

How CPEB RNA-binding proteins regulate cytoplasmic polyadenylation and translation is poorly understood. Allain and colleagues report the structures of the tandem RNA recognition motifs (RRMs) of two human paralogs (CPEB1 and CPEB4) in their free and RNA-bound states. Structural and functional studies reveal how RNA binding by CPEB proteins leads to an optimal positioning of the N-terminal and zinc-binding domains at the 3′ UTR, which favors the nucleation of ribonucleoprotein complexes for translation regulation. This study provides the molecular basis for the translational regulatory circuit established by CPEB proteins.
Keywords: translational regulation, protein–RNA interactions, CPEB1, CPEB4, cytoplasmic polyadenylation, binuclear zinc-binding domain
Abstract
Cytoplasmic changes in polyA tail length is a key mechanism of translational control and is implicated in germline development, synaptic plasticity, cellular proliferation, senescence, and cancer progression. The presence of a U-rich cytoplasmic polyadenylation element (CPE) in the 3′ untranslated regions (UTRs) of the responding mRNAs gives them the selectivity to be regulated by the CPE-binding (CPEB) family of proteins, which recognizes RNA via the tandem RNA recognition motifs (RRMs). Here we report the solution structures of the tandem RRMs of two human paralogs (CPEB1 and CPEB4) in their free and RNA-bound states. The structures reveal an unprecedented arrangement of RRMs in the free state that undergo an original closure motion upon RNA binding that ensures high fidelity. Structural and functional characterization of the ZZ domain (zinc-binding domain) of CPEB1 suggests a role in both protein–protein and protein–RNA interactions. Together with functional studies, the structures reveal how RNA binding by CPEB proteins leads to an optimal positioning of the N-terminal and ZZ domains at the 3′ UTR, which favors the nucleation of the functional ribonucleoprotein complexes for translation regulation.
The cytoplasmic polyadenylation element (CPE)-binding (CPEB) family of RNA-binding proteins are essential regulators of post-transcriptional gene expression. Initially identified as maternal mRNA regulators during early embryonic development (Richter and Lasko 2011; Weill et al. 2012), it is now clear that CPEBs are key mediators of cellular homeostasis in somatic tissues, regulating important biological processes such as cell proliferation, senescence, cell polarity, and synaptic plasticity. Consequently, when misregulated, they contribute to the development of a variety of pathological manifestations, such as tumor development, memory defects, and insulin resistance (Berger-Sweeney et al. 2006; Alexandrov et al. 2012; Ortiz-Zapater et al. 2012; Weill et al. 2012; Udagawa et al. 2013).
Accordingly, CPEB targets cover almost 20% of the human genome (Belloc et al. 2008). These CPEB-regulated mRNAs are defined by a cis-acting sequence in their 3′ untranslated region (UTR); namely, the CPE, defined by the consensus sequence 5′-UUUUA1–2U-3′ (Fox et al. 1989; McGrew et al. 1989; Richter 2007). The number of CPEs and their distance to the conserved polyadenylation site (5′-AAUAAA-3′) determine qualitatively and quantitatively whether the CPEB-regulated transcript is translationally dormant or activated by cytoplasmic polyadenylation, the subcellular localization of the mRNA, and even the alternative 3′ UTR processing (alternative splicing and alternative polyadenylation) of the pre-mRNA in the nucleus (Belloc and Mendez 2008; Eliscovich et al. 2008; Pique et al. 2008; Bava et al. 2013). Thus, the sequential functions of CPEBs in pre-mRNA processing, translational repression coupled to mRNA localization, and translational activation through cytoplasmic polyadenylation define very precise temporal and spatial patterns of gene expression that are stabilized through positive and negative feedbacks to generate bistability in CPEB-mediated cellular responses (Belloc et al. 2008).
The CPEB family of proteins is composed of four members: CPEB1–4. All four paralogs harbor a C-terminal region comprised of two RNA recognition motifs (RRMs) and a zinc-binding domain (ZZ domain) (Merkel et al. 2013) in tandem. In fact, RNA gel shift assays show that this is the region of CPEB1 responsible for binding CPE-containing RNAs (Hake and Richter 1994). While the RRMs are required for sequence-specific recognition of the CPE sequence, the ZZ domain only contributes to the affinity and does not confer specificity (Hake and Richter 1994; Huang et al. 2006). The RRMs of CPEB2–4 share 97% sequence identity between them. In contrast, the RRMs of CPEB1 share 48% pairwise sequence identity with those of CPEB2–4 (Hake et al. 1998; Fernandez-Miranda and Mendez 2012). Thus, the RNA-binding domains of CPEBs define two distinct evolutionary groups within the family, which in other species, such as Drosophila melanogaster, contain only one member in each group (Orb and Orb2). The functional differences between these two groups are unknown, but they appear to regulate overlapping populations of mRNAs and recognize the same cis-acting elements (Huang et al. 2006; Igea and Mendez 2010; Novoa et al. 2010; Pavlopoulos et al. 2011; Ortiz-Zapater et al. 2012).
In contrast to the RNA-binding domain, the N-terminal domains of CPEBs are highly variable in both length and composition across various orthologs and paralogs (Wang and Cooper 2010). This region does not contain any recognizable domain and is predicted to be intrinsically disordered. However, at least for CPEB1, it contains all of the identified regulatory motifs and most likely constitutes a docking domain for the proteins that interact with CPEBs. In particular, this region in CPEB1 contains the activation site of Aurora A kinase (Mendez et al. 2000) as well as the Cdk1 and Plk1 phosphorylation sites that trigger its ubiquitination and degradation through the PEST box (Mendez et al. 2002; Setoyama et al. 2007). Additionally, potential nuclear localization and nuclear export signals are present within the same domain as well (Ernoult-Lange et al. 2009; Lin et al. 2010). Although much less is known about the other members of the family (CPEB2–4), their N-terminal domains also appear to contain nuclear import and export signals (Kan et al. 2010) and regulatory phosphorylation sites (Igea and Mendez 2010).
All four CPEBs are differentially expressed in somatic tissues but with overlapping patterns. Thus, multiple CPEBs coexist in individual cells to coregulate overlapping populations of transcripts (Fernandez-Miranda and Mendez 2012). Distinct signal transduction pathways most probably determine the capacity of CPEBs to form translational repression or activation complexes. Hence, understanding the translational regulation circuit established by CPEBs, such as during the mitotic and meiotic cell cycles (Igea and Mendez 2010; Novoa et al. 2010), requires further knowledge about how they compete for target mRNAs and the molecular recognition mechanisms involved. In order to unveil the molecular mechanism underlying the translational regulation mediated by CPEBs, we solved the solution structures of tandem RRMs of two human CPEB paralogs, CPEB1 and CPEB4, in the free state and in complex with the CPE. Our findings reveal an unprecedented structural topology of the protein itself as well as a very distinct RNA-binding mode (reminiscent of a Venus fly trap) compared with other tandem RRMs. Furthermore, the structures also show how recognition of the CPE by CPEB1 and CPEB2–4 differs and how, in both cases, these proteins coordinate the assembly of a stable mRNA ribonucleoprotein (mRNP) complex in order to regulate translation through the 3′ UTRs of the responding mRNAs.
Results
CPEB1 and CPEB4 tandem RRMs adopt a fixed orientation in the unbound state
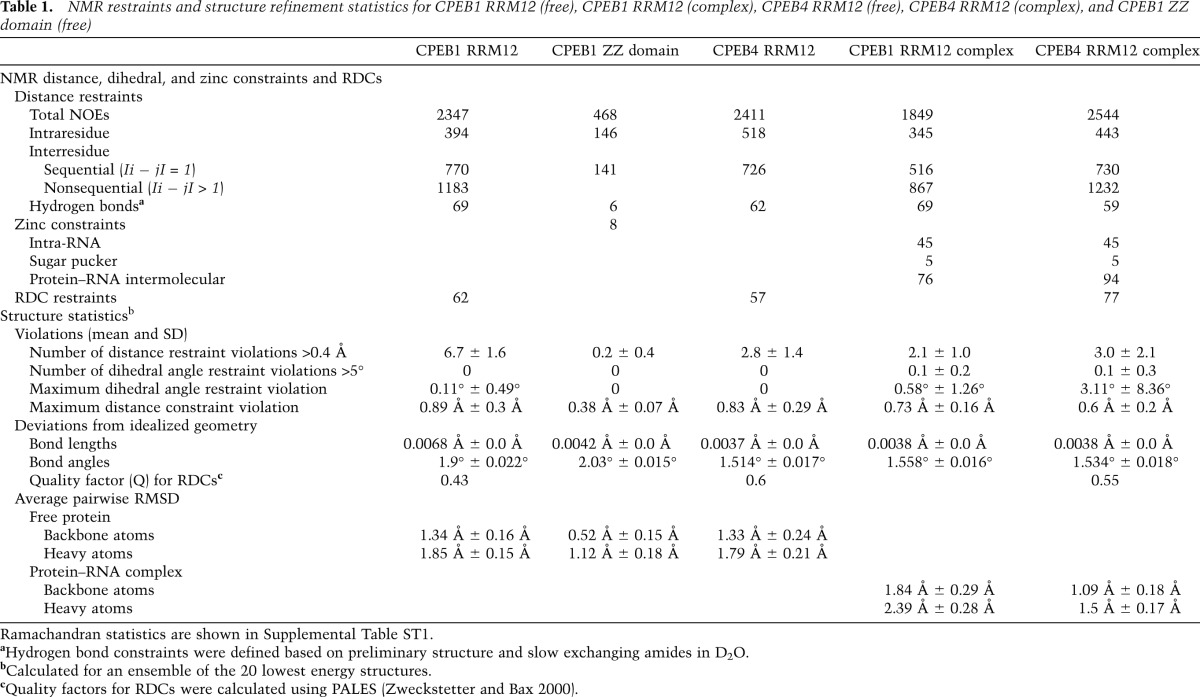
Understanding how CPEBs nucleate the assembly of mRNP complexes for translation repression or activation requires structural insights into the interactions between CPEBs and the nucleotides of the CPE element as well as into the potential conformational changes induced in CPEBs upon RNA binding. Therefore, we first determined the structures of CPEB1 and CPEB4 tandem RRMs in their free state. Following protein construct (Fig. 1A,B) and buffer optimization, tandem RRMs of these two proteins gave well-dispersed 1H–15N HSQC spectra characteristic of folded proteins. However, owing to the size and the resulting spectral overlap, structure determination by nuclear magnetic resonance (NMR) spectroscopy was not straightforward. Indeed, the initial structure calculations using the NOE assignment program ATNOS/CANDID (Herrmann et al. 2002a,b) failed to provide a meaningful ensemble of structures. We therefore pursued an iterative approach by including sets of manually assigned NOEs into the program to initially help solve the structure of each individual RRM and, later, of the full construct. In addition, residual dipolar coupling (RDC) restraints were included to improve the precision and the accuracy of the ensemble. This approach provided a precise final ensemble of structures for the tandem RRMs of CPEB1 and CPEB4 (Table1; Supplemental Table ST1; Supplemental Fig. S1A,B).
Figure 1.

Solution structures of CPEB1 and CPEB4 tandem RRMs in the free state (see also Supplemental Fig. S1). (A,B) Schematic representation of full-length CPEB1 and CPEB4 proteins. The regions corresponding to the N-terminal domain, RRM1, the inter-domain linker, RRM2, and the ZZ domain are shown by red, gray, purple, orange, and blue boxes, respectively. The same color-coding is also used for the structures. The protein is shown with the amino acid sequence of the RRMs used in these studies. (C,D) Representative structure of CPEB1 and CPEB4 tandem RRMs in ribbon representation. The structures shown below were obtained by a 90° rotation. (E–G) Two-dimensional (2D) schematic representation of CPEB1 (E), PABP (F), and CPEB4 (G) tandem RRMs. CPEB1/4 RRM1 has an insertion of two anti-parallel β strands in the β sheet (βa and βb, shown in green) compared with the canonical RRM of PABP. (E) In CPEB1, the N-terminal extension (in red) forms a parallel β strand with the β hairpin between the α2 helix and the β4 strand of RRM2. (E,G) In both CPEB1 and CPEB4, the interdomain linker (purple) forms an anti-parallel β strand with the β2 strand of RRM2.
Table 1.
NMR restraints and structure refinement statistics for CPEB1 RRM12 (free), CPEB1 RRM12 (complex), CPEB4 RRM12 (free), CPEB4 RRM12 (complex), and CPEB1 ZZ domain (free)
Both structures revealed an unprecedented arrangement of the two RRMs, which adopt a compact, V-shaped structure. RRM1 in both proteins has an extended β-sheet surface resulting from the insertion of two conserved, anti-parallel β strands between the α2 helix and the β4 strand, extending the β-sheet surface compared with a canonical RRM such as in polyA-binding protein (PABP) (βa and βb strands in Fig. 1C–G). Following RRM1, the interdomain linker (20 amino acids) adopts a similar topology in both proteins ( Fig. 1C,D, purple). The initial region of the interdomain linker (331WVLADS336) in CPEB1 adopts a short helical turn that interacts with residues of the N-terminal extension as well as with RRM2 (Fig. 1C; Supplemental Fig. S1C). Trp331 makes key interactions to position RRM2 relative to RRM1 by inserting its indole ring between the β sheet and α1 helix of RRM2 (Fig. 1C). After the helical turn, the interdomain linker folds in a β strand that runs anti-parallel to the β2 strand (RRM2) and packs against the α1 helix of RRM2. Finally, the interdomain linker runs across the RRM2 β sheet (Fig. 1C). Therefore, the interdomain linker acts as a hinge to fix the relative orientation of the two RRMs. All of the linker residues involved in such interactions are conserved in CPEB4, resulting in a similar structure and domain orientation (Fig. 1D,G; Supplemental Fig. S1D,E).
Apolar vs. polar inter-RRM interface in CPEB1 and CPEB4
In CPEB1, the interaction of Trp331 with RRM2 positions the two domains in close proximity, resulting in a number of direct contacts (Fig. 1C). Met360 (RRM2) interacts with Ile329 (RRM1), His387 (RRM2) interacts with Val285 (RRM1), and the α2–β4 loop of RRM2 interacts with the new βa–βb loop of RRM1 (Fig. 1C). Together with Trp331, these residues form a continuous hydrophobic interface between the two RRMs. Indeed, point mutation of Trp331 (interdomain linker) and Ile329 (RRM1) in CPEB1 resulted in chemical shift perturbations (CSPs) in RRM2 consistent with the structure (Supplemental Fig. S2A–D).
Similar to CPEB1, Trp154 in CPEB4 is crucial for the positioning of RRM2 relative to RRM1. However, the corresponding residues in the loops of RRM2 and the β4 strand of RRM1 are different, creating a polar surface in contrast to the apolar one of CPEB1 (Fig. 1D). Nevertheless, the relative position of the RRMs is similar to that found in CPEB1. NMR data further confirmed that CPEB4 RRMs interact in the free state, since the 1H–15N HSQC spectrum of CPEB4 RRM1 does not overlay with the spectrum of CPEB4 tandem RRMs (RRM12), and CSPs are observed on the β sheet of RRM1 (Supplemental Fig. S2E,F). Also, the correlation time (τc ≈ 15 nsec), estimated from T1/T2 relaxation rates, is consistent with the two RRMs tumbling as a single unit in solution (Supplemental Fig. S2G).
As a result of interaction, the tandem RRMs of CPEB1 and CPEB4 bury a large surface area at the interface of the two RRMs, corresponding to 1374 Å2 and 1098 Å2 in CPEB1 and CPEB4, respectively. This area is comparable with the buried surface area of other intimately interacting tandem RRMs such as PTB RRM34 (850 Å2), hnRNPL RRM34 (1400 Å2), and FIR RRM12 (900 Å2), as indicated in Barraud and Allain ( 2013). This is in contrast to other weakly interacting (more dynamic) RRMs burying a smaller surface area (400–600 Å2, such as for Prp24 RRM12 and U2AF65 RRMs), as indicated in Barraud and Allain (2013).
Novel topology of the N-terminal extension in CPEB1
In CPEB1, the N-terminal extension bridges the two RRMs by forming long-range intramolecular interactions with RRM2 (Fig. 2A). The sequence 229YKNPIYSC236, upstream of RRM1, interacts with the residues of the interdomain linker (331WVLA334), creating a hydrophobic interface between the two domains (Fig. 2A, right panel) that brings the upstream N-terminal region in proximity to the RRM2 α1 helix. This helix has an acidic character that favors its interaction with the positively charged N-terminal extension (Fig. 2B). The most upstream region of the N-terminal extension (219TWS221) interacts with the α1 helix and the last β hairpin (the α2–β4 loop) of RRM2 by forming a third parallel β strand (Fig. 2A, left panel). Most of the interactions observed are centered on Trp220, which interacts with the hydrophobic side chains of the RRM2 α1 helix (Fig. 2A). A hydrogen bond between Hε of Trp220 and the side chain oxygen of Asp372 (α1 helix of RRM2) further stabilizes this interaction. Trp220 and Asp372 are conserved in CPEB1 orthologs but not in other paralogs (Supplemental Fig. S1E). The sequence downstream from Trp220, 221SGQ223, interacts mainly with the last β hairpin of RRM2. Trp220 is essential, since a W220A mutation disrupts the fold of the protein (data not shown). Indeed, a 1H–15N heteronuclear NOE experiment indicated that both the N-terminal extension and the interdomain linker are structured (Supplemental Fig. S2H). Given that deletion of the whole N-terminal extension of CPEB1, including the region upstream of RRM1 (223QLPPRNYKNPI233), results in impaired nuclear localization of the protein, it has been proposed that this region contains noncanonical nuclear localization signals (Lin et al. 2010). However, the structure described here highlights the relevance of this region for the overall folding of CPEB1 and suggests that the failure to translocate to the nucleus may be caused by its misfolding.
Figure 2.

Intramolecular interactions of the N-terminal extension with RRM2 and structural model of CPEB1 RRM12ZZ (see also Supplemental Fig. S2). (A) Ribbon representation of a representative structure from the ensemble of CPEB1 RRM12 structures. (Left and right panels) Two enlarged views show the details from the part of the structure marked in black boxes. (B) Surface charge of CPEB1 RRM12. Negative charge is represented in red, and positive charge is represented in blue. A similar color code is also used for surface charges in other structures. The N-terminal extension with side chains in stick representation is shown in green. (C) The CPEB1 protein is shown with the amino acid sequence of the ZZ domain used in these studies. Shown below is one representative structure from the ensemble of the CPEB1 ZZ domain in light blue. The two zinc ions are shown as purple spheres. The two clusters of zinc-coordinating residues are shown in green and orange (also colored in the sequence). (D) Structural model of CPEB1 tandem RRMs and the ZZ domain.
In contrast to CPEB1, the N-terminal extension in CPEB4 (53–65) shows flexibility and does not form any intramolecular contact with RRM2 (Fig. 1D). In fact, this N-terminal extension is quite variable between CPEB orthologs and paralogs in terms of both sequence and length. Hence, it is not surprising that the N-terminal extensions of the two proteins differ in structural features.
Structure of the CPEB1 ZZ domain and structural model of CPEB1 RRMs with ZZ domain
The C-terminal region in all CPEB proteins (harboring six highly conserved cysteines and two histidines) was predicted to be a ZZ domain (Hake et al. 1998). Indeed, a recent study reported the NMR structure of the CPEB1 ZZ domain in isolation, classifying it as a ZZ domain with a potential for protein–protein interactions (Merkel et al. 2013). However, previous RNA gel shift assays have shown that this domain of CPEB1 is important for RNA binding (Hake et al. 1998). To unravel the function of this domain, we first solved the solution structure of the CPEB1 ZZ domain. The protein gave a well-dispersed 1H–15N HSQC spectrum characteristic of a folded protein, thus allowing us to obtain a precise ensemble of structures (Table1; Supplemental Fig. S2I). The domain adopts a ββαβ fold that coordinates two zinc ions in a cross-brace fashion (Fig. 2C). Indeed, the hydrophobic helical surface is suitable for interaction with other proteins (Supplemental Fig. S2J). Additionally, the β strands expose three conserved aromatic residues along with positively charged residues, which could provide an ideal binding surface for nucleic acids (Supplemental Fig. S2J). However, titrations of the CPEB1 ZZ domain (in isolation) with the consensus CPE RNA (5′-UUUUUAU-3′) did not show any CSPs (data not shown).
In our studies, the protein construct used for structure determination of the ZZ domain included the linker region between RRM2 and the ZZ domain. We observed long-range NOEs between Leu430 (present in the linker) and Tyr455 (β strand of ZZ domain). This linker region is also present in the protein construct used for the structure determination of CPEB1 tandem RRMs, where it interacts with the β-sheet surface of RRM2 (via Tyr429) (Fig. 2D). Based on this experimental evidence, we generated a structural model of CPEB1 that includes the tandem RRMs and the ZZ domain (Fig. 2D). According to this model, the ZZ domain is positioned in proximity to the β sheet of RRM2, thus implying potential interplay between the RRMs and the ZZ domain. Hence, we do not exclude the possibility that the ZZ domain binds nucleotides flanking the CPE sequence in conjunction with the RRMs.
Solution structure of CPEB4 tandem RRMs in complex with RNA
To elucidate the molecular basis of RNA recognition by CPEB proteins, we solved the solution structure of CPEB4 tandem RRMs in complex with RNA. Titration of the protein with 5′-CUUUA-3′ resulted in CSPs similar to those observed with the consensus CPE (5′-UUUUAU-3′) (Fig. 3A; Supplemental Fig. S3A). In addition, replacement of U1 by C1 resulted in better NMR data for the protein–RNA interface (Supplemental Fig. S3B), although the binding affinity was lower than that of the consensus sequence (Supplemental Fig. S3C,D). With 2544 NOE-based distance restraints (of which 94 were intermolecular) and 77 RDCs, we obtained an ensemble of structures with a root-mean-square deviation (RMSD) of 1.09 Å for backbone atoms (Table 1; Supplemental Fig. S3E). Interestingly, the close proximity of the two RRMs creates a positively charged groove that favors the binding of the short pentanucleotide RNA (Fig. 3B). All five nucleotides are bound by the protein, spanning the β sheet of RRM1 and part of RRM2, resulting in an extended conformation of RNA with all sugar puckers adopting a C2′-endo and all of the bases adopting an anti-conformation (Fig. 3C). The first four nucleotides are bound mainly by RRM1, making it the primary RNA-binding surface, whereas RRM2 binds only A5 (Fig. 3C).
Figure 3.

Structure of CPEB4 tandem RRMs in complex with the RNA (see also Supplemental Fig. S3). (A) Overlay of 1H–15N HSQC spectra of CPEB4 RRM12 in the free state (red) and bound to 5′-CUUUA-3′ (blue) at 30°C. Peaks undergoing CSPs upon RNA binding are labeled. (B) Surface charge representation of CPEB4 RRM12 in complex with RNA (yellow). (C) Stereo view of the representative structure of CPEB4 tandem RRMs in complex with RNA. Protein is shown in ribbon, while the RNA is shown in yellow stick representation. (D–F) Close-up view showing the interactions between amino acids involved in RNA interaction in CPEB4 RRM12 in complex with RNA. Protein side chains (green) involved in interactions with the RNA (yellow) are shown in stick representation. (G) In vivo functional assay to validate CPEB4 residues involved in RNA binding. The extent of RNA probe polyadenylation is plotted as a percentage of competition, quantified from three independent experiments (100% competition was assigned to the wild-type protein, and 0% competition was assigned to the MS2-negative control). Positive controls and wild-type protein are labeled in green, while mutants are labeled in red. The plotted values were obtained from the gels from Supplemental Figure S3, H and I.
CPEB4 sequence-specific interactions and their functional relevance
The structure reveals that RRM1 is the primary RNA-binding domain, which accommodates the first four nucleotides. C1, used instead of U1 to obtain better NMR data, is bound in proximity to the βb–β4 loop, precisely where the C1 base packs against the side chains of Ile144 and Lys147 (Fig. 3D). The following three nucleotides (U2, U3, and U4) interact with the aromatic residues of the two β strands (β1 and β3; described as RNP motifs in RRMs). Phe68 (RNP2) stacks with the base of U3, Tyr110 (RNP1) packs against the sugar ring of U2, and Phe112 (RNP1) stacks with the base of U4. The bases of U2, U3, and U4 are recognized by sequence-specific interactions. First, the base of U2 stacks over Gln150 and is recognized by potential hydrogen bonds with the side chains of Glu131, Lys134, and Tyr136 (all three present in the novel βa–βb strands) (Fig. 3D). The base of U3 then stacks on Phe68 and forms a hydrogen bond at the O4 position of the side chain of Arg152 (Fig. 3E), which is conserved in CPEB2–4 but not in CPEB1 (Supplemental Fig. S1E). The structure reveals additional contacts between U3 and the residues of the β1–α1 loop of RRM2, which is located above it. In particular, Arg182 contacts both the base and phosphate oxygen of U3. Finally, the ribose of U4 stacks on Pro96, with its base stacking on Phe112 with its O4 hydrogen-bonded with the conserved Lys66 (Fig. 3E). In addition, the β2–β3 loop of RRM2 covers the base of U4, with the main chain between Lys215 and Gly216 stacking over the aromatic ring. Moreover, the phosphate backbone of the U-rich sequence is further stabilized by contacts with His97, Lys108, and Phe112 of RRM1. Finally, the last nucleotide (A5) is bound by RRM2. The ribose of A5 interacts with Lys215 (β2–β3 loop), while its base stacks on Phe176 (RNP2). In addition, the C-terminal region of the protein forms a hydrophobic pocket for the A5 base, where the contacts mediated by the side chains of the conserved Tyr253 and Val254 to A5 H2 specify for an adenine at this position (Fig. 3F). Interestingly, RRM2 lacks the conserved aromatic residues on the RNP1 motif, which might explain why the protein does not bind a longer RNA sequence as compared with other tandem RRMs that harbor canonical RNP motifs in both RRMs.
In order to validate the structural results and address their functional relevance in physiological conditions, we set up an in vivo assay in Xenopus laevis oocytes (Supplemental Fig. S3F). This assay is based on the out-competition of endogenous CPEBs by expression of truncated protein variants containing only the RRMs and the ZZ domain (RRM12ZZ) from Xenopus CPEB1 (xCPEB1) and CPEB4 (xCPEB4), whose RRMs share 99% sequence identity with their human orthologs. These variants lacking the N-terminal domain can bind the RNA via the tandem RRMs but do not recruit the cofactors directly responsible for polyadenylation (such as CPSF, Symplekin, or GLD2). Therefore, in their wild-type forms, these variants constitute dominant-negative inhibitors that compete with endogenous CPEBs for binding to the RNA but fail to recruit the polyadenylation machinery (Hake and Richter 1994; Mendez et al. 2000). We followed the polyadenylation of a radioactive emi2 (early mitotic inhibitor-2) 3′ UTR probe, which is one of the well-characterized targets of CPEBs. Overexpression of either xCPEB1 or xCPEB4 RRM12ZZ resulted in the competition of the polyadenylation of the probe (Supplemental Fig. S3F,G). However, mutations of residues that affect either the structure or the RNA-binding capacity alleviated the competition ability of the mutant CPEB1 and CPEB4 truncated variants (Supplemental Fig. S3F). It should be noted that all dominant-negative variants of CPEBs require the binding to RNA, which indicates that they act through RNA binding and not through the sequestration of CPEB cofactors (Hake and Richter 1994; Mendez et al. 2000).
In the in vivo polyadenylation assay for CPEB4, the point mutations of residues interacting with U2–U4 (namely, F68A, K108A, Y110A, F112A, Y136A, K147A, Q150A, and R152A) all lead to a substantial decrease in the capacity to compete the polyadenylation of the emi2 reporter mRNA, which, according to the structural details, must originate from reduced RNA-binding capacity (Fig. 3G; Supplemental Fig. S3H). Mutations of Phe176, Lys215, and Tyr253, which interact with A5, also result in a drastic reduction in their capacity to compete (Fig. 3G; Supplemental Fig. S3H). Moreover, point mutation of the residues important for the positioning of RRM2 in close proximity to RRM1 (namely, W154A [interdomain linker] and F161A [β strand of interdomain linker]) also showed a strong decrease in the ability to compete (Fig. 3G; Supplemental Fig. S3H).
Structure of CPEB1 tandem RRMs in complex with RNA
For CPEB1 tandem RRMs, we obtained identical CSPs for the pentanucleotide RNA (5′-UUUUA-3′) and the consensus CPE sequence (5′-UUUUAU-3′) (Fig. 4A; Supplemental Fig. S4A). Protein–RNA titrations, monitored by 1H–15N HSQC spectra, showed CSPs upon RNA binding, including regions outside the RNA-binding surface (Supplemental Fig. S4B,E). This finding suggests that RNA binding triggers a potential conformational change in the protein structure. The same approach as for CPEB4 was used here, where replacement of U1 by C1 resulted in better NMR data for the protein–RNA interface, and intermolecular NOEs could then be observed (Supplemental Fig. S4C,D). We were therefore able to calculate the structure of CPEB1 RRM12 in complex with the RNA using the structure of CPEB1 RRM12 in complex with 5′-UUUUA-3′ as well as data obtained for the protein–RNA interface using both RNA sequences (5′-UUUUA-3′ and 5′-CUUUA-3′) (Fig. 4B,C). The structure was calculated using 1849 NOE-based distance restraints, including 76 intermolecular ones (Table 1). However, due to the poor stability of this protein–RNA complex, RDCs could not be measured, resulting in lower precision compared with other structures in this study (Supplemental Fig. S4F). Nevertheless, the structure provides details of the RNA recognition mode at the molecular level. Similar to the CPEB4 complex, RRM1 binds the first four nucleotides and represents the main RNA-binding surface, consistent with previous data showing a more predominant role of CPEB1 RRM1 in RNA binding (Hake et al. 1998). However, in CPEB1 tandem RRMs, the surface charge at the protein–RNA interface differs considerably compared with CPEB4 (Fig. 4B).
Figure 4.

Structural model of CPEB1 RRMs in complex with RNA (see also Supplemental Fig. S4). (A) Overlay of 1H–15N HSQC spectra of CPEB1 RRM12 in the free state (red) and in complex with 5′-UUUUA-3′ (blue) measured at 40°C. Peaks undergoing CSPs upon RNA binding are labeled. (B) Surface charge representation of CPEB1 RRM12 in complex with RNA (yellow). (C) Stereo view of the structural model of CPEB1 RRM12 in complex with 5′-UUUUA-3′. The protein is shown in ribbon representation. The RNA is shown in stick representation in yellow, and the bases are labeled. (D–F) Close-up view showing the interactions between amino acids involved in RNA interaction in CPEB1 RRM12 in complex with 5′-UUUUA-3′. Protein side chains (green) involved in interactions with the RNA (yellow) are shown in stick representation. (G) In vivo functional assay to validate CPEB1 residues involved in RNA binding. The extent of RNA probe polyadenylation is plotted as a percentage of competition, quantified from three independent experiments (100% competition was assigned to the wild-type protein, and 0% competition was assigned to the MS2-negative control). Positive controls and wild-type protein are labeled in green, while mutants are labeled in red. The plotted values were obtained from the gels from Supplemental Figure S4, G–I.
The path of the RNA in the CPEB1 RRM12 complex is similar to that observed in CPEB4. First, U1 interacts with Lys324 and Met321, which are residues equivalent to those found in CPEB4 (Fig. 4D). Second, the base of U2 stacks on Gln327 and interacts with Glu311 and Tyr313 in the novel βa–βb strands, while the ribose interacts with Tyr281 (RNP1) as in CPEB4. U3 and U4 are sandwiched between the β-sheet surface of RRM1 and the loops of RRM2, although the interacting side chains are more hydrophobic in CPEB1 than CPEB4. Specifically, the base of U3 stacks on Phe239 (RNP2) and interacts with the side chains of Ile329 (β4 strand of RRM1), Met360, and His358 (from the β1–α1 loop of RRM2) (Fig. 4E), while the base of U4 stacks on Tyr283 (RNP1) and is recognized sequence-specifically via two potential hydrogen bonds to the side chains of Lys237 and Glu265 (Fig. 4E; Supplemental Fig. S4D). Accordingly, individual mutations of Phe239, Tyr313, Gln327, Ile329, Tyr281, and Tyr283, described as interacting residues, lead to a reduction in their capacity to compete polyadenylation (Fig. 4G; Supplemental Fig. S4G).
Finally, the last nucleotide, A5, is again bound exclusively by RRM2. Similar to CPEB4, contacts with the aromatic side chains of Phe353 (RNP2) and Tyr429 can be seen. In addition, the exocyclic N6 amino group of A5 can potentially be hydrogen-bonded with Asp427 (Fig. 4F). Mutation of the A5-interacting residue Phe353 caused a decrease in its capacity to compete (Fig. 4G; Supplemental Fig. S4G). The binding pocket of A5 differs significantly between CPEB1 and CPEB4 and explains the promiscuity of CPEB1 for consensus (5′-UUUUAU-3′) and nonconsensus (5′-UUUUCAU-3′) CPEs (Pique et al. 2008). For CPEB1, at the fifth position of the CPE, the adenine might be replaced by a cytosine, the N4 amino group of which can make a similar hydrogen bond with Asp427. In addition, the C-terminal region in CPEB1 is less hydrophobic, which could accommodate the O4 of cytosine. In CPEB4, Asp427 is replaced by Lys251, and the recognition of A5 is mainly achieved via the hydrophobic contacts from the C-terminal region to H2 of A5 (Fig. 3F).
Similar to CPEB4, point mutants of the residues important for the positioning of RRM2 in close proximity to RRM1 (namely, W331A [interdomain linker] and F338A [β strand of interdomain linker]) for CPEB1 showed a strong decrease in the ability to compete (Fig. 4G; Supplemental Fig. S4G).
Conformational changes upon RNA binding
Although the protein folds of the individual RRMs in the RNA-bound form are essentially the same as those in the free state, comparison of the tertiary structure of the free and bound forms indicates that for both proteins, RNA binding results in a clear conformational change. Superimposition over RRM1 of the two structures shows that RRM2 in both complexes undergoes a rotation of ∼45° toward RRM1 in the direction required to close the RNA-binding cleft (Fig. 5A–D). This mode of binding, where the two β-sheet surfaces of the RRMs appear to close over the RNA target, is analogous to the Venus fly trap mechanism also used by many human receptors for ligand binding (Zhang et al. 2010).
Figure 5.

Conformational changes upon RNA binding (see also Supplemental Fig. S5). (A,C) Stereo view of overlay of CPEB1 (A) and CPEB4 (C) RRM12 in the free state and in complex with RNA. The structures have been overlaid on RRM1. The structure of the complex is shown in ribbon with RRM1 in black, RRM2 and the interdomain linker in green, and RNA as yellow sticks. In the free state, RRM1 is shown in gray, the interdomain linker is in purple, and RRM2 is in orange. The conformational change is depicted with the help of colored arrows. For clarity, the loops in the structures have been smoothened. (B,D) Schematic diagram depicting the conformational change upon RNA binding in CPEB1 (B) and CPEB4 (D) tandem RRMs.
Small movement of the side chains in the hinge region connecting the two domains and, more particularly, Trp331 (CPEB1) and Trp154 (CPEB4) can explain the overall movement between the two RRMs. If RNA binding results in many favorable interactions between the protein and the RNA, it also leads to breakage of a few intraprotein contacts present in the free state of the protein. In CPEB1, contacts between the new βa–βb loop in RRM1 and the α2–β4 loop in RRM2 are lost upon complexation (Supplemental Fig. S5A). Moreover, the closed conformation further stabilizes the hydrophobic contacts between the two RRMs in CPEB1. This structural change observed upon RNA binding contributes to rationalizing the large CSPs observed in regions outside the RNA-binding surface in CPEB1, in particular at the hinge region between the two RRMs (interdomain linker and N-terminal extension) (Supplemental Fig. S5E). In CPEB4, a similar conformational change is observed; however, since the two domains exhibit more flexibility at the hinge region than in CPEB1 (since the contacts from the N-terminal extension are missing), smaller CSPs occur in the region outside the RNA-binding surface (Supplemental Fig. S5E). Furthermore, RNA binding leads to the neutralization of the positively charged interdomain interface that stabilizes the RNA-bound closed conformation of CPEB4 (Supplemental Fig. S5B–D).
Discussion
Novel features in CPEB RRMs: interdomain interactions and extensions/insertions
While most of the tandem RRMs are separated by a flexible interdomain linker and therefore do not interact in the free state, such as sex-lethal (Sxl), Nucleolin, PTB RRM12, Hrp1, Npl3, and HuR (Crowder et al. 1999; Allain et al. 2000; Perez-Canadillas 2006; Vitali et al. 2006; Skrisovska and Allain 2008; Wang et al. 2013), only a few tandem RRMs show interactions; namely, PTB RRM34, Prp24, FIR, and, more recently U2AF65, hnRNPA1, and hnRNPL (Vitali et al. 2006; Bae et al. 2007; Cukier et al. 2010; Mackereth et al. 2011; Barraud and Allain 2013; Zhang et al. 2013). Such fixed orientation of two RRMs relative to each other often has a direct implication in the mechanism of action of the proteins. For example, the PTB RRM3 and PTB RRM4 interact via the α helices, which results in the positioning of β-sheet surfaces on opposite sides and complete accessibility for RNA binding (Oberstrass et al. 2005). This has functional implications in the mechanism of action of PTB via RNA looping (Lamichhane et al. 2010).
The structures of CPEB1 and CPEB4 tandem RRMs reveal yet another arrangement for the respective positions of the two RRMs. Superimposition of all of the structures of tandem RRMs in the free state clearly reveals that the CPEBs describe an unprecedented arrangement of the two RRMs (Supplemental Fig. S6A). While the interaction surfaces in the other structures involve a direct inter-RRM interaction, the fixed orientation of the RRMs in CPEB1 and CPEB4 is due primarily to the interaction between regions outside the RRM per se; namely, the interdomain linker, the region N-terminal to RRM1, and the new loop extension βa–βb of RRM1.
In addition, CPEB RRMs contain novel extensions to the canonical RRM fold. RRM1 contains an unprecedented extension downstream from helix α2, which forms a β hairpin (βa–βb) that runs anti-parallel to the β4 strand and interacts with helix α2. This small extension not only extends the β sheet to a six-stranded one (compared with a four-stranded one in PABP) (Fig. 1E–G) but, in CPEB1, also plays a key role in stabilizing the interaction with RRM2 in the free protein (Fig. 1C). Moreover, the β sheet of RRM2 is also extended via the interdomain linker, which forms an additional anti-parallel β strand with the β2 strand of RRM2 (Fig. 1C). This extension is reminiscent of the β-sheet extension found in PTB RRM2 and PTB RRM3 (Supplemental Fig. S6B,C; Oberstrass et al. 2005). Furthermore, while the additional β strand in PTB extends the RNA-binding surface for two more nucleotides, in CPEB, this interaction stabilizes the orientation of the two RRMs.
The last unusual extension is found only in CPEB1, where the region upstream of RRM1 interacts with the helical face of RRM2 (Fig. 2A). Although such an intramolecular interaction has not been reported in other RRMs, the contact found here closely resembles intermolecular interactions between small protein fragments and the hydrophobic helical face of RRMs (Kielkopf et al. 2001, 2004; Corsini et al. 2007; Konde et al. 2010; Joshi et al. 2011; Safaee et al. 2012). Although CPEB1 RRM2 cannot be classified as a UHM (U2AF homology motif), the interaction of Trp220 with the apolar helical face of RRM2 (in CPEB1) is reminiscent of the UHM–peptide interaction (Supplemental Fig. S6D,E; Kielkopf et al. 2001).
Functional implication of a unique mode of RNA recognition by tandem RRMs
Comparison of the structures of CPEB1 and CPEB4 tandem RRMs bound to RNA revealed yet another mechanism through which tandem RRMs recognize ssRNA. Indeed, superimposition of four tandem RRM structures bound to RNA over RRM1 unveils a surprisingly large range of possibilities for the position of the second RRM relative to the first, as shown in the following examples. For PABP and Sxl, the β-sheet surfaces are side by side, with RRM2 on the left of RRM1 (Deo et al. 1999; Handa et al. 1999). In TAR-DNA-binding protein (TDP-43), the RRMs are again side by side but with RRM2 on the right of RRM1 (Lukavsky et al. 2013). In the case of CPEB1 and CPEB4, the RRM2 is positioned above and almost perpendicular to RRM1 (Fig. 6A). Similar to TDP-43 but unlike all other tandem RRMs, this configuration allows binding of the RNA 5′ end by RRM1 and binding of the 3′ end by RRM2. However, more importantly, this unusual and unique topology permits RNA binding via a Venus fly trap mechanism. Indeed, upon binding, U3 and U4 are literally sandwiched between the RRMs, contacts that would not be possible if the two RRMs were placed side by side.
Figure 6.

Molecular mechanism of assembly of translation regulatory complexes by CPEB1 (see also Supplemental Fig. S6). (A) Overlay of CPEB4 RRM12 in complex with three previously characterized tandem RRM–RNA complexes: PABP (1CVJ), Sxl (1B7F), and TDP-43 (4BS2). All structures were overlaid on the RRM1 domain (shown in gray for all structures). The RRM2 domain is shown in pink, blue, orange, and green for PABP, Sxl, CPEB4, and TDP43, respectively. RNA is shown in tube representation in the corresponding RRM2 colors, with the 5′ and 3′ ends labeled in colored circles. An individual structure is shown around the overlay in the same orientation. For clarity, the loops in the structures have been smoothened. (B) Structural model of CPEB1 RRM12ZZ in complex with the RNA in stereo view. (C) A model depicting the assembly of a translational regulatory complex by CPEB1. mRNA is shown in black, with the 5′ and 3′ ends labeled in blue. A model of full-length CPEB1 is shown in ribbon representation. (Red) N-terminal domain; (gray) RRM1; (orange) RRM2; (cyan) ZZ domain. The activation phosphorylation site is shown in the N-terminal domain of CPEB1 as a green star. Other protein factors of this complex are schematically depicted, with ovals in different colors and labeled inside.
This mechanism of RNA binding might have a biological relevance to how CPEBs find their target sequence on mRNAs. Our studies demonstrate that the tandem RRMs of CPEB proteins recognize a pentanucleotide RNA sequence (5′-UUUUA-3′) in a sequence-specific fashion. RRM1 is the main RNA-binding site, which binds the first four uracils, while RRM2 binds the conserved 3′ adenine of consensus CPEs. On the basis of the unusual mode of binding described here for CPEB1 and CPEB4, we propose that CPEB proteins find their target CPE sequence by a two-step mechanism. In the first step, CPEBs are targeted to the 3′ UTRs due to the high density of poly(U)3–4 around the CPEs (Supplemental Fig. S5F). This is mediated by the RRM1 domain, which recognizes the poly(U)3–4 in a sequence-specific fashion. However, considering the weaker affinity of RRM1 in isolation for poly(U)3–4 sequences, the CPEB tandem RRMs are likely to remain bound to the mRNA in their open conformation. In the second step, the CPEBs scan the 3′ UTR for an authentic CPE, which is characterized by an adenine (or a cytosine in nonconsensus CPEs) after a stretch of four uracils. Once it encounters an adenine after a stretch of four uracils, the protein undergoes a conformational change, allowing RRM2 to bind in concert with RRM1 to the CPE with higher affinity (Supplemental Fig. S5F). This mechanism resembles in part the one used by the tandem RRMs of U2AF65 to discriminate between long and short poly(U) stretches. It is only when long poly(U) stretches are found that the tandem RRMs of U2AF65 bind the RNA; otherwise, RRM1 binding is hindered because of its interaction with RRM2 (Mackereth et al. 2011).
CPEB and paralogs
All four CPEB paralogs can assemble mRNPs that repress or activate translation. Although the precise composition of these complexes for each of the four CPEBs is still unknown, the switch from assembling a repressor to assembling an activator complex is most probably mediated by post-translational modifications in the N-terminal domain of the proteins (Fernandez-Miranda and Mendez 2012; Weill et al. 2012). Accordingly, this N-terminal domain is the most divergent region between all four CPEB paralogs, reflecting that they are most likely regulated by different signal transduction pathways and therefore mediate different cellular responses. Moreover, CPEBs appear to be coexpressed in different tissues or cell types, where they regulate overlapping mRNA populations (Fernandez-Miranda and Mendez 2012; Weill et al. 2012). In this scenario, CPEBs would compete for their target mRNAs to assemble translational repression or activation complexes. In this study, we show that the two CPEB subfamilies (CPEB1 and CPEB2–4) recognize the same cis-acting motif using a very similar RNA-binding mode despite only 48% identity in the RRMs between CPEB1 and CPEB4. This suggests that the two subparalog families can be functionally replaced by each other, as observed during mitotic and meiotic cell cycles in Xenopus oocytes (Igea and Mendez 2010; Novoa et al. 2010). However, even though the structures of the tandem RRMs of CPEB1 and CPEB4 appear overall to be very similar, we observed a number of differences that could account for the functional differences seen between the two paralog subfamilies. The most obvious one is the position of the region immediately upstream of RRM1 that interacts with RRM2 in CPEB1, while this region is unstructured in CPEB4 (Supplemental Fig. S1A,B). Consequently, RRM2 exposes a very different surface, and the spatial orientation of the N-terminal domain differs in the two paralogs. Tandem RRMs of CPEB1 and CPEB4 also present striking differences on the β-sheet surface (Supplemental Fig. S6F,G). Furthermore, the difference in the recognition of A5 between CPEB1 and CPEB4 suggests how the fifth position of the CPE might be replaced by a cytosine in nonconsensus CPEs. Molecular details at the A5-binding pocket suggest that CPEB1 might be more permissive for nonconsensus CPEs compared with CPEB2–4, in agreement with a previous report (Pique et al. 2008). In conclusion, the functional differences observed between the paralogs might reside not only in the difference in length and amino acid composition of the N-terminal domain but also in the different surfaces exposed by both RRMs in each paralog as well as their binding to RNA. However, previous studies have shown that both CPEB1 and CPEB4 can bind the CPE-containing RNA with very similar affinities. Dissociation constants of 86 nM ± 28 nM (for CPEB1) and 102 nM ± 4 nM (for CPEB4) were obtained from RNA gel shift assays using full-length CPEBs and a CPE-containing RNA probe, calculated from (Novoa et al. 2010). In the present study, the Kds obtained by NMR titration experiments are in the low micromolar range (Supplemental Fig. S3C,D). Two reasons could explain the higher Kds in our experiments. First, in contrast to previous studies, the NMR experiments were performed only with the tandem RRM constructs and not with the full-length protein, confirming that the ZZ domain contributes to RNA-binding affinity (Hake et al. 1998). Second, NMR titrations were performed with a short pentanucleotide RNA containing only one CPE, while the previously reported gel shift assays were performed with an RNA probe containing multiple CPEs (Novoa et al. 2010).
The CPEB ZZ domain is an interaction domain with a dual function
The CPEB1 ZZ domain has close structural similarity to the B-box, RING, and other binuclear ZZ domains that have been implicated in mediating interactions with other proteins rather than nucleic acids (Zheng et al. 2000). The hydrophobic region present in the α helix of the CPEB1 ZZ domain suggests its potential to interact with other proteins similar to B-box proteins (Supplemental Figs. S2J, S6H). Interestingly, this surface shows variability among CPEB paralogs, suggesting different subsets of interacting protein partners for each CPEB paralog (based on sequence alignment and homology model of the CPEB4 ZZ domain) (Supplemental Fig. S6I). However, this is most likely not the only function of the ZZ domain. In the competition assays, the RRMs require the ZZ domain to efficiently compete with endogenous CPEBs (Supplemental Fig. S3J,K), thereby indicating that this motif has a positive effect on the interaction of the protein with RNA. Similar conclusions have been obtained in vitro with recombinant CPEBs (Hake et al. 1998; Huang et al. 2006). In our model of the RRMs and the ZZ domain, the position of the ZZ domain relative to RRM2 suggests that the ZZ domain interacts with the nucleotides flanking the 3′ end of the CPE (Fig. 6B). Also, the residues in the β hairpin of the ZZ domain that potentially interact with the RRMs and the RNA are highly conserved in all of the CPEB paralogs, thus reinforcing our structural model. Altogether, the ZZ domain contributes to increasing the affinity for the target RNAs, allowing CPEB to bind to both consensus (5′-UUUUAU-3′) and nonconsensus (5′-UUUUAC-3′) CPEs (Pique et al. 2008). Therefore, we predict that the ZZ domain has a dual function, as it contributes to increasing the affinity of the RRMs for the CPEs and to promoting interactions with other protein factors in the translational repression/activation complexes assembled by CPEBs. Moreover, owing to the differences in the potential protein interaction surfaces, CPEB paralogs will recruit a different subset of interacting proteins.
Model for the assembly of translational regulation complexes by CPEB1
Our structural study on the CPEB proteins in complex with RNA, in combination with previous biochemical studies, sheds light on the assembly of translation repression/activation complexes. In the structural model of CPEB1, both the N-terminal and ZZ domains are located in proximity to RRM2 (Fig. 6B). In addition, based on the structural work of CPEB1 and CPEB4 RRMs in complex with the RNA, we show that the RNA binds with the 5′ end on RRM1 and brings the 3′ end to RRM2. This directionality in RNA binding differs from most previously characterized tandem RRM–RNA complexes and might have biological relevance. In this regard, such a binding mode brings the 3′ end of the mRNA on RRM2 near the N-terminal regulatory domain and the ZZ domain. This favors the interaction of these domains with the other protein factors, most of which are assembled at the 3′ end downstream from the CPE, leading to the assembly of the translational repression and/or activation complexes by CPEB1 (Fig. 6C). Indeed, if the 5′ end RNA would bind RRM2 in a fashion similar to other tandem RRM–RNA complexes, both the N-terminal domain and the ZZ domain would be positioned away from the 3′ end of the mRNA, where polyadenylation-specific factors should assemble. Moreover, most of the CPEs have been described upstream of the polyadenylation hexanucleotide. When CPEs are placed downstream from this hexanucleotide, they tend to be less efficient or even have a negative effect on polyadenylation under high CPEB1 levels (Pique et al. 2008). These findings are now explained by our model. However, this hypothesis would need further support through the identification of specific interacting partners of the N-terminal regulatory domain and the ZZ domain of CPEB1.
Materials and methods
Recombinant expression of CPEB protein constructs
Protein constructs corresponding to different lengths of the RNA-binding domain of CPEB1 (NP_001275748.1) and CPEB4 (AAH36899.1) were recombinantly expressed in Escherichia coli. For CPEB4, a splice variant with a short N-terminal domain was used (isoform CRA_d, 322 amino acids). His-tagged proteins from the soluble fraction were purified by Ni2+ affinity and size exclusion chromatography. Details of protein constructs, vectors, and expression protocol are described in the Supplemental Material.
Preparation of protein–RNA complexes
The unlabeled oligoribonucleotides used in this study were 5′-UUUUUAU-3′, 5′-UUUUUAAU-3′, 5′-UUUUUAAA-3′, 5′-UUUUAU-3′, 5′-UUUAU-3′, 5′-UUUUA-3′, 5′-CUUUUA-3′, and 5′-CUUUA-3′ and were purchased from Dharmacon/Thermofisher. The protein–RNA complexes were prepared by titrating RNA into protein and monitoring by NMR spectroscopy at either 30°C or 40°C.
NMR spectroscopy
All NMR experiments were performed on a Bruker AVIII-500 MHz, AVIII-600 MHz, AVIII-700 MHz, and Avance-900 MHz, all equipped with cryoprobes. Data processing was performed with TopSpin2.1/TopSpin3.0 (Bruker), and analysis was performed with Sparky (http://www.cgl.ucsf.edu/home/sparky).
For CPEB1/4 tandem RRMs, the following experiments were used for the protein backbone, aliphatic, and aromatic side chain assignments: two-dimensional (2D) 15N–1H HSQC, 2D 15N TROSY, 2D 13C–1H HSQC, three-dimensional (3D) trHNCA (Salzmann et al. 1998), 3D trHNCOCA, 3D trHNCO, 3D trHNCACO, 3D trHNCACB, 3D HCcH TOCSY, 3D hCCH TOCSY, 3D 15N, and 13C-edited NOESYs, all recorded in H2O.
To assign the resonances of unlabeled RNA, we recorded 2D 1H–1H TOCSY, 2D 1H–1H NOESY, and 2D 13C F1-filtered F2-filtered NOESY (Peterson et al. 2004). Protein–RNA intermolecular NOEs were obtained by 2D 1H–1H NOESY, 2D 1H–1H F1–13C-filtered F2–13C-edited NOESY (Zwahlen et al. 1997) and 3D 13C F1-edited, F3-filtered NOESY-HSQC spectrum (Zwahlen et al. 1997), all recorded in D2O solution. We used a mixing time (τm) of 120 msec for the NOESY spectra of free proteins and 150 msec for the protein–RNA complex. RDCs, 15N NMR relaxation, and Kd measurements are described in the Supplemental Material.
Structure calculation and refinement
The 2D 1H–1H NOESY, 3D 15N-edited NOESY, and 3D 13C-edited NOESY-HSQC were used as input spectra for the automated NOESY peak picking and NOE assignment method with ATNOS/CANDID (Herrmann et al. 2002a). The ATNOS/CANDID approach included the chemical shift list obtained from sequence-specific assignments and the NOESY spectra. Intramolecular RNA and protein–RNA intermolecular NOEs were manually assigned and calibrated based on fixed interatomic distances. Further structure calculations were then performed in CYANA3.0. In addition to the NOE-based distance constraints, RDC restraints were included in CYANA calculations. Starting from random structures, 200 preliminary structures were calculated, and the 20 conformers with the lowest target function values were energy-minimized by a restrained simulated annealing run in implicit water with the SANDER module of AMBER9 (Case et al. 2005) using the ff99 force field. For the CPEB1 ZZ domain, we recorded a conventional set of NMR experiments for the backbone and side chain assignment. Similar to RRMs, the structure calculations were performed by ATNOS/CANDID (Herrmann et al. 2002a) with further refinement in AMBER9 (Supplemental Material; Case et al. 2005). All figures of molecular structures were generated by MOLMOL (Koradi et al. 1996) or PYMOL (http://www.pymol.org).
Competition assay
Stage VI oocytes were obtained from X. laevis females as described in de Moor and Richter (1999). Polyadenylated RNA (0.036 pmol) encoding for CPEB1 or CPEB4 RRM12ZZ wild type or mutants were injected into oocytes and incubated overnight at 18°C. After 16 h, 4.6 fmol of radiolabeled emi2 3′ UTR RNA probe was injected into the oocytes. Maturation was induced by treatment with 10 µM progesterone. Total RNA was extracted (Ultraspec RNA isolation system, Biotecx) from oocytes collected at stage VI and 2 h after germinal vesicle breakdown (GVBD). The RNA from 1.5 oocytes was analyzed in 4% acryl-urea gel and visualized by autoradiography. The results were analyzed and quantified with Fiji. In order to assign a percentage of competition, for CPEB1 assays, we calculated the percentage of deadenylated probe, while for CPEB4, the distance of the median polyadenylation was taken into account. In both cases, 0% competition was assigned to MS2 control, and 100% competition was assigned to the wild-type variant. The percentage of competition shown by the mutants was normalized according to these values.
Accession numbers
The coordinates for the 20 lowest-energy structures for CPEB1 RRM12, CPEB1 RRM12 (in complex with RNA), the CPEB1 ZZ domain, CPEB4 RRM12, and CPEB4 RRM12 (in complex with RNA) are deposited in the Protein Data Bank under accession numbers 2MKH, 2MKK, 2MKE, 2MKJ, and 2MKI, respectively. The chemical shifts and structural restraints for CPEB1 RRM12, CPEB1 RRM12 (in complex with RNA), the CPEB1 ZZ domain, CPEB4 RRM12, and CPEB4 RRM12 (in complex with RNA) are deposited in the BioMagResBank under accession numbers 19775, 19778, 19771, 19777, and 19776, respectively.
Acknowledgments
We are thankful to Dr. Fred Franz Damberger, Dr. Mario Schubert, Dr. Christophe Maris, and Thea Stahel for help with NMR spectroscopy; Dr. Julien Boudet and Dr. Pierre Barraud for helpful advice and discussions for RDC measurements; and Dr. Markus Blatter and Dr. Olivier Duss for assistance in the structure calculations. This work was supported by Swiss National Science Foundation (SNF) grant 118118.
Footnotes
Supplemental material is available for this article.
Article is online at http://www.genesdev.org/cgi/doi/10.1101/gad.241133.114.
References
- Alexandrov IM, Ivshina M, Jung DY, Friedline R, Ko HJ, Xu M, O’Sullivan-Murphy B, Bortell R, Huang YT, Urano F, et al. 2012. Cytoplasmic polyadenylation element binding protein deficiency stimulates PTEN and Stat3 mRNA translation and induces hepatic insulin resistance. PLoS Genet 8: e1002457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allain FH, Gilbert DE, Bouvet P, Feigon J 2000. Solution structure of the two N-terminal RNA-binding domains of nucleolin and NMR study of the interaction with its RNA target. J Mol Biol 303: 227–241 [DOI] [PubMed] [Google Scholar]
- Bae E, Reiter NJ, Bingman CA, Kwan SS, Lee D, Phillips GN Jr, Butcher SE, Brow DA 2007. Structure and interactions of the first three RNA recognition motifs of splicing factor prp24. J Mol Biol 367: 1447–1458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barraud P, Allain FH 2013. Solution structure of the two RNA recognition motifs of hnRNP A1 using segmental isotope labeling: how the relative orientation between RRMs influences the nucleic acid binding topology. J Biol NMR 55: 119–138 [DOI] [PubMed] [Google Scholar]
- Bava FA, Eliscovich C, Ferreira PG, Minana B, Ben-Dov C, Guigo R, Valcarcel J, Mendez R 2013. CPEB1 coordinates alternative 3′-UTR formation with translational regulation. Nature 495: 121–125 [DOI] [PubMed] [Google Scholar]
- Belloc E, Mendez R 2008. A deadenylation negative feedback mechanism governs meiotic metaphase arrest. Nature 452: 1017–1021 [DOI] [PubMed] [Google Scholar]
- Belloc E, Pique M, Mendez R 2008. Sequential waves of polyadenylation and deadenylation define a translation circuit that drives meiotic progression. Biochem Soc Trans 36: 665–670 [DOI] [PubMed] [Google Scholar]
- Berger-Sweeney J, Zearfoss NR, Richter JD 2006. Reduced extinction of hippocampal-dependent memories in CPEB knockout mice. Learn Mem 13: 4–7 [DOI] [PubMed] [Google Scholar]
- Case DA, Cheatham TE 3rd, Darden T, Gohlke H, Luo R, Merz KM Jr, Onufriev A, Simmerling C, Wang B, Woods RJ 2005. The Amber biomolecular simulation programs. J Comput Chem 26: 1668–1688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corsini L, Bonnal S, Basquin J, Hothorn M, Scheffzek K, Valcarcel J, Sattler M 2007. U2AF-homology motif interactions are required for alternative splicing regulation by SPF45. Nat Struct Mol Biol 14: 620–629 [DOI] [PubMed] [Google Scholar]
- Crowder SM, Kanaar R, Rio DC, Alber T 1999. Absence of interdomain contacts in the crystal structure of the RNA recognition motifs of sex-lethal. Proc Natl Acad Sci 96: 4892–4897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cukier CD, Hollingworth D, Martin SR, Kelly G, Diaz-Moreno I, Ramos A 2010. Molecular basis of FIR-mediated c-myc transcriptional control. Nat Struct Mol Biol 17: 1058–1064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Moor CH, Richter JD 1999. Cytoplasmic polyadenylation elements mediate masking and unmasking of cyclin B1 mRNA. EMBO J 18: 2294–2303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deo RC, Bonanno JB, Sonenberg N, Burley SK 1999. Recognition of polyadenylate RNA by the poly(A)-binding protein. Cell 98: 835–845 [DOI] [PubMed] [Google Scholar]
- Eliscovich C, Peset I, Vernos I, Mendez R 2008. Spindle-localized CPE-mediated translation controls meiotic chromosome segregation. Nat Cell Biol 10: 858–865 [DOI] [PubMed] [Google Scholar]
- Ernoult-Lange M, Wilczynska A, Harper M, Aigueperse C, Dautry F, Kress M, Weil D 2009. Nucleocytoplasmic traffic of CPEB1 and accumulation in Crm1 nucleolar bodies. Mol Biol Cell 20: 176–187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Miranda G, Mendez R 2012. The CPEB-family of proteins, translational control in senescence and cancer. Ageing Res Rev 11: 460–472 [DOI] [PubMed] [Google Scholar]
- Fox CA, Sheets MD, Wickens MP 1989. Poly(A) addition during maturation of frog oocytes: distinct nuclear and cytoplasmic activities and regulation by the sequence UUUUUAU. Genes Dev 3: 2151–2162 [DOI] [PubMed] [Google Scholar]
- Hake LE, Richter JD 1994. CPEB is a specificity factor that mediates cytoplasmic polyadenylation during Xenopus oocyte maturation. Cell 79: 617–627 [DOI] [PubMed] [Google Scholar]
- Hake LE, Mendez R, Richter JD 1998. Specificity of RNA binding by CPEB: requirement for RNA recognition motifs and a novel zinc finger. Mol Cell Biol 18: 685–693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Handa N, Nureki O, Kurimoto K, Kim I, Sakamoto H, Shimura Y, Muto Y, Yokoyama S 1999. Structural basis for recognition of the tra mRNA precursor by the sex-lethal protein. Nature 398: 579–585 [DOI] [PubMed] [Google Scholar]
- Herrmann T, Guntert P, Wuthrich K 2002a. Protein NMR structure determination with automated NOE assignment using the new software CANDID and the torsion angle dynamics algorithm DYANA. J Mol Biomol 319: 209–227 [DOI] [PubMed] [Google Scholar]
- Herrmann T, Guntert P, Wuthrich K 2002b. Protein NMR structure determination with automated NOE-identification in the NOESY spectra using the new software ATNOS. J Biomol NMR 24: 171–189 [DOI] [PubMed] [Google Scholar]
- Huang YS, Kan MC, Lin CL, Richter JD 2006. CPEB3 and CPEB4 in neurons: analysis of RNA-binding specificity and translational control of AMPA receptor GluR2 mRNA. EMBO J 25: 4865–4876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Igea A, Mendez R 2010. Meiosis requires a translational positive loop where CPEB1 ensues its replacement by CPEB4. EMBO J 29: 2182–2193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi A, Coelho MB, Kotik-Kogan O, Simpson PJ, Matthews SJ, Smith CW, Curry S 2011. Crystallographic analysis of polypyrimidine tract-binding protein–Raver1 interactions involved in regulation of alternative splicing. Structure 19: 1816–1825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kan MC, Oruganty-Das A, Cooper-Morgan A, Jin G, Swanger SA, Bassell GJ, Florman H, van Leyen K, Richter JD 2010. CPEB4 is a cell survival protein retained in the nucleus upon ischemia or endoplasmic reticulum calcium depletion. Mol Cell Biol 30: 5658–5671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kielkopf CL, Rodionova NA, Green MR, Burley SK 2001. A novel peptide recognition mode revealed by the X-ray structure of a core U2AF35/U2AF65 heterodimer. Cell 106: 595–605 [DOI] [PubMed] [Google Scholar]
- Kielkopf CL, Lucke S, Green MR 2004. U2AF homology motifs: protein recognition in the RRM world. Genes Dev 18: 1513–1526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konde E, Bourgeois B, Tellier-Lebegue C, Wu W, Perez J, Caputo S, Attanda W, Gasparini S, Charbonnier JB, Gilquin B, et al. 2010. Structural analysis of the Smad2–MAN1 interaction that regulates transforming growth factor-β signaling at the inner nuclear membrane. Biochemistry 49: 8020–8032 [DOI] [PubMed] [Google Scholar]
- Koradi R, Billeter M, Wuthrich K 1996. MOLMOL: a program for display and analysis of macromolecular structures. J Mol Graph 14: 51–55, 29–32 [DOI] [PubMed] [Google Scholar]
- Lamichhane R, Daubner GM, Thomas-Crusells J, Auweter SD, Manatschal C, Austin KS, Valniuk O, Allain FH, Rueda D 2010. RNA looping by PTB: evidence using FRET and NMR spectroscopy for a role in splicing repression. Proc Natl Acad Sci 107: 4105–4110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin CL, Evans V, Shen S, Xing Y, Richter JD 2010. The nuclear experience of CPEB: implications for RNA processing and translational control. RNA 16: 338–348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukavsky PJ, Daujotyte D, Tollervey JR, Ule J, Stuani C, Buratti E, Baralle FE, Damberger FF, Allain FH 2013. Molecular basis of UG-rich RNA recognition by the human splicing factor TDP-43. Nat Struct Mol Biol 20: 1443–1449 [DOI] [PubMed] [Google Scholar]
- Mackereth CD, Madl T, Bonnal S, Simon B, Zanier K, Gasch A, Rybin V, Valcarcel J, Sattler M 2011. Multi-domain conformational selection underlies pre-mRNA splicing regulation by U2AF. Nature 475: 408–411 [DOI] [PubMed] [Google Scholar]
- McGrew LL, Dworkin-Rastl E, Dworkin MB, Richter JD 1989. Poly(A) elongation during Xenopus oocyte maturation is required for translational recruitment and is mediated by a short sequence element. Genes Dev 3: 803–815 [DOI] [PubMed] [Google Scholar]
- Mendez R, Murthy KG, Ryan K, Manley JL, Richter JD 2000. Phosphorylation of CPEB by Eg2 mediates the recruitment of CPSF into an active cytoplasmic polyadenylation complex. Mol Cell 6: 1253–1259 [DOI] [PubMed] [Google Scholar]
- Mendez R, Barnard D, Richter JD 2002. Differential mRNA translation and meiotic progression require Cdc2-mediated CPEB destruction. EMBO J 21: 1833–1844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merkel DJ, Wells SB, Hilburn BC, Elazzouzi F, Perez-Alvarado GC, Lee BM 2013. The C-terminal region of cytoplasmic polyadenylation element binding protein is a ZZ domain with potential for protein-protein interactions. J Mol Biol 425: 2015–2026 [DOI] [PubMed] [Google Scholar]
- Novoa I, Gallego J, Ferreira PG, Mendez R 2010. Mitotic cell-cycle progression is regulated by CPEB1 and CPEB4-dependent translational control. Nat Cell Biol 12: 447–456 [DOI] [PubMed] [Google Scholar]
- Oberstrass FC, Auweter SD, Erat M, Hargous Y, Henning A, Wenter P, Reymond L, Amir-Ahmady B, Pitsch S, Black DL, et al. 2005. Structure of PTB bound to RNA: specific binding and implications for splicing regulation. Science 309: 2054–2057 [DOI] [PubMed] [Google Scholar]
- Ortiz-Zapater E, Pineda D, Martinez-Bosch N, Fernandez-Miranda G, Iglesias M, Alameda F, Moreno M, Eliscovich C, Eyras E, Real FX, et al. 2012. Key contribution of CPEB4-mediated translational control to cancer progression. Nat Med 18: 83–90 [DOI] [PubMed] [Google Scholar]
- Pavlopoulos E, Trifilieff P, Chevaleyre V, Fioriti L, Zairis S, Pagano A, Malleret G, Kandel ER 2011. Neuralized1 activates CPEB3: a function for nonproteolytic ubiquitin in synaptic plasticity and memory storage. Cell 147: 1369–1383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Canadillas JM 2006. Grabbing the message: structural basis of mRNA 3′UTR recognition by Hrp1. EMBO J 25: 3167–3178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson RD, Theimer CA, Wu H, Feigon J 2004. New applications of 2D filtered/edited NOESY for assignment and structure elucidation of RNA and RNA–protein complexes. J Biomol NMR 28: 59–67 [DOI] [PubMed] [Google Scholar]
- Pique M, Lopez JM, Foissac S, Guigo R, Mendez R 2008. A combinatorial code for CPE-mediated translational control. Cell 132: 434–448 [DOI] [PubMed] [Google Scholar]
- Richter JD 2007. CPEB: a life in translation. Trends Biochem Sci 32: 279–285 [DOI] [PubMed] [Google Scholar]
- Richter JD, Lasko P 2011. Translational control in oocyte development. Cold Spring Harb Perspect Biol 3: a002758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Safaee N, Kozlov G, Noronha AM, Xie J, Wilds CJ, Gehring K 2012. Interdomain allostery promotes assembly of the poly(A) mRNA complex with PABP and eIF4G. Mol Cell 48: 375–386 [DOI] [PubMed] [Google Scholar]
- Salzmann M, Pervushin K, Wider G, Senn H, Wuthrich K 1998. TROSY in triple-resonance experiments: new perspectives for sequential NMR assignment of large proteins. Proc Natl Acad Sci 95: 13585–13590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Setoyama D, Yamashita M, Sagata N 2007. Mechanism of degradation of CPEB during Xenopus oocyte maturation. Proc Natl Acad Sci 104: 18001–18006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skrisovska L, Allain FH 2008. Improved segmental isotope labeling methods for the NMR study of multidomain or large proteins: application to the RRMs of Npl3p and hnRNP L. J Mol Biol 375: 151–164 [DOI] [PubMed] [Google Scholar]
- Udagawa T, Farny NG, Jakovcevski M, Kaphzan H, Alarcon JM, Anilkumar S, Ivshina M, Hurt JA, Nagaoka K, Nalavadi VC, et al. 2013. Genetic and acute CPEB1 depletion ameliorate fragile X pathophysiology. Nat Med 19: 1473–1477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitali F, Henning A, Oberstrass FC, Hargous Y, Auweter SD, Erat M, Allain FH 2006. Structure of the two most C-terminal RNA recognition motifs of PTB using segmental isotope labeling. EMBO J 25: 150–162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang XP, Cooper NG 2010. Comparative in silico analyses of cpeb1-4 with functional predictions. Bioinform Biol insights 4: 61–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Zeng F, Liu Q, Liu H, Liu Z, Niu L, Teng M, Li X 2013. The structure of the ARE-binding domains of Hu antigen R (HuR) undergoes conformational changes during RNA binding. Acta Crystallogr D Biol Crystallogr 69: 373–380 [DOI] [PubMed] [Google Scholar]
- Weill L, Belloc E, Bava FA, Mendez R 2012. Translational control by changes in poly(A) tail length: recycling mRNAs. Nat Struct Mol Biol 19: 577–585 [DOI] [PubMed] [Google Scholar]
- Zhang F, Klebansky B, Fine RM, Liu H, Xu H, Servant G, Zoller M, Tachdjian C, Li X 2010. Molecular mechanism of the sweet taste enhancers. Proc Natl Acad Sci 107: 4752–4757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Zeng F, Liu Y, Zhao Y, Lv H, Niu L, Teng M, Li X 2013. Crystal structures and RNA-binding properties of the RNA recognition motifs of heterogeneous nuclear ribonucleoprotein L: insights into its roles in alternative splicing regulation. J Biol Chem 288: 22636–22649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng N, Wang P, Jeffrey PD, Pavletich NP 2000. Structure of a c-Cbl–UbcH7 complex: RING domain function in ubiquitin–protein ligases. Cell 102: 533–539 [DOI] [PubMed] [Google Scholar]
- Zwahlen C, Legault P, Vincent SJF, Greenblatt J, Konrat R, Kay LE 1997. Methods for measurement of intermolecular NOEs by multinuclear NMR spectroscopy: application to a bacteriophage λ N-peptide/boxB RNA complex. J Am Chem Soc 119: 6711–6721 [Google Scholar]
- Zweckstetter M, Bax A 2000. Prediction of sterically induced alignment in a dilute liquid crystalline phase: aid to protein structure determination by NMR. J Am Chem Soc 122: 3791–3792 [Google Scholar]