

Abstract
Protein abundance and phosphorylation convey important information about pathway activity and molecular pathophysiology in diseases including cancer, providing biological insight, informing drug and diagnostic development, and guiding therapeutic intervention. Analyzed tissues are usually collected without tight regulation or documentation of ischemic time. To evaluate the impact of ischemia, we collected human ovarian tumor and breast cancer xenograft tissue without vascular interruption and performed quantitative proteomics and phosphoproteomics after defined ischemic intervals. Although the global expressed proteome and most of the >25,000 quantified phosphosites were unchanged after 60 min, rapid phosphorylation changes were observed in up to 24% of the phosphoproteome, representing activation of critical cancer pathways related to stress response, transcriptional regulation, and cell death. Both pan-tumor and tissue-specific changes were observed. The demonstrated impact of pre-analytical tissue ischemia on tumor biology mandates caution in interpreting stress-pathway activation in such samples and motivates reexamination of collection protocols for phosphoprotein analysis.
Genomic analysis of thousands of tumor samples from multiple cancer types is currently being carried out by consortia such as the Cancer Genome Atlas (TCGA)1 and the International Cancer Genome Consortium. These studies are yielding new insights into the biology of breast (1), brain (2), colon (3), lung (4), ovarian (5), and endometrial cancers (6, 7). In clinical trial settings, tumor biopsies are also being characterized to identify the molecular basis for drug response and to identify therapeutic targets (8). There is growing recognition that proteomic characterization of genomically annotated samples could provide complementary as well as unique information on cancer biology and signaling that is inaccessible through DNA and RNA analysis alone. In response to this perspective, reverse-phase protein array (RPPA) technology (9) has been incorporated into the analysis pipeline for TCGA samples. RPPA is a highly parallelized, chip-based dot-blot approach employing very well-characterized anti-protein and anti-phosphopeptide antibodies. The power of proteomics in the context of TCGA has recently been highlighted by the ability of RPPA to identify new molecular taxonomies in breast cancer subtypes not discernible via genomics (1). A limitation of RPPA is that only around 200 antibodies are currently demonstrated to work well in this methodology.
We hypothesized that concurrent and complementary evaluation of the functional proteomes (including posttranslational modifications) of tumors will improve our ability to diagnose, treat, and prevent cancers by enabling a better understanding of the molecular basis of these diseases, especially when integrated and analyzed together with the comprehensive genomic characterization information. Recent advances in the preparation of samples and their quantitative analysis by means of mass-spectrometry-based proteomics and phosphoproteomics now yield datasets covering nearly half of the predicted proteome and identifying many thousands of phosphopeptides (10–14). These advances have sparked efforts utilizing state-of-the-art proteomics to characterize the proteomes and define changes in the posttranslational modification landscape of tumor samples. The National Cancer Institute's Clinical Proteomic Tumor Analysis Consortium is therefore characterizing the proteomes of large numbers of breast, colon, and ovarian tumor samples that have been genomically characterized as part of the TCGA program.
A concern faced by the Clinical Proteomic Tumor Analysis Consortium program is that the samples provided by TCGA for proteomic analysis were not obtained with proteomic studies in mind. Importantly, the total duration of ischemia prior to sample freezing is generally unknown in TCGA samples but frequently spans tens of minutes to over an hour. The total ischemia duration comprises a period of warm ischemia corresponding to the time from blood vessel ligation to surgical excision and then cold ischemia corresponding to the delay time to freezing post-excision (usually a mix of transportation time to pathology and pathological analyses). Until now, the effect of cold ischemia on the stability of proteins and phosphosites has primarily been studied only for selected candidate proteins and phosphosites for which high-quality antibodies were available. A study employing AQUA, a quantitative immunofluorescence technique, found slightly increased expression of hypoxia inducible factor but no changes in protein abundance in the four breast cancer biomarker proteins ER, PR, HER2, and Ki67 over a time span of up to 7 h, though signal reduction occurred in a subset of samples at longer intervals of up to 48 h of cold ischemia (15). In another study employing immunohistochemistry, the breast cancer biomarker proteins ER, PR, and Her2 were found to be stable for up to 2 h at room temperature (16). Evidence from RPPA studies evaluating the time course of changes induced by cold ischemia has shown that even when the total protein levels and levels of many phosphoproteins are constant, the phosphorylation stoichiometry of specific proteins can change, in some cases significantly (9, 17). For example, Espina et al. used 55 phosphosite-specific antibodies in RPPA format to show that in various tissues, including breast, ovary, colon, and endometrium, cold ischemia can cause up to a 2-fold variation in phosphorylation abundance and that these processes can be augmented by phosphatase inhibitors and reduced by kinase inhibitors (17) or heat stabilization (18). However, because of clinical sample handling limitations, the earliest time points evaluated in this study occurred between 4 and 40 min after tissue resection with an average time to cryopreservation of 19.3 min (17). The duration of blood vessel ligation, a common procedure in nearly all surgeries, was not noted, and so the actual total ischemia times (warm ischemia plus cold ischemia) might have been longer. Hennessy et al. analyzed the effects of cold ischemia in breast cancer tumor samples using RPPA and found that 21 of 82 proteins and phosphoproteins demonstrated time-dependent instability at room temperature between 6 and 24 h of cold ischemia but exhibited few effects at earlier time points (9). Gundish et al. used both RPPA and LC-MS/MS to analyze a time course of cold ischemia in normal liver tissue from mouse and rat (34). No significant changes were observed in the phosphoproteome after up to 60 min of cold ischemia, in agreement with the findings of Hennessy et al. In the LC-MS/MS study by Gundisch et al., approximately 1700 phosphosites were quantified, but no statistically significant alterations of individual phosphosites after over 60 min of cold ischemia were found. A few other unbiased mass-spectrometry-based proteomic studies of cold ischemia have been reported. These studies identified a small number of proteins that changed in abundance (primarily as a result of degradation, presumably via proteolysis) after long (> 3 h) periods of cold ischemia (19, 20). For example, Li et al., using two-dimensional fluorescence difference gel electrophoresis in conjunction with mass spectrometry, identified 26 proteins that changed over 48 h of cold ischemia time, chiefly as a result of degradation (20). The sample collection and analysis approaches used in most of these studies did not permit assessment of changes within the one-hour interval investigated in our study.
Before embarking on analyses of posttranslational modifications in TCGA-profiled tumor samples, we wanted to better understand the potential effects of ischemia on the proteome and phosphoproteome of tumor tissue. Here we describe our time-course studies of patient-derived xenografts of human breast cancer tumors and patient-derived ovarian cancer tumors. Tumor tissues were excised prior to vascular ligation in order to accurately define the ischemia time, and they were prepared centrally using a cryopulverization method that maintained the tissue in a frozen state. Samples were analyzed using a common sample-processing workflow for global, quantitative proteome and phosphoproteome analysis using iTRAQ chemical mass tag labeling for quantification (11, 21) and state-of-the-art high-performance, multidimensional LC-MS/MS on high-performance mass spectrometers for data collection.
EXPERIMENTAL PROCEDURES
Preparation of Ovarian Tumor Samples for Proteomic Analysis
After obtaining consent to Institutional Review Board–approved protocols, we collected tissue from high-grade serous ovarian carcinoma tumors from four patients. Immediately after resection, the tumors were dissected into four contiguous and adjacent strips, each no larger than 10 mm × 3 mm × 3 mm. Tumor strips were placed into cryovials and frozen in liquid nitrogen at specified time points.
Generation and Preparation of Xenograft Tumor Samples for Proteomic Analysis
All human tissues for these experiments were processed in compliance with NIH regulations and institutional guidelines approved by the Institutional Review Board at Washington University. All animal procedures were reviewed and approved by the Institutional Animal Care and Use Committee at Washington University in St. Louis, MO. Patient-derived xenograft (PDX) tumors from established basal (WHIM6) and luminal (WHIM20) breast cancer subtypes were raised subcutaneously in 8-week-old NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ mice (Jackson Labs, Bar Harbor, ME) as previously described (22, 23). Tumors from each animal were harvested by surgical excision at ∼1.5 cm3, rapidly divided into four pieces, and snap-frozen by immersion in a liquid nitrogen bath at times 0 (∼30 s), 5, 30, and 60 min post-excision.
Protein Extraction, Digestion, Labeling, and Mass Spectrometry Analysis of Peptides from Patient-derived Ovarian Tumors and Breast Cancer PDX Models
Proteins extracted from tumor tissues (∼50-mg tissue weight for ovarian and ∼100-mg tissue weight for breast cancer xenograft) were reduced, alkylated, and subjected to enzyme digestion. Peptides were labeled with 4-plex iTRAQ reagents and separated using an off-line high pH (7.5 or 10) reversed-phase column. Fractions were collected and concatenated (14, 24) into 24 fractions, and 5% of each fraction was analyzed via LC-MS/MS for quantitative global proteomics measurement. The remainder (95%) of each of the 24 fractions was further concatenated into 12 fractions, and phosphopeptides were enriched using immobilized metal (Fe3+) affinity chromatography prior to LC-MS/MS analysis.
Peptides from ovarian cancer samples were analyzed using a nanoLC system coupled to an LTQ-Orbitrap-Velos MS (Thermo Scientific) at Pacific Northwest National Laboratories. Peptides from breast cancer PDX samples were analyzed using a nanoLC system coupled to a Q Exactive MS (Thermo Scientific) at the Broad Institute. All mass spectrometry data were analyzed using the Spectrum Mill software package, v4.1 beta (Agilent Technologies, Santa Clara, CA). Peptide identifications and quantification information were further combined into protein and phosphosite tables. Statistical analysis of cold-ischemia regulated proteins and phosphosites via kinetic modeling and moderated F-test was performed using the R software environment (version 2.15.2).
RPPA Analysis
Protein expression or phosphorylation was measured via RPPA as previously described (9).
More detailed experimental methods are presented in the supplemental “Materials and Methods” section.
RESULTS
Experimental Design to Study Effects of Cold Ischemia on the Proteome and Phosphoproteome of Ovarian and Breast Cancer Samples Using Quantitative LC-MS/MS
In this study we analyzed the effect of delayed freezing on protein and phosphoprotein stability in tumor samples derived from four ovarian cancer patients and two breast cancer PDXs (one basal and one luminal) (22). Blood vessels were not ligated prior to tumor excision, which eliminated effects of warm ischemia. Individual tumor samples were cut into four sections, which were subsequently stored at room temperature for predefined time intervals (Fig. 1A). Upon partitioning, the first section was frozen immediately with a time span of less than 1 min from excision to freezing in liquid nitrogen. Later time-point sections were left at room temperature and frozen after 5, 30, or 60 min in order to investigate the kinetics of changes in the proteome and phosphoproteome caused by delayed freezing.
Fig. 1.

Quantitative proteome and phosphoproteome analysis of human ovarian tumors and xenograft breast tumors subjected to controlled ischemia. A, experimental design to study effects of post-excision delay time before freezing across four time points. After excision, tumor samples were cut into four equal pieces and incubated for the indicated times at room temperature before freezing. A total of four different ovarian tumors and three pooled breast cancer xenograft samples for the basal and for the luminal subtype were analyzed. All of these samples were biologically distinct and can be considered as biological replicates. B, quantitative proteomics and phosphoproteomics workflow using 4-plex iTRAQ labeling. Tumor samples were cryofractured and proteins were extracted with urea lysis buffer prior to digestion into peptides using trypsin. Peptide samples derived at four different ischemic time points were labeled using iTRAQ reagents, mixed equably, and separated using high-pH reversed-phase chromatography. Fractions were combined in a noncontiguous way into 24 fractions for proteome analysis (5% of the total material) and 12 fractions for phosphoproteome analysis (95% of the total material). Ovarian cancer samples were analyzed on an LTQ-Orbitrap Velos, and xenograft breast cancer samples were analyzed on a Q Exactive mass spectrometer. Phosphosite and protein identification and quantification were achieved using Spectrum Mill.
The ovarian tumors were obtained from four individual cancer patients with high-grade serous ovarian carcinoma, and the four time-point samples per patient were derived from four contiguous, adjacent portions of the same tumor. Although the use of surgically excised tumors was desirable because it recapitulated a typical biospecimen collection scenario, it was recognized that intratumor heterogeneity (e.g. genetic differences or differences in non-tumor elements) could result in proteomic variations not related to ischemic time. To address this concern, as well as to extend our analysis to another tumor type, we carried out the same ischemia time-course study in xenografts from two distinct molecular phenotypes of breast cancer, ER+ (luminal) and ER− (basal-like). Tumor xenografts have less cellular infiltration and therefore are less heterogeneous than tumors excised from patients. Additionally, because the total tumor material was not limiting for the breast cancer xenograft samples, we pooled 5 to 10 xenografts (derived from the same luminal and basal-like tumors) at each time point to dampen the effects of biological variability. We generated three individual pools for the basal and the luminal breast cancer xenograft samples which can be considered as biological replicates, as each individually grown xenograft tumor was present only in one pooled sample.
For analysis of the proteome and phosphoproteome, frozen tumor samples were cryofractured, and proteins were extracted with an 8 m urea buffer and digested into peptides using trypsin (Fig. 1B). For relative quantification across the four ischemia time points of each tumor, peptide samples were chemically labeled with iTRAQ reagents and subsequently combined into a single sample. iTRAQ labeling covalently attaches isotope-coded isobaric mass tags to primary amine groups at the N termini of peptides and lysine side chains. Upon fragmentation in the mass spectrometer, reporter ions at defined masses are released, and the ratios of these reporter ions to one another enables relative quantification (21). After iTRAQ labeling, peptide samples were fractionated using basic reverse-phase liquid chromatography to reduce the overall sample complexity per fraction and thus increase the depth of coverage (24). Basic reverse-phase liquid chromatography fractions were combined in a noncontiguous manner into 24 proteome and 12 phosphoproteome fractions, with 5% of the total material contributing to the proteome analysis and 95% to the phosphoproteome analysis. Phosphopeptides were further enriched using immobilized metal affinity chromatography.
Cold Ischemia Induces Significant Changes in Protein Phosphorylation but No Changes in Overall Protein Abundance
Our quantitative LC-MS/MS platform was extensively tested within the Clinical Proteomic Tumor Analysis Consortium program to ensure the reproducibility of peptide identification and quantification for proteome and phosphoproteome analysis. Average Pearson correlations for the process replicate analysis of breast cancer xenograft tumors were r = 0.808 on the proteome level and r = 0.812 on the phosphoproteome level. Using our platform, nearly 15,000 different proteins and up to 26,000 distinct phosphosites were reproducibly quantified in at least two tumor samples (Table I, supplemental Tables S1 and S2). Among these were 86 phospho-tyrosine (pTyr) sites and 202 pTyr sites that were reproducibly measured in the ovarian and breast cancer tumor samples, respectively. The total protein and phosphosite counts were in general higher for xenograft than for human ovarian tumor samples, in part because the xenograft samples contained additional proteins from mouse stroma (no more than 20% based on genomic analysis), but also because the breast tumor samples were analyzed using a more sensitive mass spectrometer (see supplemental “Materials and Methods”). To identify consistent ischemia-induced alterations, we employed two complementary statistical tests. The first approach assumed that most of the changes were unidirectional and governed by the first-order chemical kinetics rule. In this method we used a nonlinear regression with three degrees of freedom that fit initial abundance, final abundance, and a kinetic constant. The second approach did not rely on an explicit assumption that the changes followed a particular pattern. Instead, each time point was treated as an independent group in an analysis of variance, with statistical significance assessed using a moderated F-test. This allowed us to capture additional trends that were not unidirectional. Proteins and phosphosites with a Benjamini–Hochberg corrected p value of <0.01 in either test (i.e. the union of significant proteins/phosphosites) were considered to be regulated by ischemia. In this analysis, we used a relatively stringent p value of 0.01 (compared with the usual p value of 0.05) for statistical significance in order to minimize false positive results. When the number of samples studied is relatively small, as in this study, borderline p values can change in significance as more samples are analyzed. As with any statistical analysis, we expected that the list of phosphosites ranked as significant would include some false positives and false negatives. We provide tables of p values for all proteins and phosphosites (supplemental Tables S1 and S2) to facilitate assessment of significance not based on a specific p value threshold.
Table I. Number of quantified and ischemia-regulated phosphosites and proteins in ovarian and breast cancer tumors.
| Number of tumor, samples | Total | Average per tumor sample | Overlap in at least n − 1 samples | Kinetics-based regression testa | Moderated F-testa | Union of both testsa | Percent regulatedb | |
|---|---|---|---|---|---|---|---|---|
| Quantified phosphorylation sites | (Up/down) | (Up/down) | (Up/down) | (Up/down) | ||||
| Ovarian cancer | 4 | 23,607 | 13,156 | 9443 | 307/97 | 386/63 | 432/111 | 4.6/1.2 |
| Basal breast cancer | 3 | 38,366 | 27,668 | 26211 | 1252/948 | 1156/633 | 1493/1027 | 5.7/3.9 |
| Luminal breast cancer | 3 | 34,327 | 25,814 | 25102 | 4154/820 | 4220/962 | 4977/1139 | 19.8/4.5 |
| Quantified proteins | ||||||||
| Ovarian cancer | 4 | 9498 | 7550 | 6985 | 0/0 | 0/0 | 0/0 | 0/0 |
| Basal breast cancer | 3 | 17,158 | 14,989 | 14,970 | 0/0 | 0/0 | 0/0 | 0/0 |
| Luminal breast cancer | 3 | 14,224 | 12,641 | 12,679 | 0/0 | 0/0 | 0/0 | 0/0 |
a Significant regulation at a kinetics-based regression test or moderated F-test false discovery rate p < 0.01.
b Percent regulated phosphosites and proteins within overlap dataset.
We did not detect a single significantly regulated protein with altered abundance levels at up to 60 min of ischemia time for any of the ovarian tumor samples or for the luminal and basal-like PDX samples. In contrast, significant ischemia-induced changes in the phosphoproteome were observed for all analyzed tumor types (Table I). The kinetics-based regression and the moderated F-test analysis (analysis of variance) identified a largely overlapping set of regulated phosphorylation sites for a given tumor; only a small number of changes were identified uniquely by only one of the statistical tests (supplemental Fig. S1). Response to cold ischemia was asymmetrically distributed, with more phosphorylation sites being up-regulated than down-regulated for all of the tumors (Fig. 2). The largest changes in ischemia-induced phosphorylation were observed for the luminal breast cancer PDX, which exhibited 19.8% up-regulated and 4.5% down-regulated phosphorylation sites at a 1% false discovery rate (Table I). The ischemic behavior of ovarian and basal-like breast cancer samples, which have been proposed to be molecularly similar (1), was less dramatic, with 4.6% and 5.7% up-regulated and 1.2% and 3.9% down-regulated phosphorylation sites, respectively. For these three tumor types, only the tails of the density distribution of Log2 ratios increased with ischemic time. In contrast, the global median of the luminal breast cancer phosphoproteome was shifted to increased abundance levels (Fig. 2). The effects of cold ischemia on pTyr sites were comparable to those observed on pSer/pThr sites in the whole phosphoproteome analysis. In the ovarian cancer and basal-like breast cancer samples, 8% of all pTyr sites were up-regulated and 2% to 4% were down-regulated. Ischemia-regulated pTyr sites were more frequent in the luminal tumors, with 13.6% up-regulated and 5.4% down-regulated sites.
Fig. 2.
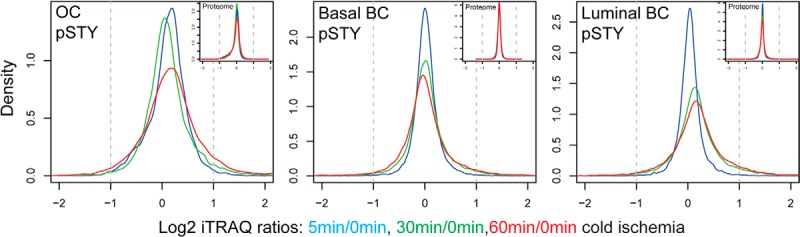
Global proteome analysis revealed no changes in protein abundance level, but specific alterations in the phosphoproteome induced by ischemia were observed. Density plots are shown for averaged phosphosite iTRAQ ratios (labeled with “pSTY”) and protein iTRAQ ratios (small insets) for the ovarian cancer (OC) samples and the basal-like and luminal breast cancer (BC) samples. Only phosphosites/proteins were plotted that were quantified in at least three ovarian tumors and at least two basal-like or luminal tumor samples.
Kinetic Analysis of Phosphoproteome Dynamics Reveals Phosphosites That Change as Early as 5 min After Sample Excision
To identify temporal trends, we used fuzzy c-means clustering (25) to assign membership of all regulated phosphorylation sites from all three tumor types in nine different clusters with different temporal profiles. These clusters were further combined into three up- (U) and three down- (D) regulated groups (Fig. 3A). In addition, we calculated the median half-activation times for all sites that belonged to one of these groups to determine at what time these sites had reached 50% of their maximum alteration (saturation). The early groups U1 and D1 showed T1/2 values of 3 and 2 min, and the late groups U3 and D3 had values of of 46 and 34.4 min, respectively. The number of phosphorylation sites grouped into the different temporal clusters varied across all three tumor types (Fig. 3B). Enrichment analysis of Gene Ontology Biological Process (GO BP) terms for the up-regulated clusters revealed activation of MAPK cascade and Ras signaling pathways for the early group U1 (Fig. 3C). Specific activation of transcriptional regulators was observed for the medium group U2, and regulation of chromatin modification and assembly (among other biological processes) was noted for the late group U3. In the down-regulated clusters D1–D3, regulation of Rho, Ras, and GTPase signal transduction and regulation of cytoskeleton organization were among the more highly enriched GO BP terms. Fewer GO BP terms were enriched for groups D1–D3 because of the smaller number of down-regulated phosphorylation sites. The response kinetics in the different tumor types were slightly offset, with some kinases appearing in different temporal clusters for the different tumor types (supplemental Fig. S2).
Fig. 3.

Temporal dynamics of phosphorylation changes resulting from cold ischemia. A, fuzzy c-means clustering of temporal profiles for all regulated phosphosites observed in the ovarian and breast cancer samples. We detected six up-regulated (U) clusters, which were further grouped pairwise into early (U1), middle (U2), and late clusters (U3), and three down-regulated (D1, D2, D3) clusters using a fuzzyfication parameter m = 1.6. Phosphosites were assigned to each cluster with a membership value α > 0.7. T1/2 indicates the median over all half-maximum time points for all phosphosites within a cluster. Half-maximum time points were determined via first-order kinetic modeling analysis. B, number of regulated phosphosites assigned to each cluster for the ovarian and breast cancer tumors. C, gene enrichment analysis of regulated human phosphoproteins across early, middle, and late clusters. We used DAVID Bioinformatics Resources 6.7 (39) to test for enrichment of GO BPs in each cluster relative to a list of all proteins containing nonregulated sites using a modified Fisher's exact test (EASE score). GO BP categories with p < 0.01 and a minimum occurrence of ≥10 genes/proteins were called significant. p values were −Log10 transformed, and the transformed values for each annotation were plotted as a heat map in Gene-E.
Common Phosphoproteome Changes Induced by Cold Ischemia Are Observed Independent of Tumor Type
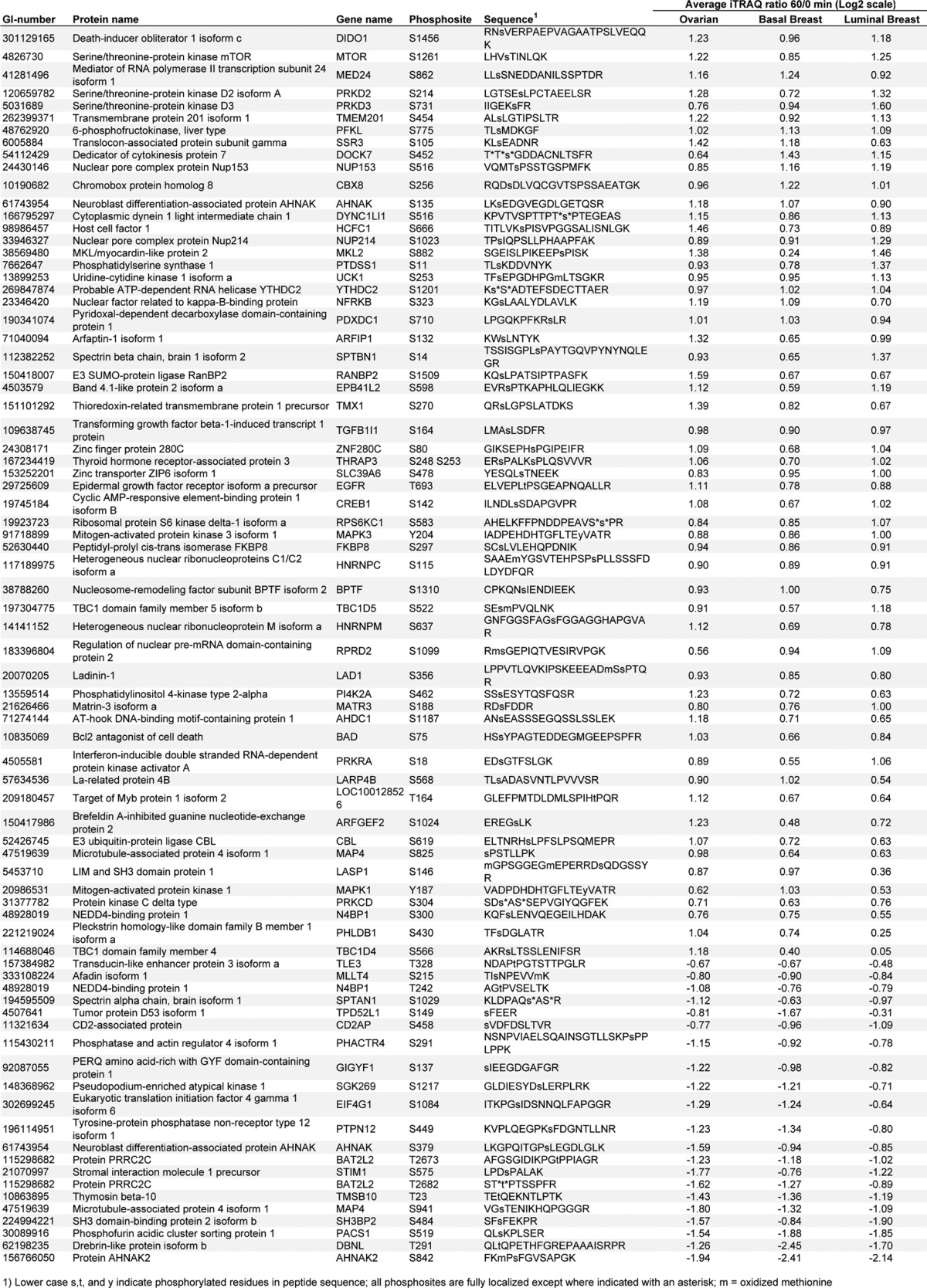
To define the set of ischemia-regulated phosphorylation sites in common across the ovarian and the two breast cancer samples, we plotted all time-point ratios measured in at least two different tumor types and calculated intertumor Pearson correlation coefficients (Fig. 4A). The intertumor type correlation increased with longer ischemia time and reached r values of 0.6 at 30 and 60 min. The intratumor type correlation (which for breast constituted replicate analyses of differing pools) for the 60-min time-point ratios was r = 0.66 for the basal-like breast tumors, r = 0.80 for the luminal breast tumors, and r = 0.39 for the ovarian tumor samples. As expected, we observed a higher degree of biological variability among the individual ovarian tumor samples than among the pooled replicate samples for the basal-like and luminal breast xenograft samples (supplemental Fig. S3). Averaging values across samples conserved commonly regulated effects and improved the intertumor relative to the intratumor correlation for ovarian cancer. Across all three tumor types we found 137 phosphorylation sites to be commonly up-regulated and 21 to be down-regulated (Figs. 4B and 4C; Table II). These 158 commonly regulated phosphorylation sites were identified as regulated with a false discovery rate less than 0.01 in each individual tumor type. Therefore, it is very unlikely that any of the sites observed to be regulated in common across the three different tumor types were false positives.
Fig. 4.

Ischemia induced common phosphorylation events in ovarian and basal-like/luminal breast cancer tumor samples. A, scatter plots of averaged phosphosite ratios (5:0, 30:0, and 60:0) over at least three ovarian tumors (OC) and at least two basal-like (BA) or luminal (LU) breast cancer samples. X and Y indicate which tumor type is plotted on the respective axis. Intertumor Pearson correlation coefficients are indicated for each tumor type comparison. B, C, Venn diagrams of the overlap of up-regulated (B) and down-regulated (C) phosphosites between ovarian and basal-like/luminal breast cancer samples.
Table II. Commonly regulated phosphorylation sites that were significantly altered as a result of cold ischemia in ovarian cancer as well as in basal and luminal breast cancer.

We also analyzed the enrichment of KEGG pathways among all cold-ischemia-regulated phosphoproteins for each tumor type and found that most enriched pathways were detected in at least two of the tumor types (supplemental Fig. S4). Activation patterns of MAPK and the ErbB signaling pathway were common to all three tumor types. Among the most enriched tumor-specific biological processes were the activation of RIG-1-like receptor signaling pathway in basal breast cancer, up-regulation of mTOR signaling, WNT signaling and adherens junctions in luminal breast cancer, and up-regulation of vascular smooth muscle contraction in ovarian cancer. Among the three tumor types evaluated, the luminal breast cancer samples exhibited the greatest number and percentage of uniquely regulated phosphosites. This indicates that there might be tissue-specific changes across lineages that should ideally be assessed prior to the analysis of patient samples in each tumor lineage.
Cold Ischemia Induces Changes in Kinase Phosphorylation Status Across All Major Kinase Subfamilies
Next, we investigated how different protein kinase subfamilies were affected by cold ischemia induced by delayed freezing of tumor tissue. We quantified phosphorylation sites on more than 360 human protein kinase isoforms in this study. In total, 14 protein kinases with regulated phosphorylation sites were detected in all three tumor types (Fig. 5A): EGFR (pT693), ARAF (T181, S186), Erk1/MAPK3 (Y204), Erk2/MAPK1 (T185, Y187), p38a/MAPK14 (Y182), PKCD (S304), PRKD2 (S214), PRKD3 (S731), NEK7 (S187), NEK9 (S332), RSK2 (T577), RSKL1 (S583), mTOR (S1261), and Sgk269/PEAK1 (S1217). These commonly regulated kinases, as well as tumor-specifically regulated kinases, were evenly distributed among all major kinase subfamilies (Fig. 5A). We found that 48% of all kinases contained a regulated site for the luminal breast tumors, in contrast to 17% and 29% for the ovarian and basal breast tumors,respectively. This is consistent with the overall higher level of ischemia-induced changes observed in the luminal subtype.
Fig. 5.

Stress-response kinases were affected by cold ischemia. A, kinases detected with and without regulated phosphosites were mapped in a dendrogram of the human kinome (40) using KinomeCluster. Ischemia-regulated kinases were observed across all major kinase subfamilies. B, protein–protein interaction and kinase-substrate network of phosphoproteins commonly regulated in all analyzed ovarian and breast cancer samples. High confidence, experimentally validated protein–protein interaction information was obtained from the STRING protein interaction database (26). Kinase–substrate relationships were derived from the PhosphoSite database (27). Protein–protein interaction and site-specific kinase–phosphosite relationships were illustrated using Cytoscape (41), with blue edges indicating protein–protein interactions and red dashed arrows indicating kinase–substrate relationships. Up-regulated phosphosites are shown in green, and down-regulated sites are in red. Kinases are depicted as squares, whereas all other proteins appear as circles.
Common Ischemia-regulated Phosphoproteins Belong to an EGFR/MAPK Signaling Module
We further analyzed the set of 139 commonly regulated phosphoproteins for modules of proteins that physically interact with each other or have been described as kinase–substrate pairs. Using curated protein–protein interaction information from the STRING database (26) and site-specific kinase–substrate relationship information from the PhosphoSitePlus database (27), we were able to group 30 of these ischemia-regulated phosphoproteins into a functional protein network (Fig. 5B). This network was dominated by proteins involved in EGFR and MAPK signaling. The EGFR was up-regulated at T693, a site involved in receptor internalization (28) and suppression of EGF-induced JNK activation (29). Distal to EGFR, the adaptor protein SHC1 and the phosphatase PTPN12 were detected, which bind to EGFR and are involved in MAPK activation. P38/MAPK14, Erk1/MAPK3, and Erk2/MAPK1 were up-regulated on phosphosites in their activation loops, and sites on proteins downstream of Erk1/MAPK3 such as stathmin 1 and capicua were also up-regulated. As EGFR T693 phosphorylation has a negative effect on its downstream signaling and p38/MAPK14, Erk1/MAPK3, and Erk2/MAPK1 can directly phosphorylate the EGFR at this site (28), we suggest that phosphorylation of the T693 site in EGFR could be involved in negative-feedback regulation of the EGFR due to cold ischemia.
We also mapped regulated phosphosites to the KEGG database JNK and p38 MAPK pathways (supplemental Fig. S5). The p38/MAPK14 pathway was up-regulated in all tumor types, whereas the JNK pathway was observed to be up-regulated only in the luminal breast cancer tumors. Of note, JNK is also known as stress-activated kinase or SAPK, and p38 has also been characterized as a stress-response kinase. Interestingly, DNA and RNA sequencing analysis of the luminal breast tumor revealed a splice site mutation in the stress-pathway kinase MAP3K1 that generates an out-of-frame transcript (22). MAP3K1 phosphorylation was not observed to be up-regulated in the luminal PDX, in contrast to the basal line. However, contrary to expectations for loss of MAP3K1 function, the JNK pathway was strongly activated by ischemia (supplemental Fig. S5). This suggests a MAP3K1-independent activation mechanism for the JNK pathway that could increase cell survival when ischemia-like conditions occur in an evolving, growing tumor, similar to events proposed in cardiac myocytes (30). Specific phosphorylation of apoptosis-inducing caspases (CASP2/3/9) was also only observed in the luminal breast cancer tumors.
Among the other commonly regulated phosphoproteins (Fig. 5B), a group of proteins was found that functioned in mRNA export (NUP214), translation (EIF4G1), and poly(A)-tail shortening (PARN, TNKS1BP1). Pleiotropic regulators of mitotic progression such as Nek7 and Nek9 were also regulated on phosphosites by ischemia, as were transcriptional regulators (TRIM28) and pro-apoptotic regulators (BAD).
Comparison of Results Obtained via Quantitative Mass-spectrometry-based Proteomics and RPPA
Because RPPA measurements are widely used to investigate cancer-relevant signaling pathways, we compared the results obtained via quantitative LC-MS/MS with results obtained via RPPA on the same samples of all tumors in the current study. The RPPA studies used 40 phosphosite-specific antibodies, 10 of which were to pTyr sites. We successfully identified 25 and quantified 23 of the 40 phosphosites measured based on these antibodies in our phosphoproteome analysis using LC-MS/MS, including 7 of 10 pTyr sites. We observed a good correlation of results for the ovarian, basal-like breast, and luminal breast cancers, with Pearson correlation values of r = 0.79, 0.83, and 0.75, respectively (supplemental Fig. S6, supplemental Table S3). Of the 15 phosphosites detected via RPPA but not LC-MS/MS, 10 were located in surrounding sequences that upon tryptic digestion would yield peptides ill suited to mass spectrometric identification—that is, peptides shorter than 5 amino acids or longer than 30 amino acids with only a single basic residue. Lack of detection of the remaining 5 (of 40) phosphosites via LC-MS/MS might have been due to greater sensitivity of the phosphoantibodies for these sites or unexpected cross-reactivity of the antibodies leading to false positive identification in RPPA. Protein-level quantification via RPPA for 87 proteins across all tumor types revealed, in agreement with our MS data, no changes at the protein level due to cold ischemia (supplemental Table S3). Across all tumor types, 85 of these proteins were identified with two or more peptides, and 80 proteins were quantified in at least two tumors with mass spectrometry.
DISCUSSION
In this study, we systematically analyzed the effect of up to one hour of cold ischemia time on the stability of the proteome and phosphoproteome of ovarian and breast cancer tumor samples. Cold ischemia is a complex physiological perturbation that integrates the effects of tissue stress, hypoxia, hypoglycemia, acidosis, hypothermia, and electrolyte disturbance. It is of special relevance in tissue profiling as both a pervasive and a comparatively modifiable preanalytical variable. The one-hour end point we chose to study is particularly pertinent because it represents a convergence of three important parameters: the cold ischemic time allowed in TCGA tumor tissue acquisition protocols, the upper limit of cold ischemic time recommended by the American Society of Clinical Oncology guidelines for breast cancer tumor marker assessment (31), and the upper bound on intervals typically reported for those aspects of sample processing that contribute to routine cold ischemia, including specimen transport to pathology and macroscopic evaluation by a pathologist (19).
The extent of the proteome and phosphoproteome detected and quantified in our study is the largest in any ischemia study and among the largest in any proteomics experiment to date, owing to advances in sample preparation, use of the latest generation LC-MS instrumentation, and statistically rigorous data analysis methods. Our comprehensive proteome analysis, which greatly expands on prior studies of the consequences of cold ischemia on protein expression (15, 16), revealed that none of the >10,000 proteins detected and quantified changed in abundance as a result of cold ischemia in a time span of up to one hour post-excision. However, because of the inability of conventional “bottom-up” MS-based proteomics technology to analyze the integrity of intact proteins (as samples are digested to peptides prior to analysis), we cannot exclude the possibility that limited proteolysis such as apoptotic regulation by caspases occurs at early time points of cold ischemia. Protein stability across this level of coverage, which approaches the anticipated scale of the expressed proteome, provides compelling evidence that the results of quantitative global proteomics are robust to the common vagaries of post-excision tissue handling in surgical and pathology suites. These findings are similar to those obtained in a study on mRNA integrity in breast cancer tissue in which no substantial effects on mRNA yield/integrity or transcript expression were detected after either 40 min or three hours of cold ischemia (32).
Although the global proteome is remarkably stable, the impact of cold ischemia on the phosphoproteome is more pronounced, with some alterations occurring within 5 min of removal of the tissue from its blood supply, the earliest post-baseline time point investigated in this study. Between 6% and 24% of the more than 25,000 distinct, chiefly pSer or pThr phosphosites we identified were observed to be regulated by cold ischemia in one or more of the three tumor types we analyzed. Importantly, our study therefore identified a large subset of the phosphoproteome that remains relatively stable and can be accurately measured, even in samples subjected to a full hour of cold ischemia. These results suggest that phosphoproteome analysis of the TCGA tumor samples can proceed with an acceptable expected yield of biologically relevant phosphoprotein data.
We observed a common phosphosite signature of cold ischemia response in the ovarian and breast cancer tumors consisting of 137 up- and 21 down-regulated sites that underwent up to 6-fold alterations in abundance (Table II, supplemental Fig. S7). The strong increase in phosphorylation across tumor types induced by cold ischemia mainly involved the activation of MAPK stress-response pathways, apoptosis, and transcriptional regulation. Among the ischemia-regulated proteins were proteins such as p38/MAPK14 (Y182) and Erk1/MAPK3 (Y204) that have previously been shown via RPPA to change as a result of ischemia (17). Although some phosphosites were observed to decrease in common across all three tumor types examined, the signature of increased phosphorylation was far stronger, suggesting that kinase activation is a more pervasive response than loss of phosphorylation due to increased phosphatase activity. Minor differences in the kinetics of protein phosphorylation were observed between the different tumor types, which might have been due to different baseline tissue oxygenation or vascularization. The common phosphosite signature of cold ischemia response we observed, or one derived from it, could, in principle, be used to identify tumor samples that underwent strong cold ischemic stress conditions, and potentially to estimate the duration of the shock period. Such information could then inform the interpretation of experimental results. The generalizability of the signature to other tumors and tissues has not yet been demonstrated, but it is readily testable. We are developing targeted MS-based assays (multiple reaction monitoring MS or similar) employing stable-isotope-labeled internal standard peptides for a number of the ischemia-marker candidates to facilitate such analyses (33). Furthermore, although warm ischemia was not explicitly addressed in this study, the nature of the tissue insult is in many respects comparable to that of cold ischemia, and it is plausible that our data could provide the foundation of a global ischemia signature. The importance of establishing such quality standards for samples used for proteomic and phosphoproteomic analysis is clear.
Our results are in partial agreement with a previous report by Espina and colleagues, who found through RPPA analysis that apoptosis, hypoxia, and proliferation-related proteins increase transiently within the initial 30 min post-excision (17). The effects observed in our study were larger in amplitude and did not return to baseline, possibly because of tissue-specific differences or the longer total ischemia time in the RPPA study. In a recent LC-MS/MS-based analysis of delayed freezing of mouse and rat livers, Gundisch et al. found that most phosphoproteome changes are diffuse and unpredictable and can be minimized when samples are kept on ice prior to fixation (34). The lack of a common cold-ischemia response in this study may be explained by different stress-response mechanisms in healthy and diseased tissues. Our results also contrast with those of Pinhel and coworkers (35), who reported that delaying time to fixation post-resection significantly decreases the immunoreactivity of p-Akt and p-ERK1/2, two potentially useful diagnostic markers of receptor tyrosine kinase pathway activation. This discrepancy is likely due to the very long latency to effective formalin fixation of the resection samples analyzed in that study. The increased phosphorylation of Erk1 we observed was supported by both LC-MS/MS and RPPA analyses, adding confidence regarding the activation of MAPK pathways by cold ischemia.
An important potential caveat to our study is that significant regional heterogeneity in particular signaling molecules across the tumor could, if present, contribute to apparent differences in the effects of ischemia on phosphosite stability. Of necessity, different portions of tumor material were analyzed at each of the time points. Furthermore, heterogeneity in protein expression across tumors has long been observed in immunohistochemistry and, more recently, with other approaches including exome sequencing (36). Although regional heterogeneity is a legitimate concern, it is unlikely that it was a critical determinant in our study. We did not observe substantial regional variability in the total number of proteins and phosphosites identified across time points in our study. This might be due to the fact that the volume of the samples analyzed at each time point was relatively large, and for the breast cancer samples it included regions pooled across multiple tumors, reducing concerns with microheterogeneity. Alternatively, undersampling by the LC-MS/MS system because of the complexity of the tumor samples might have reduced our ability to detect such differences. However, the kinetics of change observed in different phosphosites were more consistent with the monotonic perturbation of increased ischemia time than with the random effects expected of heterogeneity. Notably, a prior direct analysis of intratumoral heterogeneity via RPPA failed to detect major changes across different parts of the tumor (9).
In addition to common stress responses, we also observed tumor-specific responses to cold ischemia. The variability in the stability of phosphosites in luminal breast, basal breast, and serous ovarian cancers, together with coordinate regulation of many phosphosites, suggests that phosphosite stability will be pathway, context, and lineage dependent. For example, we observed very strong induction of protein phosphorylation in a luminal breast tumor that contained a splice site mutation in the gene for the stress kinase MAP3K1. This correlation of a mutated gene to a strong phosphoproteome phenotype might indicate that the full extent of ischemic response is dependent on the genetic repertoire of each tumor, a suggestion further supported by the greater biological variability observed in the responses of individual ovarian tumors. The effects of specific genetic events can be more clearly studied with a larger number of genomically annotated PDX models, which would also permit additional perturbations to be studied, including those induced by therapeutic approaches. Together, these observations argue for caution and circumspection in the interpretation of phosphoproteomic data from TCGA samples, as well as from samples populating the vast majority of valuable “collaborative” repositories collected with conventional constraints on cold ischemia time.
Because tissues undergoing excision are alive and able to mount coordinated responses to insults, variation in ischemia time can either mask or artifactually amplify underlying pathways of importance. For example, proteins in stress-response pathways, including those we have shown to be regulated by cold ischemia, have been implicated in cancer progression and drug resistance (37, 38). Certainly, phosphoprotein measurements used to guide clinical decision making should be specifically studied for stability within the parameters of their particular sample-acquisition protocol. Although somewhat greater latitude may be allowed for research specimens, at a minimum our results suggest that the total duration of cold ischemia (and, by extension, warm ischemia) should be carefully noted for samples to be analyzed using any proteomic methods. Whenever possible, cancer and other tissue specimens should be frozen as soon after resection as possible, as cold-ischemia-induced changes in the phosphoproteome become evident after just a few minutes. If warm ischemic effects are at all comparable to the cold ischemic effects described here, biopsies taken preoperatively or intraoperatively before the interruption of the vascular supply might be needed for accurate characterization of the tumor phosphoproteome. If minimally ischemic acquisition and immediate freezing are beyond institutional capacity or fall outside clinical sample handling protocols, the commonly regulated ischemia phosphorylation sites identified in this study may be monitored, potentially through the use of targeted MS-based assays (33), to identify samples that might have been subjected to increased ischemic stress. A metric based on targeted measurement of this set would allow suspect samples to be flagged, though it would not necessarily provide guidance as to which specific phosphosites remained interpretable. The utility of the set of phosphosite markers we have identified as commonly perturbed in ischemic breast and ovarian cancer will remain uncertain until additional studies of other tumor types are able to establish whether such a tool would be of general use.
Supplementary Material
Footnotes
Author contributions: P.M., F.Y., T.L., D.M., V.A.P., M.A.G.illette, K.R.C., D.A.L., R.T., S.A.D., H.R., D.W.C., D.L., F.W., K.D.R., G.B.M., R.D.S., A.G.P., M.E., and S.A.C. designed research; P.M., F.Y., T.L., D.M., V.A.P., J.W.Q., M.A. G.ritsenko, R.J.M., D.A.L., P.E., J.E.S., S.A.D., K.V.R., R.T.K., S.L., N.O., and F.D. performed research; P.M., F.Y., D.M., V.A.P., and K.R.C. contributed new reagents or analytic tools; P.M., F.Y., T.L., D.M., V.A.P., M.A. G.illette, K.R.C., J.W.Q., S.A.D., K.V.R., D.F., and S.A.C. analyzed data; P.M., F.Y., T.L., D.M., V.A.P., M.A. G.illette, K.R.C., D.A.L., R.T., S.A.D., K.V.R., D.F., D.L., F.W., K.D.R., G.B.M., R.D.S., A.G.P., M.E., and S.A.C. wrote the paper.
* This work was supported, in whole or in part, by grants from the NCI, National Institutes of Health (Grant Nos. U24CA160034 to S.A.C. and A.G.P., U24CA160019 to R.D.S. and K.D.R., U24CA160035 to M.J.E. and R.R.T., U24CA159988 to D.L., and U24CA160036 to D.C.), as part of the NCI Clinical Proteomics Tumor Analysis Consortium. The PDX models were developed through grants to Matthew J. Ellis by Susan G. Komen for the Cure (Grant Nos. BCTR0707808 and KG090422). The Siteman Cancer Center Tissue Procurement Core is supported by Grant No. P30CA091842 from NCI, National Institutes of Health. Tissue procurement core was supported Grant No. 3P50 CA68438 from NCI, National Institutes of Health. The HAMLET Core was supported by CTSA grant UL1 RR024992. The ovarian cancer sample collection was supported by the Chia Family Foundation. The RPPA analysis was supported by MD Anderson Cancer Center Support Grant (CCSG) CA016672 from National Institutes of Health and Grant Nos. PO1CA099031, U54CA112970, KG081694, and P30 CA16672 to Gordon Mills.
 This article contains supplemental material.
This article contains supplemental material.
1 The abbreviations used are:
- used are: TCGA
- the Cancer Genome Atlas
- iTRAQ
- isobaric tags for relative and absolute quantification
- PDX
- patient-derived xenograft
- RPPA
- reverse-phase protein array
- pTyr
- phospho-tyrosine
- GO BP
- Gene Ontology Biological Process
- EGFR
- epidermal growth factor receptor.
REFERENCES
- 1. Cancer Genome Atlas Network (2012) Comprehensive molecular portraits of human breast tumours. Nature 490, 61–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Cancer Genome Atlas Research Network (2008) Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 455, 1061–1068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Cancer Genome Atlas Network (2012) Comprehensive molecular characterization of human colon and rectal cancer. Nature 487, 330–337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Cancer Genome Atlas Research Network (2012) Comprehensive genomic characterization of squamous cell lung cancers. Nature 489, 519–525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Cancer Genome Atlas Research Network (2011) Integrated genomic analyses of ovarian carcinoma. Nature 474, 609–615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Cancer Genome Atlas Research Network (2013) Integrated genomic characterization of endometrial carcinoma. Nature 497, 67–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Salvesen H. B., Carter S. L., Mannelqvist M., Dutt A., Getz G., Stefansson I. M., Raeder M. B., Sos M. L., Engelsen I. B., Trovik J., Wik E., Greulich H., Bø T. H., Jonassen I., Thomas R. K., Zander T., Garraway L. A., Oyan A. M., Sellers W. R., Kalland K. H., Meyerson M., Akslen L. A., Beroukhim R. (2009) Integrated genomic profiling of endometrial carcinoma associates aggressive tumors with indicators of PI3 kinase activation. Proc. Natl. Acad. Sci. U.S.A. 106, 4834–4839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ellis M. J., Ding L., Shen D., Luo J., Suman V. J., Wallis J. W., Van Tine B. A., Hoog J., Goiffon R. J., Goldstein T. C., Ng S., Lin L., Crowder R., Snider J., Ballman K., Weber J., Chen K., Koboldt D. C., Kandoth C., Schierding W. S., McMichael J. F., Miller C. A., Lu C., Harris C. C., McLellan M. D., Wendl M. C., DeSchryver K., Allred D. C., Esserman L., Unzeitig G., Margenthaler J., Babiera G. V., Marcom P. K., Guenther J. M., Leitch M., Hunt K., Olson J., Tao Y., Maher C. A., Fulton L. L., Fulton R. S., Harrison M., Oberkfell B., Du F., Demeter R., Vickery T. L., Elhammali A., Piwnica-Worms H., McDonald S., Watson M., Dooling D. J., Ota D., Chang L. W., Bose R., Ley T. J., Piwnica-Worms D., Stuart J. M., Wilson R. K., Mardis E. R. (2012) Whole-genome analysis informs breast cancer response to aromatase inhibition. Nature 486, 353–360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hennessy B. T., Lu Y., Gonzalez-Angulo A. M., Carey M. S., Myhre S., Ju Z., Davies M. A., Liu W., Coombes K., Meric-Bernstam F., Bedrosian I., McGahren M., Agarwal R., Zhang F., Overgaard J., Alsner J., Neve R. M., Kuo W. L., Gray J. W., Borresen-Dale A. L., Mills G. B. (2010) A technical assessment of the utility of reverse phase protein arrays for the study of the functional proteome in non-microdissected human breast cancers. Clin. Proteomics 6, 129–151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wisniewski J. R., Dus K., Mann M. (2013) Proteomic workflow for analysis of archival formalin-fixed and paraffin-embedded clinical samples to a depth of 10 000 proteins. Proteomics Clin. Appl. 7, 225–233 [DOI] [PubMed] [Google Scholar]
- 11. Mertins P., Udeshi N. D., Clauser K. R., Mani D. R., Patel J., Ong S. E., Jaffe J. D., Carr S. A. (2012) iTRAQ labeling is superior to mTRAQ for quantitative global proteomics and phosphoproteomics. Mol. Cell. Proteomics 11, M111.014423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Huttlin E. L., Jedrychowski M. P., Elias J. E., Goswami T., Rad R., Beausoleil S. A., Villén J., Haas W., Sowa M. E., Gygi S. P. (2010) A tissue-specific atlas of mouse protein phosphorylation and expression. Cell 143, 1174–1189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lundby A., Secher A., Lage K., Nordsborg N. B., Dmytriyev A., Lundby C., Olsen J. V. (2012) Quantitative maps of protein phosphorylation sites across 14 different rat organs and tissues. Nat. Commun. 3, 876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Mertins P., Qiao J. W., Patel J., Udeshi N. D., Clauser K. R., Mani D. R., Burgess M. W., Gillette M. A., Jaffe J. D., Carr S. A. (2013) Integrated proteomic analysis of post-translational modifications by serial enrichment. Nat. Methods 10, 634–637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Neumeister V. M., Anagnostou V., Siddiqui S., England A. M., Zarrella E. R., Vassilakopoulou M., Parisi F., Kluger Y., Hicks D. G., Rimm D. L. (2012) Quantitative assessment of effect of preanalytic cold ischemic time on protein expression in breast cancer tissues. J. Natl. Cancer Inst. 104, 1815–1824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Yildiz-Aktas I. Z., Dabbs D. J., Bhargava R. (2012) The effect of cold ischemic time on the immunohistochemical evaluation of estrogen receptor, progesterone receptor, and HER2 expression in invasive breast carcinoma. Mod. Pathol. 25, 1098–1105 [DOI] [PubMed] [Google Scholar]
- 17. Espina V., Edmiston K. H., Heiby M., Pierobon M., Sciro M., Merritt B., Banks S., Deng J., VanMeter A. J., Geho D. H., Pastore L., Sennesh J., Petricoin E. F., 3rd, Liotta L. A. (2008) A portrait of tissue phosphoprotein stability in the clinical tissue procurement process. Mol. Cell. Proteomics 7, 1998–2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ahmed M. M., Gardiner K. J. (2011) Preserving protein profiles in tissue samples: differing outcomes with and without heat stabilization. J. Neurosci. Methods 196, 99–106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gündisch S., Hauck S., Sarioglu H., Schott C., Viertler C., Kap M., Schuster T., Reischauer B., Rosenberg R., Verhoef C., Mischinger H. J., Riegman P., Zatloukal K., Becker K. F. (2012) Variability of protein and phosphoprotein levels in clinical tissue specimens during the preanalytical phase. J. Proteome Res. 11, 5748–5762 [DOI] [PubMed] [Google Scholar]
- 20. Li J., Kil C., Considine K., Smarkucki B., Stankewich M. C., Balgley B., Vortmeyer A. O. (2013) Intrinsic indicators for specimen degradation. Lab. Invest. 93, 242–253 [DOI] [PubMed] [Google Scholar]
- 21. Ross P. L., Huang Y. N., Marchese J. N., Williamson B., Parker K., Hattan S., Khainovski N., Pillai S., Dey S., Daniels S., Purkayastha S., Juhasz P., Martin S., Bartlet-Jones M., He F., Jacobson A., Pappin D. J. (2004) Multiplexed protein quantitation in Saccharomyces cerevisiae using amine-reactive isobaric tagging reagents. Mol. Cell. Proteomics 3, 1154–1169 [DOI] [PubMed] [Google Scholar]
- 22. Li S., Shen D., Shao J., Crowder R., Liu W., Prat A., He X., Liu S., Hoog J., Lu C., Ding L., Griffith O. L., Miller C., Larson D., Fulton R. S., Harrison M., Mooney T., McMichael J. F., Luo J., Tao Y., Goncalves R., Schlosberg C., Hiken J. F., Saied L., Sanchez C., Giuntoli T., Bumb C., Cooper C., Kitchens R. T., Lin A., Phommaly C., Davies S. R., Zhang J., Kavuri M. S., McEachern D., Dong Y. Y., Ma C., Pluard T., Naughton M., Bose R., Suresh R., McDowell R., Michel L., Aft R., Gillanders W., DeSchryver K., Wilson R. K., Wang S., Mills G. B., Gonzalez-Angulo A., Edwards J. R., Maher C., Perou C. M., Mardis E. R., Ellis M. J. (2013) Endocrine therapy resistant ESR1 variants revealed by genomic characterization of breast cancer derived xenografts. Cell Reports 4, 1116–1130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ding L., Ellis M. J., Li S., Larson D. E., Chen K., Wallis J. W., Harris C. C., McLellan M. D., Fulton R. S., Fulton L. L., Abbott R. M., Hoog J., Dooling D. J., Koboldt D. C., Schmidt H., Kalicki J., Zhang Q., Chen L., Lin L., Wendl M. C., McMichael J. F., Magrini V. J., Cook L., McGrath S. D., Vickery T. L., Appelbaum E., Deschryver K., Davies S., Guintoli T., Lin L., Crowder R., Tao Y., Snider J. E., Smith S. M., Dukes A. F., Sanderson G. E., Pohl C. S., Delehaunty K. D., Fronick C. C., Pape K. A., Reed J. S., Robinson J. S., Hodges J. S., Schierding W., Dees N. D., Shen D., Locke D. P., Wiechert M. E., Eldred J. M., Peck J. B., Oberkfell B. J., Lolofie J. T., Du F., Hawkins A. E., O'Laughlin M. D., Bernard K. E., Cunningham M., Elliott G., Mason M. D., Thompson D. M., Jr., Ivanovich J. L., Goodfellow P. J., Perou C. M., Weinstock G. M., Aft R., Watson M., Ley T. J., Wilson R. K., Mardis E. R. (2010) Genome remodelling in a basal-like breast cancer metastasis and xenograft. Nature 464, 999–1005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wang Y., Yang F., Gritsenko M. A., Wang Y., Clauss T., Liu T., Shen Y., Monroe M. E., Lopez-Ferrer D., Reno T., Moore R. J., Klemke R. L., Camp D. G., 2nd, Smith R. D. (2011) Reversed-phase chromatography with multiple fraction concatenation strategy for proteome profiling of human MCF10A cells. Proteomics 11, 2019–2026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kumar L., M E. F. (2007) Mfuzz: a software package for soft clustering of microarray data. Bioinformation 2, 5–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Franceschini A., Szklarczyk D., Frankild S., Kuhn M., Simonovic M., Roth A., Lin J., Minguez P., Bork P., von Mering C., Jensen L. J. (2013) STRING v9.1: protein-protein interaction networks, with increased coverage and integration. Nucleic Acids Res. 41, D808–D815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hornbeck P. V., Kornhauser J. M., Tkachev S., Zhang B., Skrzypek E., Murray B., Latham V., Sullivan M. (2012) PhosphoSitePlus: a comprehensive resource for investigating the structure and function of experimentally determined post-translational modifications in man and mouse. Nucleic Acids Res. 40, D261–D270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Winograd-Katz S. E., Levitzki A. (2006) Cisplatin induces PKB/Akt activation and p38(MAPK) phosphorylation of the EGF receptor. Oncogene 25, 7381–7390 [DOI] [PubMed] [Google Scholar]
- 29. Bagowski C. P., Stein-Gerlach M., Choidas A., Ullrich A. (1999) Cell-type specific phosphorylation of threonines T654 and T669 by PKD defines the signal capacity of the EGF receptor. EMBO J. 18, 5567–5576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Dougherty C. J., Kubasiak L. A., Prentice H., Andreka P., Bishopric N. H., Webster K. A. (2002) Activation of c-Jun N-terminal kinase promotes survival of cardiac myocytes after oxidative stress. Biochem. J. 362, 561–571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Harris L., Fritsche H., Mennel R., Norton L., Ravdin P., Taube S., Somerfield M. R., Hayes D. F., Bast R. C., Jr.; American Society of Clinical Oncology (2007) American Society of Clinical Oncology 2007 update of recommendations for the use of tumor markers in breast cancer. J. Clin. Oncol. 25, 5287–5312 [DOI] [PubMed] [Google Scholar]
- 32. Hatzis C., Sun H., Yao H., Hubbard R. E., Meric-Bernstam F., Babiera G. V., Wu Y., Pusztai L., Symmans W. F. (2011) Effects of tissue handling on RNA integrity and microarray measurements from resected breast cancers. J. Natl. Cancer Inst. 103, 1871–1883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Gillette M. A., Carr S. A. (2013) Quantitative analysis of peptides and proteins in biomedicine by targeted mass spectrometry. Nat. Methods 10, 28–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Gündisch S., Grundner-Culemann K., Wolff C., Schott C., Reischauer B., Machatti M., Groelz D., Schaab C., Tebbe A., Becker K. F. (2013) Delayed times to tissue fixation result in unpredictable global phosphoproteome changes. J. Proteome Res. 12, 4424–4434 [DOI] [PubMed] [Google Scholar]
- 35. Pinhel I. F., Macneill F. A., Hills M. J., Salter J., Detre S., A'hern R., Nerurkar A., Osin P., Smith I. E., Dowsett M. (2010) Extreme loss of immunoreactive p-Akt and p-Erk1/2 during routine fixation of primary breast cancer. Breast Cancer Res. 12, R76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Gerlinger M., Rowan A. J., Horswell S., Larkin J., Endesfelder D., Gronroos E., Martinez P., Matthews N., Stewart A., Tarpey P., Varela I., Phillimore B., Begum S., McDonald N. Q., Butler A., Jones D., Raine K., Latimer C., Santos C. R., Nohadani M., Eklund A. C., Spencer-Dene B., Clark G., Pickering L., Stamp G., Gore M., Szallasi Z., Downward J., Futreal P. A., Swanton C. (2012) Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N. Engl. J. Med. 366, 883–892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Gauthier M. L., Berman H. K., Miller C., Kozakeiwicz K., Chew K., Moore D., Rabban J., Chen Y. Y., Kerlikowske K., Tlsty T. D. (2007) Abrogated response to cellular stress identifies DCIS associated with subsequent tumor events and defines basal-like breast tumors. Cancer Cell 12, 479–491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Schiff R., Reddy P., Ahotupa M., Coronado-Heinsohn E., Grim M., Hilsenbeck S. G., Lawrence R., Deneke S., Herrera R., Chamness G. C., Fuqua S. A., Brown P. H., Osborne C. K. (2000) Oxidative stress and AP-1 activity in tamoxifen-resistant breast tumors in vivo. J. Natl. Cancer Inst. 92, 1926–1934 [DOI] [PubMed] [Google Scholar]
- 39. Huang da W., Sherman B. T., Lempick i R. A. (2009) Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 4, 44–57 [DOI] [PubMed] [Google Scholar]
- 40. Manning G., Whyte D. B., Martinez R., Hunter T., Sudarsanam S. (2002) The protein kinase complement of the human genome. Science 298, 1912–1934 [DOI] [PubMed] [Google Scholar]
- 41. Shannon P., Markiel A., Ozier O., Baliga N. S., Wang J. T., Ramage D., Amin N., Schwikowski B., Ideker T. (2003) Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.