

Abstract
Aberrant expression, activation, and stabilization of epidermal growth factor receptor (EGFR) are causally associated with several human cancers. Post-translational modifications and protein-protein interactions directly modulate the signaling and trafficking of the EGFR. Activated EGFR is internalized by endocytosis and then either recycled back to the cell surface or degraded in the lysosome. EGFR internalization and recycling also occur in response to stresses that activate p38 MAP kinase. Mass spectrometry was applied to comprehensively analyze the phosphorylation, ubiquitination, and protein-protein interactions of wild type and endocytosis-defective EGFR variants before and after internalization in response to EGF ligand and stress. Prior to internalization, EGF-stimulated EGFR accumulated ubiquitin at 7 K residues and phosphorylation at 7 Y sites and at S1104. Following internalization, these modifications diminished and there was an accumulation of S/T phosphorylations. EGFR internalization and many but not all of the EGF-induced S/T phosphorylations were also stimulated by anisomycin-induced cell stress, which was not associated with receptor ubiquitination or elevated Y phosphorylation. EGFR protein interactions were dramatically modulated by ligand, internalization, and stress. In response to EGF, different E3 ubiquitin ligases became maximally associated with EGFR before (CBL, HUWE1, and UBR4) or after (ITCH) internalization, whereas CBLB was distinctively most highly EGFR associated following anisomycin treatment. Adaptin subunits of AP-1 and AP-2 clathrin adaptor complexes also became EGFR associated in response to EGF and anisomycin stress. Mutations preventing EGFR phosphorylation at Y998 or in the S1039 region abolished or greatly reduced EGFR interactions with AP-2 and AP-1, and impaired receptor trafficking. These results provide new insight into spatial, temporal, and mechanistic aspects of EGFR regulation.
Receptor tyrosine kinases such as the epidermal growth factor receptor (EGFR)1 are aberrantly activated by mutation and/or over-expression in numerous human cancers (1, 2). Ligand-activated EGFR, similar to many receptor tyrosine kinases, is normally subject to clathrin-mediated endocytosis (CME) involving internalization and followed by sorting through the endosomal compartment (reviewed in 3). From endosomes, and as a function of which ligand is bound, the receptor may be recycled back to the cell surface or down-regulated as a consequence of trafficking to lysosomes for proteolytic degradation (4, 5). The EGFR also undergoes CME-mediated internalization and recycling back to the plasma membrane in response to cellular stresses that activate p38 MAPK, for example in response to the chemotherapeutic agent cisplatin, the antibiotic anisomycin, and the cytokine tumor necrosis factor-α (TNFα) (6–8). Various oncogenic mutations in the EGFR, as well as hetero-dimerization with other ErbB family members impairs EGFR down-regulation (9). This leads to aberrant, sustained EGFR signaling, which elicits cellular responses central to the cancer cell phenotype including cell proliferation, survival, motility/migration, and invasion (reviewed in 10).
EGFR signaling and trafficking involve an overlapping set of factors that have been extensively reviewed (10, 11). These processes are products of EGFR protein-protein interactions and post-translational modifications (PTMs) including phosphorylation, ubiquitinylation, and lysine acetylation (12). Extracellular binding of ligand induces EGFR dimerization and trans-autophosphorylation at intracellular tyrosine residues, which serve as binding sites for various enzymes and adaptor proteins (11). These receptor-binding proteins are involved in signaling and/or receptor trafficking, and also lead to further modulation of receptor PTMs. For example, binding of the E3 ubiquitin ligase CBL at EGFR pY1069 (13–15) or indirectly through the adaptor protein Grb2, which binds primarily at pY1092 (16), are both involved in EGFR ubiquitinylation and down-regulation (17). Although not an exclusive mechanism, EGFR internalization mainly involves clathrin and the AP-2 clathrin adaptor complex (12, 18–22) in addition to Grb2 (18, 23, 24). EGFR internalization and recycling in response to stress-induced p38 MAPK activation requires AP-2, but not Grb2 (18), and is reportedly independent of receptor kinase activity, tyrosine phosphorylation, and ubiquitination (6–8). Trafficking of endocytosed EGFR to the lysosome, but not the initial internalization step itself, requires CBL (25, 26), and is associated with ubiquitination at up to six lysine residues within the EGFR kinase domain (14). Additionally, ubiquitin-interacting endocytosis factors including Hrs, STAM, and STAM2 become tyrosine phosphorylated in response to EGFR activation (27), and EGFR ubiquitination is required for endosomal sorting (3). Gill and colleagues identified in the EGFR a region spanning residues 997–1046 as conferring endocytic function to otherwise endocytosis-defective EGF receptors truncated after the kinase domain (28). Consistent with this, EGFR phosphorylation sites linked with receptor trafficking are present within or proximal to this part of the receptor. For example, EGFR phosphorylation at S991 and Y998 accumulate with relatively slow kinetics following stimulation of cells with EGF (29). Phosphorylation-defective variants Y998F and S991A are impaired for ligand-stimulated down-regulation relative to wild type (WT) EGFR, but remain proficient for rapid EGFR-to-ERK signaling (29). Non-phosphorylated Y998 was cited as part of an AP-2 binding motif (Y998RAL) (22), while a nearby di-leucine motif (LL1034/35) also serves as an AP-2 binding site (22, 30). Phosphorylations at EGFR S1039 and T1041 occur downstream of p38 MAPK in response to anisomycin-induced cell stress, and are also phosphorylated at lower levels as part of the normal cellular response to EGFR activation by EGF (29). The adaptor protein Odin (ANKS1A) becomes tyrosine phosphorylated prior to EGFR internalization following EGF treatment of cells, and functions as an effector of EGFR recycling (31). Therefore, in response to diverse extracellular signals a multitude of reversible PTMs and interacting proteins govern EGFR internalization, trafficking, and ultimately, stability and signaling. However, our understanding of spatial-temporal and mechanistic relationships of individual EGFR PTMs and protein interactions, and their biological consequences are largely qualitative and incomplete.
The objective of the current study was to characterize and compare aspects of the initial, pre- and post-internalization stages of EGFR endocytosis in response to EGF and cell stress. A battery of methods was applied to identify and absolutely or relatively quantify EGFR phosphorylation, ubiquitination, and protein-protein interactions. These included fluorescence microscopic imaging, and quantitative LC-MS/MS including targeted measurements by selected reaction monitoring (SRM), and comprehensive quantification by using ultra high resolution MS. These were applied with an established model system based on human HEK293 cells engineered to express defined levels of wild type and various phosphorylation-defective EGFR variants tagged with the Flag epitope. The comprehensive analysis revealed distinctive patterns of EGFR modifications and interactions that correlated with receptor activation and internalization. Generally, EGF-stimulated EGFR tyrosine phosphorylations and lysine ubiquitinations, which were maximal prior to internalization, decreased 15-min after receptor internalization was initiated, whereas S/T phosphorylations increased. A subset of EGF-stimulated S/T phosphorylations including pS991 and pS1039 and proximal S/T residues accumulated to an even greater extent in response to anisomycin. EGFR variants with amino acid substitutions at these positions were largely impaired for AP-1 and AP-2 interactions, showed altered patterns of ubiquitination, and resistance to EGF-stimulated receptor down-regulation. These results provide new insight into the dynamics and molecular events associated with EGFR function.
EXPERIMENTAL PROCEDURES
Unless otherwise stated, reagents were purchased from Sigma-Aldrich.
Cell Culture and HEK293 T-REx Stable Cell Lines
Tetracycline inducible stable HEK293 T-REx cell lines designed to express C-terminal Flag tagged wild type and mutant EGFR were created using a pcDNA5.1 FRT/TO plasmid according to the manufacturer's instructions (Invitrogen, Carlsbad, CA). They were maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% bovine serum and 200 μg/ml hygromycin B (32).
Western Blotting and Immunoprecipitation
For Western blot analysis and immunoprecipitation (IP), HEK293 T-REx cells were incubated with 1 μg/ml tetracycline (Tet) for 24 h in serum plus medium, changed to serum-free medium without Tet for 4 h, treated with or without indicated reagent for the indicated durations, then rinsed with ice-cold phosphate-buffered saline (PBS) and lysed in TGH buffer (1% (v/v) Triton X-100, 1% sodium deoxycholate, 10% (v/v) glycerol, 50 mm HEPES, pH 7.4, 150 mm NaCl, 2 mm EGTA, 2 mm EDTA, 50 mm NaF, protease inhibitors, and 1 mm Na3VO4) (22). Whole cell lysates (15 μg protein/sample) were resolved by SDS-PAGE, and then probed with primary and secondary antibodies as described previously (29, 31). For IP and LC-MS/MS analysis, clarified whole-cell lysates (10 mg protein/sample) were extracted with immobilized anti-flag M2 beads (25 μl, Sigma (St. Louis, MI)). After 3 h incubation, the beads were washed 3 times with TGH buffer and twice with HPLC water. The proteins bound on M2 beads were eluted with 150 μl of 0.15% of trifluoroacetic acid (TFA), neutralized to 100 mm NH4HCO3, and digested with trypsin. The tryptic peptides were purified by 200 μl C18 stage tips (Thermo Scientific, Rockford, IL) and analyzed by Q-Exactive LC-MS/MS (Thermo Fisher, San Jose, CA) (29). All results reflect at least three independent biological replicates.
Immunofluorescence Staining and Confocal Microscopy
HEK-EGFR T-REx cells were grown on poly-l-lysine-coated coverslips and incubated with 1 μg/ml tetracycline, but without serum for 24 h. Cells were treated with EGF (50 ng/ml) for 1 h at 4 °C. For EGF treatment at 4 °C samples, cells were fixed immediately. For EGF treatment at 37 °C samples, cells were washed free of unbound ligand, and exposed to pre-warmed ligand-free medium at 37 °C for 15 min. For anisomycin treatment cells, no 4 °C pre-incubation time was carried out. The cells were incubated with 10 μm anisomycin at 37 °C for 30 min or at 4 °C for 1 h. After treatment, cells were fixed with freshly prepared 3.7% paraformaldehyde, permeabilized and detected by using anti-EGFR Alexa Fluor488 conjugated mouse antibody (Santa Cruz Biotechnology, Santa Cruz, CA). Confocal microscopy was performed using a Zeiss LSM 510 META laser-scanning microscope (Carl Zeiss Inc., Thornwood, NY). Antibody to Rab4A was from Santa Cruz (d-20; sc-312). Additional details on experimental protocols were published previously (29, 31).
We note that according to the above protocol, during the 37 °C incubation that was permissive for receptor endocytosis, the actual extracellular EGF concentration was substantially less than 50 ng/ml, but was not precisely measured. In all other experiments in which EGFR was monitored by Western blot or by AP-MS, the EGF concentration employed was 10 ng/ml; 1 ng/ml was tested but abandoned because responses were generally too low and variable to achieve statistical significance.
Cleavable Biotin Internalization and Biotin-EGF Binding Assays
To measure EGFR recycling, cells were washed with PBS (pH 8.0) and incubated with Sulfo-NHS-S-S-Biotin (0.5 mg/ml; Pierce, Rockford, IL) in PBS (pH 8.0) for 20 min at 22 °C. Following EGF treatment with or without 10 μm monensin, cells were placed on ice, washed, and residual surface-exposed biotin adducts were removed by three sequential 8-min incubations with ice-cold glutathione cleavage solution. Cells were then washed and lysed. Biotinylated proteins were affinity purified by using streptavidin (SA)-coated beads (Thermo Fisher, San Jose, CA), and recovered biotinylated EGFR was quantified by anti-EGFR Western blotting (8, 31).
EGFR internalization rates were determined by using the method of de Wit et al. (33, 34) with slight modification. Briefly, cells were plated at 20,000/well in 96-well plates, grown for 24 h, and then shifted to 1 μg/ml tetracycline and 50 μg/ml PEI-containing media for 24 h. To measure surface EGFR, cultures were washed and treated with 50 ng/ml biotin-labeled EGF (Invitrogen Molecular Probes) in PBS containing Ca2+/Mg2+/glucose for 1 h at 4 °C, then washed with PBS and fixed in PBS containing 3% paraformaldehyde, 0.25% glutaraldehyde, and 0.25% Triton X-100. To measure internalized EGFR, cultures were washed and treated with 50 ng/ml biotin-labeled EGF and 10 μm monensin in PBS with Ca2+/Mg2+/glucose for 15 min at 37 °C, followed by an acid wash (125 mm NaCl, and 25 mm acetic acid, pH 3) to strip off surface-bound EGF, followed by fixation. Fixed cells were treated with 50 mm glycine, blocked, and incubated with streptavidin-HRP, and detected by TMB solution (Calbiochem, Billerica, MA).
Selected Reaction Monitoring (SRM) Mass Spectrometry and LC-MS/MS Analysis
SRM-MS analysis of EGFR was carried out as described previously (29, 31). After data acquisition on a triple quadrupole instrument (TSQ Vantage, Thermo Fisher, San Jose, CA), Skyline software (35) was used for SRM data acquisition and total ion current calculations. Collision energy was calculated based on the precursor ion charge state and mass-to-charge ratio using the standard equations CE = 0.03*m/z + 2.905 and CE = 0.038*m/z + 2.281 for doubly and triply charged precursors, respectively. A synthetic heavy isotope containing peptide (IPLENLQIIR) was used as a standard peptide for EGFR quantification.
For LC-MS/MS analysis, the tryptic peptides from anti-flag IP complexes were separated on a 50-cm Easy-Spray column with a 75-μm inner diameter packed with 2 μm C18 resin (Thermo Scientific, Odense Denmark). The peptides were eluted over 120 min (250 nl/min) using a 0 to 40% acetonitrile gradient in 0.1% formic acid with an EASY nLC 1000 chromatography system operating at 50 °C (Thermo-Fisher Scientific). The LC was coupled to a Q Exactive mass spectrometer (36) by using a nano-ESI source (Thermo Fisher Scientific). Mass spectra were acquired in a data-dependent mode with an automatic switch between a full scan and up to 10 data-dependent MS/MS scans. Target value for the full scan MS spectra was 3,000,000 with a maximum injection time of 120 ms and a resolution of 70,000 at m/z 400. The ion target value for MS/MS was set to 1,000,000 with a maximum injection time of 120 ms and a resolution of 17,500 at m/z 400. Repeat sequencing of peptides was kept to a minimum by dynamic exclusion of sequenced peptides for 20 s (37).
Acquired raw files were analyzed by using MaxQuant software (38)(version 1.3.0.5) for quantification, and X! Tandem (The GPM, thegpm.org; version CYCLONE; 2010.12.01.1) and Scaffold (version Scaffold_3.4.3, Proteome Software Inc., Portland, OR) for further validation. The modified peptides validated by inspection of MS/MS spectra in Scaffold are shown in supplemental Fig. S6. The Andromeda probabilistic search engine (39) was used to search peak lists against the Swiss-Prot database (2012.12 version, 20226 entries) after addition of variant EGFR protein sequences relevant to the study. The default search parameters were used as describe by Deeb et al. (37). The search included cysteine carbamidomethylation as a fixed modification, N-terminal acetylation, methionine oxidation phospho-serine, -threonine and -tyrosine, and Gly-Gly addition to lysine as variable modifications. The second peptide identification option in Andromeda was enabled. For statistical evaluation of the data obtained, the posterior error probability and false discovery rate were used. The false discovery rate was determined by searching a reverse database. A false discovery rate of 0.01 for proteins and peptides was permitted. Two miscleavages were allowed, and a minimum of seven amino acids per identified peptide were required. Peptide identification was based on a search with an initial mass deviation of the precursor ion of up to 6 ppm, and the allowed fragment mass deviation was set to 20 ppm. To match identifications across different replicates and adjacent fractions, the “match between runs” option in MaxQuant was enabled within a time window of 2 min. For the determination of protein levels, at least two unmodified peptides were required for LFQ calculation. Phosphorylation site localization probabilities were required to exceed 75% (supplemental Table S1).
Bioinformatics analysis was completed by using Perseus tools within the MaxQuant environment. LFQ intensity for EGFR was used to normalize EGFR-associated proteins and modified peptides across samples. When required, missing values were imputed with a width of 0.3, and downshift of 1.8. For heat map analysis, the normalized intensities of peptides or LFQ intensities of proteins, extracted from MaxQuant, were compared with maximal intensities of corresponding peptides or proteins and expressed in a range from 0 to 1. Values are means of three biological replicates for each sample. Log10 maximum intensities were used in heat maps. For unsupervised clustering of peptides and volcano plot analysis, normalized peptide or LFQ protein intensities were log2 transformed, and the missing values were replaced by data imputation in Perseus. The Z-score of peptides or proteins were clustered with Euclidean average or correlation average by K-means process by using Perseus (perseus-framework.org/). The logarithmic ratio of protein intensities between two samples and the negative logarithmic p values of the Welch t test performed from triplicates between two samples were calculated for Volcano plot by Perseus and exported to Microsoft Excel for plotting. All values are means of three biological replicate for each sample.
The mass spectrometry proteomics data describe in this paper have been deposited to the ProteomeXchange Consortium j(http://www.proteomexchange.org) via the PRIDE partner repository with the dataset identifier PXD000788.
Statistical Analysis
Statistical significance was determined by using Student's t test. p values less than 0.05 were considered to be statistically significant.
RESULTS
Effect of Temperature, Ligand, and Stress on EGFR Phosphorylation
In order to facilitate the isolation and analysis of EGFR from cells expressing physiologically relevant amounts of wild type (WT) and variant receptors, tetracycline-inducible EGFR-Flag-expressing cell lines were established based on HEK293. This line is an established model system for the analysis of EGFR signaling, endocytosis/trafficking, and stability (29, 31, 32, 40). The Western blot (WB) in Fig. 1A shows that the expression level of EGFR in stably transfected HEK293 cells gradually increased during a 48 h incubation period after cell treatment with 1 μg/ml tetracycline (Tet). There was no significant difference in EGFR expression when a range of Tet concentrations (0.1 to 1 μg/ml) was used (data not shown). SRM-MS was used to quantify EGFR expression levels as described previously (31). This showed that the EGFR protein expression level was ∼2+E5 copies/cell after a 24 h Tet treatment (Fig. 1B). This is similar to the amount of EGFR expressed in Hela cells, another frequently used EGFR model system (41). In non-small cell lung carcinoma EGFR protein is expressed at concentrations ranging from 8.6+E4 per cell in tumors lacking EGFR gene amplification to 3.6+E6 copies per cell in tumors with EGFR amplification (42). Hence, the EGFR cell density used in this study is comparable to other model systems and endogenous pathophysiological levels. Subsequent experiments were conducted with cells 24 h post-Tet-treatment unless otherwise stated. EGFR expression was measured to be very similar when assessed by Western blot (Fig. 1A) and SRM-MS (Fig. 1B; and Supplementary Information supplemental Fig. S1). After Tet withdrawal, Flag-epitope-tagged receptors were degraded gradually with a half-life of roughly 24 h (Fig. 1C). Within 4 h of Tet withdrawal EGFR degradation was minimal and unless stated otherwise all experiments described in this report were carried out within this time frame.
Fig. 1.

Induction and stability of EGFR expression in HEK-EGFR T-REx cells. A, Western blot (WB) analysis of whole cell lysates (WCL) from HEK-EGFR T-REx cells with indicated antibody. Tetracycline (Tet) inducible HEK293 T-REx cells stably transfected with the EGFR-Flag pcDNA vector were treated with 1 μg/ml Tet for the indicated times. B, Quantification of EGFR by Selected Reaction Monitoring Mass Spectrometry (SRM). SRM quantifications were made from WCL of HEK-EGFR T-REx cells within a linear range relative to a spiked-in, stable isotope-containing standard peptide (IPLENLQIIR) of identical sequence (supplemental Fig. S1B). Error bars denote standard deviation from three biological replicates. C, EGFR degradation after Tet withdrawal. The indicated antibodies were used for WB analysis of WCL from HEK-EGFR T-REx cells after 24 h Tet treatment and then without Tet for the indicated times.
Immunofluorescence microscopy was used to compare EGFR localization following EGF treatment at 4 °C and 37 °C, and in response to the protein synthesis inhibitor and cell stress response inducer, anisomycin (Aniso) (8, 43) (Fig. 2A, and see bright field images in supplemental Fig. S2A). In serum-deprived, untreated control cells (Cont), EGFR were mainly localized at the cell periphery, consistent with a mostly plasma membrane localization (Fig. 2A), and this pattern was retained when cells were exposed to EGF but maintained at ice temperature (EGF4C, Fig. 2A), as expected (23, 29, 44). This apparent plasma membrane staining pattern was also observed when cells were incubated at 4 °C with or without anisomycin (supplemental Fig. S2B). By contrast, as expected (18, 29, 31), when cells were treated with anisomycin (Aniso) at 37 °C or when EGF-treated cells were warmed to 37 °C for 15 min (EGF37C), EGFR were effectively internalized (Fig. 2A). As a control, the punctate EGFR staining pattern associated with internalized EGFR was found to colocalize with Rab4A, a marker of the recycling and degradative endosomal compartment (45) (supplemental Fig. 2C). As described below, under these experimental conditions EGFR became associated with the AP-2 clathrin adaptor complex, suggesting EGFR endocytosis occurred by CME, however, the extent to which EGFR may have internalized by clathrin-independent mechanisms was not measured.
Fig. 2.

EGFR trafficking and phosphorylation after cell treatment with EGF or anisomycin at 4 °C and 37 °C. A, Immunofluorescence imaging of EGFR in HEK-EGFR T-REx cells following the indicated treatments. After 24 h Tet induction, serum-deprived HEK-EGFR T-REx cells were treated with EGF (50 ng/ml) for 1 h at 4 °C, washed free of unbound ligand, and then provided with pre-warmed ligand-free medium at 37 °C for 0 min (EGF4C) or 15 min (EGF37C), or the cells were incubated with anisomycin (10 μm) at 37 °C for 30 min (Aniso). Control cells were HEK-EGFR T-REx without any treatment (Cont). The cells were fixed and stained with anti-EGFR AF488 antibodies (white pseudo-color) and DAPI (blue for nuclear staining). Yellow arrows point to membrane EGFR, and red arrows point to internalized EGFR. Scale bars are 7 μm. B, WB analysis of WCL from HEK-EGFR T-REx cells following the indicated treatments. HEK-EGFR T-REx cells were untreated (Cont) or treated at 37 °C with EGF (10 ng/ml) for 15 min (EGF37C) or with anisomycin (10 μm) for 30 min (Aniso); or the cells were incubated at 4 °C for 1 h without any reagent treatment (Cont4C) or with EGF (EGF4C) or anisomycin (Aniso4C). Results shown are representative of three independent experiments. C, Heat map showing relative amounts of site-specific EGFR phosphorylation in HEK293 T-REx (HEK) cells and HEK-EGFR T-REx cells following the indicated treatments. Flag-tagged EGFR was isolated by immunoprecipitation and analyzed by LC-MS/MS. Phosphopeptides were quantified according to integrated extracted ion currents with MaxQuant software. Values shown are means of three biological replicates. The order in the vertical direction was established by manually clustering according to conditions associated with maximum phosphorylation, and with Y phosphorylations highlighted in gray. Values for peptides of same sequence and number of phosphor-sites were summed and labeled with an asterisk. The intensity bars on the far right represent Log10 of maximum intensities, and covering a range from 6 to 10.
Analysis of total cellular protein-pY and EGFR-pY1092 by Western blot showed that EGF treatment (EGF37C) effectively stimulated EGFR as evidenced by its phosphorylation at Y1092 and the appearance of cellular pY-containing proteins (Fig. 2B). When EGF treatment was performed at 4 °C (EGF4C), the signals were slightly higher. Consistent with our previous findings (31), MAP kinases ERK1 and ERK2 were not phosphorylated when the EGF-treated cells were held at 4 °C, but became activated when the EGF treated cells were warmed to 37 °C (Fig. 2B). Levels of stress-activated MAP kinase p38 expression were similar under all test conditions (Fig. 2B), and phosphorylated p38 (pp38) was slightly elevated in all cells exposed to the 4 °C condition (Fig. 2B, lanes 3, 5, and 7). However, the pp38 signal was more pronounced in cells treated with EGF at 37 °C, and was markedly elevated following anisomycin treatment at 37 °C (Aniso; Fig. 2B). Therefore, the EGF4C, EGF37C, and Aniso conditions were established as respective models for EGF-activated, pre- and post-internalization EGFR, and for stress response-associated EGFR internalization.
EGFR phosphorylation and ubiquitination were measured by affinity purification-mass spectrometry (AP-MS) essentially as described previously (29). In these experiments peptides were measured by LC-MS/MS analysis of trypsin-digested anti-Flag samples, and did not involve targeted enrichment of modified peptides. In order to integrate and display the large dataset, which represents three biological replicates and encompassing 30 EGFR phosphorylation sites associated with seven cell states, a heat map was generated (see supplemental Table S1, which includes all peptide MS information including phosphorylation site localization probabilities). The measured EGFR phosphorylations encompass 7 pY, 19 pS, and 4 pT sites, all of which have been reported previously, except, to the best of our knowledge, pS227. The MS/MS spectra for phospho-peptides were verified by manual inspection and shown in supplemental Fig. S6. Fig. 2C shows a heat map representing EGFR phosphorylations associated with five different cell states. Each phosphorylation site is presented in a relative manner in proportion to the maximum value measured at that site, and with limited summing of values obtained from phospho-isomers as indicated by asterisks (see the legend to Fig. 2, and Experimental Procedures). Relative levels of phosphorylation at a given site are comparable in the horizontal direction as a function of the indicated cell conditions and treatments. To the right of the heat map “Intensity” bars provide an indication of the magnitude of the MS signals associated with the indicated phospho-peptides. However, because each peptide, including phospho-isomers, has a distinctive MS response rate, the intensity values provide only a crude representation of peptide abundance. To simplify the result, only phosphopeptides with MS intensity values greater than 6.5 on a log10 scale are shown in Fig. 2C. The data were manually organized in the vertical direction with Y phosphorylations clustered at the top, and attempting to group together phosphorylations that correlate positively with the same treatment conditions. EGFR recovered from control (Cont) and various 4 °C samples (i.e. Cont4C, Aniso4C, and EGF4C) generally had very little S/T phosphorylation (Fig. 2C and supplemental Table S1). As expected, phosphorylated EGFR peptides were not detected by analysis of negative control cells not expressing ectopic EGFR-Flag.
Each of the “stimulated” cell states was characterized by a distinctive set of phosphorylations (Fig. 2C). All the detected pY sites were maximally phosphorylated in response to EGF prior to internalization (EGF4C). Curiously, a lone pS site at EGFR residue 1104 was observed to follow the pY pattern of maximum phosphorylation under the EGF4C condition, albeit with a modest MS intensity of 2E+7. These sites were also phosphorylated, but to a lesser extent post receptor internalization (EGF37C). Six phosphopeptides, encompassing 11 pS/pT sites on the EGFR reached maximum levels under the post-internalization condition (EGF37C). At this time point (i.e. after 15 min at 37 °C) EGFR pY levels had diminished considerably, although pY1016, the Grb2-binding site at pY1092, and pY1197 persisted to a greater extent than the other pY sites, consistent with previous observations that the latter two sites reach relatively high stoichiometry (46). The values for pY1092 and pY1197 under the EGF37C condition were, respectively, 77% and 40% of their EGF4C values. These results are in agreement with the anti-pY Western analysis (Fig. 2B). AP-MS analysis revealed slight increases in EGFR phosphorylation at Y1092 (1.8-fold increase) and Y1197 (1.6-fold increase) following anisomycin treatment at 37 °C, which were not easily discernable by Western analysis. EGFR from unstimulated cells (Cont), or cells incubated at 4 °C for 1 h (Cont4C) or treated with anisomycin at 4 °C for 1 h (Aniso4C) had fewer phosphorylation sites and signals (Fig. 2C and supplemental Table S1). Peptides including pY998 and pY1069 were not detected. Y1069 is preceded by a trypsin cut site (arginine) that may have been hindered by adjacent phosphorylation at Y1069. Y998 phosphorylation accumulates with slow kinetics at 37 °C (29), and is generally a more prominent phosphorylation site in tumor tissue than cultured cells (47) and hence would be expected to be only a minor modification at the 15 min time point used in this study (29). Also, the lower amount of EGFR (fivefold fewer copies per cell) and EGF (10-fold less) used in this study compared with Tong et al. (29) may be additional factors that limited the detection of these phosphorylations.
There were 23 sites of S or T phosphorylation identified in the seven samples, with nine showing highest signals in the EGF37C sample (Fig. 2C and supplemental Table S1). Some of these phosphorylations were also associated with the other treatment conditions. They mapped to various intracellular regions of the EGFR such as T693 and S695 in the juxtamembrane region, T725 near the amino-terminal end of the kinase domain, and S1190/T1191 near the extreme carboxyl terminus. Overall, EGFR from cells treated with anisomycin (Aniso) had the highest pS/pT signals. Eleven of 23 identified pS/T sites had their highest phosphorylation signals following anisomycin treatment (Fig. 2C and supplemental Table S1). These sites are distributed throughout the EGFR intracellular region including the noted juxtamembrane sites (pT693, and pS695); pS991 and pS995, which are proximal to Y998 and located immediately carboxyl to the kinase domain; pS1039, pT1041, and pS1042, which map to a S/T-rich region associated with stress-induced, p38 MAPK-dependent phosphorylation (8, 29); and pS1070 and pS1071, which are stress-induced phosphorylation sites adjacent to the CBL-binding site at Y1069 (48). Four out of 5 multiply phosphorylated peptides such as pT693pS695 and pS1039pT1041pS1042 had their greatest signals in Aniso samples indicating simultaneous rather than discrete modification of proximal residues during the cell stress response.
The EGFR Interactome as a Function of EGF, Internalization, and Stress
EGFR protein-protein interactions were measured and analyzed by using an AP-MS protocol (49), essentially as described previously (29). Approximately 300 proteins were considered EGFR associated by having greater than sixfold binding (p < 0.05) in EGFR complexes than controls (supplemental Fig. S3A; see supplemental Table S2 for associated protein MS information). Seventy-four proteins were selected for further analysis (Fig. 3 and supplemental Fig. S3B) based on their known EGFR interactions as listed in the databases HAPPI (50) or HPRD (51), and/or known to be involved in endocytosis/trafficking, or associated with the processes of phosphorylation or ubiquitination. EGFR amount was used to normalize the quantification of associated proteins (see arrow in Fig. 3 and supplemental Fig. S3B). The heat map shown in Fig. 3, organized in a supervised manner similar to the phosphorylation analysis in Fig. 2C, showed that there were overlapping groups of EGFR binding proteins defined by their maximal EGFR association pre- and post-internalization, or in response to anisomycin. Unsupervised hierarchical clustering of the 74 proteins across the range of cell states gave essentially the same result (supplemental Fig. S3B). In agreement with the observed maximal EGFR Y phosphorylation in the EGF-stimulated, pre-internalization condition, various signaling proteins containing pY-binding domains were recovered in highest amounts in the EGF4C samples, including for example GRB2, CBL, SOS1/2, SHC1, phospholipase C-γ1 (PLCG1), CSK, and VAV. Exceptions to this were two SH2 domain containing proteins, the Src-family tyrosine kinase YES1, and the transcription factor STAT1. Both these proteins displayed EGF-stimulated EGFR association under the EGF4C condition, but unlike the other known pY-binding proteins, which measured as maximum in the EGF4C samples, they did not. YES1, which was also EGFR-associated under basal, serum-deprived conditions, was most enriched in the EGF37C samples. STAT1 association was stimulated by EGF at 4 °C and decreased post-internalization (EGF37C), but was most prevalent following the anisomycin treatment.
Fig. 3.
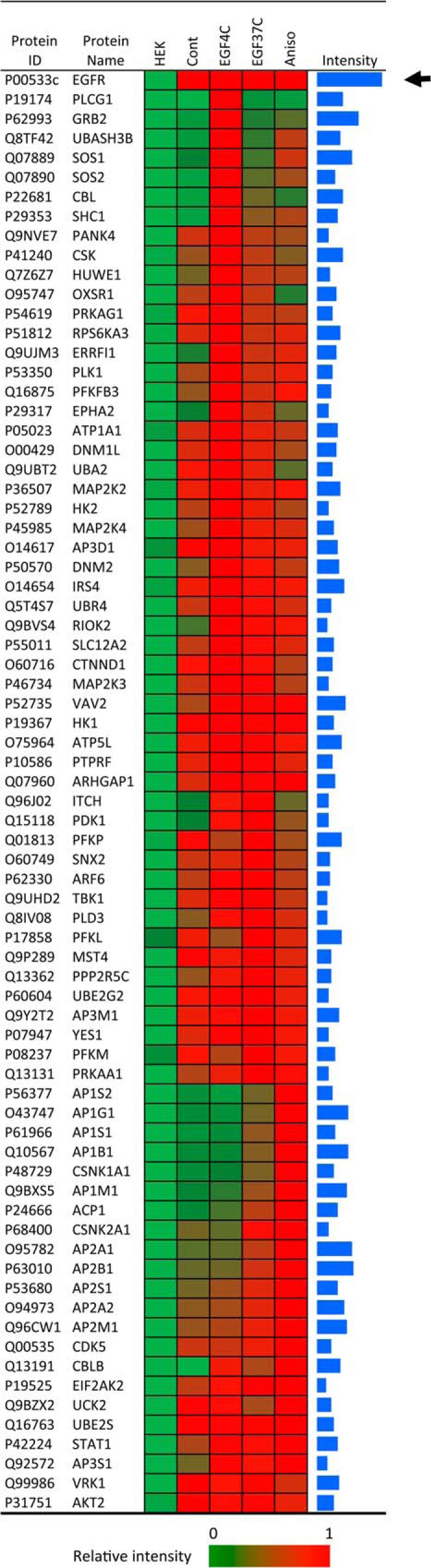
The EGFR interactome before and after ligand and stress-induced internalization. Heat map showing relative amounts of EGFR (see arrow) and EGFR-associated proteins in HEK-EGFR T-REx cells following the indicated treatments and AP-MS analysis. 74 known EGFR associated proteins from immunoprecipitated wild type EGFR after indicated treatment were grouped according to the condition associated with their maximum EGFR association. The intensity bars on the far right represent Log10 of maximum intensities, and covering a range from 6 to 10.
Five E3 ubiquitin ligases were measured as EGFR associated (Fig. 3). The observed EGF-stimulated strong binding of CBL at 4 °C is consistent with the assumption that the known CBL binding site containing pY1069 accumulated under the EGF4C condition as expected. CBL association with EGFR was strongly stimulated by EGF at 4 °C and greatly diminished after 15 min at 37 °C, and with even less receptor-association detected in response to anisomycin. Like CBL, HUWE1 and UBR4 were maximally associated with EGF-stimulated EGFR prior to internalization. UBR4 was also receptor-associated to an extent prior to EGF treatment. ITCH was unique in that it peaked in its EGFR association after EGF-stimulated receptor internalization. Curiously, while CBLB displayed EGF-stimulated EGFR association at 4 °C that diminished following the 37 °C incubation similar to CBL, it was distinctly different in that it displayed maximal EGFR association in response to anisomycin (Fig. 3). The enzymes PDK1, PLD3, and PRKAA1 displayed EGF-stimulated EGFR association at 4 °C and accumulated to greater amounts under the EGF37C condition, but showed only minor increases in EGFR association in response to anisomycin. Ten clathrin adaptor complex subunits were detected as inducibly EGFR associated. In all cases their binding in response to EGF was greater in the EGF37C samples than seen with EGF4C, but the maximal association of these proteins was in response to anisomycin, and is described further in the context of Fig. 6, below.
Fig. 6.

AP complex association and ubiquitination of wild type and variant EGFR before and after EGF- and stress-induced internalization. Shown are heat maps associated with WT and variant EGFR and the indicated cell treatments. A, Association of adaptin proteins; B, Relative amounts of EGFR-associated poly-ubiquitin-derived peptides and EGFR-derived peptides containing diglycine-modified lysine, as denoted by K-GG or K(gl), and indicating the position of the modified residue; C, Relative amount of five EGFR-associated E3 ubiquitin ligases. MaxQuant was used for protein or peptide quantification, and log10 maximum intensity values within the range from 6 to 10 are shown as blue bars.
EGFR Ubiquitination as a Function of EGF, Internalization, and Stress
The EGFR IP samples from EGF-stimulated cells were found to contain EGFR- and polyubiquitin-derived peptides containing diglycine-modified lysine, which is a signature of ubiquitin modification (e.g. 52) (Fig. 4). A ubiquitin modification at EGFR K970 is shown in Fig. 4A as a representative example of the characteristic 114 Da di-glycine lysine modification detected by tandem MS. The detection of ubiquitin peptides conjugated at position 11, 48, and 63 indicated that EGFR or EGFR-associated proteins were polyubiquitinated after EGF treatment (Fig. 4B). Compared with the K11 and K48 conjugates, ubiquitination at ubiquitin K63 showed the highest MS intensity signal, consistent with Huang et al. (53). EGFR was found ubiquitinated at 7 positions (K716, K737, K757, K867, K875, K970, and K1061; Fig. 4B) in response to EGF at 4 °C, which includes six previously documented sites within the kinase domain, and K1061 located in carboxyl tail region. The highest ubiquitination signals were associated with EGFR after EGF treatment at 4 °C (Fig. 4B), indicating that ligand activated EGFR became highly ubiquitinated before internalization. The EGFR-derived diglycine peptides measured ∼1.6-fold greater in the EGF4C than the EGF37C samples suggesting an overall decline in EGFR ubiquitination following internalization during the 15 min incubation at 37 °C. There was detectable EGFR ubiquitination in resting cells (Cont), with the major ubiquitination site being K875 and with the major polyubiquitin linkage at ubiquitin K48 (see Cont, Fig. 4B). The K48 linkage has been associated mainly with receptor signaling rather than degradation (14, 26, 53). Ubiquitination at EGFR K875 increased only 2.5-fold after EGF treatment at 37 °C (relative to Cont), whereas other sites such as K867 had ∼20-fold increases. By contrast, there was no statistically significant ubiquitination above control levels in response to anisomycin treatment.
Fig. 4.

EGFR ubiquitination before and after EGFR internalization. A, Tandem mass spectrum of the EGFR-derived peptide of sequence ELIIEFSK970MAR, containing diglycine-modified lysine, denoted gl after the modified reside, as a mark of ubiquitination; associated with EGF treatment at 37 °C. The y and b series of ions are indicated. The sequence of the peptide derived from this spectrum is shown at the top of the panel. B, Heat map showing relative amount of three ubiquitin peptides and 7 EGFR ubiquitination sites from EGFR IP complexes with indicated treatment. MaxQuant was used to quantify peptides and proteins. The intensity bars on the far right represent Log10 of maximum intensities, and covering a range from 6 to 10.
EGFR Interactions with Adaptins as a Function of Receptor Phosphorylation and Cell State
As shown in Fig. 2C anisomycin induced considerable phosphorylation at EGFR S1039 and simultaneously at neighboring residues including T1041 and S1042. An MS/MS spectrum of a triply phosphorylated peptide containing all three modifications is shown in Fig. 5A. Pronounced phosphorylation of these residues in response to EGF was reported previously in the context of an endocytosis-defective EGFR variant defective for phosphorylation at position Y998 (29), and these phosphorylation sites map to a region required for stress-induced EGFR recycling (8).
Fig. 5.
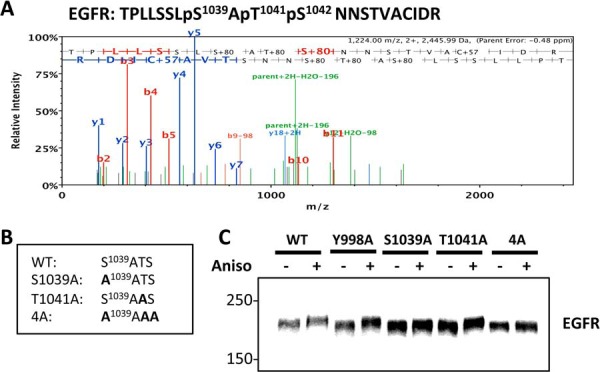
Altered SDS-PAGE mobility of EGFR variants as a measure of anisomycin-induced S/T phosphorylation. A, Tandem mass spectrum of the triple phosphorylated EGFR peptide TPLLSSLpS1039ApT1041pS1042NNSTVACIDR associated with EGF treatment at 37 °C. The y and b series of ions are indicated. The sequence of the peptide derived from the spectrum is shown at the top of the panel. B, The amino acid sequence spanning EGFR residues 1039 through 1042 are shown for wild type and the indicated EGFR variant proteins. C, Western blot analysis of WCL from cells expressing the indicated WT and variant EGFR proteins. HEK293 T-Rex cells were treated with Tet for 24 h for EGFR induction, and then treated without or with anisomycin (10 μm, 30 min) as indicated. The upward band shift associated with anisomycin treatment is an indication of S/T phosphorylation.
To further investigate the relationships between these phosphorylations and EGFR trafficking and protein-protein interactions, a series of phosphorylation-defective variants were made (Fig. 5B). Alanine substitution at 1039 and 1041 yielded the respective variants S1039A and T1041A, and the 4A variant was generated by changing the wild type (WT) sequence beginning at 1039 from SATS to AAAA. These EGFR variants, and a phosphorylation-defective variant Y998A were expressed in HEK293 T-REx cells (supplemental Fig. S4A and Fig. 5C). After anisomycin treatment, WT EGFR, and the Y998A and T1041A variants showed pronounced upward band shifts, consistent with the interpretation these receptors became highly S/T phosphorylated. This effect was diminished in S1039A, and missing in the 4A variant (Fig. 5C). K-means clustering analysis (of 20 samples with three biological replicates) identified the same major correlations between these treatments and EGFR phosphorylation, with ligand-activated EGFR phosphorylation before internalization being associated with receptor pY sites, and post-internalization and stress-induced EGFR phosphorylations comprised of maximum S/T phosphorylations (supplemental Fig. S4B).
Given the important role of the clathrin adaptor complexes in endocytosis and intracellular trafficking (12, 20–22, 30), we further examined the interaction of AP-1 and AP-2 subunits with WT and phosphorylation-defective EGFR variants as a function of EGF and anisomycin treatments (Fig. 6A). The association of the ten different AP-1 and AP-2 adaptin subunits to WT EGFR was most pronounced following anisomycin treatment, and this was essentially abolished in both the Y998A and 4A variants. Compared with the anisomycin-induced binding, a lesser amount of EGFR binding with the adaptins was observed under the EGF37C condition, and this too was abolished in the Y998A and 4A variants, although with some residual binding observed between AP2A2 and AP2M1 with the 4A variant (Fig. 6A; see also supplemental Fig. S4C, supplemental Table S3 and supplemental Table S4). A basal level of association between AP-2 subunits and WT EGFR was observed in the absence of cell stimulation by EGF or stress (Cont), and this was absent in the Y998A variant, but largely unperturbed in the 4A variant. Similar amounts of AP-3 complex proteins were bound with EGFR under different conditions including without EGF treatment (Cont, Fig. 3, EGF4C group).
The loss of adaptin interactions with the variants prompted an examination of their ubiquitination and endocytosis. Consistent with the findings of WT EGFR, the variants were not appreciably ubiquitinated following anisomycin treatment (Fig. 6B). However EGF-stimulated receptor ubiquitination was greatly decreased in the Y998A variant, and to a lesser extent in the 4A variant (Fig. 6B). Also, with the Y998A variant, there was a pronounced decrease in the recovery of K63 but not K48 polyubiquitin moieties in the affinity purified EGFR complexes (Fig. 6B).
As shown in Fig. 6C, the binding of the E3 ubiquitin ligases was examined as a function of the phosphorylation site mutations. In response to EGF at 4 °C, compared with WT EGFR, both CBL and CBLB binding was slightly diminished in the Y998A variant, but actually slightly increased in the 4A variant. The anisomycin-stimulated binding of CBLB to WT EGFR was greatly decreased in both the Y998A and 4A variants. Binding of ITCH, which as described above was maximal under the EGFR37C condition (Fig. 3), was unaffected by the Y998A substitution, but abolished in the 4A variant.
Defective EGFR Down-regulation with Phosphorylation-defective EGFR Variants Y998A and 4A
Given the altered ubiquitination and adaptin interactions observed with the EGFR phosphorylation-defective variants, their endocytosis was examined. Immunofluorescence analysis showed that as expected WT EGFR displayed intracellular punctate staining characteristic of the endosome compartment following treatment of cells at 37 °C with EGF for 15 min or anisomycin for 30 min (Fig. 7A). By contrast, under these conditions the Y998A and 4A variant EGF receptors remained mostly aligned with the cell periphery (Fig. 7A; also, see bright field images in supplemental Fig. S5A). These results implied EGFR variants had reduced EGFR internalization or possibly accelerated EGFR recycling. A time-course Western analysis of EGFR abundance following EGF stimulation indicated a more rapid down-regulation of WT EGFR than Y998A and 4A EGFR (Fig. 7B). The level of WT was decreased to approximately one-half within 60 min of EGF addition, which is consistent with the interpretation that internalized receptors (Fig. 7A) were destined for degradation. However, the variants Y998A and 4A did not decrease to a T1/2 value until ∼180 min indicating a relative impairment in down-regulation.
Fig. 7.

Deminished internalization and degradation of EGFR variants Y998A and 4A EGFR. A, Immunostaining of WT and variant EGFR in HEK293 T-REx cells associated with the indicated EGF and anisomycin treatments. The experiment was carried out as described for Fig. 2A. B, EGFR degradation after EGF treatment. WB analysis was carried out on WCL from HEK293 T-REx cells expressing the indicated WT or variant EGFR. Cells were incubated with 1 μg/ml Tet for 24 h to induce EGFR expression, and then treated with 100 ng/ml EGFR as indicated. C, Measurement of cell surface localized EGFR. Cells expressing WT EGFR or the indicated variant receptor were incubated with 50 ng/ml biotinylated EGF at 4 °C for 1 h. The bound biotinylated EGF, representing the amount of surface EGFR, was measured by HRP-Streptavidin. D, Measurement of internalized EGFR. Following incubation with biotinylated EGF and monensin at 37 °C for 15 min, whole cells were washed with acid to remove extracellular ligand, and then measured for internalized biotinylated EGF as a measure of internalized EGFR. E, Using monensin and EGFR covalently modified with a cleavagle form of biotin to indirectly measure EGFR recycling. Following EGF treatment for 15 min, biotin moities were cleaved from cell surface-exposed biotinylated EGFR, and then internalized biotinylated EGFR were captured with streptavidin beads. EGFR was quantified by anti-Flag WB. The monensin-stimulated increase in intracellular EGFR was taken as a measure of the recycling fraction of EGFR. Values shown are means of three biological replicates. Asterisks denote p < 0.05, n = 3.
Next, biotinylated EGF was used to quantify EGFR localization after EGF treatment (33, 34). To measure surface EGFR, biotinylated EGF was incubated with WT, Y998A and 4A EGFR cells at 4 °C for 1 h. HRP-conjugated streptavidin was used to quantify biotinylated EGF. The results showed that WT and mutant EGFR cells had comparable amounts of EGF binding at 4 °C (Fig. 7C), confirming that the different cell lines expressed similar amounts of cell surface-exposed EGFR. To measure internalized EGFR, cells were treated with EGF and the recycling inhibitor monensin (54). As expected, monensin- but not vehicle-treated cells showed a dramatic accumulation of internalized receptor in response to EGF (supplemental Fig. S5B). Acid washing was used to strip away extracellular biotin-EGF, which allowed the quantification of endocytosed biotin-EGF. As shown in Fig. 7D, compared with cells expressing WT EGFR, cells expressing the Y998A and 4A variants accumulated significantly less internalized biotin-EGF. To further test whether the EGFR variants had altered EGFR recycling, EGF-induced internalization of biotinylated EGFR was measured. EGFR at the cell surface was covalently modified with cleavable biotin, and then cells were treated with or without monensin and EGF as described previously (8, 31). After first eliminating surface-exposed biotin by cleavage, internalized biotin labeled EGFR (i.e. following EGF stimulation) was captured by using streptavidin affinity purification and detected by Western blot (supplemental Fig. S5C). Internalized biotinylated EGFR captured as a function of monensin thus represents a population of internalized receptors that would have recycled back to the cell surface (supplemental Fig. S5C). By this analysis there was no significant difference in the recycling of the Y998A and 4A variants compared with WT (n = 3; Fig. 7E). Therefore, compared with WT EGFR, the Y998A and 4A EGFR variants had reduced binding to clathrin adaptor complexes, reduced EGFR internalization, and reduced EGF-stimulated degradation.
DISCUSSION
Knowledge of the molecular events that regulate and signify EGFR internalization and stability may reveal new strategies to treat EGFR-driven tumors. The objective of this study was an integrated analysis of EGFR phosphorylation, ubiquitination, and protein-protein interactions before and after ligand-stimulated internalization, and compared with receptors internalized as a consequence of anisomycin-induced cellular stress. To complete this study experimental conditions were established as models of pre- and post-internalized EGFR. EGF receptors of cells exposed to EGF at 4 °C were retained at the plasma membrane and did not activate ERK, but became fully catalytically activated, accumulated tyrosine phosphorylation in the carboxyl-terminal tail region, and associated with known major binding partners including Shc1 and Grb2. Subsequent incubation of EGF-treated cells at 37 °C allowed receptor internalization and activation of ERK. Other approaches to impede EGFR internalization have been developed including expression of a K44A dynamin variant (55), tamoxifen-induced depletion of dynamin (56), and by using a small molecule inhibitor of dynamin GTPase activity (57). For pY sites that were maximally phosphorylated at 4 °C, their average decrease in phosphorylation after the 37 °C incubation (∼twofold) is similar to the dynamic range measured in other studies that have aimed to capture early peaks in EGF-stimulated EGFR phosphorylation at 37 °C (58, 59) (Fig. 2C and supplemental Fig. S4B). This suggests the EGF4C protocol did not result in an over-accumulation of receptor tyrosine phosphorylation, and is consistent with, but does not prove a model wherein EGFR is capable of reaching maximum tyrosine phosphorylation rapidly after ligand stimulation, prior to internalization. Anisomycin activated p38 MAPK, as evidenced by p38 activation loop phosphorylation, and triggered EGFR internalization comparable to that induced by EGF. These results support our HEK293 cell model and experimental protocol as a valid system for the analysis of the dynamics and molecular features of ligand- and stress-induced EGFR internalization.
In terms of EGFR phosphorylation dynamics, our results are consistent with the comprehensive study by Olsen et al. (58), which showed in a cell model that most EGFR S/T phosphorylations increase gradually after an initial wave of Y phosphorylation, and plateau ∼20 min after EGF treatment. Inducible phosphorylation of EGFR S/T sites was closely related to receptor internalization, with only very little detected prior to internalization. An exception to this was EGFR residue S1104, which became maximally phosphorylated in response to EGF prior to EGFR internalization. This indicates that the EGFR signaling network includes an EGFR serine kinase(s) activated as part of an immediate/early response to EGFR activation. Our study did not address the identity of the kinase(s) responsible for this modification. However, in a previous study we identified EGFR sites S1039 and T1041 as p38 MAPK-dependent phosphorylation sites modified in response to EGF and stress (29). The loss of the EGFR gel shift phenomenon in the EGFR 4A variant in which phosphorylation sites at/near S1039 were changed to Ala suggests this is a major region of EGFR phosphorylation associated with stress-induced internalization. This is further supported by the detection of peptides from this region that were multiply phosphorylated and associated with strong MS intensities in response to anisomycin (Fig. 2C), and because a deletion encompassing this region was found defective for stress-induced EGFR recycling (8). However, our data indicate EGFR phosphorylation at/near S1039 also plays a role in the cellular response to EGF. This is because the EGFR 4A variant was resistant to EGF-induced internalization and down-regulation compared with the wild type receptor (Fig. 7). We conclude that EGFR phosphorylation at/near S1039 is positively correlated with the extent of EGFR recycling, hence more prominently phosphorylated in response to anisomycin than EGF, as seen in Fig. 2C. This is consistent with our observation that EGFR Y998F, which is subject to elevated recycling in response to EGF (supplemental Fig. S5B), accumulates high levels of pS1039 and pT1041, compared with WT EGFR (29).
When EGF receptors were activated by ligand at 4 °C, the condition that resulted in the greatest EGFR pY accumulation, maximal association was measured between the receptor and various signaling proteins containing pY-binding domains (e.g. SH2, PTB). These interactions are canonical (60), and clearly do not require receptor internalization. In addition to pY-mediated protein-protein interactions, EGFR trafficking involves cycles of ubiquitin attachment and removal, and involvement of different E3 ubiquitin ligases and de-ubiquinating enzymes (3, 5, 61). CBL and CBLB are structurally related paralogous gene products that are widely co-expressed and thought to play redundant cellular roles (62). Our analysis revealed distinctive differences in their EGFR associations, including the maximal association of CBLB in response to anisomycin, which was not associated with receptor ubiquitination or Y phosphorylation (Fig. 3). This indicates that CBLB association does not require receptor Y phosphorylation, and does not necessarily lead to EGFR ubiquitination. Hence CBLB may function to some extent in the general absence of elevated tyrosine kinase activity and without catalyzing substrate (EGFR) ubiquitination. ITCH is known to become tyrosine phosphorylated after EGFR stimulation, and to associate with CBL (63). However, because we observed that ITCH association with EGFR increased post-internalization during which time CBL association decreased, it is unlikely the ITCH association was mediated solely by CBL. Our data suggest that ITCH may function after the other E3 ubiquitin ligases, because it was uniquely more highly EGFR associated post-internalization.
We note that our AP-MS protocol does not distinguish between proteins associated directly or indirectly with the EGFR. In addition, while captured EGFR complexes were enriched for both K48 and K63 types of polyubiquitin chains in response to EGF, our data do not indicate which proteins were modified, except for those cases where we measured directly EGFR-derived peptides bearing the di-glycine signature of ubiquitination. Curiously, among the EGFR ubiquitination sites, K875 was distinctive in three respects. First, it was apparent under basal conditions (i.e. prior to EGF addition); second, the magnitude of its EGF-stimulated increase was ∼10-fold less than the other EGFR ubiquitination sites (Fig. 4); and third, it accumulated to maximum levels under the EGF37C condition in the context of the 4A variant. This suggests K875 modification may be associated with EGFR trafficking at a stage demarcated by the 4A mutation, which, as noted above, is associated with EGFR recycling.
EGFR association with the heterotetrameric clathrin adaptor complexes AP-1 and AP-2 has been observed previously (64). Herein, AP-1 and AP-2 were strongly associated with EGFR internalization in response to anisomycin stress and/or EGF (Fig. 6). This is consistent with the known role of AP-2 in endocytosis, and indicates overlap in the molecular mechanism of EGFR internalization by ligand and stress. AP-1, not typically associated with CME, is involved in trafficking between the trans-Golgi network and endosome compartment (65). Our data suggest this trafficking axis is associated with EGFR following both EGF-stimulated and stress-induced EGFR internalization. The EGFR variants 4A, and especially Y998A, were strikingly defective for interaction with these complexes, and in the case of Y998A showed diminished EGF-stimulated ubiquitination. These data are generally consistent with previous studies linking Y998 and the region encompassing S1039 and proximal S/T sites in EGFR trafficking (8, 22, 28, 29). A model emerges wherein phosphorylation in these EGFR tail regions modulates protein interactions and ubiquitination modifications that ultimately govern the balance between recycling and lysosome trafficking, and hence net stability of internalized EGF receptors. Because we have not observed Y998 to become phosphorylated in response to anisomycin, we conclude the phenotype of the Y998A substitution is not caused by a defect in Y998 phosphorylation. We conclude clathrin AP adaptor complex binding involves a mechanism compatible with the tyrosine (or phenylalanine, as shown in supplemental Fig. S5) but not an alanine side chain at this position. Lastly, because phosphorylation at Y998 accumulates relatively slowly following EGF-induced EGFR activation (29, 58), we further speculate this modification may curtail or modulate clathrin AP adaptor complex interactions as a mechanism to limit the duration or extent of receptor down-regulation induced by ligand, and thereby preserve a pool of receptors. The testing of such models may uncover additional factors involved in the modulation of receptor signaling, trafficking, and stability, which in turn may reveal new strategies to treat EGFR-driven tumors.
Supplementary Material
Acknowledgments
We thank Drs. Brian Raught and Lei Li for helpful discussions.
Footnotes
Author contributions: J.T. and M.F.M. designed research; J.T. and P.T. performed research; J.T., P.T., and M.F.M. analyzed data; J.T. and M.F.M. wrote the paper.
* This work was supported by the Canada Research Chairs Program, and a grant from the Canadian Institutes of Health Research to M.F.M.
 This article contains supplemental Figs. S1 to S6 and Tables S1 to S4.
This article contains supplemental Figs. S1 to S6 and Tables S1 to S4.
1 The abbreviations used are:
- EGFR
- epidermal growth factor receptor
- AP
- clathrin adaptor proteins
- AP-MS
- affinity purification-mass spectrometry
- CME
- clathrin-mediated endocytosis
- IP
- immunoprecipitation purification
- SRM
- selected reaction monitoring
- WCL
- whole cell lysate.
REFERENCES
- 1. Yewale C., Baradia D., Vhora I., Patil S., Misra A. (2013) Epidermal growth factor receptor targeting in cancer: a review of trends and strategies. Biomaterials 34, 8690–8707 [DOI] [PubMed] [Google Scholar]
- 2. Zaczek A., Brandt B., Bielawski K. P. (2005) The diverse signaling network of EGFR, HER2, HER3, and HER4 tyrosine kinase receptors and the consequences for therapeutic approaches. Histol. Histopathol. 20, 1005–1015 [DOI] [PubMed] [Google Scholar]
- 3. Goh L. K., Sorkin A. (2013) Endocytosis of receptor tyrosine kinases. Cold Spring Harb. Pers. Biol. 5, a017459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Roepstorff K., Grovdal L., Grandal M., Lerdrup M., van Deurs B. (2008) Endocytic downregulation of ErbB receptors: mechanisms and relevance in cancer. Histochem. Cell Biol. 129, 563–578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Zwang Y., Yarden Y. (2009) Systems biology of growth factor-induced receptor endocytosis. Traffic 10, 349–363 [DOI] [PubMed] [Google Scholar]
- 6. Vergarajauregui S., San Miguel A., Puertollano R. (2006) Activation of p38 mitogen-activated protein kinase promotes epidermal growth factor receptor internalization. Traffic 7, 686–698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Winograd-Katz S. E., Levitzki A. (2006) Cisplatin induces PKB/Akt activation and p38(MAPK) phosphorylation of the EGF receptor. Oncogene 25, 7381–7390 [DOI] [PubMed] [Google Scholar]
- 8. Zwang Y., Yarden Y. (2006) p38 MAP kinase mediates stress-induced internalization of EGFR: implications for cancer chemotherapy. EMBO J. 25, 4195–4206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Mosesson Y., Mills G. B., Yarden Y. (2008) Derailed endocytosis: an emerging feature of cancer. Nat. Rev. Cancer 8, 835–850 [DOI] [PubMed] [Google Scholar]
- 10. Lemmon M. A., Schlessinger J. (2010) Cell signaling by receptor tyrosine kinases. Cell 141, 1117–1134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Schlessinger J. (2002) Ligand-induced, receptor-mediated dimerization and activation of EGF receptor. Cell 110, 669–672 [DOI] [PubMed] [Google Scholar]
- 12. Goh L. K., Huang F., Kim W., Gygi S., Sorkin A. (2010) Multiple mechanisms collectively regulate clathrin-mediated endocytosis of the epidermal growth factor receptor. J. Cell Biol. 189, 871–883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Levkowitz G., Waterman H., Ettenberg S. A., Katz M., Tsygankov A. Y., Alroy I., Lavi S., Iwai K., Reiss Y., Ciechanover A., Lipkowitz S., Yarden Y. (1999) Ubiquitin ligase activity and tyrosine phosphorylation underlie suppression of growth factor signaling by c-Cbl/Sli-1. Mol. Cell 4, 1029–1040 [DOI] [PubMed] [Google Scholar]
- 14. Huang F., Kirkpatrick D., Jiang X., Gygi S., Sorkin A. (2006) Differential regulation of EGF receptor internalization and degradation by multiubiquitination within the kinase domain. Mol. Cell 21, 737–748 [DOI] [PubMed] [Google Scholar]
- 15. Waterman H., Katz M., Rubin C., Shtiegman K., Lavi S., Elson A., Jovin T., Yarden Y. (2002) A mutant EGF-receptor defective in ubiquitylation and endocytosis unveils a role for Grb2 in negative signaling. EMBO J. 21, 303–313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lowenstein E. J., Daly R. J., Batzer A. G., Li W., Margolis B., Lammers R., Ullrich A., Skolnik E. Y., Bar-Sagi D., Schlessinger J. (1992) The SH2 and SH3 domain-containing protein GRB2 links receptor tyrosine kinases to ras signaling. Cell 70, 431–442 [DOI] [PubMed] [Google Scholar]
- 17. Sorkin A., Goh L. K. (2009) Endocytosis and intracellular trafficking of ErbBs. Exp. Cell Res. 315, 683–696 [DOI] [PubMed] [Google Scholar]
- 18. Grandal M. V., Grovdal L. M., Henriksen L., Andersen M. H., Holst M. R., Madshus I. H., van Deurs B. (2012) Differential roles of Grb2 and AP-2 in p38 MAPK- and EGF-induced EGFR internalization. Traffic 13, 576–585 [DOI] [PubMed] [Google Scholar]
- 19. Hinrichsen L., Harborth J., Andrees L., Weber K., Ungewickell E. J. (2003) Effect of clathrin heavy chain- and alpha-adaptin-specific small inhibitory RNAs on endocytic accessory proteins and receptor trafficking in HeLa cells. J. Biol. Chem. 278, 45160–45170 [DOI] [PubMed] [Google Scholar]
- 20. Johannessen L. E., Pedersen N. M., Pedersen K. W., Madshus I. H., Stang E. (2006) Activation of the epidermal growth factor (EGF) receptor induces formation of EGF receptor- and Grb2-containing clathrin-coated pits. Mol. Cell. Biol. 26, 389–401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Rappoport J. Z., Simon S. M. (2009) Endocytic trafficking of activated EGFR is AP-2 dependent and occurs through preformed clathrin spots. J. Cell Sci. 122, 1301–1305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sorkin A., Mazzotti M., Sorkina T., Scotto L., Beguinot L. (1996) Epidermal growth factor receptor interaction with clathrin adaptors is mediated by the Tyr974-containing internalization motif. J. Biol. Chem. 271, 13377–13384 [DOI] [PubMed] [Google Scholar]
- 23. Wang Z., Moran M. F. (1996) Requirement for the adapter protein GRB2 in EGF receptor endocytosis. Science 272, 1935–1939 [DOI] [PubMed] [Google Scholar]
- 24. Jiang X., Huang F., Marusyk A., Sorkin A. (2003) Grb2 regulates internalization of EGF receptors through clathrin-coated pits. Mol. Biol. Cell 14, 858–870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Duan L., Miura Y., Dimri M., Majumder B., Dodge I. L., Reddi A. L., Ghosh A., Fernandes N., Zhou P., Mullane-Robinson K., Rao N., Donoghue S., Rogers R. A., Bowtell D., Naramura M., Gu H., Band V., Band H. (2003) Cbl-mediated ubiquitinylation is required for lysosomal sorting of epidermal growth factor receptor but is dispensable for endocytosis. J. Biol. Chem. 278, 28950–28960 [DOI] [PubMed] [Google Scholar]
- 26. Huang F., Goh L. K., Sorkin A. (2007) EGF receptor ubiquitination is not necessary for its internalization. Proc. Natl. Acad. Sci. U.S.A. 104, 16904–16909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Steen H., Kuster B., Fernandez M., Pandey A., Mann M. (2002) Tyrosine phosphorylation mapping of the epidermal growth factor receptor signaling pathway. J. Biol. Chem. 277, 1031–1039 [DOI] [PubMed] [Google Scholar]
- 28. Chang C. P., Lazar C. S., Walsh B. J., Komuro M., Collawn J. F., Kuhn L. A., Tainer J. A., Trowbridge I. S., Farquhar M. G., Rosenfeld M. G., Wiley H. S., Gill G. N. (1993) Ligand-induced internalization of the epidermal growth factor receptor is mediated by multiple endocytic codes analogous to the tyrosine motif found in constitutively internalized receptors. J. Biol. Chem. 268, 19312–19320 [PubMed] [Google Scholar]
- 29. Tong J., Taylor P., Peterman S. M., Prakash A., Moran M. F. (2009) Epidermal growth factor receptor phosphorylation sites Ser991 and Tyr998 are implicated in the regulation of receptor endocytosis and phosphorylations at Ser1039 and Thr1041. Mol. Cell. Proteomics 8, 2131–2144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Huang F., Jiang X., Sorkin A. (2003) Tyrosine phosphorylation of the beta2 subunit of clathrin adaptor complex AP-2 reveals the role of a di-leucine motif in the epidermal growth factor receptor trafficking. J. Biol. Chem. 278, 43411–43417 [DOI] [PubMed] [Google Scholar]
- 31. Tong J., Sydorskyy Y., St-Germain J. R., Taylor P., Tsao M. S., Moran M. F. (2013) Odin (ANKS1A) modulates EGF receptor recycling and stability. PLoS One 8, e64817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ernst A., Avvakumov G., Tong J., Fan Y., Zhao Y., Alberts P., Persaud A., Walker J. R., Neculai A. M., Neculai D., Vorobyov A., Garg P., Beatty L., Chan P. K., Juang Y. C., Landry M. C., Yeh C., Zeqiraj E., Karamboulas K., Allali-Hassani A., Vedadi M., Tyers M., Moffat J., Sicheri F., Pelletier L., Durocher D., Raught B., Rotin D., Yang J., Moran M. F., Dhe-Paganon S., Sidhu S. S. (2013) A strategy for modulation of enzymes in the ubiquitin system. Science 339, 590–595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. de Wit R., Hendrix C. M., Boonstra J., Verkleu A. J., Post J. A. (2000) Large-scale screening assay to measure epidermal growth factor internalization. J. Biomol. Screen. 5, 133–140 [DOI] [PubMed] [Google Scholar]
- 34. Frey M. R., Dise R. S., Edelblum K. L., Polk D. B. (2006) p38 kinase regulates epidermal growth factor receptor downregulation and cellular migration. EMBO J. 25, 5683–5692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. MacLean B., Tomazela D. M., Shulman N., Chambers M., Finney G. L., Frewen B., Kern R., Tabb D. L., Liebler D. C., MacCoss M. J. (2010) Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics 26, 966–968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Michalski A., Damoc E., Hauschild J. P., Lange O., Wieghaus A., Makarov A., Nagaraj N., Cox J., Mann M., Horning S. (2011) Mass spectrometry-based proteomics using Q Exactive, a high-performance benchtop quadrupole Orbitrap mass spectrometer. Mol. Cell. Proteomics 10, M111 011015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Deeb S. J., D'Souza R. C., Cox J., Schmidt-Supprian M., Mann M. (2012) Super-SILAC allows classification of diffuse large B-cell lymphoma subtypes by their protein expression profiles. Mol. Cell. Proteomics 11, 77–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Cox J., Mann M. (2008) MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 26, 1367–1372 [DOI] [PubMed] [Google Scholar]
- 39. Cox J., Neuhauser N., Michalski A., Scheltema R. A., Olsen J. V., Mann M. (2011) Andromeda: a peptide search engine integrated into the MaxQuant environment. J. Proteome Res. 10, 1794–1805 [DOI] [PubMed] [Google Scholar]
- 40. Tong J., Taylor P., Jovceva E., St-Germain J., Jin L., Nikolic A., Gu X., Li Z., Trudel S, Moran M. (2008) Tandem Immunoprecipitation of phosphotyrosine-mass spectrometry (TIPY-MS) indicates C19orf19 becomes tyrosine phosphorylated and associated with activated epidermal growth factor receptor. J. Proteome Res. 7, 1067–1077 [DOI] [PubMed] [Google Scholar]
- 41. Nagaraj N., Wisniewski J. R., Geiger T., Cox J., Kircher M., Kelso J., Paabo S., Mann M. (2011) Deep proteome and transcriptome mapping of a human cancer cell line. Mol. Sys. Biol. 7, 548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Hembrough T., Thyparambil S., Liao W. L., Darfler M. M., Abdo J., Bengali K. M., Taylor P., Tong J., Lara-Guerra H., Waddell T. K., Moran M. F., Tsao M. S., Krizman D. B., Burrows J. (2012) Selected reaction monitoring (SRM) analysis of epidermal growth factor receptor (EGFR) in formalin fixed tumor tissue. Clin. Proteomics 9, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Singhirunnusorn P., Ueno Y., Matsuo M., Suzuki S., Saiki I., Sakurai H. (2007) Transient suppression of ligand-mediated activation of epidermal growth factor receptor by tumor necrosis factor-alpha through the TAK1-p38 signaling pathway. J. Biol. Chem. 282, 12698–12706 [DOI] [PubMed] [Google Scholar]
- 44. Moehren G., Markevich N., Demin O., Kiyatkin A., Goryanin I., Hoek J. B., Kholodenko B. N. (2002) Temperature dependence of the epidermal growth factor receptor signaling network can be accounted for by a kinetic model. Biochemistry 41, 306–320 [DOI] [PubMed] [Google Scholar]
- 45. McCaffrey M. W., Bielli A., Cantalupo G., Mora S., Roberti V., Santillo M., Drummond F., Bucci C. (2001) Rab4 affects both recycling and degradative endosomal trafficking. FEBS Lett. 495, 21–30 [DOI] [PubMed] [Google Scholar]
- 46. Batzer A. G., Rotin D., Urena J. M., Skolnik E. Y., Schlessinger J. (1994) Hierarchy of binding sites for Grb2 and Shc on the epidermal growth factor receptor. Mol. Cell. Biol. 14, 5192–5201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Stover D., Caldwell J., Marto J., Root K., Mestan J., Stumm M., Ornatsky O., Orsi C., Radosevic N., Liao L., Fabbro D., Moran M. F. (2004) Differential phosphoprofiles of EGF and EGFR kinase inhibitor-treated human tumor cells and mouse xenografts. Clin. Proteomics 1, 69–80 [Google Scholar]
- 48. Adachi S., Natsume H., Yamauchi J., Matsushima-Nishiwaki R., Joe A. K., Moriwaki H., Kozawa O. (2009) p38 MAP kinase controls EGF receptor downregulation via phosphorylation at Ser1046/1047. Cancer Lett. 277, 108–113 [DOI] [PubMed] [Google Scholar]
- 49. Hubner N. C., Mann M. (2011) Extracting gene function from protein-protein interactions using Quantitative BAC InteraCtomics (QUBIC). Methods 53, 453–459 [DOI] [PubMed] [Google Scholar]
- 50. Chen J. Y., Mamidipalli S., Huan T. (2009) HAPPI: an online database of comprehensive human annotated and predicted protein interactions. BMC Genomics 10 Suppl 1, S16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Peri S., Navarro J. D., Kristiansen T. Z., Amanchy R., Surendranath V., Muthusamy B., Gandhi T. K., Chandrika K. N., Deshpande N., Suresh S., Rashmi B. P., Shanker K., Padma N., Niranjan V., Harsha H. C., Talreja N., Vrushabendra B. M., Ramya M. A., Yatish A. J., Joy M., Shivashankar H. N., Kavitha M. P., Menezes M., Choudhury D. R., Ghosh N., Saravana R., Chandran S., Mohan S., Jonnalagadda C. K., Prasad C. K., Kumar-Sinha C., Deshpande K. S., Pandey A. (2004) Human protein reference database as a discovery resource for proteomics. Nucleic Acids Res. 32, D497–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Jeram S. M., Srikumar T., Pedrioli P. G., Raught B. (2009) Using mass spectrometry to identify ubiquitin and ubiquitin-like protein conjugation sites. Proteomics 9, 922–934 [DOI] [PubMed] [Google Scholar]
- 53. Huang F., Zeng X., Kim W., Balasubramani M., Fortian A., Gygi S. P., Yates N. A., Sorkin A. (2013) Lysine 63-linked polyubiquitination is required for EGF receptor degradation. Proc. Natl. Acad. Sci. U.S.A. 110, 15722–15727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Stein B. S., Bensch K. G., Sussman H. H. (1984) Complete inhibition of transferrin recycling by monensin in K562 cells. J. Biol. Chem. 259, 14762–14772 [PubMed] [Google Scholar]
- 55. Vieira A. V., Lamaze C., Schmid S. L. (1996) Control of EGF receptor signaling by clathrin-mediated endocytosis. Science 274, 2086–2089 [DOI] [PubMed] [Google Scholar]
- 56. Sousa L. P., Lax I., Shen H., Ferguson S. M., De Camilli P., Schlessinger J. (2012) Suppression of EGFR endocytosis by dynamin depletion reveals that EGFR signaling occurs primarily at the plasma membrane. Proc. Natl. Acad. Sci. U.S.A 109, 4419–4424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Omerovic J., Hammond D. E., Prior I. A., Clague M. J. (2012) Global snapshot of the influence of endocytosis upon EGF receptor signaling output. J. Proteome Res. 11, 5157–5166 [DOI] [PubMed] [Google Scholar]
- 58. Olsen J. V., Blagoev B., Gnad F., Macek B., Kumar C., Mortensen P., Mann M. (2006) Global, in vivo, and site-specific phosphorylation dynamics in signaling networks. Cell 127, 635–648 [DOI] [PubMed] [Google Scholar]
- 59. Wolf-Yadlin A., Hautaniemi S., Lauffenburger D. A., White F. M. (2007) Multiple reaction monitoring for robust quantitative proteomic analysis of cellular signaling networks. Proc. Natl. Acad. Sci. U.S.A. 104, 5860–5865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Koch C. A., Anderson D., Moran M. F., Ellis C., Pawson T. (1991) SH2 and SH3 domains: elements that control interactions of cytoplasmic signaling proteins. Science 252, 668–674 [DOI] [PubMed] [Google Scholar]
- 61. Akimov V., Rigbolt K. T., Nielsen M. M., Blagoev B. (2011) Characterization of ubiquitination dependent dynamics in growth factor receptor signaling by quantitative proteomics. Mol. Biosyst. 7, 3223–3233 [DOI] [PubMed] [Google Scholar]
- 62. Mohapatra B., Ahmad G., Nadeau S., Zutshi N., An W., Scheffe S., Dong L., Feng D., Goetz B., Arya P., Bailey T. A., Palermo N., Borgstahl G. E., Natarajan A., Raja S. M., Naramura M., Band V., Band H. (2013) Protein tyrosine kinase regulation by ubiquitination: critical roles of Cbl-family ubiquitin ligases. Biochim. Biophys. Acta 1833, 122–139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Courbard J. R., Fiore F., Adelaide J., Borg J. P., Birnbaum D., Ollendorff V. (2002) Interaction between two ubiquitin-protein isopeptide ligases of different classes, CBLC and AIP4/ITCH. J. Biol. Chem. 277, 45267–45275 [DOI] [PubMed] [Google Scholar]
- 64. Sorkina T., Bild A., Tebar F., Sorkin A. (1999) Clathrin, adaptors and eps15 in endosomes containing activated epidermal growth factor receptors. J. Cell Sci. 112 (Pt 3), 317–327 [DOI] [PubMed] [Google Scholar]
- 65. McPherson P. S. (2010) Proteomic analysis of clathrin-coated vesicles. Proteomics 10, 4025–4039 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.