mol Cell Biol 29: 3124–3133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy D, Zhang Z, Lu Z, Hsieh CL, Lieber MR 2010. Competition between the RNA transcript and the nontemplate DNA strand during R-loop formation in vitro: a nick can serve as a strong R-loop initiation site. Mol Cell Biol 30: 146–159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salisbury J, Hutchison KW, Graber JH 2006. A multispecies comparison of the metazoan 3′-processing downstream elements and the CstF-64 RNA recognition motif. BMC Genomics 7: 55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos-Pereira JM, Herrero AB, García-Rubio ML, Marín A, Moreno S, Aguilera A 2013. The Npl3 hnRNP prevents R-loop-mediated transcription-replication conflicts and genome instability. Genes Dev 27: 2445–2458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shalem O, Sanjana NE, Hartenian E, Shi X, Scott DA, Mikkelsen TS, Heckl D, Ebert BL, Root DE, Doench JG, et al. 2014. Genome-scale CRISPR–Cas9 knockout screening in human cells. Science 343: 84–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw NN, Arya DP 2008. Recognition of the unique structure of DNA:RNA hybrids. Biochimie 90: 1026–1039 [DOI] [PubMed] [Google Scholar]
- Skourti-Stathaki K, Proudfoot NJ, Gromak N 2011. Human senataxin resolves RNA/DNA hybrids formed at transcriptional pause sites to promote Xrn2-dependent termination. Mol Cell 42: 794–805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein H, Hausen P 1969. Enzyme from calf thymus degrading the RNA moiety of DNA–RNA hybrids: effect on DNA-dependent RNA polymerase. Science 166: 393–395 [DOI] [PubMed] [Google Scholar]
- Steinmetz EJ, Conrad NK, Brow DA, Corden JL 2001. RNA-binding protein Nrd1 directs poly(A)-independent 3′-end formation of RNA polymerase II transcripts. Nature 413: 327–331 [DOI] [PubMed] [Google Scholar]
- Steinmetz EJ, Warren CL, Kuehner JN, Panbehi B, Ansari AZ, Brow DA 2006. Genome-wide distribution of yeast RNA polymerase II and its control by Sen1 helicase. Mol Cell 24: 735–746 [DOI] [PubMed] [Google Scholar]
- Stirling PC, Chan YA, Minaker SW, Aristizabal MJ, Barrett I, Sipahimalani P, Kobor MS, Hieter P 2012. R-loop-mediated genome instability in mRNA cleavage and polyadenylation mutants. Genes Dev 26: 163–175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strasser K, Masuda S, Mason P, Pfannstiel J, Oppizzi M, Rodriguez-Navarro S, Rondon AG, Aguilera A, Struhl K, Reed R, et al. 2002. TREX is a conserved complex coupling transcription with messenger RNA export. Nature 417: 304–308 [DOI] [PubMed] [Google Scholar]
- Sugimoto N, Nakano S, Katoh M, Matsumura A, Nakamuta H, Ohmichi T, Yoneyama M, Sasaki M 1995. Thermodynamic parameters to predict stability of RNA/DNA hybrid duplexes. Biochemistry 34: 11211–11216 [DOI] [PubMed] [Google Scholar]
- Sun Q, Csorba T, Skourti-Stathaki K, Proudfoot NJ, Dean C 2013. R-loop stabilization represses antisense transcription at the Arabidopsis FLC locus. Science 340: 619–621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suraweera A, Lim Y, Woods R, Birrell GW, Nasim T, Becherel OJ, Lavin MF 2009. Functional role for senataxin, defective in ataxia oculomotor apraxia type 2, in transcriptional regulation. Hum Mol Genet 18: 3384–3396 [DOI] [PubMed] [Google Scholar]
- Sze CI, Su M, Pugazhenthi S, Jambal P, Hsu LJ, Heath J, Schultz L, Chang NS 2004. Down-regulation of WW domain-containing oxidoreductase induces Tau phosphorylation in vitro. A potential role in Alzheimer’s disease. J Biol Chem 279: 30498–30506 [DOI] [PubMed] [Google Scholar]
- Terns MP, Terns RM 2011. CRISPR-based adaptive immune systems. Curr Opin Microbiol 14: 321–327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas M, White RL, Davis RW 1976. Hybridization of RNA to double-stranded DNA: formation of R-loops. Proc Natl Acad Sci 73: 2294–2298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuduri S, Crabbe L, Conti C, Tourriere H, Holtgreve-Grez H, Jauch A, Pantesco V, De Vos J, Thomas A, Theillet C, et al. 2009. Topoisomerase I suppresses genomic instability by preventing interference between replication and transcription. Nat Cell Biol 11: 1315–1324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ursic D, Himmel KL, Gurley KA, Webb F, Culbertson MR 1997. The yeast SEN1 gene is required for the processing of diverse RNA classes. Nucleic Acids Res 25: 4778–4785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ursic D, Chinchilla K, Finkel JS, Culbertson MR 2004. Multiple protein/protein and protein/RNA interactions suggest roles for yeast DNA/RNA helicase Sen1p in transcription, transcription-coupled DNA repair and RNA processing. Nucleic Acids Res 32: 2441–2452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahba L, Amon JD, Koshland D, Vuica-Ross M 2011. RNase H and multiple RNA biogenesis factors cooperate to prevent RNA:DNA hybrids from generating genome instability. Mol Cell 44: 978–988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahba L, Gore SK, Koshland D 2013. The homologous recombination machinery modulates the formation of RNA–DNA hybrids and associated chromosome instability. eLife 2: e00505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Yang H, Shivalila CS, Dawlaty MM, Cheng AW, Zhang F, Jaenisch R 2013. One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering. Cell 153: 910–918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T, Wei JJ, Sabatini DM, Lander ES 2014. Genetic screens in human cells using the CRISPR–Cas9 system. Science 343: 80–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westover KD, Bushnell DA, Kornberg RD 2004. Structural basis of transcription: nucleotide selection by rotation in the RNA polymerase II active center. Cell 119: 481–489 [DOI] [PubMed] [Google Scholar]
- Wongsurawat T, Jenjaroenpun P, Kwoh CK, Kuznetsov V 2012. Quantitative model of R-loop forming structures reveals a novel level of RNA–DNA interactome complexity. Nucleic Acids Res 40: e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu B, Clayton DA 1996. RNA–DNA hybrid formation at the human mitochondrial heavy-strand origin ceases at replication start sites: an implication for RNA–DNA hybrids serving as primers. EMBO J 15: 3135–3143 [PMC free article] [PubMed] [Google Scholar]
- Yamane A, Resch W, Kuo N, Kuchen S, Li Z, Sun HW, Robbiani DF, McBride K, Nussenzweig MC, Casellas R 2011. Deep-sequencing identification of the genomic targets of the cytidine deaminase AID and its cofactor RPA in B lymphocytes. Nat Immunol 12: 62–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, Wang H, Shivalila CS, Cheng AW, Shi L, Jaenisch R 2013. One-step generation of mice carrying reporter and conditional alleles by CRISPR/Cas-mediated genome engineering. Cell 154: 1370–1379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, McBride KM, Hensley S, Lu Y, Chedin F, Bedford MT 2014. Arginine methylation facilitates the recruitment of TOP3B to chromatin to prevent R loop accumulation. Mol Cell 53: 484–497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu K, Chedin F, Hsieh CL, Wilson TE, Lieber MR 2003. R-loops at immunoglobulin class switch regions in the chromosomes of stimulated B cells. Nat Immunol 4: 442–451 [DOI] [PubMed] [Google Scholar]
- Yüce Ö, West SC 2013. Senataxin, defective in the neurodegenerative disorder ataxia with oculomotor apraxia 2, lies at the interface of transcription and the DNA damage response. Mol Cell Biol 33: 406–417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaitsev EN, Kowalczykowski SC 2000. A novel pairing process promoted by Escherichia coli RecA protein: inverse DNA and RNA strand exchange. Genes Dev 14: 740–749 [PMC free article] [PubMed] [Google Scholar]